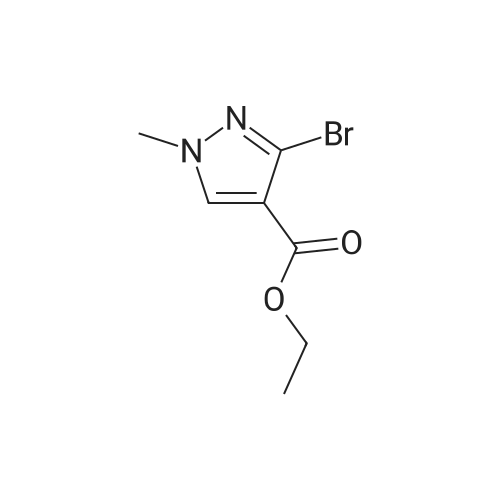
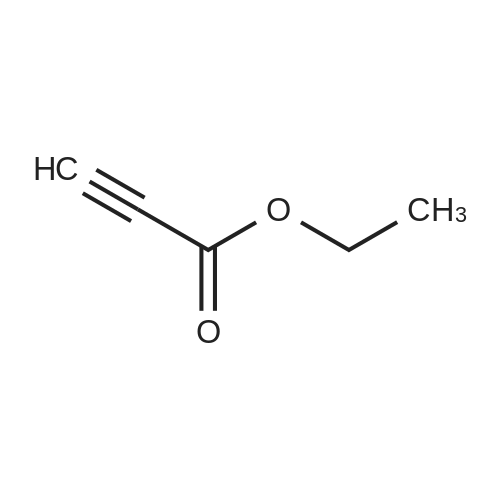
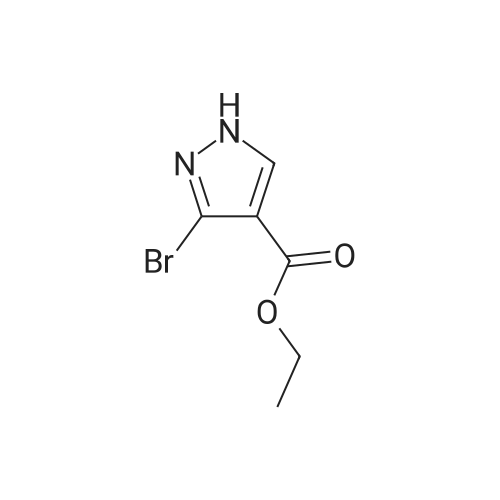
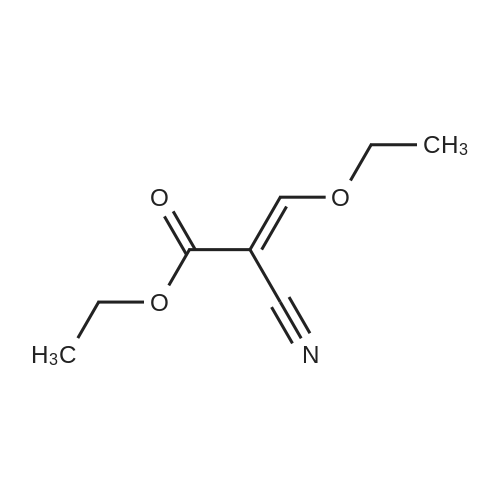
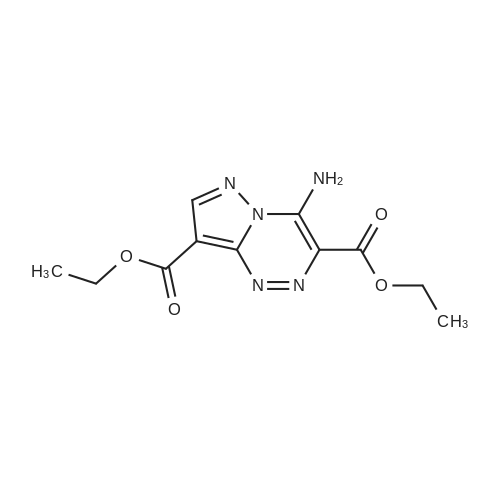
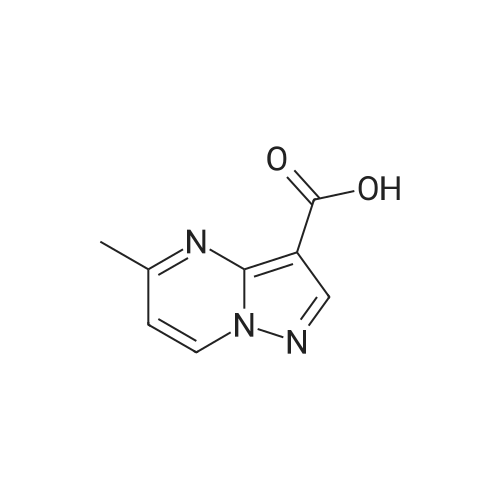
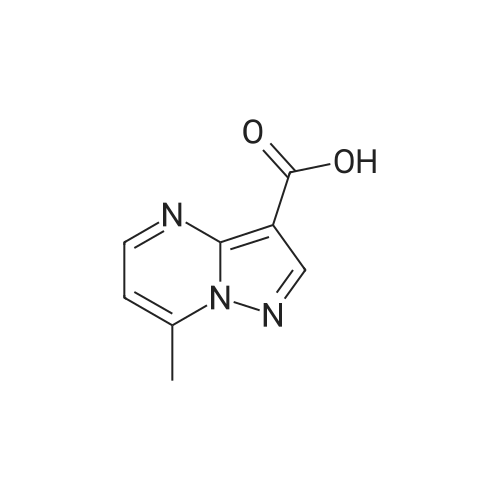
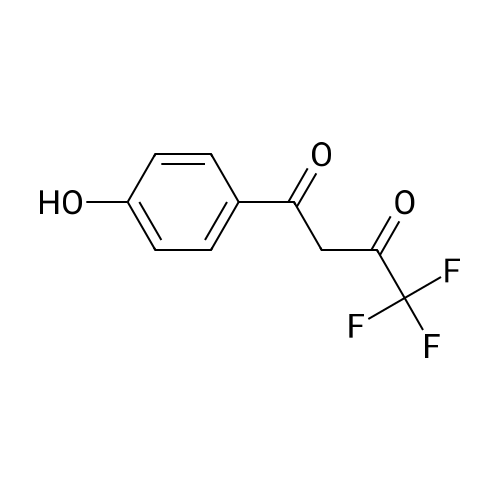
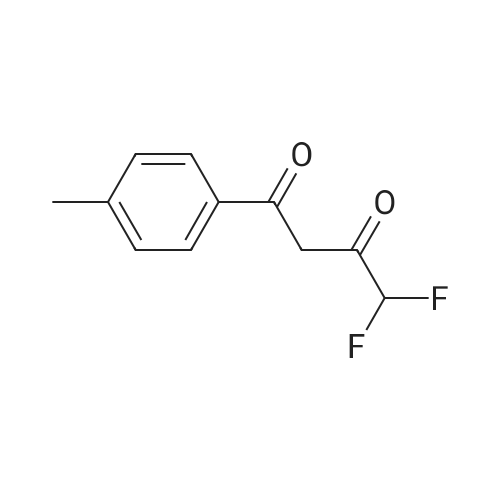
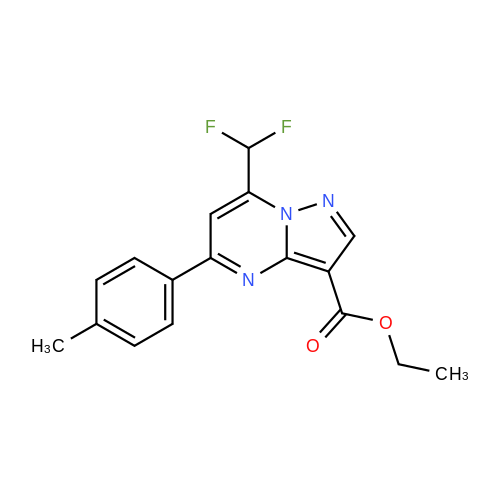
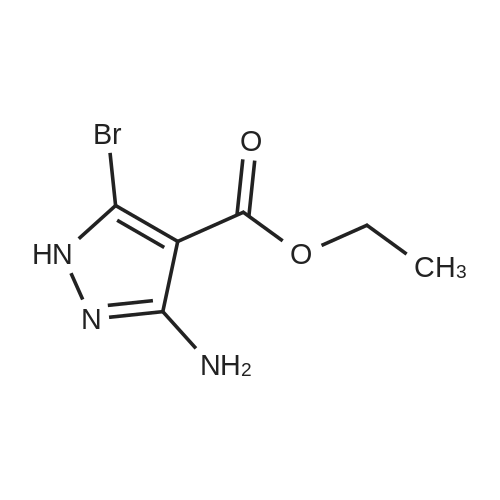
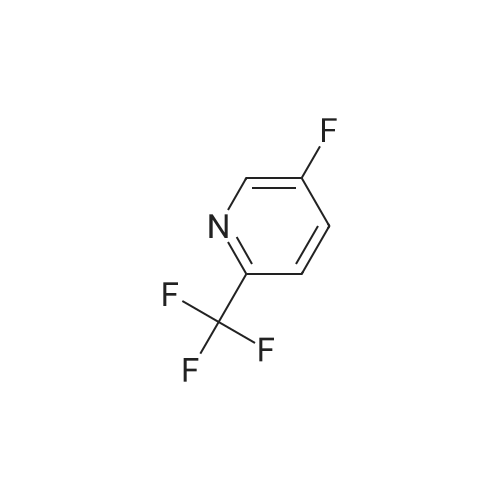
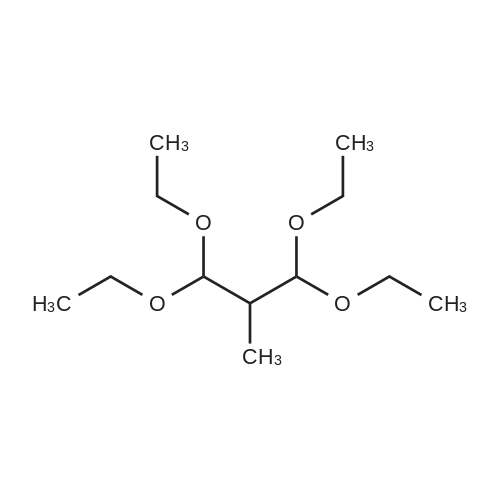
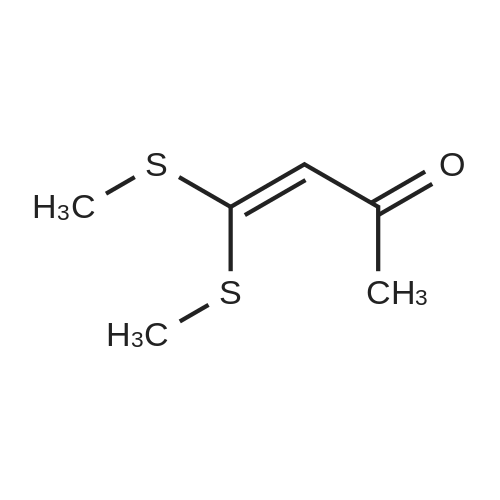
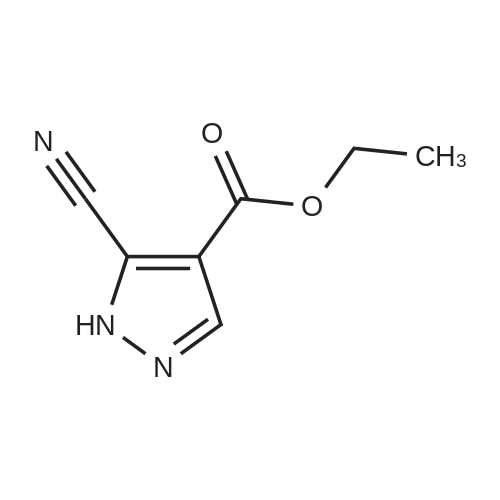
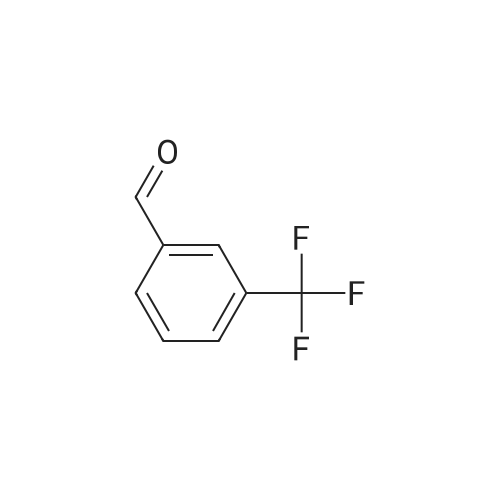
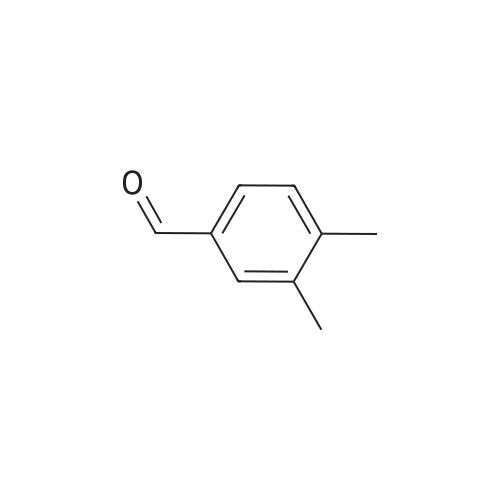
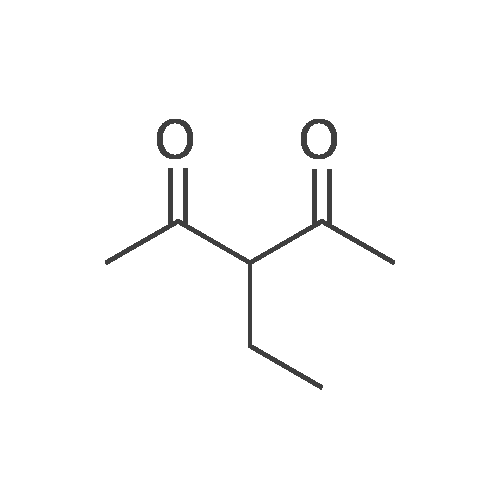
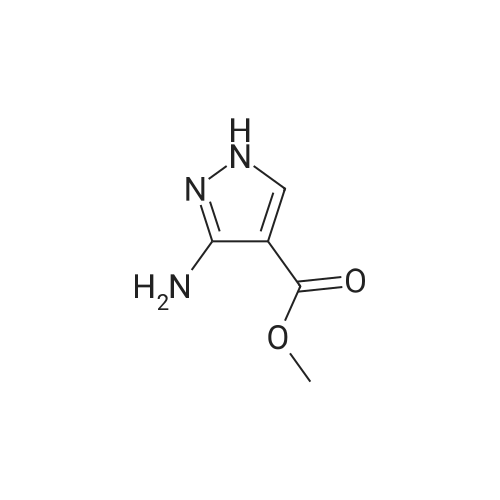
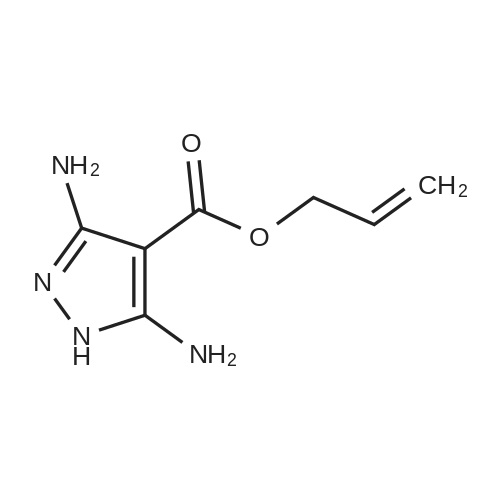
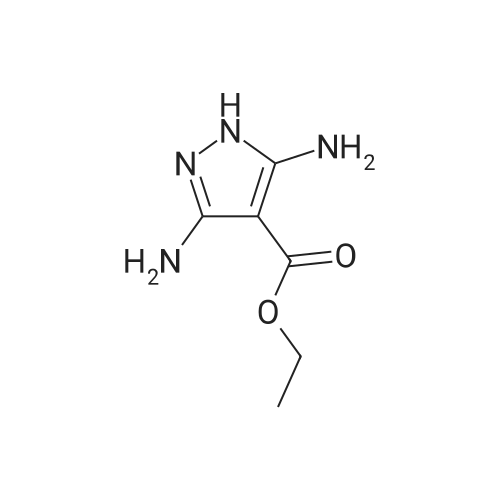
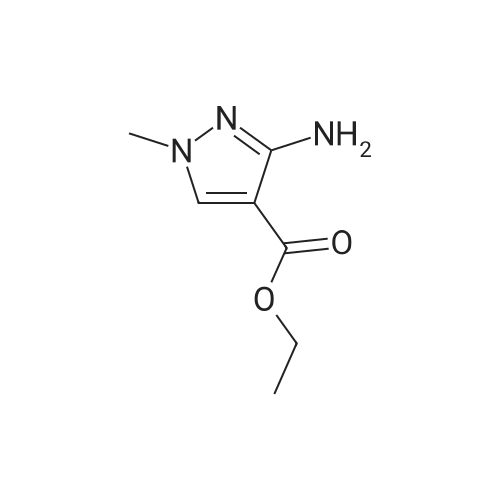
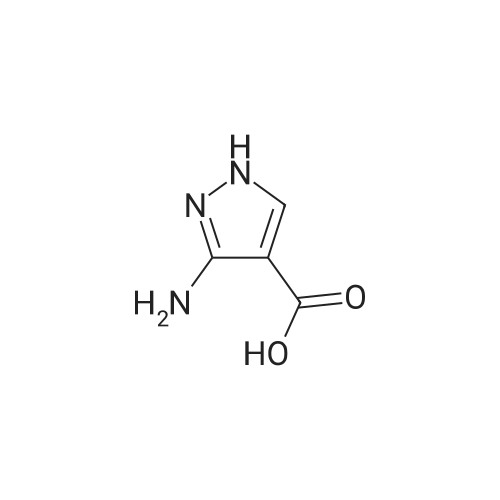
| 1: 19.9 mg
2: 61.7 mg |
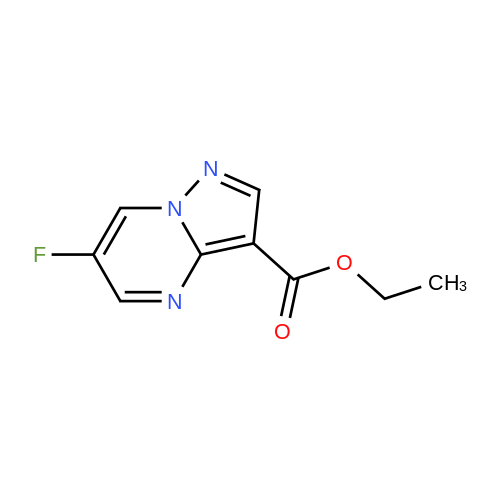
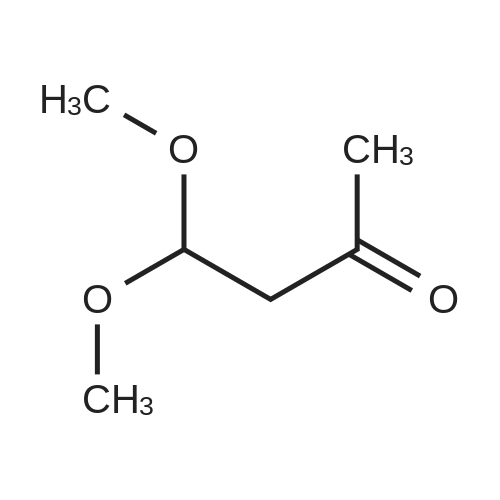
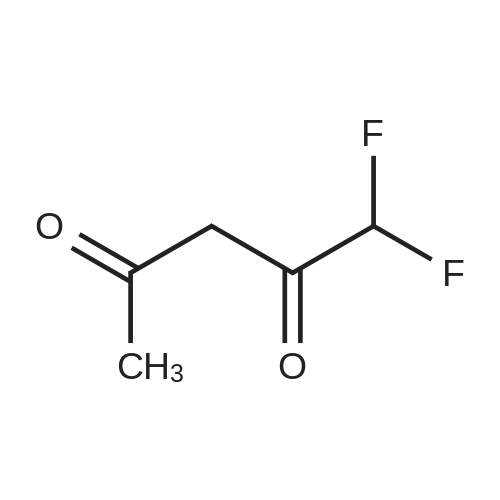
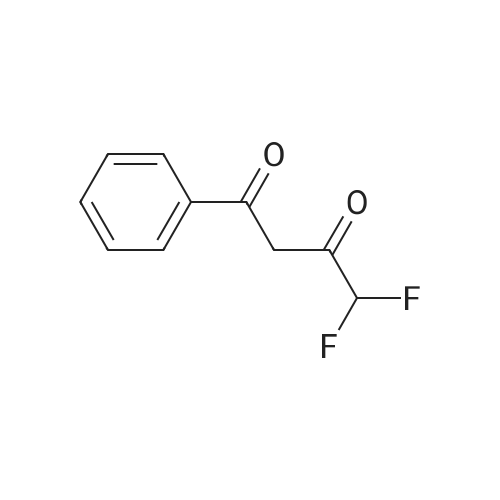
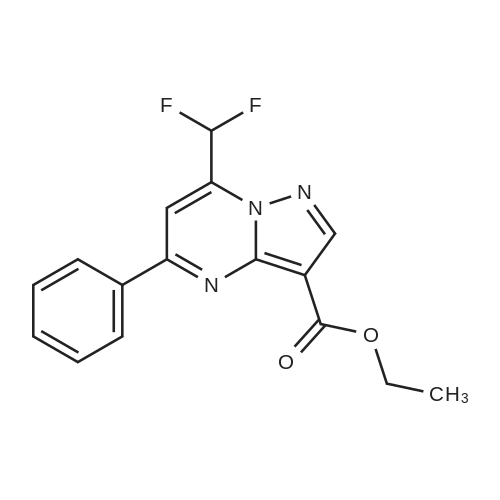
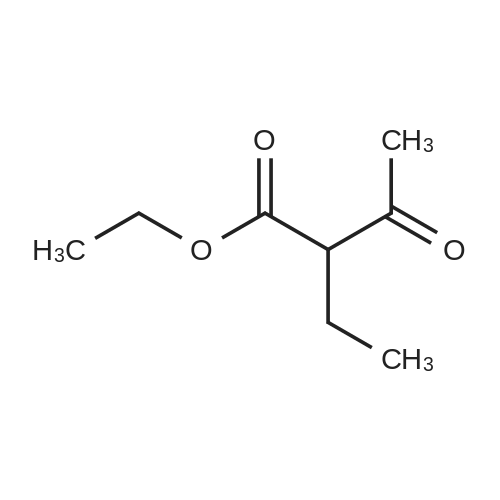
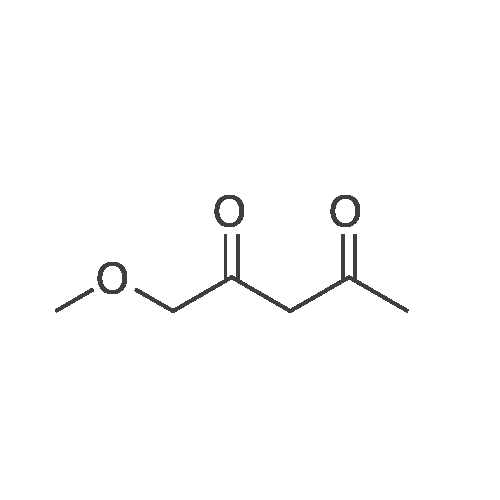
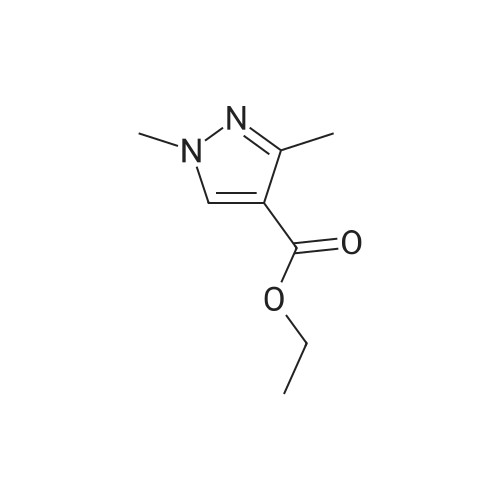
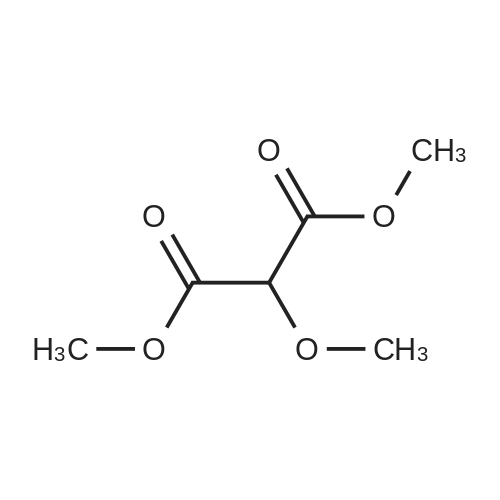
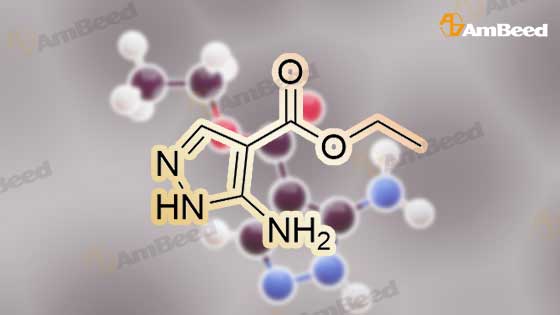
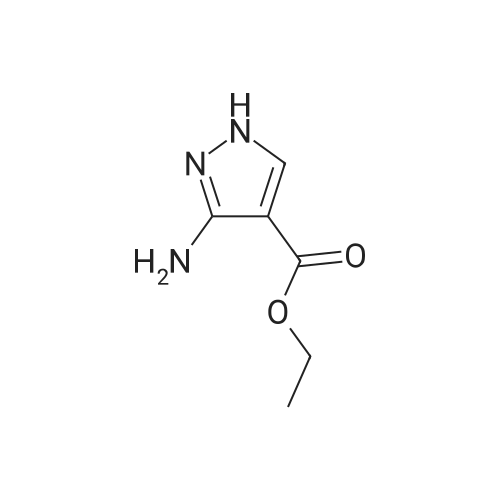
Stage #1: 1-methoxypentane-2,4-dione; ethyl 5-amino-1H-pyrazole-4-carboxylate With acetic acid at 110℃; for 1h; Microwave irradiation;
Stage #2: With water; lithium hydroxide In tetrahydrofuran; methanol at 20℃; for 72h;
Stage #3: 8-amino-2-(2-chloro-4,6-difluorophenyl)-2-azaspiro[4.5]decan-1-one With benzotriazol-1-yloxyl-tris-(pyrrolidino)-phosphonium hexafluorophosphate; N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 20℃; for 12h; |
241; 242 N-[(trans)-2-(2-chloro-4,6-difluorophenyl)-1-oxo-2-azaspiro[4.5]dec-8-yl]-7-(methoxymethyl)-5-methylpyrazolo[1,5-a]pyrimidine-3-carboxamide
A mixture of ethyl 5-(methoxymethyl)-7-methylpyrazolo[1 ,5-a]pyrim idine-3-carboxylate(isomer 1) and ethyl 7-(methoxymethyl)-5-methylpyrazolo[1 ,5-a]pyrimidine-3-carboxylate(isomer 2) (1.35 g, 5.42 mmol, ratio of isomers ca 1:1) was stirred in a mixture of lithiumhydroxide (27 ml, 27 mmol, 1 M aqueous solution), tetrahydrofuran (35 ml) and methanol(9.4 ml) for 3 days at room temperature. For work-up, aqueous hydrochloric acid (2 M) was added and the precipitate formed was collected by filtration. The filtrated was extracted with ethyl acetate (3x), washed with brine, filtrated through a silicon filter and concentrated to give the title compounds as mixture of isomers (ca 1:1) (820 mg, 69%) which was used in the nextstep without further purification. Benzotriazol-1 -yl-oxytripyrrolidinophosphonium hexafluorophosphate (218 mg, 419 limol)and N,N-diisopropylethylamine (300 iii, 1 .7 mmol) were added to a mixture of 8-amino-2-(2-chloro-4,6-difluorophenyl)-2-azaspiro[4.5]decan-1-one (isomer 1) (200 mg, 55 % purity, 349iimol) and a mixture of regioisomers of 5-(methoxymethyl)-7-methylpyrazolo[1 5-a]pyrim idine-3-carboxylic acid (isomer 1) and 7-(methoxymethyl)-5-methylpyrazolo[1 ‘5- a]pyrimidine-3-carboxylic acid (isomer 2) (92.8 mg, 419 limol) in N,N-dimethylformamide (3.9 ml) and the mixture was stirred at room temperature for 12 h. For work-up, the mixture was concentrated and the residue was purified by preparative HPLC [Instrument: WatersAutopurificationsystem; column: Waters XBrigde 018 5i lOOx3Omm; eluent A: water + 0.2 Vol-% aqueous ammonia (32%), eluent B: acetonitrile; gradient: 0.00-0.50 mm 38% B (25->70m1/min), 0.51-5.50 mm 38-60% B (70m1/min), DAD scan: 21 0-400 nm] to give N-[(trans)2-(2-chloro-4, 6-dif luorophenyl)-1 -oxo-2-azaspiro[4.5]dec-8-yl]-7-(methoxymethyl)-5-methylpyrazolo[1,5-a]pyrimidine-3-carboxamide (19.9 mg, 11% yield, R = 4.54 -4.79 mm)and N-[(trans)-2-(2-chloro-4,6-difluorophenyl)- 1 -oxo-2-azaspiro[4.5]dec-8-yl]-5-(methoxymethyl)-7-methylpyrazolo[1 ,5-a]pyrim idine-3-carboxam ide (61 .7 mg, 34% yield, R = 3.78-4.03 mm)N-[(trans)-2-(2-chloro-4,6-difluorophenyl)- 1 -oxo-2-azaspiro[4.5]dec-8-yl]-7-(methoxym ethyl)5-methylpyrazolo[1 , 5-a]pyrim idine-3-carboxam ide:LC-MS (Method 2): R = 1.24 mm; MS (ESIpos): m/z = 518 [M÷H]1HNMR (400 MHz, DMSO-d6) 6 [ppm]: 1.469 (0.51), 1.481 (0.55), 1.499 (0.61), 1.510(0.58), 1.671 (1.16), 1.681 (1.29), 1.694 (1.25), 1.710 (0.75), 1.722 (0.45), 1.994 (0.66),2.003 (0.68), 2.013 (0.46), 2.025 (0.65), 2.035 (0.59), 2.059 (0.55), 2.197 (0.88), 2.214(1.81), 2.232 (1.04), 2.327 (0.43), 2.518 (1.24), 2.523 (0.88), 2.669 (0.47), 2.693 (11.36),3.521 (16.00), 3.540 (0.41), 3.547 (0.75), 3.633 (0.68), 3.640 (0.43), 3.651 (0.50), 3.657(0.56), 4.961 (3.83), 4.964 (3.79), 7.165 (2.56), 7.504 (0.44), 7.511 (0.63), 7.527 (0.60),7.535 (1.09), 7.542 (0.87), 7.546 (0.77), 7.549 (0.58), 7.554 (0.66), 7.558 (0.88), 7.564 (0.87), 7.568 (0.73), 7.571 (0.43), 7.575 (0.49), 7.983 (0.96), 8.002 (0.92), 8.504 (4.99),8.514 (0.41). Isolated as side product in the synthesis of N-[(trans)-2-(2-chloro-4,6-difluorophenyl)-1 -oxo-2- azaspiro[4. 5]dec-8-yI]-7-(methoxymethyl)-5-methylpyrazolo[1 ,5-a]pyrim idine-3-carboxamide:(61 .7 mg, 34% yield, R = 3.78-4.03 mm)LC-MS (Method 2): R = 1.18 mm; MS (ESIpos): m/z = 518 [M÷H][ppm]:1.694(0.55),2.804(0.45),7.536(0.81),(0.51), 1.4831.722 (0.44),(1.74), 2.2243.527 (0.58),(6.91), 7.2427.547 (0.71),(0.48), 7.960(0.56), 1.4931.994 (0.61),(0.99), 2.5183.532 (0.47),(2.03), 7.2447.550 (0.54),(0.89), 7.9791HNMR (400 MHz, DMSO-d6) 6 (0.53), 1.672 (1.08), 1.682 (1.21), 2.003 (0.64), 2.026 (0.60), 2.035(1.05), 2.523 (0.69), 2.802 (5.97),3.551 (0.69), 3.636 (0.64), 3.654(2.05), 7.512 (0.57), 7.528 (0.53),7.554 (0.63), 7.558 (0.80), 7.565(0.85), 8.560 (4.95).1 .453 (0.47), 1 .464(1.17), 1.710 (0.74),2.190 (0.84), 2.207(6.34), 3.463 (16.00),3.660 (0.54), 4.665(0.99), 7.543 (0.80),7.568 (0.68), 7.575 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping