* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Tetrahedron, 1992, vol. 48, # 36, p. 7479 - 7488
[2] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[3] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[4] Tetrahedron, 1992, vol. 48, # 36, p. 7479 - 7488
[5] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[6] Synthesis, 2005, # 10, p. 1619 - 1624
2
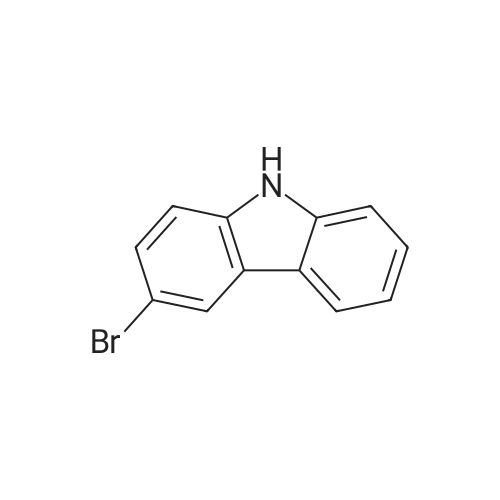
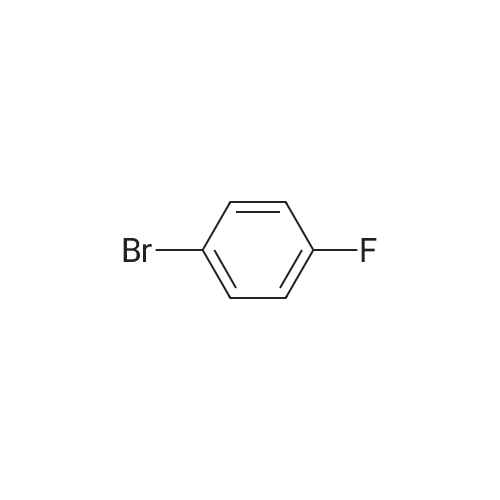
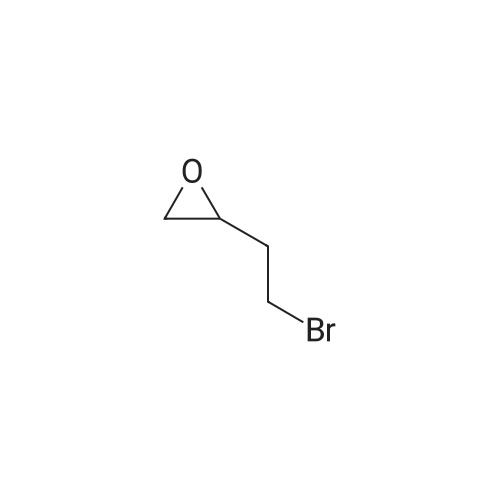
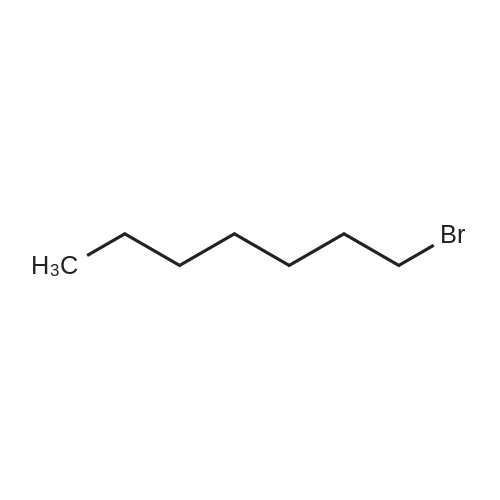
[ 86-74-8 ]
[ 6825-20-3 ]
Yield
Reaction Conditions
Operation in experiment
96%
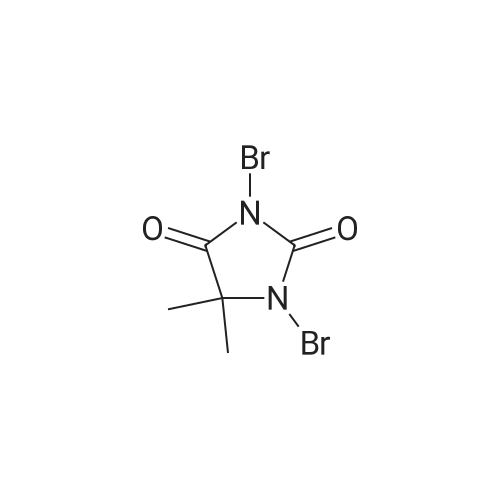
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione In ethanol at 20 - 25℃; for 2 h;
The detailed steps of the present invention are as follows:among them,The amount of charge is as follows:Carbazole: 167.2 g,Dibromohydantoin:300. lg,Anhydrous B.Alcohol: 1800 ml.167. 2 g of carbazole (lmol) and 1000 ml of absolute ethanol were added to a 2000 ml reaction flask and stirred for 20 minutes.So as to disperse uniformly.Dibromohydantoin was then added in batches''285 g (1. 05 mol) was added and the reaction temperature was controlled at 20-25 ° C, about 2 hoursWhen added, after adding a large number of white solid precipitation. And reacted at this temperature for 1 hour. The reaction was monitored by HPLC. When the monobrominated product was less than 1percent, the reaction was stopped and the reaction was stopped at 20-25 ° CThe filter cake was added to a 1000 ml three-necked flask, and 800 ml of absolute ethanol was added. The mixture was heated to reflux (80 ° C) for 1 hour, cooled slowly to 20-25 ° C,Dried at 100 ° C under normal pressure to obtain 312 g of a white powdery solid. Figure 3 shows the 3, 6-dibromo carbazole HPLC spectrum andTable 3 shows that the 3,6-dibromocarbazole content was 99.00percent, and the GC spectrum of 3,6-dibromo carbazole as shown in Figure 4 and the results shown in Table 4The yield of 3, 6-dibromocarbazole was 99.66percent, and the yield of 3, 6-dibromocarbazole was 96.0percent.
90%
With N-Bromosuccinimide In dichloromethane; N,N-dimethyl-formamide at 20℃;
A solution of NBS (42.72g, 0.24mol) in 200mL DMF was added slowly to a suspension of carbazole (20g, 0.12mol) in dichloromethane (1.2L). The reaction mixture was stirred at room temperature and the progress of the reaction was controlled by GCMS analysis. Then the solution was washed with water (3×200mL), the organic layer was dried under magnesium sulfate and filtered. The solvent was evaporated and the residue was dissolved in acetone (100mL) and precipitated with hexane (600mL). 3,6-dibromocarbazole was filtered and dried under vacuum to afford 35g (yield 90percent) of product.1H NMR (300MHz, CDCl3): δ 7.31 (d, 2H, JHH=8.6Hz, H1), 7.52 (d, 2H, JHH=8.6Hz, H2), 8.13 (d, 2H, JHH=1.9Hz, H4), 10.37 (s, 1H, H9). 13C NMR (75HMz, CDCl3): δ 112.2 (C1), 112.6 (C3), 123.3 (C4), 124.1 (Ci, N–C=C–), 129.3 (C2), 138.3 (Ci, N–C=C–). MS (EI): m/z (rel. intensity – percent): 327+ (49), 325 (100), 326 (20), 246 (16), 165 (28), 138 (11), 83 (11).
90%
With N-Bromosuccinimide In dichloromethane for 4 h;
9.7 g (58 mmol) of the carbazole was dissolved in 600 ml of dichloromethane, and 21.7 g (121.8 mmol) of N-bromosuccinimide was added dropwise over 10 minutes. After 4 hours, the solvent was removed in vacuo, The residue was subjected to ethanol and water, Filtered, washed with ethanol and dried to give 17.0 g (90percent) of a colorless powder.
88%
With N-Bromosuccinimide; silica gel In dichloromethane at 20℃; for 24 h;
A mixture of carbazole (1.00 g, 6.00 mmol) and silica gel(8.00 g, 133.33 mmol) was added to CH2Cl2(60 mL) at roomtemperature, N-bromosuccinimide (1.07 g, 12.00 mmol),added to the above system, the mixture was reacted for 24 hat room temperature, then filtering and evaporating off thesolvent, and the residue was extracted with n-hexane/ethylacetate (10:1, v/v) to generate a white solid. 1.71 g, yield:88percent.Mp: 204–206 °C. 1H NMR (400 MHz, DMSO-d6, δ,ppm): 11.60 (s, 1H, –NH), 8.44 (d, J = 8.8 Hz, 2H), 7.54(d, J = 8.9 Hz, 2H), 7.48 (d, J = 8.7 Hz, 2H). 13C NMR(101 MHz, DMSO-d6, δ, ppm): 139.3, 129.2, 123.9, 123.8,113.8, 111.5. Anal. calcd for C12H7Br2N:C, 44.35; H, 2.17;Br, 49.17; N, 4.31. Found: C, 44.28; H, 2.15; Br, 49.14; N,4.29.
85%
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0 - 20℃; for 6 h; Flash photolysis
To a 500 mL three-necked flask was added carbazole (24.7 g, 0.1 mol)Dimethylformamide 200 mL,Stir to dissolve,NBS (49.84 g, 0.28 mol) was dissolved in 120 mL of DMF,Ice bath to 0 ,Slowly drop the NBS solution,Reaction to light,After the dropwise addition, the temperature was allowed to rise to room temperature and the reaction was continued for 6 hours.The reaction solution was added dropwise to the water to precipitate, and the crude product was collected by suction filtration. The filtrate was recrystallized from absolute ethanol and dried to give white needles. Yield: 85percent
83%
With N-Bromosuccinimide In tetrahydrofuran at 20℃; for 4 h;
4-3: Synthesis of 3,6-dibromo-9H-carbazole: Carbazole (5.0 g, 29.9 mmol) was dissolved in THF (50 mL), and then N-bromosuccinimide (11.18 g, 62.80mmol) was added thereto to prepare a mixed solution. Then, the mixed solution was reacted at room temperature for 4hours. After the reaction of the mixed solution, a solvent was removed, and the reacted mixed solution was extractedwith water and ethyl acetate to obtain an extract. The extract was dried by magnesium sulfate (MgSO4), and then areaction product was separated by column chromatography (n-hexane/EtOAc = 4/1). The yield ofthe reaction productwas 83percent (8.1 g).1HNMR(300 MHz,CDCl3) 8.13-(s, -Ar, 2H), 7.53(d, -Ar, 2H), 7.32-7.3(d, -Ar, 2H)
80%
With N-Bromosuccinimide In tetrahydrofuran for 4 h; Inert atmosphere; Cooling with ice
In the 100mL single-mouth bottle,1.00 g (5.99 mmol) of carbazole was added, dissolved in tetrahydrofuran, and the jar was placed in an ice bath and protected from light.Then 2.67g (15mmol) of NBS was dissolved in tetrahydrofuran and slowly poured into a constant pressure dropping funnel. Into a 100ml single-mouth bottle,An ice bath was stirred under nitrogen for 4 h.The reaction was poured into 100 mL of water.Extract three times with ethyl acetate, The resulting liquid spins dry,With ethyl acetate:Column chromatography of petroleum ether = 1:200 gave 1.56 g (4.8 mmol) of a white solid, Compound 2, yield 80percent.
76%
With N-Bromosuccinimide; silica gel In dichloromethane
Synthesis of Compound 2:; Compound 2 was obtained by dibromination of compound 1.This dibromination was carried out by making this compound (2.01 g, 12.0 mmol) react with N-succinimide (4.20 g, 23.8 mmol), in the presence of silica preactivated at 120° C. (40.50 g, 0.063-0.2 nm) in order to protonate the nitrogen atom of compound 1, and in dichloromethane (350 ml), as described by Smith et al. in Tetrahedron, 1992, 48, 36, 7479-7488, [4].The yield of the reaction was 76percent.
73%
With N-Bromosuccinimide; dibenzoyl peroxide In dichloromethane at 20℃;
Carbazole (16.72g, 100mmol), NBS (59.4g, 210mmol), BPO (2.42g, 10mmol), followed by stirring at room temperature, insert the methylenechloride (300mL). When the reaction is complete, methylene chloride and water, and extracted using an aqueous solution of NaHCO3 was obtained organic layer was dried with Na2SO4, and the product was recrystallized 23.73g (73percent) and was concentrated.
68%
With N-Bromosuccinimide In N,N-dimethyl-formamide for 0.5 h; Cooling with ice
3,6-Dibromo-9H-carbazole (1), a solution of N-bromosuccinimide (0.21 mol) in DMF (80 mL) was slowly added to a solution of carbazole (0.1 mol) in DMF (20 mL) in an ice bath. After reaction for 30 min, the mixture was poured into ice water (1 L), and the crude product was collected by filtration to give a blue powder. Recrystallization from EtOH/H2O afforded blue crystals with a yield of 68percent. 1H NMR (500 MHz, CDCl3): δ 8.14 (d, J = 1.9 Hz, 2H), 8.12 (s, 1H), 7.53 (d, J = 8.6, 1.9 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H).
64%
With N-Bromosuccinimide; silica gel In dichloromethane at 20℃; for 12 h; Darkness
9H- carbazole (1) (2.00g, 12.0mmol) in dichloromethane (100ml)To, N- bromosuccinimide (4.30g, 24.0mmol) and silica gel(SiO2) was added (20.0g).The dark, was stirred at room temperature for 12 hours, filtered, washed twice with water, dried over anhydrous sodium sulfate, filtered, using a rotary evaporator, the solvent was distilled off under reduced pressure at room temperature from the filtrate, the crude product I got things. The resulting crude product was recrystallized from dichloromethane, 3,6-dibromo-9H-carbazole and (2b) 2.50 g (yield: 64percent) was obtained.
61.5%
With N-Bromosuccinimide In dichloromethane; N,N-dimethyl-formamide for 3 h; Inert atmosphere
Step 1: In a 1000 mL three-necked flask equipped with a mechanical stirrer, 4 g (0.024 mol)Carbazole and 240 mL of dichloromethane, under an argon atmosphere at 5 mL / minAt a rate of dropwise addition of N-bromosuccinimideN, N '-dimethylformamide solution,Wherein N-bromosuccinimide is8.54 g (0.048 mol), N, N'-dimethylformamide solution was 40 mL; stirred for 3 hours,The solution was washed four times with 280 mL of distilled water,The organic phase was dried over anhydrous magnesium sulfate; the solvent in the organic phase was distilled off under reduced pressure at room temperature,To give a pale gray crude product; mixed with acetone and n-hexane(In which acetone is 30 mL and n-hexane is 400 mL) to recrystallize the light gray crude product;The final product was vacuum dried at 70 ° C for 10 hours to give a gray bulk crystal, I.e., 3,6-dibromocarbazole; the resulting product was 4.9 g in a yield of 61.5percent
55%
With N-Bromosuccinimide In N,N-dimethyl-formamide at 0 - 20℃; for 2 h; Inert atmosphere
Synthesis of 3,6-dibromocarbazole. Carbazole (1 .0 g, 6 mmol) was added to anhydrous DMF (15 mL) at room temperature while stirring. The resulting mixture was stirred at room temperature for another 15 minutes. N- bromosuccinimide (2.3 g, 13 mmol) in anhydrous DMF (10 mL) was added drop wise to carbazole-DMF mixture at 0°C in inert atmosphere. The resulting solution was allowed to reach to the room temperature and continued stirring for another 2 hours. This solution was poured to Dl water. The milky white precipitate formed was filtered and dissolved in DCM . DCM layer was washed with water several times. Then DCM layer was dried with anhydrous sodium sulfate and solvent was removed under high vacuum. Resultant white solid was recrystallized using ethanol to get light brown silvery flake-like 3,6-dibromocarbazole crystals (1 .1 g, 55percent yield) . MS, ESI - m/z 323 (M+); 1 H NMR (Deuterated DMSO, 5 00 MHz, ppm) : δ 1 1 .49 (S, 1 H), 8.08 -8.10 (D, 2H), 7.72 (S, 2H), 7.32-7.34 (D, 2H).
47%
With N-Bromosuccinimide In N,N-dimethyl-formamide at 20℃; Cooling with ice
N-N-dimethylformamide (DMF) (20 ml) dissolved with carbazole (10 g, 59.8 mmol) was stirred under ice-The solution was added dropwise to a solution of N-bromosuccinimide (21.28 g, 120 mmol) in DMF (30 mL) with a constant pressure dropping funnelin. After completion of the drop, the ice bath was removed and the reaction was conducted overnight at room temperature. The reaction solution was then washed with deionized water and analyzedThe solid was filtered off and the product was recrystallized from ethanol. Yield: 47percent.
Reference:
[1] Molecular Crystals and Liquid Crystals, 2012, vol. 567, # 1, p. 102 - 109,8
[2] Organic and Biomolecular Chemistry, 2016, vol. 14, # 39, p. 9406 - 9415
[3] Patent: CN105523990, 2016, A, . Location in patent: Paragraph 0012;0013;0016
[4] Journal of Materials Chemistry, 2008, vol. 18, # 12, p. 1296 - 1301
[5] Organic Letters, 2011, vol. 13, # 12, p. 3186 - 3189
[6] Journal of Organometallic Chemistry, 2014, vol. 750, p. 150 - 161
[7] Dalton Transactions, 2018, vol. 47, # 12, p. 4040 - 4044
[8] Patent: KR2018/76551, 2018, A, . Location in patent: Paragraph 0157-0160
[9] Chemical Papers, 2018, vol. 72, # 11, p. 2955 - 2963
[10] Electrochimica Acta, 2016, vol. 196, p. 140 - 152
[11] Journal of Nanoscience and Nanotechnology, 2011, vol. 11, # 2, p. 1373 - 1376
[12] Patent: CN106588869, 2017, A, . Location in patent: Paragraph 0057; 0058; 0059; 0060
[13] Patent: EP2711359, 2014, A1, . Location in patent: Paragraph 0041; 0043; 0092; 0093; 0106; 0107; 0149; 0150
[14] RSC Advances, 2013, vol. 3, # 30, p. 12091 - 12095
[15] Macromolecules, 2014, vol. 47, # 9, p. 2875 - 2882
[16] Chemical Communications, 2017, vol. 53, # 82, p. 11334 - 11337
[17] Macromolecules, 2006, vol. 39, # 24, p. 8303 - 8310
[18] Organic and Biomolecular Chemistry, 2015, vol. 13, # 30, p. 8335 - 8348
[19] Patent: CN107759597, 2018, A, . Location in patent: Paragraph 0027-0029
[20] New Journal of Chemistry, 2013, vol. 37, # 3, p. 632 - 639
[21] Patent: US2008/146816, 2008, A1, . Location in patent: Page/Page column 4
[22] Electrochimica Acta, 2012, vol. 65, p. 104 - 114
[23] Chemical Communications, 2015, vol. 51, # 6, p. 1046 - 1049
[24] Organic and Biomolecular Chemistry, 2016, vol. 14, # 10, p. 2961 - 2968
[25] Patent: KR2015/115226, 2015, A, . Location in patent: Paragraph 0158; 0159; 0160
[26] Chemistry - An Asian Journal, 2013, vol. 8, # 5, p. 1015 - 1022
[27] Chinese Journal of Catalysis, 2016, vol. 37, # 4, p. 533 - 538
[28] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1992, vol. 28, # 11, p. 1260 - 1263[29] Khimiya Geterotsiklicheskikh Soedinenii, 1992, # 11, p. 1477 - 1480
[30] Molecular Crystals and Liquid Crystals Science and Technology Section A: Molecular Crystals and Liquid Crystals, 2001, vol. 369, p. 17 - 35
[31] Patent: JP2016/20322, 2016, A, . Location in patent: Paragraph 0071; 0072; 0073
[32] Patent: CN105461613, 2016, A, . Location in patent: Paragraph 0046-0047
[33] Patent: WO2016/112027, 2016, A1, . Location in patent: Paragraph 0156
[34] Organic Letters, 2013, vol. 15, # 1, p. 164 - 167
[35] Polymer, 2011, vol. 52, # 10, p. 2189 - 2197
[36] Patent: CN106046054, 2016, A, . Location in patent: Paragraph 0040; 0041; 0042
[37] Journal of the American Chemical Society, 2017, vol. 139, # 12, p. 4559 - 4567
[38] Angewandte Chemie, 1901, vol. 14, p. 784
[39] Diss. <Braunschweig 1926>3 22,
[40] Collection of Czechoslovak Chemical Communications, 1957, vol. 22, p. 64,71
[41] Chemische Berichte, 1925, vol. 58, p. 2375
[42] Journal of the Chemical Society, 1927, p. 1215
[43] Fortschr. Teerfarbenfabr. Verw. Industriezweige, vol. 11, p. 1194
[44] Tetrahedron, 1992, vol. 48, # 36, p. 7479 - 7488
[45] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[46] Patent: WO2010/43693, 2010, A1, . Location in patent: Page/Page column 14
[47] Journal of the American Chemical Society, 2010, vol. 132, # 49, p. 17599 - 17610
[48] Organic and Biomolecular Chemistry, 2011, vol. 9, # 20, p. 6913 - 6916
[49] Organic Electronics: physics, materials, applications, 2011, vol. 12, # 1, p. 125 - 135
[50] Polymer, 2011, vol. 52, # 3, p. 804 - 813
[51] Chemistry - A European Journal, 2012, vol. 18, # 48, p. 15512 - 15522
[52] Organic Electronics: physics, materials, applications, 2013, vol. 14, # 3, p. 730 - 743
[53] Journal of Materials Chemistry A, 2013, vol. 1, # 6, p. 2297 - 2306
[54] RSC Advances, 2013, vol. 3, # 41, p. 18821 - 18834
[55] Journal of Polymer Science, Part A: Polymer Chemistry, 2014, vol. 52, # 2, p. 272 - 286
[56] Journal of Polymer Science, Part A: Polymer Chemistry, 2014, vol. 52, # 8, p. 1172 - 1184
[57] Journal of Materials Chemistry C, 2015, vol. 3, # 31, p. 8142 - 8151
[58] Journal of Materials Chemistry C, 2016, vol. 4, # 6, p. 1271 - 1280
[59] Science China Chemistry, 2017, vol. 60, # 10, p. 1356 - 1366
[60] Organic Electronics: physics, materials, applications, 2014, vol. 15, # 1, p. 266 - 275
[61] Journal of the American Chemical Society, 2014, vol. 136, # 18, p. 6672 - 6683
[62] Russian Journal of General Chemistry, 2014, vol. 84, # 7, p. 1313 - 1319[63] Zh. Obshch. Khim., 2014, vol. 84, # 7, p. 1117 - 1123,7
[64] Dyes and Pigments, 2015, vol. 114, # C, p. 239 - 252
[65] RSC Advances, 2015, vol. 5, # 74, p. 59960 - 59969
[66] Dyes and Pigments, 2016, vol. 133, p. 25 - 32
[67] Electrochimica Acta, 2016, vol. 210, p. 673 - 680
[68] Dyes and Pigments, 2017, vol. 137, p. 24 - 35
[69] Patent: KR2015/144421, 2015, A, . Location in patent: Paragraph 0317; 0318; 0319
[70] Reactive and Functional Polymers, 2017, vol. 110, p. 47 - 54
[71] Polyhedron, 2017, vol. 124, p. 125 - 130
[72] Patent: US4061655, 1977, A,
3
[ 122-39-4 ]
[ 6825-20-3 ]
Reference:
[1] Russian Chemical Bulletin, 1993, vol. 42, # 1, p. 214[2] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1993, # 1, p. 234 - 235
[3] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1988, vol. 27, # 1-12, p. 402 - 403
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 13, p. 4626 - 4634
6
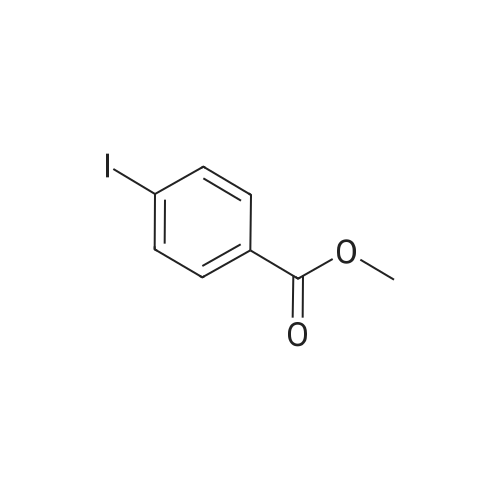
[ 16292-17-4 ]
[ 6825-20-3 ]
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 13, p. 4626 - 4634
7
[ 4051-56-3 ]
[ 6825-20-3 ]
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 13, p. 4626 - 4634
8
[ 99234-92-1 ]
[ 6825-20-3 ]
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 13, p. 4626 - 4634
9
[ 5149-08-6 ]
[ 6825-20-3 ]
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 13, p. 4626 - 4634
10
[ 86-74-8 ]
[ 1592-95-6 ]
[ 6825-20-3 ]
Reference:
[1] Tetrahedron, 1992, vol. 48, # 36, p. 7479 - 7488
[2] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[3] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[4] Tetrahedron, 1992, vol. 48, # 36, p. 7479 - 7488
[5] Canadian Journal of Chemistry, 1992, vol. 70, # 3, p. 870 - 876
[6] Synthesis, 2005, # 10, p. 1619 - 1624
11
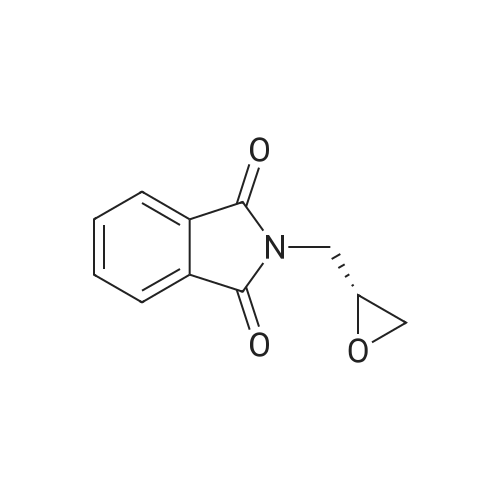
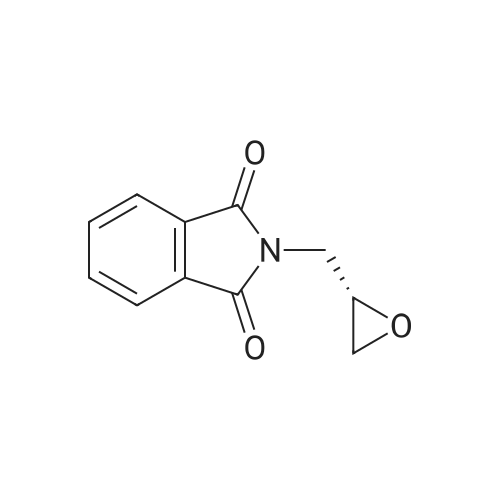
[ 912850-81-8 ]
[ 6825-20-3 ]
Reference:
[1] Gazzetta Chimica Italiana, 1892, vol. 22 II, p. 570,572[2] Gazzetta Chimica Italiana, 1895, vol. 25 II, p. 395,398,399
12
[ 77-48-5 ]
[ 86-74-8 ]
[ 6825-20-3 ]
Reference:
[1] Anales de la Asociacion Quimica Argentina (1921-2001), 1951, vol. 39, p. 184,186
13
[ 7726-95-6 ]
[ 108-24-7 ]
[ 86-74-8 ]
[ 64-19-7 ]
[ 6825-20-3 ]
Reference:
[1] Chemische Berichte, 1925, vol. 58, p. 2375
[2] Journal of the Chemical Society, 1927, p. 1215
[3] Chem. Zentralbl., 1914, vol. 85, # II, p. 182
[4] Fortschr. Teerfarbenfabr. Verw. Industriezweige, vol. 11, p. 1194
14
[ 228091-77-8 ]
[ 67-64-1 ]
[ 6825-20-3 ]
Reference:
[1] Chemische Berichte, 1925, vol. 58, p. 2375
15
[ 7647-01-0 ]
[ 228091-77-8 ]
[ 6825-20-3 ]
Reference:
[1] Chemische Berichte, 1925, vol. 58, p. 2375
With tetrabutylammomium bromide; potassium hydroxide In acetone for 3 h; Reflux
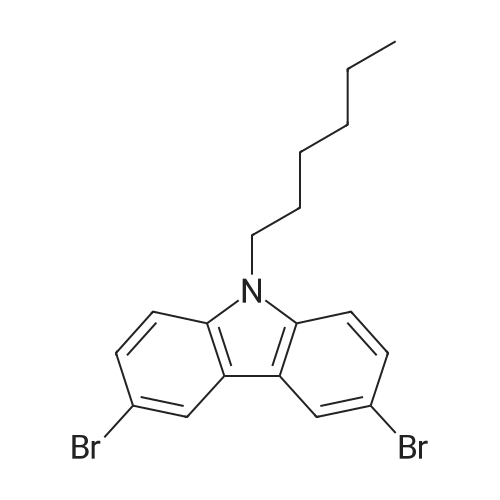
General procedure: Hexyl bromide (1.45mL, 15.7mmol), tetrabutylammonium bromide (0.16g, 0.5mmol) and potassium hydroxide (0.83g, 14.8mmol) were added to a solution of 3,6-dibromocarbazole (6.18g, 9.2mol) in acetone (previously dried under 3 sieves). The reaction mixture was stirred for 3hand refluxed. Progress of the reaction was controlled by GCMS analysis. Then the mixture was cooled to room temperature and the solvent was evaporated. The residue was dissolved in diethyl ether (100mL), filtered and washed with water (3×10mL). The organic layer was dried under magnesium sulfate and filtered. Then the solvent was evaporated and the residue was stirred with pentane (100mL) for 1h, filtered and dried under vacuum. 6.1g (yield 78percent) of 3,6-dibromo-N-hexylcarbazole was obtained as white solid. The product was crystallized by dissolving in small amount of hot hexane and left to cool at room temperature.1H NMR (300MHz, CDCl3): δ 0.86 (t, 3H, JHH=7.0Hz, H6′), 1.21–1.38 (m, 6H, H3′, H4′, H5′), 1.81 (quint, 2H, JHH=7.3Hz, H2′) 4.24 (t, 2H, JHH=7.2Hz, H1′), 7.27 (d, 2H, JHH=8.7Hz, H2), 7.55 (d, 2H, JHH=8.7Hz, H1), 8.14 (d, 2H, JHH=1.9Hz, H4). 13C NMR (75MHz, CDCl3): δ 13.9 (C6′), 22.5 (C5’), 26.9 (C3′), 28.8 (C2′), 31.5 (C4′), 43.3 (C1′), 110.4 (C1), 111.9 (C3), 123.2 (C4), 123.4 (Ci, N–C=C–), 129.0 (C2), 139.3 (Ci, N–C=C–). MS (EI): m/z (rel. intensity – percent): 411+ (31), 409 (58), 408 (9), 341 (10), 340 (53), 338 (100), 337 (34), 258 (20), 152 (10). Crystal Data: C18H19Br2N; formula weight Mr=409.16; orthorhombic; Pca21; a=20.3019(16) , b=4.5582(4) , c=18.4012(18) , V=1702.8(3) 3, Z=4, dx=1.60gcm−3, λ=0.71073 (MoKα), μ=4.75mm−1; reflections: collected 27,069, unique 3018 (Rint=10.58percent), with I>2σ(I) 1827; R=6.43percent, wR2=13.82percent, S=1.03, max/min Δρ: 0.79/−0.33e−3. Perspective view of the molecule 4 is presented below. Thermal ellipsoids are drawn at the 50percent probability level, hydrogen atoms are represented by spheres of arbitrary radii.
35%
With potassium hydroxide In acetone at 20℃; Inert atmosphere; Reflux
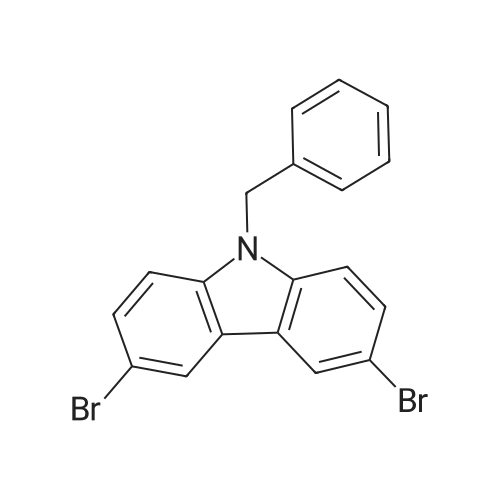
3,6-Dibromocarbazole 2 (112.0 g, 0.344 mol), benzyl bromide (41 ml,0.344 mol), and nBu4NHSO4 were mixed in 200 ml of acetone and stirred at room temperature under nitrogen until dissolved. KOH (19.3 g, 0.344 mol) was then added to the above transparent solution and the resultant mixture was refluxed for 4 hours, where a white precipitate was observed. The hot mixture was concentrated to remove the majority of acetone. Upon cooling, additional white precipitate appeared. The precipitate was filtered, dissolved in methyl-f-butyl ether, washed with water, dried over Na2SO4, and concentrated to yield a major amount of pure N-benzyl-3,6- dibromocarbazole 3. The filtrate was also washed with water and extracted with methyl-f-butyl ether, dried over Na2SO4, concentrated, and purified over 12Og Siψ2 with hexane and hexane:ethylacetate (95:5) eluents to yield a minor amount of pure N-benzyl-3,6-dibromocarbazole 3. The combined yield from crystallization and chromatography was 49 g (35percent) of N-benzyl-3,6-dibromocarbazole 3.
Reference:
[1] Journal of Materials Chemistry, 2007, vol. 17, # 14, p. 1433 - 1438
[2] Journal of Organometallic Chemistry, 2014, vol. 750, p. 150 - 161
[3] Dalton Transactions, 2018, vol. 47, # 12, p. 4040 - 4044
[4] Patent: WO2010/43693, 2010, A1, . Location in patent: Page/Page column 14; 15
[5] Electrochimica Acta, 2016, vol. 210, p. 673 - 680
18
[ 6825-20-3 ]
[ 100-44-7 ]
[ 118599-27-2 ]
Reference:
[1] Journal of Organic Chemistry, 1989, vol. 54, # 4, p. 965 - 968
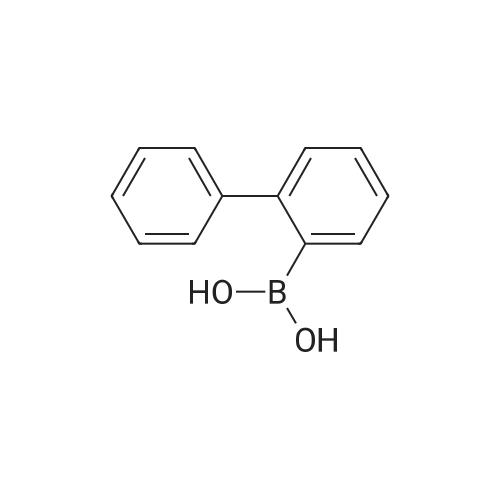
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 80℃; for 16 h; Inert atmosphere
It was dissolved in 3,6-dibromo-9H-carbazole(100 g, 308 mmol) in a nitrogen environment in 700 mL tetrahydrofuran(THF), here the phenylboronic acid(45.1 g, 370 mmol) and put tetrakis(triphenylphosphine) palladium (10.7 g, 9.24mmol) was stirred. Into a potassium carbonate (213 g, 1,540 mmol) in a saturated water was heated to reflux at 80 ° C for 16 hours. After the reaction was completed, the reaction solution into water and extracted with DCM then water was removedwith anhydrous MgSO4, filter and was concentrated under reduced pressure. Thus the resulting residue was separated and purified by flash column chromatography to obtain the compound I-23(74.8 g, 76percent).
75%
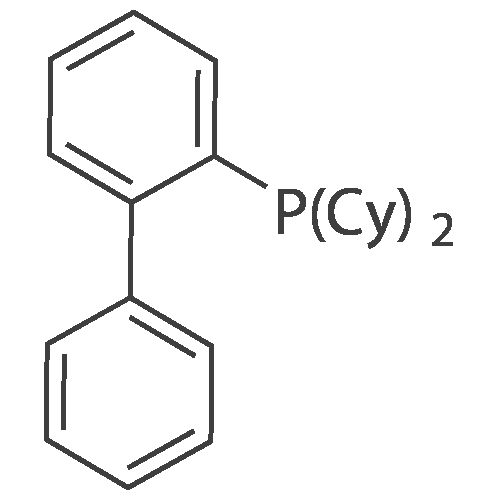
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; tris-(dibenzylideneacetone)dipalladium(0); potassium phosphate monohydrate In water; tolueneInert atmosphere; Reflux
Synthesis of 3,6-diphenyl-9H-carbazole 3,6-Dibromo-9H-carbazole (10.0 g, 30.8 mmol), phenylboronic acid (8.25 g, 67.7 mmol) Pd2(dba)3 (0.564 g, 0.615 mmol), dicyclohexyl(2',6'-dimethoxy-[1,1'-biphenyl]-2-yl)phosphine (SPhOS) (1.011 g, 2.462 mmol), and potassium phosphate hydrate (28.3 g, 123 mmol) were dissolved in the mixture of toluene (350 mL) and water (40 mL) in a three-necked flask. The mixture was degassed by bubbling nitrogen, then it washeated to reflux overnight. After completion of the reaction, the mixture partitioned between ethyl acetate and water. The aqueous layer was washed 3 times with ethyl acetate and the combined organic layers were washed with brine and water. The crude compound was purified by column chromatography on silica gel, eluted with hexane/DCM 1/1 (v/v) mixture. The target compound was obtained as a white solid (7.4 g, 75 percent yield).
74%
With potassium carbonate In tetrahydrofuranInert atmosphere; Reflux
Example 10; a) 3.09 g (9.23 mmol) of 3,6-dibromocarbazole and 2.78 g (22.2 mmol) of phenylboronic acid are stirred in 140 ml of THF under argon. 12.76 g (92.30 mmol) of potassium carbonate in 46 ml of water are added. 0.27 g (0.23 mmol) of palladium tetrakis(triphenylphosphine) are added under argon and the reaction is stirred under reflux over night. At room temperature, a 1 percent aqueous solution of NaCN is added to the medium which is boiled for 20 min. At room temperature, ethyl acetate is added and the organic phase is washed with water, dried over magnesium sulfate, filtered and evaporated under reduced pressure. The crude material is purified by chromatography over silica gel (cyclohexane/ethyl acetate 4:1 ) to give 2.17 g of a white solid (74percent). 1H NMR (CDCI3, 300 MHz) 8.34 (d, J = 1.8 Hz, 2H), 8.11 (s, 1 H), 7.74- 7.68 (m, 6H), 7.52-7.45 (m, 6H), 7.37-7.32 (m, 2H).
74%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In toluene at 80℃; for 8 h; Inert atmosphere
In the nitrogen atmosphere, 3,6-dibromocarbazole (5 g, 15.4 mmol) was added sequentially to the flask, Phenylboronic acid (4.1 g, 33.9 mmol) (Triphenylphosphine) palladium (0.7 g, 0.6 mmol) Toluene (45 ml), 2M sodium carbonate (45 ml), And the mixture was stirred at 80 ° C for 8 hours. After separating the organic phase, Concentrated under reduced pressure in an evaporator. The resulting residue was purified by silica gel column chromatography, 3,6-diphenylcarbazole (3.6 g, yield 74percent) was obtained.
74%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate In toluene at 80℃; for 8 h; Inert atmosphere
In the nitrogen atmosphere,3,6-dibromocarbazole (5 g, 15.4 mmol) was added sequentially to the flask,Phenylboronic acid (4.1 g, 33.9 mmol)(Triphenylphosphine) palladium (0.7 g, 0.6 mmol)Toluene (45 ml),2M sodium carbonate (45 ml),And stirred at 80 ° C for 8 hours.After separating the organic phase,Concentrated under reduced pressure in an evaporator.The resulting residue was purified by silica gel column chromatography,3,6-diphenylcarbazole (3.6 g, yield 74percent) was obtained.
73%
With potassium carbonate In toluene for 10 h; Heating / reflux
Preparation of 3,6-diphenylcarbazole Reaction 5As depicted in Reaction 5, 15g (46 mmol) of 3,6-dibromocarbazole, 14g (92 mmol) of phenylboronic acid, K2CO3 (2 mol), Pd(PPh3)4 and toluene were refluxed for 10 hours. The reaction mixture was extracted with methylene chloride. The solvent was removed, and then the residue was purified by column chromatography (eluent: hexane) to give the title compound. Yield: 73percent. MS (El) (Calcd. for C24H17N: 319.14, Found: 319).
68%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 80℃;
Carbazole (16.72g, 100mmol), NBS (59.4g, 210mmol), BPO (2.42g, 10mmol), methylene chloride (300 ml) at room temperature is injected into the medal and then 2000. When reaction is completed methylene chloride and water, NaHCO 3 extracted using aqueous solution obtained after the organic layer Na 2 SO 4 dried to, for the products and re-crystallized concentrating 23.73g (73percent)obtained. Said product obtained (23.73g, 73.02mmol), phenylboronic acid (19.6g, 160.64mmol), Pd (PPh 3) 4, (8.44g, 7.3mmol), K 2 CO 3 (60.55g, 438.12mmol), THF (320 ml), water (160 ml) in 80 °C agitation return current doesn't have any error frames, turns on the light. When reaction is completed for reducing temperature a high temperature to the normal temperature after methylene chloride and a water extraction of the organic layer obtained after MgSO 4 dried to, for the products and re-crystallized concentrating 15.9g (68percent)obtained
63%
With potassium carbonate In 1,2-dimethoxyethane; water at 80℃; for 3.5 h;
As an example of a material according to the invention, a synthetic method of a compound represented by formula (59) 9-[4-(3, 6-diphenyl-N-carbazolyl)] phenyl- 10-phenylanthracene (hereinafter referred to as DPCzPA) below will be described. [0209][0210] This compound is prepared in accordance with the synthetic method shown below. Note that 9-phenyl-10-(4-bromophenyl) anthracene is prepared in the manner shown in Example 1. [0211]First, a synthetic method of 3,6-dibromocarbazole will be shown below. A mixture of 6.5 g (20.0 mmol) 3,6-dibromocarbazole, 5.0 g (41.0 mmol) of phenylboronic acid, 93 mg (0.40 mmol) of palladium acetate, 6.9 g (5.2 mmol) of potassium carbonate, water (25 mL), 610 mg of tri(ortho-tolyl) phosphine, and dimethoxyethane (50 mL) is heated to reflux at 80 0C for 3.5 three hours. After the reaction, the solution is rinsed with water, aqueous layer is extracted with toluene, and it is rinsed together with the organic layer using saturated salt solution, and thereafter EPO <DP n="67"/>dried with magnesium sulfate. After natural filtration, the filtrate is condensed to obtain 4.1 g of 3,6-di(2-phenyl-phenyl)-carbazoleas a white solid at a yield of 63 percent. A synthetic scheme of 3,6-di(2-phenyl-phenyl)-carbazoleis shown below.
63%
With potassium carbonate In 1,2-dimethoxyethane; water at 80℃; for 3.5 h;
[Step 2] A synthesis method of 3,6-diphenylcarbazole is described.; A synthesis scheme of 3,6-diphenylcarbazole is represented by (a-2). [Show Image] 6.5 g of 3,6-dibromocarbazole (20 mmol), 5.0 g of phenylboron acid (41 mmol), 93mg of palladium(II) acetate (0.40 mmol), 610mg of tri(ortho-tolyl)phosphine (1.9mmol) were put into a 200mL three-necked flask, and then, the inside of the flask was substituted by nitrogen. Into the mixture, 50 ml of ethyleneglycol dimethylether (abbreviation : DME), and 25 mL of a pottassium carbonate water solution (2.0 mol/L) were added. This mixture was refluxed for 3.5 hours at 80 °C. After the reaction, the reaction mixture was washed with water and a water layer was extracted with toluene. The extracted solution and an organic layer were washed with a saturated saline, and dried by magnesium sulphate. The mixture was naturally filtrated. The filtrate was condensed, and 4.1 g of an objective matter, a white powder solid was obtained at the yield 63 percent.
63%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In 1,4-dioxane; water at 100℃; for 17 h; Inert atmosphere
A mixture of 3,6-dibromo-9H- carbazole (3.0 g, 9.2 mmol) (Aldrich), phenylboronic acid (3.0 g, 25 mmol) (Aldrich), tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) (0.3 g, 0.26 mmol) (Frontier Scientific, Logan, UT) and potassium carbonate (6.5 g, 47 mmol) (Aldrich) in dioxane/water (75 mL/15 mL) (Aldrich) was degassed with bubbling argon (Airgas, San Marcos, CA) for 60 min and heated at about 100 °C on a hot plate with a silicone oil bath for about 16 hours. Upon cooling to room temperature, the whole mixture was mixed with ethyl acetate (Aldrich) and rinsed with brine. Then, the organic mixture was dried over Na2S04 (Aldrich), loaded on silica gel (Grade 135) and purified by flash column using eluents of ethyl acetate/hexanes (10percent to 30percent) (Aldrich). After removal of solvents, a white solid (Compound OSC-3) was obtained (2.1 g, in 63percent yield). Confirmed by 1 HNMR (Jeol Instruments, Peabody, MA).
63%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In 1,4-dioxane; water at 100℃; for 16 h;
A mixture of 3,6-dibromo-9H-carbazole (3.0 g, 9.2 mmol), phenylboronic acid (3.0 g, 25 mmol), Pd(PPh3)4 (0.3 g, 0.26 mmol) and potassium carbonate (6.5 g, 47 mmol) in dioxane/water (75 mL/15 ml) was degassed and heated at about 100 °C for about 16 hours. Upon cooling to room temperature, the whole was worked up with ethylacetate/brine, the organic was dried over Na2So4, loaded on silica gel and purified by flash column using eluents of ethyl acetate/hexane (10percent to 30percent). After removal of solvents, a white solid (Compound 11 ) was obtained (2.1 g, in 63percent yield).
48%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In tetrahydrofuran; water at 80℃; for 10 h; Inert atmosphere
30 g (92.3 mmol) of 3,6-dibromo-9H-carbazole was dissolved in 0.3 L of tetrahydrofuran (THF) in a nitrogenenvironment, 28 g (231 mmol) of phenyl boronic acid and 3.2 g (2.8 mmol) of tetrakis(triphenylphosphine)palladium wereadded thereto, and the mixture was agitated. 40.8 g (277 mmol) of potassium carbonate saturated in water was addedthereto, and the mixture was heated and refluxed at 80 °C for 10 hours. When the reaction was terminated, water wasadded to the reaction solution, and the mixture was extracted with dichloromethane (DCM) and treated with anhydrousMgSO4 to remove moisture and then, filtered and concentrated under a reduced pressure. The obtained residue wasseparated and purified through flash column chromatography, obtaining a compound I-4 (14.3 g, 48 percent). HRMS (70 eV, EI+): m/z calcd for C24H17N: 319.1361, found: 319.1. HRMS (70 eV, EI+): m/z calcd for C24H17N: 319.1361, found: 319.1.
33%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In water; N,N-dimethyl-formamide at 80℃; for 4 h; Inert atmosphere
General procedure: Under nitrogen protection system, 3-bromo -9H- carbazole 1.6mmol, tetrakistriphenylphosphine palladium 0.64mmol, anhydrous carbonatePotassium1.92mmol phenylboronic acid 1.92mmol, was dissolved in N, N- dimethylformamide: H2O = (15ml: 2ml) mixed solvent,80 stirring for 4h, after completion of the reaction, cooled to room temperature, the mixed solution was extracted with dichloromethane and water, the organic phase was dried over anhydrous magnesiumDried and evaporated, evaporated and the product after column chromatography to obtain 3-phenyl -9H- carbazole (A-1) 1.44mmol, 90percent yield. The method of the same A-1, by column chromatography to obtain a white solid product 3,6-diphenyl -9H- carbazole (A-2), producingRate of 33percent.
38 g
Stage #1: With sodium carbonate In 1,4-dioxane; water for 1 h; Inert atmosphere Stage #2: With tris-(dibenzylideneacetone)dipalladium(0); tri-tert-butyl phosphine In 1,4-dioxane; waterReflux; Inert atmosphere
3,6-dibromocarbazole (50 g, 0.154 mol), phenylboronic acid (41 g, 0.336 mol), 150ml water, 80g sodium carbonate, and 600ml dioxane were charged into a 2 liter pot with magnetic stirrer, reflux condenser and nitrogen inlet, and sparged with nitrogen for one hour. Pd2DBA3 (6g, 0.0066mol) and tri-t-butylphosphine (3 g, 0.0148mol) was quickly added from the drybox. The reaction was refluxed overnight. The next day water was added to the reaction mixture and methylene chloride extractions were preabsorbed to 141 g of activated silica and purified by column chromatography using methylene chloride/hexanes yielding 38 grams of product 3,6-diphenylcarbazole.
Reference:
[1] Patent: KR2015/6758, 2015, A, . Location in patent: Paragraph 0362-0364
[2] Patent: EP2818468, 2014, A2, . Location in patent: Paragraph 0102; 0103
[3] Patent: WO2010/46259, 2010, A1, . Location in patent: Page/Page column 55
[4] Patent: TWI554499, 2016, B, . Location in patent: Page/Page column 89
[5] Patent: TW2016/38086, 2016, A, . Location in patent: Page/Page column 41; 42
[6] Patent: WO2008/120899, 2008, A1, . Location in patent: Page/Page column 29
[7] Patent: KR2015/115226, 2015, A, . Location in patent: Paragraph 0158; 0159; 0160
[8] Patent: WO2006/104221, 2006, A1, . Location in patent: Page/Page column 63-64
[9] Patent: EP1905768, 2008, A1, . Location in patent: Page/Page column 36
[10] Patent: WO2015/195837, 2015, A1, . Location in patent: Paragraph 0060
[11] Patent: JP2016/526025, 2016, A, . Location in patent: Paragraph 0104; 0123
[12] Patent: EP2889296, 2015, A1, . Location in patent: Paragraph 0168-0171
[13] Journal of the American Chemical Society, 2017, vol. 139, # 12, p. 4559 - 4567
[14] Patent: CN105906547, 2016, A, . Location in patent: Paragraph 0139; 0140; 0141; 0142; 0143; 0144
[15] Patent: US6670054, 2003, B1, . Location in patent: Page column 27
[16] Patent: EP1820801, 2007, A1, . Location in patent: Page/Page column 89
[17] Patent: US2005/84711, 2005, A1,
[18] Patent: WO2013/142633, 2013, A1, . Location in patent: Paragraph 28; 29
With copper(l) iodide; caesium carbonate In N,N-dimethyl-formamide at 220℃; for 0.5 h; Microwave irradiation
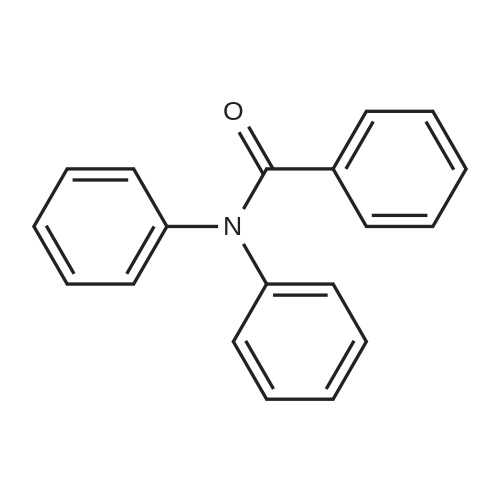
General procedure: 9H-Carbazole (1.0 mmol), Cs2CO3 (1.0 mmol), iodobenzene (1.1 mmol), CuI (0.1 mmol), and DMF (2 mL) were added to a 5-mL vial. The vial was sealed with a crimp cap and placed in a Biotage initiator microwave cavity. After irradiation at 220 °C for the appropriate time and subsequent cooling, the reaction mixture was diluted with saturated aqueous ammonium chloride. Products were isolated by extraction into ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated. Products were purified by silica gel column chromatography using a hexane/ethyl acetate solvent. N-Phenyl-carbazole (2a)22 was obtained (96percent yield) as a white solid.
Reference:
[1] Tetrahedron, 2011, vol. 67, # 26, p. 4820 - 4825
[2] Bulletin of the Korean Chemical Society, 2011, vol. 32, # 7, p. 2461 - 2464
[3] RSC Advances, 2015, vol. 5, # 64, p. 51512 - 51523
26
[ 6825-20-3 ]
[ 460-00-4 ]
[ 73087-83-9 ]
Yield
Reaction Conditions
Operation in experiment
66%
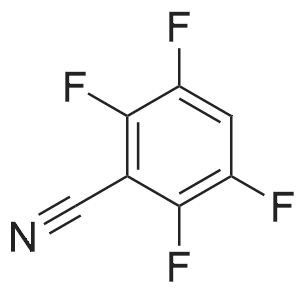
With caesium carbonate In N,N-dimethyl-formamide at 150℃; for 24 h;
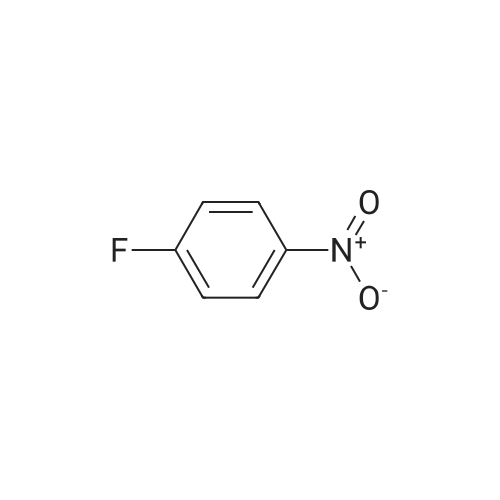
General procedure: A mixture of a fluorinated aryl halide (2.0 mmol), a carbazole (0.5 mmol), and a base (2.0 mmol) in solvent (2 mL) was allowed to react under air atmosphere. The reaction mixture was heated to the specified temperature for 24 h. After reaction completion, the mixture was added to brine (15 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined extract was concentrated under reduced pressure and the product was isolated by short chromatography on a silica gel (200–300 mesh) column.
With copper(l) iodide; 18-crown-6 ether; potassium carbonate In DMPU (1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone) at 170℃; for 20 h;
Synthesis of Intermediate E; 3.25 g (10.0 mmol) of 3, 6-dibromocarbazole, 10.2 g (50.0 mmole) iodobenzene, 190 mg (1.0 mmole) of Cul, 132 mg (0.5 mmole) of 18-C-6, 2.76 g (20.0mmole) of K2CO3 were dissolved in 50 mL of DMPU, and stirred at 170°C for 20 hours. The mixture was cooled to room temperature and 50 mL of diethylether was added thereto. Then the mixture was washed with plenty of water and ammonium hydroxide solution. A collected organic layer was dried over MgSO4 to evaporate the solvent. The residue was purified using silica gel column chromatography to obtain 3.40 g of white solid Intermediate E (Yield: 85 percent). (NMR (CDCl3, 400MHz) δ (ppm) 7.92 (m, 2H), 7.55-7.47 (m, 6H), 7.36-7.16 (m, 3H); 13C NMR (CDCl3, 100MHz) δ (ppm) 142.6, 137.6, 130.2, 129.8, 127.4, 127.0, 122.8, 122.5, 115.3, 111.3).
75%
With tris-(dibenzylideneacetone)dipalladium(0); triphenylphosphine; sodium t-butanolate In toluene at 100℃; for 24 h;
To a round bottom flask Sub 2-1-6 (6.5g, 20mmol), Sub 2-1-2-1 (4.1g, 20mmol),Pd2 (dba) 3 (0.9g, 1mmol), PPh3 (0.5g, 2mmol ), NaOt-Bu (5.8g, 60mmol), were addedto toluene (210mL), respectively, and refluxed under stirring for 24 hours at 100 ° C.The organic layer was dried and the ether was extracted with water over MgSO4 andconcentrated and to the resulting organic silicagel column and recrystallized from a Sub2-1-7-1 6.0g (yield: 75percent) was obtained.
3.27 g
With palladium diacetate; tris-(o-tolyl)phosphine; sodium t-butanolate In toluene
2.5 g (14.9 mmol) ofcarbazole was used (utilized) to perform an NI3S bromination to synthesize 2,5-dibromocar- bazole. Then, the 2,5-dibromocarbazole and iodinated phenyl were used (utilized) to synthesize 3.72 g (9.28 mmol) of Intermediate E through a buchwald reaction.
Reference:
[1] Patent: EP1921082, 2008, A1, . Location in patent: Page/Page column 35-36
[2] Journal of Nanoscience and Nanotechnology, 2011, vol. 11, # 2, p. 1373 - 1376
[3] Patent: KR2015/93995, 2015, A, . Location in patent: Paragraph 0149; 0153; 0182-0184
[4] RSC Advances, 2015, vol. 5, # 64, p. 51512 - 51523
[5] Patent: KR2015/144421, 2015, A, . Location in patent: Paragraph 0317; 0318; 0319
28
[ 591-50-4 ]
[ 6825-20-3 ]
[ 4043-87-2 ]
[ 57103-20-5 ]
Yield
Reaction Conditions
Operation in experiment
90.5%
With copper(l) iodide; caesium carbonate In N,N-dimethyl-formamide at 110℃; for 12 h; Inert atmosphere
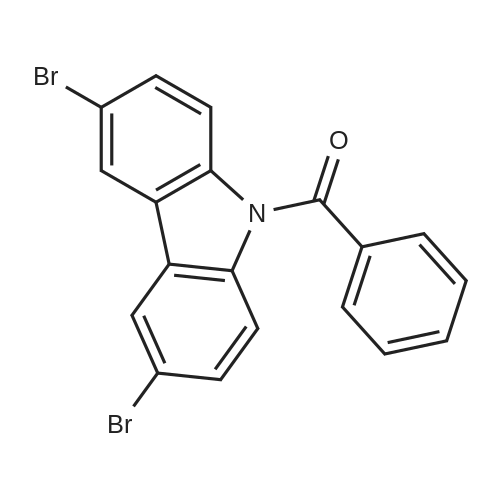
A.3,6-dibromocarbazole (200 mg, 0.62 mmol) was dissolved in DMF solvent,Then iodobenzene (188.3 mg, 0.92 mmol) was added,DL-piperidinecarboxylic acid (11.88 mg, 0.09 mmol),Cuprous oxide (8.27 mg, 0.04 mmol),Cesium carbonate (299.75 mg, 0.92 mmol),Under nitrogen protection,The reaction system was placed at 110 ° C under reflux for 12 h,After the reaction is complete,Ethyl acetate extraction,Organic layer washed three times,Dried over anhydrous sodium sulfate,The solvent is evaporated under reduced pressure;The crude product was purified by column chromatography with ethyl acetate and petroleum ether mixed solvent (20: 1)To obtain compound 3,6-dibromo-9-phenylcarbazole (white solid, 225 mg, yield 90.5percent).
Reference:
[1] Patent: CN106883182, 2017, A, . Location in patent: Page/Page column 11
29
[ 6825-20-3 ]
[ 1235481-90-9 ]
Reference:
[1] Journal of the American Chemical Society, 2011, vol. 133, # 5, p. 1428 - 1437
[2] Tetrahedron Letters, 2013, vol. 54, # 33, p. 4429 - 4431
[3] Patent: US9095571, 2015, B2,
[4] Patent: US9095571, 2015, B2,
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione; In ethanol; at 20 - 25℃; for 2.0h;
The detailed steps of the present invention are as follows:among them,The amount of charge is as follows:Carbazole: 167.2 g,Dibromohydantoin:300. lg,Anhydrous B.Alcohol: 1800 ml.167. 2 g of carbazole (lmol) and 1000 ml of absolute ethanol were added to a 2000 ml reaction flask and stirred for 20 minutes.So as to disperse uniformly.Dibromohydantoin was then added in batches''285 g (1. 05 mol) was added and the reaction temperature was controlled at 20-25 C, about 2 hoursWhen added, after adding a large number of white solid precipitation. And reacted at this temperature for 1 hour. The reaction was monitored by HPLC. When the monobrominated product was less than 1%, the reaction was stopped and the reaction was stopped at 20-25 CThe filter cake was added to a 1000 ml three-necked flask, and 800 ml of absolute ethanol was added. The mixture was heated to reflux (80 C) for 1 hour, cooled slowly to 20-25 C,Dried at 100 C under normal pressure to obtain 312 g of a white powdery solid. Figure 3 shows the 3, 6-dibromo carbazole HPLC spectrum andTable 3 shows that the 3,6-dibromocarbazole content was 99.00%, and the GC spectrum of 3,6-dibromo carbazole as shown in Figure 4 and the results shown in Table 4The yield of 3, 6-dibromocarbazole was 99.66%, and the yield of 3, 6-dibromocarbazole was 96.0%.
90%
With N-Bromosuccinimide; In dichloromethane; N,N-dimethyl-formamide; at 20℃;
A solution of NBS (42.72g, 0.24mol) in 200mL DMF was added slowly to a suspension of carbazole (20g, 0.12mol) in dichloromethane (1.2L). The reaction mixture was stirred at room temperature and the progress of the reaction was controlled by GCMS analysis. Then the solution was washed with water (3×200mL), the organic layer was dried under magnesium sulfate and filtered. The solvent was evaporated and the residue was dissolved in acetone (100mL) and precipitated with hexane (600mL). 3,6-dibromocarbazole was filtered and dried under vacuum to afford 35g (yield 90%) of product.1H NMR (300MHz, CDCl3): delta 7.31 (d, 2H, JHH=8.6Hz, H1), 7.52 (d, 2H, JHH=8.6Hz, H2), 8.13 (d, 2H, JHH=1.9Hz, H4), 10.37 (s, 1H, H9). 13C NMR (75HMz, CDCl3): delta 112.2 (C1), 112.6 (C3), 123.3 (C4), 124.1 (Ci, N-C=C-), 129.3 (C2), 138.3 (Ci, N-C=C-). MS (EI): m/z (rel. intensity - %): 327+ (49), 325 (100), 326 (20), 246 (16), 165 (28), 138 (11), 83 (11).
90%
With N-Bromosuccinimide; In dichloromethane; for 4.0h;
9.7 g (58 mmol) of the carbazole was dissolved in 600 ml of dichloromethane, and 21.7 g (121.8 mmol) of N-bromosuccinimide was added dropwise over 10 minutes. After 4 hours, the solvent was removed in vacuo, The residue was subjected to ethanol and water, Filtered, washed with ethanol and dried to give 17.0 g (90%) of a colorless powder.
88%
With N-Bromosuccinimide; silica gel; In dichloromethane; at 20℃; for 24.0h;
A mixture of carbazole (1.00 g, 6.00 mmol) and silica gel(8.00 g, 133.33 mmol) was added to CH2Cl2(60 mL) at roomtemperature, N-bromosuccinimide (1.07 g, 12.00 mmol),added to the above system, the mixture was reacted for 24 hat room temperature, then filtering and evaporating off thesolvent, and the residue was extracted with n-hexane/ethylacetate (10:1, v/v) to generate a white solid. 1.71 g, yield:88%.Mp: 204-206 C. 1H NMR (400 MHz, DMSO-d6, delta,ppm): 11.60 (s, 1H, -NH), 8.44 (d, J = 8.8 Hz, 2H), 7.54(d, J = 8.9 Hz, 2H), 7.48 (d, J = 8.7 Hz, 2H). 13C NMR(101 MHz, DMSO-d6, delta, ppm): 139.3, 129.2, 123.9, 123.8,113.8, 111.5. Anal. calcd for C12H7Br2N:C, 44.35; H, 2.17;Br, 49.17; N, 4.31. Found: C, 44.28; H, 2.15; Br, 49.14; N,4.29.
85%
With N-Bromosuccinimide; In N,N-dimethyl-formamide; at 0 - 20℃; for 6.0h;Flash photolysis;
To a 500 mL three-necked flask was added carbazole (24.7 g, 0.1 mol)Dimethylformamide 200 mL,Stir to dissolve,NBS (49.84 g, 0.28 mol) was dissolved in 120 mL of DMF,Ice bath to 0 ,Slowly drop the NBS solution,Reaction to light,After the dropwise addition, the temperature was allowed to rise to room temperature and the reaction was continued for 6 hours.The reaction solution was added dropwise to the water to precipitate, and the crude product was collected by suction filtration. The filtrate was recrystallized from absolute ethanol and dried to give white needles. Yield: 85%
83%
With N-Bromosuccinimide; In tetrahydrofuran; at 20℃; for 4.0h;
4-3: Synthesis of 3,6-dibromo-9H-carbazole: Carbazole (5.0 g, 29.9 mmol) was dissolved in THF (50 mL), and then N-bromosuccinimide (11.18 g, 62.80mmol) was added thereto to prepare a mixed solution. Then, the mixed solution was reacted at room temperature for 4hours. After the reaction of the mixed solution, a solvent was removed, and the reacted mixed solution was extractedwith water and ethyl acetate to obtain an extract. The extract was dried by magnesium sulfate (MgSO4), and then areaction product was separated by column chromatography (n-hexane/EtOAc = 4/1). The yield ofthe reaction productwas 83% (8.1 g).1HNMR(300 MHz,CDCl3) 8.13-(s, -Ar, 2H), 7.53(d, -Ar, 2H), 7.32-7.3(d, -Ar, 2H)
83%
With N-Bromosuccinimide; In tetrahydrofuran; at 20℃; for 4.0h;
The carbazole (5.0 g, 29.9 mmol) was dissolved in THF (50 ml) and N-bromosuccinimide (11.18 g, 62.80 mmol) was added. After reacting at room temperature for 4 hours, the solvent was distilled off and extracted with water and ethyl acetate. The extract was dried over MgSO4 and then purified by column chromatography (n-hexane / EtOAc = 4/1) to give the title compound. The yield was 83% (8.1 g).
83%
With N-Bromosuccinimide; In dichloromethane; for 12.0h;Cooling with ice; Darkness;
Completely dissolve carbazole (1.67 g, 10 mmol) with 300 ml of dichloromethane solution, add 30 g of 100-200 mesh silica gel, and add N-bromosuccinimide in three batches under ice bath (0 C). (NBS, 3.92g, 22mmol), reacted in the dark for 12 hours; then the reaction mixture was filtered with suction, and the filter cake was washed with dichloromethane for 5 times. The organic phase was collected, dried and concentrated. The ether (5/100, v/v) was recrystallized three times to give a white solid. The yield was 83%.The results of 1H NMR, 13C NMR, MS and elemental analysis indicated that the obtained compound was the target product, and the chemical reaction equation was as follows.
80%
With N-Bromosuccinimide; In tetrahydrofuran; for 4.0h;Inert atmosphere; Cooling with ice;
In the 100mL single-mouth bottle,1.00 g (5.99 mmol) of carbazole was added, dissolved in tetrahydrofuran, and the jar was placed in an ice bath and protected from light.Then 2.67g (15mmol) of NBS was dissolved in tetrahydrofuran and slowly poured into a constant pressure dropping funnel. Into a 100ml single-mouth bottle,An ice bath was stirred under nitrogen for 4 h.The reaction was poured into 100 mL of water.Extract three times with ethyl acetate, The resulting liquid spins dry,With ethyl acetate:Column chromatography of petroleum ether = 1:200 gave 1.56 g (4.8 mmol) of a white solid, Compound 2, yield 80%.
76%
With N-Bromosuccinimide; silica gel; In dichloromethane;
Synthesis of Compound 2:; Compound 2 was obtained by dibromination of compound 1.This dibromination was carried out by making this compound (2.01 g, 12.0 mmol) react with N-succinimide (4.20 g, 23.8 mmol), in the presence of silica preactivated at 120 C. (40.50 g, 0.063-0.2 nm) in order to protonate the nitrogen atom of compound 1, and in dichloromethane (350 ml), as described by Smith et al. in Tetrahedron, 1992, 48, 36, 7479-7488, [4].The yield of the reaction was 76%.
73%
With N-Bromosuccinimide; dibenzoyl peroxide; In dichloromethane; at 20℃;
Carbazole (16.72g, 100mmol), NBS (59.4g, 210mmol), BPO (2.42g, 10mmol), followed by stirring at room temperature, insert the methylenechloride (300mL). When the reaction is complete, methylene chloride and water, and extracted using an aqueous solution of NaHCO3 was obtained organic layer was dried with Na2SO4, and the product was recrystallized 23.73g (73%) and was concentrated.
73%
With N-Bromosuccinimide; dibenzoyl peroxide; In dichloromethane; at 20℃;
Carbazole (16.72 g, 100 mmol), NBS (59.4 g, 210 mmol), BPO (2.42 g, 10 mmol) and methylene chloride 300 mL were added and stirred at room temperature. After the reaction was completed, the obtained organic layer was extracted with methylene chloride, water, and aqueous NaHCO3, dried, concentrated with Na2SO4, and recrystallized to obtain 23.73 g (73%) of the product. The product obtained (23.73 g, 73.02 mmol), phenylboronic acid (19.6 g, 160.64 mmol), Pd (PPh3) 4 (8.44 g, 7.3 mmol), K2CO3 (60.55 g, 438.12 mmol), THF (320 mL), H2O (160 mL) was added and stirred and refluxed at 80 C. After the reaction was completed, the temperature was lowered to room temperature, extracted with methylene chloride and water, and the obtained organic layer was dried over MgSO4, concentrated, and recrystallized to obtain 15.9 g (68%) of the product.
68%
With N-Bromosuccinimide; In N,N-dimethyl-formamide; for 0.5h;Cooling with ice;
3,6-Dibromo-9H-carbazole (1), a solution of N-bromosuccinimide (0.21 mol) in DMF (80 mL) was slowly added to a solution of carbazole (0.1 mol) in DMF (20 mL) in an ice bath. After reaction for 30 min, the mixture was poured into ice water (1 L), and the crude product was collected by filtration to give a blue powder. Recrystallization from EtOH/H2O afforded blue crystals with a yield of 68%. 1H NMR (500 MHz, CDCl3): delta 8.14 (d, J = 1.9 Hz, 2H), 8.12 (s, 1H), 7.53 (d, J = 8.6, 1.9 Hz, 2H), 7.32 (d, J = 8.6 Hz, 2H).
64%
With N-Bromosuccinimide; silica gel; In dichloromethane; at 20℃; for 12.0h;Darkness;
9H- carbazole (1) (2.00g, 12.0mmol) in dichloromethane (100ml)To, N- bromosuccinimide (4.30g, 24.0mmol) and silica gel(SiO2) was added (20.0g).The dark, was stirred at room temperature for 12 hours, filtered, washed twice with water, dried over anhydrous sodium sulfate, filtered, using a rotary evaporator, the solvent was distilled off under reduced pressure at room temperature from the filtrate, the crude product I got things. The resulting crude product was recrystallized from dichloromethane, 3,6-dibromo-9H-carbazole and (2b) 2.50 g (yield: 64%) was obtained.
61.5%
With N-Bromosuccinimide; In dichloromethane; N,N-dimethyl-formamide; for 3.0h;Inert atmosphere;
Step 1: In a 1000 mL three-necked flask equipped with a mechanical stirrer, 4 g (0.024 mol)Carbazole and 240 mL of dichloromethane, under an argon atmosphere at 5 mL / minAt a rate of dropwise addition of N-bromosuccinimideN, N '-dimethylformamide solution,Wherein N-bromosuccinimide is8.54 g (0.048 mol), N, N'-dimethylformamide solution was 40 mL; stirred for 3 hours,The solution was washed four times with 280 mL of distilled water,The organic phase was dried over anhydrous magnesium sulfate; the solvent in the organic phase was distilled off under reduced pressure at room temperature,To give a pale gray crude product; mixed with acetone and n-hexane(In which acetone is 30 mL and n-hexane is 400 mL) to recrystallize the light gray crude product;The final product was vacuum dried at 70 C for 10 hours to give a gray bulk crystal, I.e., 3,6-dibromocarbazole; the resulting product was 4.9 g in a yield of 61.5%
55%
With N-Bromosuccinimide; In N,N-dimethyl-formamide; at 0 - 20℃; for 2.0h;Inert atmosphere;
Synthesis of 3,6-dibromocarbazole. Carbazole (1 .0 g, 6 mmol) was added to anhydrous DMF (15 mL) at room temperature while stirring. The resulting mixture was stirred at room temperature for another 15 minutes. N- bromosuccinimide (2.3 g, 13 mmol) in anhydrous DMF (10 mL) was added drop wise to carbazole-DMF mixture at 0C in inert atmosphere. The resulting solution was allowed to reach to the room temperature and continued stirring for another 2 hours. This solution was poured to Dl water. The milky white precipitate formed was filtered and dissolved in DCM . DCM layer was washed with water several times. Then DCM layer was dried with anhydrous sodium sulfate and solvent was removed under high vacuum. Resultant white solid was recrystallized using ethanol to get light brown silvery flake-like 3,6-dibromocarbazole crystals (1 .1 g, 55% yield) . MS, ESI - m/z 323 (M+); 1 H NMR (Deuterated DMSO, 5 00 MHz, ppm) : delta 1 1 .49 (S, 1 H), 8.08 -8.10 (D, 2H), 7.72 (S, 2H), 7.32-7.34 (D, 2H).
47%
With N-Bromosuccinimide; In N,N-dimethyl-formamide; at 20℃;Cooling with ice;
N-N-dimethylformamide (DMF) (20 ml) dissolved with carbazole (10 g, 59.8 mmol) was stirred under ice-The solution was added dropwise to a solution of N-bromosuccinimide (21.28 g, 120 mmol) in DMF (30 mL) with a constant pressure dropping funnelin. After completion of the drop, the ice bath was removed and the reaction was conducted overnight at room temperature. The reaction solution was then washed with deionized water and analyzedThe solid was filtered off and the product was recrystallized from ethanol. Yield: 47%.
With N-Bromosuccinimide; In acetonitrile; at 20℃;Inert atmosphere; Cooling with ice;
A 2 L dry round bottom flask was charged with carbazole 1 (90.7 g, 0.543 mol) and 1 L dry acetonitrile and stirred for one hour at room temperature under nitrogen, where most of the carbazole dissolved. N- bromosuccinimide (193.1 g, 1.084 mol) was added to the suspension neat in portions, where the reaction was exothermic. The mixture was immediately cooled in an ice-water bath following the addition. The mixture was left to be stirred overnight at room temperature. The resulting white precipitate was filtered, washed with acetonitrile, and dried to yield 112.O g (63%) 3,6-dibromocarbazole 2.
With N-Bromosuccinimide; silica gel;
2.5 g (14.9 mmol) ofcarbazole was used (utilized) to perform an NBS bromination to synthesize 2,5-dibromocar- bazole. Then, the 2,5-dibromocarbazole and iodinated phenyl were used to synthesize 3.72 g (9.28 mmol) of Intermediate E through a buchwald reaction.
With bromine; In carbon disulfide;
A. 3,6-dibromocarbazole Carbazole (50 g) is slurried in 1200 ml of carbon disulfide and heated on a steam bath. A solution of bromine (34 ml) in 130 ml of carbon disulfide is added dropwise to the refluxing mixture over thirty minutes. After refluxing an additional five minutes, the reaction mixture is chilled in an ice bath. The resultant precipitate is collected by filtration and recrystallized from ethanol to yield 33.3 g of the title compound, melting point 210-213 C.
With tetrabutylammomium bromide; sodium hydroxide In toluene at 120℃;
95%
With tetrabutylammomium bromide; potassium hydroxide In acetone for 6h; Reflux;
95%
With tetrabutylammomium bromide; potassium hydroxide In water; toluene for 24h; Heating;
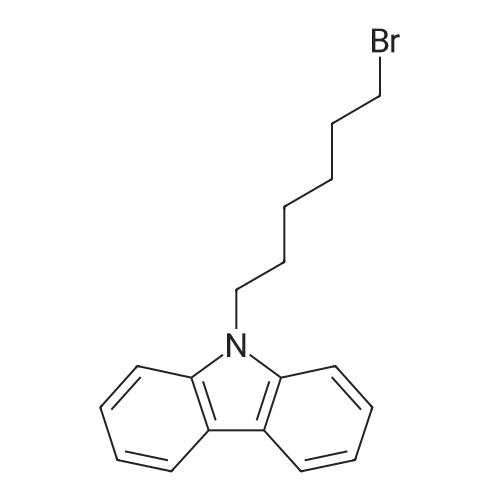
5 Example 5 3,6-dibromo-N-octylcarbazole (M-3)
3,6-dibromocarbazole (16.25 g, 0.05 mol) was added to a 250 mL three-necked flask,Toluene (100 mL),Tetrabutylammonium bromide (0.8 g, 3.5 mmol)Stirring dissolved,A solution of 50 wt% KOH (11 mL) was slowly added dropwise,Followed by addition of n-C8H17Br (19.3 g, 0.1 mol)After heating for 24 h,The reaction was quenched with water and the organic phase was washed with water. The aqueous phase was extracted with CH2Cl2 and washed twice. The organic phases were combined and dried over anhydrous MgSO4. The solvent was removed by distillation under reduced pressure to give a pale yellow solid,Recrystallization from petroleum ether gave a white powdery solid. Yield: 95%.
93%
With sodium hydroxide In dimethyl sulfoxide at 20℃; for 4h;
2.2.2. Synthesis of 3,6-dibromo-9-octyl-9H-carbazole (III) [61]
3,6-dibromocarbazole was dissolved in DMSO (50 mL). Then this mixture was added 1-bromooctane (1.5 eq.) and 50% NaOH solution. The mixture was stirred at room temperature for 4 h. Ether (100 mL) and deionized water (100 mL) were washed and dried over MgSO 4 . Crude product filtration white solid was recrys- tallized with ethanol to obtain pure product III. Isolated yield: (11.25 g 93%) mp. 86-87 °C FT-IR (KBr): 3073, 2957, 2922, 2851, 1623, 1592, 1469, 1434, 1347, 1283, 1221, 1144, 1051, 1012, 865, 798, 643, 562 cm -1 ( Fig. 1 ). 1 H NMR (500 MHz, CDCl 3 , δppm): 8.06 (qs, 2H, Cz-H4, H5), 7.47 (dd, J = 8.7, J = 2.1 Hz, 2H, Cz- H2, H7), 7.19 (d, J = 9.0 Hz, 2H, Cz-H1, H8), 4.16 (t, J = 7.4 Hz, 2H), 1.81-1.78 (m, 2 H ), 1.24-1.15 (m, 10 H ), 0,78 (t, J = 7.2 Hz, 3 H ). 13 C NMR (125 MHz, CDCl 3 , δppm): 139.36, 129.07, 123.48, 123.33, 111.97, 110.49, 43.43, 31.84, 29.41, 29.23, 28.95, 27.31, 22.69, 14.18.
91%
Stage #1: 3,6-dibromo-9H-carbazole With tetrabutylammomium bromide In dimethyl sulfoxide at 20℃;
Stage #2: With sodium hydroxide In dimethyl sulfoxide at 20℃; for 0.166667h;
Stage #3: 1-bromo-octane In dimethyl sulfoxide
3 Synthesis of intermediate 2
General procedure: Add 2,7-dibromoindole (6.48 g, 20 mmol), DMSO (200 mL), and appropriate amount of TBAB to a 500 mL three-necked flask.After stirring for a few minutes at room temperature,Sodium hydroxide solution (50 mL, 50% by weight) was slowly added to the reaction system.Continue magnetic stirring at room temperature, after 10 minutes,Inject brominated n-octane (8 mL, 46 mmol) with a syringe.Stir overnight. Stop the reaction,The reaction solution was adjusted to neutral with HCl solution.Then extracted with CH2Cl2 (200 mL×6),Saturated saline (200 mL × 6),Dry over anhydrous magnesium sulfate,Rotate the solvent,The residue was subjected to silica gel column chromatography [eluent, petroleum ether]Purified white solid2,7-dibromo-9,9-dioctylfluorene 9.82 g,The yield was 90%.
87%
With potassium carbonate In acetonitrile for 20h; Reflux; Inert atmosphere;
4.2. Synthesis and characterizations of L1
To a solution of 3,6- Dibromo carbazole (3.25 g, 10 mmol) and 1-bromooctane (2.12 g, 11 mmol) in acetonitrile (100 mL), K2CO3 powder (2.76 g, 20 mmol) was added. The resultant suspension was refluxed for 20 h under nitrogen. After cooling to 25 C, the acetonitrile was concentrated in vacuo to give a grey residue, which was purified by column chromatography (neutral Al2O3) eluting with a1:1 mixture of dichloromethane and petroleum ether to give 1, as a white solid: (3.80 g, 8.7 mmol, 87%). 1H NMR (400 MHz, CDCl3)d (ppm): 8.13 (s, 2H), 7.57 (d, J 4.0 Hz, 2H), 7.28 (d, J 4.0 Hz, 2H),4.23 (t, J 6.0 Hz, 2H), 1.84e1.80 (m, 2H), 1.33e1.23 (m, 10H), 0.89(t, J 3.7 Hz, 3H). 13C NMR (101 MHz, CDCl3) d (ppm): 139.23,128.99, 123.47, 123.24, 111.98, 110.39, 43.34, 31.78, 29.33, 29.16,28.81, 27.24, 22.67, 14.11. MS: Found For: 436.0786 [M-H] (Calcd.For: 437.2201) (Fig. S26). To a solution of 1 (0.44 g, 1.0 mmol) and 4-([2,2': 60, 200- terpyridyl]-4'-) - benzene boric acid (0.92 g,2.60 mmol) in THF (100 mL), aqueous NaOH (200 mg, 5.0 mmol) was added. The mixture was degassed for 10 min, then Pd(PPh3)4 (130 mg, 0.11 mmol) was added. After refluxing for 48 h under N2, the solvent was removed in vacuo to give a residue that was dissolved in dichloromethane and washed with water. The organic layer was dried over anhydrous MgSO4, concentrated in vacuo togive a residue that was purified by flash column chromatography (neutral Al2O3) eluting with dichloromethane and petroleum ether (1:1, v:v) to give L1, as a white solid: (0.67 g, 75%).
80%
Stage #1: 3,6-dibromo-9H-carbazole With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide In water; dimethyl sulfoxide for 0.5h;
Stage #2: 1-bromo-octane In water; dimethyl sulfoxide for 8h;
3,6-Dibromo-9-octyl-9H-carbazole (2)
According to the literature procedure,[40] to a solution of 3,6-dibromo-9H-carbazole (3.25 g, 10 mmol) and benzyltriethylammoniumchloride (0.2 g, 0.84 mmol) in DMSO (80 mL) was added aqueous sodium hydroxide (20 mL, 2 M), and the mixture was stirred for 30 min. Then, 1-bromooctane (2.1 g, 11 mmol) was added dropwise. The resulting mixture was stirred for 8h. After adjusting the solution pH value to 7 by addition of hydrochloric acid solution, the mixture was extracted with ethyl acetate, dried over anhydrous MgSO4, and concentrated under vacuum. The residue was purified by silica gel column chromatography using petroleum ether to give the desired 2 as a white solid (3.5 g, 80 %). δH (CDCl3, 600 MHz) 8.08 (2H, s), 7.52 (2H, d, J 8.7), 7.21 (2H, d, J 8.7), 4.15 (2H, t, J 7.3), 1.82-1.72 (2H, m), 1.37-1.11 (10H, m), 0.88 (3H, t, J 7.1). δC (CDCl3, 151 MHz) 139.25, 128.97, 123.39, 123.20, 111.91, 110.36, 43.30, 31.80, 29.34, 29.19, 28.87, 27.25, 22.64, 14.13.
41%
Stage #1: 3,6-dibromo-9H-carbazole With potassium carbonate In N,N-dimethyl-formamide at 20℃;
Stage #2: 1-bromo-octane In N,N-dimethyl-formamide at 20℃; for 96h;
Representative synthesis of 3,6-dibromo-N-hexyl carbazole(2b)
General procedure: 3,6-Dibromocarbazole (2.0 g, 6.2 mmol), and potassium carbonate (0.94 g, 6.8 mmol) were dissolved in 20 mL DMF under ambient conditions forming an orange solution. 1-Bromohexane (0.91 mL, 6.5 mmol) was added to the solution resultingin an immediate colour change. The resulting pink solution was stirred at room temperature for 4 days, yielding a white solid on the inner surface of the flask. The solid and solution were poured into 400 mL of distilled water and apink crude solid was isolated by vacuum filtration. The product was isolatedfrom unreacted carbazole by recrystallization from hot ethanol as 0.92 g whitesolid (43 % yield
With sodium hydride In tetrahydrofuran; N,N-dimethyl-formamide at 20℃; for 24h;
Stage #1: 3,6-dibromo-9H-carbazole With potassium <i>tert</i>-butylate In tetrahydrofuran at 20℃; Inert atmosphere; Schlenk technique;
Stage #2: 3-bromomethylheptane In tetrahydrofuran Reflux; Inert atmosphere; Schlenk technique;
80%
With tetrabutylammomium bromide; potassium hydroxide In water; toluene at 110℃; for 4h;
77%
Stage #1: 3,6-dibromo-9H-carbazole With tetrabutylammomium bromide; potassium hydroxide In toluene for 1h;
Stage #2: 3-bromomethylheptane In toluene at 20℃; for 24h; Reflux;
75%
With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 24h; Inert atmosphere;
Synthesis of 3,6-dibromo-9-(2-ethylhexyl)carbazole
Analogously to the description in [S1], a solution of 3,6-dibromocarbazole (1.00 g, 3.10 mmol) and NaH (0.20 g, 4.5 mmol) in DMF (5 mL) was degassed with nitrogen for 15 min. 2-Ethylhexyl bromide (0.90 g, 4.5 mmol) was added by a syringe. The mixture was stirred at room temperature for 24 h. Water was added, and the organic layer was extracted with CH2Cl2 (30 mL × 3). After the combined organic layers were dried over MgSO4, the solvent was removed under reduced pressure. Column chromatography (SiO2, hexane) gave the desired product as colorless liquid (1.00 g, 75%). 1H NMR (300 MHz, CDCl3): δ 8.08 (d, J = 1.7 Hz, 2H), 7.53 (dd, J = 1.8, 8.7 Hz, 2H), 7.20 (d, J = 8.7 Hz, 2H), 4.02 (d, J = 7.5 Hz, 2H), 2.15-1.83 (m, 1H), 1.41-1.08 (m, 8H), 1.00-0.69 ppm (m, 6H). 13C NMR (75 MHz, CDCl3): δ 140.49, 129.72, 124.12, 123.93, 112.64, 111.38, 48.23, 39.93, 31.54, 29.35, 24.93, 23.61, 14.61, 11.46 ppm. IR (neat): ν = 2958, 2928, 2869, 1847, 1719, 1622, 1588, 1548, 1470, 1437, 1380, 1363, 1343, 1285, 1147, 1057, 1018, 965, 911, 867, 834, 795, 742, 713, 665, 644, 614 cm-1.
56%
With potassium carbonate In N,N-dimethyl-formamide at 80℃; for 40h;
56%
Stage #1: 3,6-dibromo-9H-carbazole With potassium carbonate In N,N-dimethyl-formamide at 80℃; Inert atmosphere;
Stage #2: 3-bromomethylheptane In N,N-dimethyl-formamide at 80℃; for 16h; Inert atmosphere;
Synthesis of N-(2-Ethylhexyl)-3,6-dibromocarbazole
A mixture of 3,6-dibromocarbazole (18.46 mmol) and K2CO3 (37 mmol) in DMF(20 ml) was refluxed in N2 atmosphere and heated to 80 C. 2-Ethylhexylbromide(28 mmol) was added dropwise. After complete addition, the reaction was continued for16 hrs. The product was isolated by diethylether/water extraction. The organic layer wasdried with MgSO4, then filtered and concentrated. The viscous liquid N-(2-ethylhexyl)-3,6-dibromocarbazole was obtained by column chromatography (eluent: hexane). Yield56%. Figure 1 shows 1H and 13C NMR spectra of N-2-ethylhexyl-3,6-dibromocarbazole.1H NMR (300MHz in CDCl3, d): 8.11 (d, 2H, Ar-H), 7.48 (d, 2H, Ar-H), 7.22 (d, 2H,Ar-H), 4.18 (m, 2H, -N-CH2-), 1.231.44 (m, 9H, -CH-, -CH2-), 0.91 (m, 6H, -CH3).13C NMR (60MHz in CDCl3, d): 139.7, 129.0, 123.4, 123.2, 112.1, 110.7, 47.6, 39.5,31.1, 29.0, 24.6, 23.3, 14.3, 11.1.
33%
With potassium hydroxide In N,N-dimethyl-formamide at 20℃; for 96h;
Representative synthesis of 3,6-dibromo-N-hexyl carbazole(2b)
General procedure: 3,6-Dibromocarbazole (2.0 g, 6.2 mmol), and potassium carbonate (0.94 g, 6.8 mmol) were dissolved in 20 mL DMF under ambient conditions forming an orange solution. 1-Bromohexane (0.91 mL, 6.5 mmol) was added to the solution resultingin an immediate colour change. The resulting pink solution was stirred at room temperature for 4 days, yielding a white solid on the inner surface of the flask. The solid and solution were poured into 400 mL of distilled water and apink crude solid was isolated by vacuum filtration. The product was isolatedfrom unreacted carbazole by recrystallization from hot ethanol as 0.92 g whitesolid (43 % yield).
With potassium hydroxide In tetrahydrofuran for 2h; Heating;
3.02 g (77.2%)
With potassium carbonate In N,N-dimethyl-formamide
12.1 3,6-dibromo-9-(2-ethylhexyl)-carbazole (M10)
Step 1 3,6-dibromo-9-(2-ethylhexyl)-carbazole (M10) To a solution of 3,6-dibromocarbazole (3.0 g, 8.95 mmol) in 25 mL of dried DMF was added potassium carbonate (2.47 g, 17.9 mmol). The mixture was stirred and degassed for 1 h, after which 1-bromo-2-ethylhexane (2.70 g, 13.4 mmol) was added dropwise, followed by refluxing for 2 days. The reaction mixture was poured into 100 mL of distilled water and extracted with chloroform. The combined organic layer was dried with anhydrous magnesium sulfate. After removal of the solvent and unreacted 2-ethylhexyl bromide under reduced pressure, the residue was purified by column chromatography on silica gel using hexane and ethyl acetate as the eluent to afford 3,6-dibromo-9-(2-ethylhexyl)-carbazole as a waxy solid. Yield: 3.02 g (77.2%), M.P.: 62.4-63.6° C.
With potassium hydroxide In acetone for 12h; Reflux;
With sodium hydride In N,N-dimethyl-formamide at 0 - 20℃; for 12h;
With tetrabutylammomium bromide; potassium hydroxide In water at 20℃; for 0.5h;
92.35%
Stage #1: 3,6-dibromo-9H-carbazole With potassium hydroxide In dimethyl sulfoxide at 20℃; for 1h;
Stage #2: 1-bromo-hexane In dimethyl sulfoxide at 20℃; for 10h;
Synthesis of Intermediate 2:
Add the raw material 3,6-dibromocarbazole (10.0 g, 30.97 mmol) to the 200 mL single-necked bottle in turn,Potassium hydroxide (6.934 g, 123.88 mmol)And 60 mL of dimethyl sulfoxide (DMSO) solvent. After stirring at room temperature for 1 hour, 1-bromohexane (6.607 g, 40.26 mmol) was added,Then the reaction was stirred at room temperature.Through TLC spot monitoring, the reaction was stopped after 10 hours of reaction. The reaction solution was extracted with dichloromethane, washed with water ten times, dried with anhydrous ethanol, and evaporated under reduced pressure to spin out the organic solvent. The crude product was purified by column chromatography, using petroleum ether as the eluent to isolate 11.64 g of white fluffy solid intermediate 2,The yield was 92.35%.
92.35%
Stage #1: 3,6-dibromo-9H-carbazole With potassium hydroxide In dimethyl sulfoxide at 20℃; for 1h;
Stage #2: 1-bromo-hexane In dimethyl sulfoxide at 20℃;
Synthesis of Intermediate 2
The raw materials 3,6-dibromocarbazole (10.0 g, 30.97 mmol), potassium hydroxide (6.934 g, 123.88 mmol) and 60 mL of dimethyl sulfoxide (DMSO) solvent were sequentially added to a 200 mL single-mouth flask. After stirring for 1 hour at room temperature, 1-bromohexane (6.607 g, 40.26 mmol) was added, and then the reaction was stirred at room temperature. Monitoring by TLC dot plate, the reaction was stopped after 10 hours of reaction. The reaction solution was extracted with dichloromethane, washed with water ten times, dried with absolute ethanol, and evaporated under reduced pressure to spin out the organic solvent. The crude product was purified by column chromatography and separated with petroleum ether as the eluent to obtain 11.64 g of white fluffy solid intermediate 2 with a yield of 92.35%.
89%
With tetrabutylammomium bromide; potassium hydroxide In water; toluene at 110℃; for 4h;
87%
Stage #1: 3,6-dibromo-9H-carbazole With sodium hydride In tetrahydrofuran for 0.5h; Inert atmosphere;
Stage #2: 1-bromo-hexane In tetrahydrofuran at 110℃; for 12h; Inert atmosphere;
85%
With potassium hydroxide In tetrahydrofuran for 2h; Heating;
83%
Stage #1: 3,6-dibromo-9H-carbazole With potassium hydroxide In N,N-dimethyl-formamide for 0.5h; Inert atmosphere;
Stage #2: 1-bromo-hexane In N,N-dimethyl-formamide at 20℃; for 12h; Inert atmosphere;
2.1.1 Synthesis of 3,6-dibromo-9 hexyl-9H carbazole(3)
To a solution of 3,6-dibromo-9H-carbazole (0.200g, 0.615mmol) in dry N,N-dimethylformamide was added KOH (0.105g, 1.845mmol) and allowed to stir for 30min. To this, 1-bromo hexane (0.962mL, 0.799mmol) dissolved in dry N,N-dimethylformamide was added drop wise and allowed to room temperature with stirring for 12h. After complete consumption of the dibromocarbazole (TLC), crushed ice was added and stirred well. The precipitate formed filtered and dried. Then, the obtained crude solid was subjected to silica gel chromatography using a mixture of ethyl acetate and n-hexane as an eluent to obtain pure product 3 (83%). 1H NMR (CDCl3, 300MHz) δ 8.13 (s, 2H), 7.55-7.52 (m, 2H), 7.25-7.27 (m, 2H), 4.24-4.22 (t, 2H, J=6.929Hz), 1.79-1.84 (m, 2H), 1.24-1.35 (m, 6H), 0.873-0.87(m, 3H). 13C NMR (CDCl3, 300MHz) δ 139.2, 128.9, 123.3, 123.1, 111.8, 110.3, 43.2, 31.4, 28.7, 26.8, 22.4, 13.9. MS (ESI) m/z: 411 (M+2)+.
80%
With sodium hydride In tetrahydrofuran for 12h; Inert atmosphere; Reflux;
78%
With tetrabutylammomium bromide; potassium hydroxide In acetone for 3h; Reflux;
2.2 2.2.2 3,6-Dibromo-N-hexylcarbazole (4)
Hexyl bromide (1.45mL, 15.7mmol), tetrabutylammonium bromide (0.16g, 0.5mmol) and potassium hydroxide (0.83g, 14.8mmol) were added to a solution of 3,6-dibromocarbazole (6.18g, 9.2mol) in acetone (previously dried under 3 sieves). The reaction mixture was stirred for 3hand refluxed. Progress of the reaction was controlled by GCMS analysis. Then the mixture was cooled to room temperature and the solvent was evaporated. The residue was dissolved in diethyl ether (100mL), filtered and washed with water (3×10mL). The organic layer was dried under magnesium sulfate and filtered. Then the solvent was evaporated and the residue was stirred with pentane (100mL) for 1h, filtered and dried under vacuum. 6.1g (yield 78%) of 3,6-dibromo-N-hexylcarbazole was obtained as white solid. The product was crystallized by dissolving in small amount of hot hexane and left to cool at room temperature.1H NMR (300MHz, CDCl3): δ 0.86 (t, 3H, JHH=7.0Hz, H6′), 1.21-1.38 (m, 6H, H3′, H4′, H5′), 1.81 (quint, 2H, JHH=7.3Hz, H2′) 4.24 (t, 2H, JHH=7.2Hz, H1′), 7.27 (d, 2H, JHH=8.7Hz, H2), 7.55 (d, 2H, JHH=8.7Hz, H1), 8.14 (d, 2H, JHH=1.9Hz, H4). 13C NMR (75MHz, CDCl3): δ 13.9 (C6′), 22.5 (C5’), 26.9 (C3′), 28.8 (C2′), 31.5 (C4′), 43.3 (C1′), 110.4 (C1), 111.9 (C3), 123.2 (C4), 123.4 (Ci, N-C=C-), 129.0 (C2), 139.3 (Ci, N-C=C-). MS (EI): m/z (rel. intensity - %): 411+ (31), 409 (58), 408 (9), 341 (10), 340 (53), 338 (100), 337 (34), 258 (20), 152 (10). Crystal Data: C18H19Br2N; formula weight Mr=409.16; orthorhombic; Pca21; a=20.3019(16) , b=4.5582(4) , c=18.4012(18) , V=1702.8(3) 3, Z=4, dx=1.60gcm-3, λ=0.71073 (MoKα), μ=4.75mm-1; reflections: collected 27,069, unique 3018 (Rint=10.58%), with I>2σ(I) 1827; R=6.43%, wR2=13.82%, S=1.03, max/min Δρ: 0.79/-0.33e-3. Perspective view of the molecule 4 is presented below. Thermal ellipsoids are drawn at the 50% probability level, hydrogen atoms are represented by spheres of arbitrary radii.
70.3%
With potassium hydroxide In tetrahydrofuran for 2h; Reflux;
70%
Stage #1: 3,6-dibromo-9H-carbazole With sodium hydroxide In tetrahydrofuran; water at 20℃; for 1.5h;
Stage #2: 1-bromo-hexane In tetrahydrofuran; water at 55℃; for 12h;
70%
With sodium hydroxide In dimethyl sulfoxide at 110℃; for 6h;
2.2. Synthesis of 3, 6 -Dibromo-9-hexyl-9H-carbazole (HC) (2)
3,6-dibromo-Carbazole (1) (5g, 1eq.), 1-bromohexane (1.7 eq.), sodium hydroxide (8.4 eq.) were added to dimethyl sulfoxide (40 ml) taken in a round-bottomed flask. Reaction temperature 110 °C was maintained for 6 h and then cooled to room temperature. The reaction mixture was extracted with ethyl acetate (EA)/water mixture and the organic layer was collected and dried in sodium sulfate. The solvent was evaporated and the resulting crude prod- uct was purified by column chromatography on silica gel (60-120 mesh) by using hexane as a solvent to give a white solid with a 70% yield. FT-IR (KBr, cm -1 ): 3077, 2930 (s, CH aromatic), 2850 (m, aliphatic CH), 2291, 1638, 1598 (s, aromatic C = C), 1441 (s, alkane C-H bend), 1239 (aromatic CH), 1909, 1746 (w, aromatic CH bend), 846 (s, aromatic CH bend), 714 (s, C-Br). 1 H NMR (500 MHz, CDCl 3, ppm) 8.14 (s, 2H), 7.55 (d, J = 8.7 Hz, 2H), 7.27 (d, J = 9.0 Hz, 2H), 4.24 (t, J = 7.2 Hz, 2H), 1.86 -1.79 (m, 2H), 1.37 -1.23 (m, 6H), 0.85 (t, J = 6.8 Hz, 3H). 13 C NMR (126 MHz, CDCl 3, ppm) 138.98, 129.03, 123.94, 123.29, 111.95, 110.41, 42.75, 31.66, 28.83, 26.89, 13.76. (See supporting information, Figure S1, S2, S3, S12). Cross peaks of the coupling are marked in the COSY spectrum (S12).
43%
Stage #1: 3,6-dibromo-9H-carbazole With potassium carbonate In N,N-dimethyl-formamide at 20℃;
Stage #2: 1-bromo-hexane In N,N-dimethyl-formamide at 20℃; for 96h;
Representative synthesis of 3,6-dibromo-N-hexyl carbazole(2b)
3,6-Dibromocarbazole (2.0 g, 6.2 mmol), and potassium carbonate (0.94 g, 6.8 mmol) were dissolved in 20 mL DMF under ambient conditions forming an orange solution. 1-Bromohexane (0.91 mL, 6.5 mmol) was added to the solution resultingin an immediate colour change. The resulting pink solution was stirred at room temperature for 4 days, yielding a white solid on the inner surface of the flask. The solid and solution were poured into 400 mL of distilled water and apink crude solid was isolated by vacuum filtration. The product was isolatedfrom unreacted carbazole by recrystallization from hot ethanol as 0.92 g whitesolid (43 % yield). 1H-NMR (toluene-d8, 27 °C, 500 MHz); δ (ppm) =7.88 (d, JHH= 1.5 Hz, 2 H, Ar-H 2,7), 7.43 (dd, JHH= 1.5Hz, 9 Hz, 2 H, Ar-H 4,5), 6.74 (d, JHH= 9 Hz, 2 H, Ar-H 1,8), 3.57(t, JHH= 7.3 Hz, 2 H, NC-H), 1.38 (pentet, JHH= 2.0 Hz, 2H, HC-H), 1.15- 1.00 (m, 6 H, HC-H), 0.83 (t, JHH= 7.5 Hz, 3 H, H2C-H).13C-NMR (toluene-d8, 27 °C, 125 MHz); δ (ppm) = 139.2 (C3, C6),128.9 (C1′, C8′), 123.5 (C1, C8), 123.4 (C4′, C5′), 112.0 (C4, C5), 110.0 (C2,C7), 42.6 (N-CH2), 31.4 (-CH2-), 28.6 (-CH2-),26.7 (-CH2-), 22.5 (-CH2-), 13.8 (-CH3). HRMS (m/z);[M]+ calculated for C18H19N79Br2,406.98843; found 406.98946; error (ppm), -1.2. Elemental Analysis:calculated: 52.84 %C, 4.68 %H, and 3.42 % N; found: 52.86 %C, 4.70 %H, and 3.43%N.
With sodium hydroxide
With tetrabutylammomium bromide; potassium hydroxide In water; toluene
With tetrabutylammomium bromide; potassium hydroxide In water; toluene at 110℃; Inert atmosphere;
2
3,6-dibromocarbazole (10 mmol, 3.24 g),Tetrabutylammonium bromide (201mg),Potassium hydroxide aqueous solution (1.74 g, 7 mL) and 50 mL of toluene solvent were added to a 250 mL two-necked flask.Stir slowly at room temperature,Vacuuming through nitrogen, under a nitrogen atmosphere,Add bromohexane (12 mmol, 1.98 g) and continue to vacuum and pass nitrogen.Slowly heat up to 110 ° C reflux,Stop the reaction after the reaction is completed,Cool to room temperature. Extract with water and dichloromethane,Collect organic layers,Add an appropriate amount of anhydrous magnesium sulfate to remove excess water.Filter and condense the liquid to concentratePurification by column chromatography gave product M3.
With tetrabutylammomium bromide; potassium hydroxide; In acetone; for 3h;Reflux;
General procedure: Hexyl bromide (1.45mL, 15.7mmol), tetrabutylammonium bromide (0.16g, 0.5mmol) and potassium hydroxide (0.83g, 14.8mmol) were added to a solution of 3,6-dibromocarbazole (6.18g, 9.2mol) in acetone (previously dried under 3 sieves). The reaction mixture was stirred for 3hand refluxed. Progress of the reaction was controlled by GCMS analysis. Then the mixture was cooled to room temperature and the solvent was evaporated. The residue was dissolved in diethyl ether (100mL), filtered and washed with water (3×10mL). The organic layer was dried under magnesium sulfate and filtered. Then the solvent was evaporated and the residue was stirred with pentane (100mL) for 1h, filtered and dried under vacuum. 6.1g (yield 78%) of 3,6-dibromo-N-hexylcarbazole was obtained as white solid. The product was crystallized by dissolving in small amount of hot hexane and left to cool at room temperature.1H NMR (300MHz, CDCl3): delta 0.86 (t, 3H, JHH=7.0Hz, H6?), 1.21-1.38 (m, 6H, H3?, H4?, H5?), 1.81 (quint, 2H, JHH=7.3Hz, H2?) 4.24 (t, 2H, JHH=7.2Hz, H1?), 7.27 (d, 2H, JHH=8.7Hz, H2), 7.55 (d, 2H, JHH=8.7Hz, H1), 8.14 (d, 2H, JHH=1.9Hz, H4). 13C NMR (75MHz, CDCl3): delta 13.9 (C6?), 22.5 (C5?), 26.9 (C3?), 28.8 (C2?), 31.5 (C4?), 43.3 (C1?), 110.4 (C1), 111.9 (C3), 123.2 (C4), 123.4 (Ci, N-C=C-), 129.0 (C2), 139.3 (Ci, N-C=C-). MS (EI): m/z (rel. intensity - %): 411+ (31), 409 (58), 408 (9), 341 (10), 340 (53), 338 (100), 337 (34), 258 (20), 152 (10). Crystal Data: C18H19Br2N; formula weight Mr=409.16; orthorhombic; Pca21; a=20.3019(16) , b=4.5582(4) , c=18.4012(18) , V=1702.8(3) 3, Z=4, dx=1.60gcm-3, lambda=0.71073 (MoKalpha), mu=4.75mm-1; reflections: collected 27,069, unique 3018 (Rint=10.58%), with I>2sigma(I) 1827; R=6.43%, wR2=13.82%, S=1.03, max/min Deltarho: 0.79/-0.33e-3. Perspective view of the molecule 4 is presented below. Thermal ellipsoids are drawn at the 50% probability level, hydrogen atoms are represented by spheres of arbitrary radii.
35%
With potassium hydroxide;tetra(n-butyl)ammonium hydrogensulfate; In acetone; at 20℃;Inert atmosphere; Reflux;
3,6-Dibromocarbazole 2 (112.0 g, 0.344 mol), benzyl bromide (41 ml,0.344 mol), and nBu4NHSO4 were mixed in 200 ml of acetone and stirred at room temperature under nitrogen until dissolved. KOH (19.3 g, 0.344 mol) was then added to the above transparent solution and the resultant mixture was refluxed for 4 hours, where a white precipitate was observed. The hot mixture was concentrated to remove the majority of acetone. Upon cooling, additional white precipitate appeared. The precipitate was filtered, dissolved in methyl-f-butyl ether, washed with water, dried over Na2SO4, and concentrated to yield a major amount of pure N-benzyl-3,6- dibromocarbazole 3. The filtrate was also washed with water and extracted with methyl-f-butyl ether, dried over Na2SO4, concentrated, and purified over 12Og Sitheta2 with hexane and hexane:ethylacetate (95:5) eluents to yield a minor amount of pure N-benzyl-3,6-dibromocarbazole 3. The combined yield from crystallization and chromatography was 49 g (35%) of N-benzyl-3,6-dibromocarbazole 3.
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 80℃; for 16h;Inert atmosphere;
It was dissolved in 3,6-dibromo-9H-carbazole(100 g, 308 mmol) in a nitrogen environment in 700 mL tetrahydrofuran(THF), here the phenylboronic acid(45.1 g, 370 mmol) and put tetrakis(triphenylphosphine) palladium (10.7 g, 9.24mmol) was stirred. Into a potassium carbonate (213 g, 1,540 mmol) in a saturated water was heated to reflux at 80 C for 16 hours. After the reaction was completed, the reaction solution into water and extracted with DCM then water was removedwith anhydrous MgSO4, filter and was concentrated under reduced pressure. Thus the resulting residue was separated and purified by flash column chromatography to obtain the compound I-23(74.8 g, 76%).
75%
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; tris-(dibenzylideneacetone)dipalladium(0); potassium phosphate monohydrate; In water; toluene;Inert atmosphere; Reflux;
Synthesis of 3,6-diphenyl-9H-carbazole 3,6-Dibromo-9H-carbazole (10.0 g, 30.8 mmol), phenylboronic acid (8.25 g, 67.7 mmol) Pd2(dba)3 (0.564 g, 0.615 mmol), dicyclohexyl(2',6'-dimethoxy-[1,1'-biphenyl]-2-yl)phosphine (SPhOS) (1.011 g, 2.462 mmol), and potassium phosphate hydrate (28.3 g, 123 mmol) were dissolved in the mixture of toluene (350 mL) and water (40 mL) in a three-necked flask. The mixture was degassed by bubbling nitrogen, then it washeated to reflux overnight. After completion of the reaction, the mixture partitioned between ethyl acetate and water. The aqueous layer was washed 3 times with ethyl acetate and the combined organic layers were washed with brine and water. The crude compound was purified by column chromatography on silica gel, eluted with hexane/DCM 1/1 (v/v) mixture. The target compound was obtained as a white solid (7.4 g, 75 % yield).
75.5%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 120℃;
3,6-dibromo-9H-carbazole (5 g, 15.3 mmol) and phenylboronic acid (5.63 g, 46.15 mmol) was dissolved in tetrahydrofuran (THF) (60 ml) into a 2-necked round bottom flask, Tetrakis(triphenylphosphine)palladium(0), Pd(PPh3)4) (0.89 g, 0.77 mmol), K2CO3 (8.3 g) was dissolved in distilled water (30 ml) at 2M, and the mixture was stirred at 120 C. Then, after the reaction was completed, the reaction product was extracted with CH2Cl2 and water, and the organic layer was dried over MgSO4 and concentrated. The concentrated reaction product was purified by silica gel column chromatography to obtain 3.7 g of 3,6-diphenyl-9H-cabazole (hereinafter referred to as Sub 2) (yield: 75.5%).
74%
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In tetrahydrofuran;Inert atmosphere; Reflux;
Example 10; a) 3.09 g (9.23 mmol) of 3,6-dibromocarbazole and 2.78 g (22.2 mmol) of phenylboronic acid are stirred in 140 ml of THF under argon. 12.76 g (92.30 mmol) of potassium carbonate in 46 ml of water are added. 0.27 g (0.23 mmol) of palladium tetrakis(triphenylphosphine) are added under argon and the reaction is stirred under reflux over night. At room temperature, a 1 % aqueous solution of NaCN is added to the medium which is boiled for 20 min. At room temperature, ethyl acetate is added and the organic phase is washed with water, dried over magnesium sulfate, filtered and evaporated under reduced pressure. The crude material is purified by chromatography over silica gel (cyclohexane/ethyl acetate 4:1 ) to give 2.17 g of a white solid (74%). 1H NMR (CDCI3, 300 MHz) 8.34 (d, J = 1.8 Hz, 2H), 8.11 (s, 1 H), 7.74- 7.68 (m, 6H), 7.52-7.45 (m, 6H), 7.37-7.32 (m, 2H).
74%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In toluene; at 80℃; for 8h;Inert atmosphere;
In the nitrogen atmosphere, 3,6-dibromocarbazole (5 g, 15.4 mmol) was added sequentially to the flask, Phenylboronic acid (4.1 g, 33.9 mmol) (Triphenylphosphine) palladium (0.7 g, 0.6 mmol) Toluene (45 ml), 2M sodium carbonate (45 ml), And the mixture was stirred at 80 C for 8 hours. After separating the organic phase, Concentrated under reduced pressure in an evaporator. The resulting residue was purified by silica gel column chromatography, 3,6-diphenylcarbazole (3.6 g, yield 74%) was obtained.
74%
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In toluene; at 80℃; for 8h;Inert atmosphere;
In the nitrogen atmosphere,3,6-dibromocarbazole (5 g, 15.4 mmol) was added sequentially to the flask,Phenylboronic acid (4.1 g, 33.9 mmol)(Triphenylphosphine) palladium (0.7 g, 0.6 mmol)Toluene (45 ml),2M sodium carbonate (45 ml),And stirred at 80 C for 8 hours.After separating the organic phase,Concentrated under reduced pressure in an evaporator.The resulting residue was purified by silica gel column chromatography,3,6-diphenylcarbazole (3.6 g, yield 74%) was obtained.
73%
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In toluene; for 10h;Heating / reflux;
Preparation of 3,6-diphenylcarbazole Reaction 5As depicted in Reaction 5, 15g (46 mmol) of 3,6-dibromocarbazole, 14g (92 mmol) of phenylboronic acid, K2CO3 (2 mol), Pd(PPh3)4 and toluene were refluxed for 10 hours. The reaction mixture was extracted with methylene chloride. The solvent was removed, and then the residue was purified by column chromatography (eluent: hexane) to give the title compound. Yield: 73%. MS (El) (Calcd. for C24H17N: 319.14, Found: 319).
69%
With palladium diacetate; potassium carbonate; triphenylphosphine; In tetrahydrofuran; water;Reflux;
First, 100 g (310 mmol) of 3,6-dibromocarbazole (CAS 6825-20-3) E1 together with 189 g (1550 mmol) of phenylboronic acid (CAS 98-80-6) E2And 430 g (3.1 mol) of potassium carbonate was introduced into a 41 four-necked flask and dissolved in 1000 ml of tetrahydrofuran and 300 ml of water.After the mixture had been degassed for 30 minutes, 140 mg (0.62 mmol) of palladium acetate and 650 mg of triphenylphosphine were added, and the mixture was heated overnight under reflux.700 ml of water was then added to the batch and the aqueous phase was extracted several times with dichloromethane.The combined organic phases were dried over sodium sulfate and the solvent mixture was removed in vacuo.The resulting residue was dissolved in 1.5 mL of dichloromethane and filtered.The solvent was again removed in vacuo and the solid was washed with stirring with 600 mL of ethanol.Filtration and drying gave 68 g (0.21 mol, 69%) of desired material.
68%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 80℃;
Carbazole (16.72g, 100mmol), NBS (59.4g, 210mmol), BPO (2.42g, 10mmol), methylene chloride (300 ml) at room temperature is injected into the medal and then 2000. When reaction is completed methylene chloride and water, NaHCO 3 extracted using aqueous solution obtained after the organic layer Na 2 SO 4 dried to, for the products and re-crystallized concentrating 23.73g (73%)obtained. Said product obtained (23.73g, 73.02mmol), phenylboronic acid (19.6g, 160.64mmol), Pd (PPh 3) 4, (8.44g, 7.3mmol), K 2 CO 3 (60.55g, 438.12mmol), THF (320 ml), water (160 ml) in 80 C agitation return current doesn't have any error frames, turns on the light. When reaction is completed for reducing temperature a high temperature to the normal temperature after methylene chloride and a water extraction of the organic layer obtained after MgSO 4 dried to, for the products and re-crystallized concentrating 15.9g (68%)obtained
63%
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In 1,2-dimethoxyethane; water; at 80℃; for 3.5h;
As an example of a material according to the invention, a synthetic method of a compound represented by formula (59) 9-[4-(3, 6-diphenyl-N-carbazolyl)] phenyl- 10-phenylanthracene (hereinafter referred to as DPCzPA) below will be described. [0209][0210] This compound is prepared in accordance with the synthetic method shown below. Note that 9-phenyl-10-(4-bromophenyl) anthracene is prepared in the manner shown in Example 1. [0211]First, a synthetic method of 3,6-dibromocarbazole will be shown below. A mixture of 6.5 g (20.0 mmol) 3,6-dibromocarbazole, 5.0 g (41.0 mmol) of phenylboronic acid, 93 mg (0.40 mmol) of palladium acetate, 6.9 g (5.2 mmol) of potassium carbonate, water (25 mL), 610 mg of tri(ortho-tolyl) phosphine, and dimethoxyethane (50 mL) is heated to reflux at 80 0C for 3.5 three hours. After the reaction, the solution is rinsed with water, aqueous layer is extracted with toluene, and it is rinsed together with the organic layer using saturated salt solution, and thereafter EPO <DP n="67"/>dried with magnesium sulfate. After natural filtration, the filtrate is condensed to obtain 4.1 g of 3,6-di(2-phenyl-phenyl)-carbazoleas a white solid at a yield of 63 %. A synthetic scheme of 3,6-di(2-phenyl-phenyl)-carbazoleis shown below.
63%
With potassium carbonate;palladium diacetate; Tri(p-tolyl)phosphine; In 1,2-dimethoxyethane; water; at 80℃; for 3.5h;
[Step 2] A synthesis method of 3,6-diphenylcarbazole is described.; A synthesis scheme of 3,6-diphenylcarbazole is represented by (a-2). [Show Image] 6.5 g of 3,6-dibromocarbazole (20 mmol), 5.0 g of phenylboron acid (41 mmol), 93mg of palladium(II) acetate (0.40 mmol), 610mg of tri(ortho-tolyl)phosphine (1.9mmol) were put into a 200mL three-necked flask, and then, the inside of the flask was substituted by nitrogen. Into the mixture, 50 ml of ethyleneglycol dimethylether (abbreviation : DME), and 25 mL of a pottassium carbonate water solution (2.0 mol/L) were added. This mixture was refluxed for 3.5 hours at 80 C. After the reaction, the reaction mixture was washed with water and a water layer was extracted with toluene. The extracted solution and an organic layer were washed with a saturated saline, and dried by magnesium sulphate. The mixture was naturally filtrated. The filtrate was condensed, and 4.1 g of an objective matter, a white powder solid was obtained at the yield 63 %.
63%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 100℃; for 17h;Inert atmosphere;
A mixture of 3,6-dibromo-9H- carbazole (3.0 g, 9.2 mmol) (Aldrich), phenylboronic acid (3.0 g, 25 mmol) (Aldrich), tetrakis(triphenylphosphine)palladium (Pd(PPh3)4) (0.3 g, 0.26 mmol) (Frontier Scientific, Logan, UT) and potassium carbonate (6.5 g, 47 mmol) (Aldrich) in dioxane/water (75 mL/15 mL) (Aldrich) was degassed with bubbling argon (Airgas, San Marcos, CA) for 60 min and heated at about 100 C on a hot plate with a silicone oil bath for about 16 hours. Upon cooling to room temperature, the whole mixture was mixed with ethyl acetate (Aldrich) and rinsed with brine. Then, the organic mixture was dried over Na2S04 (Aldrich), loaded on silica gel (Grade 135) and purified by flash column using eluents of ethyl acetate/hexanes (10% to 30%) (Aldrich). After removal of solvents, a white solid (Compound OSC-3) was obtained (2.1 g, in 63% yield). Confirmed by 1 HNMR (Jeol Instruments, Peabody, MA).
63%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 100℃; for 16h;
A mixture of 3,6-dibromo-9H-carbazole (3.0 g, 9.2 mmol), phenylboronic acid (3.0 g, 25 mmol), Pd(PPh3)4 (0.3 g, 0.26 mmol) and potassium carbonate (6.5 g, 47 mmol) in dioxane/water (75 mL/15 ml) was degassed and heated at about 100 C for about 16 hours. Upon cooling to room temperature, the whole was worked up with ethylacetate/brine, the organic was dried over Na2So4, loaded on silica gel and purified by flash column using eluents of ethyl acetate/hexane (10% to 30%). After removal of solvents, a white solid (Compound 11 ) was obtained (2.1 g, in 63% yield).
48%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; at 80℃; for 10h;Inert atmosphere;
30 g (92.3 mmol) of 3,6-dibromo-9H-carbazole was dissolved in 0.3 L of tetrahydrofuran (THF) in a nitrogenenvironment, 28 g (231 mmol) of phenyl boronic acid and 3.2 g (2.8 mmol) of tetrakis(triphenylphosphine)palladium wereadded thereto, and the mixture was agitated. 40.8 g (277 mmol) of potassium carbonate saturated in water was addedthereto, and the mixture was heated and refluxed at 80 C for 10 hours. When the reaction was terminated, water wasadded to the reaction solution, and the mixture was extracted with dichloromethane (DCM) and treated with anhydrousMgSO4 to remove moisture and then, filtered and concentrated under a reduced pressure. The obtained residue wasseparated and purified through flash column chromatography, obtaining a compound I-4 (14.3 g, 48 %). HRMS (70 eV, EI+): m/z calcd for C24H17N: 319.1361, found: 319.1. HRMS (70 eV, EI+): m/z calcd for C24H17N: 319.1361, found: 319.1.
33%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In water; N,N-dimethyl-formamide; at 80℃; for 4h;Inert atmosphere;
General procedure: Under nitrogen protection system, 3-bromo -9H- carbazole 1.6mmol, tetrakistriphenylphosphine palladium 0.64mmol, anhydrous carbonatePotassium1.92mmol phenylboronic acid 1.92mmol, was dissolved in N, N- dimethylformamide: H2O = (15ml: 2ml) mixed solvent,80 stirring for 4h, after completion of the reaction, cooled to room temperature, the mixed solution was extracted with dichloromethane and water, the organic phase was dried over anhydrous magnesiumDried and evaporated, evaporated and the product after column chromatography to obtain 3-phenyl -9H- carbazole (A-1) 1.44mmol, 90% yield. The method of the same A-1, by column chromatography to obtain a white solid product 3,6-diphenyl -9H- carbazole (A-2), producingRate of 33%.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In 1,2-dimethoxyethane; ethanol; water; for 18h;Heating / reflux;
EXAMPLE I Synthesis of 3,6-diphenyl carbazole In a 250 milliliter round bottom flask there were added 3,6-dibromocarbazole (5 grams), 50 milliliters of 1,2-dimethoxyethane, phenylboric acid (4.8 grams) dissolved in ethanol, and sodium carbonate (4.2 grams) dissolved in 20 milliliters of water.. After the resulting solution was saturated with argon, 0.49 gram of tetrakis-(triphenylphosphine) palladium was added.. The reaction mixture was heated to reflux and stirred for 18 hours.. The reaction flask was removed from the heat and cooled to room temperature, about 22 C. to about 25 C. The resulting solution was transferred to a separatory funnel, and the organic layer, which contained product, was separated from the aqueous phase.. After removal of the organic solvents by evaporation, the residue was subjected to column chromatography on silica gel to yield 3.5 grams of 3,6-diphenyl carbazole as colorless powder product.. Its chemical structure was confirmed by Proton IR analysis.
According to an ordinary process, 3,6-diphenylcarbazole was synthesized by reacting 3,6-dibromocarbazole and phenylboronic acid.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; toluene;
Manufacturing Example 8 A solution was prepared by adding 12.7 g of 3,6-dibromocarbazole, 10.0 g of phenylboronic acid, and 2.61 g of tetrakis(triphenylphosphine)palladium into 40 ml of ethanol and 170 ml of toluene. The resulting solution was blended with 90 ml of a 22% aqueous solution of sodium carbonate, and the mixture was heated under reflux in a nitrogen atmosphere for 5 hours. Insoluble materials were removed therefrom by performing hot filtration using a filter aid. Thereafter, the organic layer was separated from the aqueous layer, and the solvent was distilled off under a reduced pressure. After washing with water and drying were performed, a light brown powder was prepared. The resulting powder was subjected to a column chromatography treatment (eluant: toluene), followed by washing with hexane. Thus, 6.0 g of a colorless needle crystal of 3,6-diphenylcarbazole was produced. The physical properties thereof were as follows. melting point: 180.5 C. to 181.5 C. elemental analysis value (%): measured value/calculated value C 90.44/90.24 H 5.25/5.38 N 4.31/4.39 infrared absorption spectrum (KBr pellet method) NH stretching vibration: 3423, 3378 cm-1
38 g
3,6-dibromocarbazole (50 g, 0.154 mol), phenylboronic acid (41 g, 0.336 mol), 150ml water, 80g sodium carbonate, and 600ml dioxane were charged into a 2 liter pot with magnetic stirrer, reflux condenser and nitrogen inlet, and sparged with nitrogen for one hour. Pd2DBA3 (6g, 0.0066mol) and tri-t-butylphosphine (3 g, 0.0148mol) was quickly added from the drybox. The reaction was refluxed overnight. The next day water was added to the reaction mixture and methylene chloride extractions were preabsorbed to 141 g of activated silica and purified by column chromatography using methylene chloride/hexanes yielding 38 grams of product 3,6-diphenylcarbazole.
3,6-Bis(5-indolyl)carbazole-2',2'-dicarboxylic acid diethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.07 g (46%)
With cesium fluoride;palladium diacetate; In 1,4-dioxane; hexane; ethyl acetate;
Step 3: A representative procedure for the mono-phasic bis-cross coupling reaction of bis-arylhalides with boronic acids is a modification of cross coupling reactions described by Buchwald, et al (J. Am. Chem. Soc. 1998, 120, 9722-9723). 3,6-Dibromocarbazole (1.52 g, 4.69 mmol), 2.73 g (11.72 mmol) 2-ethoxycarbonylindole-5-boronic acid, and 5.34 g (35.16 mmol) CsF were dissolved in 50 mL anhydrous dioxane under argon. Pd(OAc)2 (106 mg, 0.47 mmol) and 245 mg (0.70 mmol) dicyclohexylphosphinobiphenyl were dissolved in 20 mL dioxane under argon and added to the above mixture. The resulting mixture was stirred and refluxed under argon overnight, cooled to room temperature, diluted with 200 mL EtOAc and filtered through celite. The filtrate was washed with 3*75 mL H2O. The organic phase was dried over sodium sulfate and the solvent was evaporated. The residue was purified by flash-column chromatography using hexane/EtOAc (7:3-6:4) as eluent to yield 1.07 g (46%) of 3,6-Bis(5-indolyl)carbazole-2',2'-dicarboxylic acid diethyl ester. 1H NMR-1.41 (t, J=6.9 Hz, 6H), 4.41 (q, J=7.2 Hz, 4H), 7.26 (d, J=1.8 Hz, 2H), 7.59 (s, 2H), 7.62 (s, 2H), 7.77 (m, 2H), 7.80 (m, 2H), 8.08 (d, J=0.9 Hz, 2H), 8.61 (d, J=1.2 Hz, 2H), 11.34 (s, 1H), 11.96 (s, 2H).
EXAMPLE I Synthesis of 3,6-Diphenyl Carbazole In a 250 milliliter round bottom flask there were added 3,6-dibromocarbazole (5 grams), 50 milliliters of 1,2-dimethoxyethane, phenylboric acid (4.8 grams) dissolved in ethanol, and sodium carbonate (4.2 grams) dissolved in 20 milliliters of water. After the resulting solution was saturated with argon, 0.49 gram of tetrakis-(triphenylphosphine) palladium was added. The reaction mixture was heated to reflux and stirred for 18 hours. The reaction flask was removed from the heat and cooled to room temperature, about 22° C. to about 25° C. The resulting solution was transferred to a separatory funnel, and the organic layer, which contained product, was separated from the aqueous phase. After removal of the organic solvents by evaporation, the residue was subjected to column chromatography on silica gel to yield 3.5 grams of 3,6-diphenyl carbazole as colorless powder product. Its chemical structure was confirmed by Proton IR analysis.
Stage #1: 3,6-dibromo-9H-carbazole; copper(l) cyanide In N,N-dimethyl-formamide at 150℃; for 48h; Inert atmosphere;
Stage #2: With hydrogenchloride; iron(III) chloride In water; N,N-dimethyl-formamide for 2h; Inert atmosphere;
31%
Stage #1: 3,6-dibromo-9H-carbazole; copper(l) cyanide With 1-methyl-pyrrolidin-2-one at 140℃;
Stage #2: With hydrogenchloride; iron(III) chloride In water for 1h;
In <i>N</i>-methyl-acetamide; aqueous ethylenediamine
B B.
B. 3,6-dicyanocarbazole A slurry of 3,6-dibromocarbazole (33.3 g) and cuprous cyanide (39 g) in 50 ml of dimethylformamide is heated at 160° C for seven days. The reaction mixture is poured into 1 liter of aqueous ethylenediamine (20%, v/v) and the mixture is stirred for fifteen minutes and filtered. The precipitate is slurried in 1 liter of aqueous ethylenediamine (20%, v/v) and stirred for about 16 hours. The precipitate is collected by filtration and recrystallized from dimethylformamide to yield 15 g of 3,6-dicyanocarbazole, melting point 360° C.
With 4-methyl-morpholine at 200℃;
In N,N-dimethyl-formamide at 150℃; for 72h;
4.1 1) Synthesis of compound 4B
In the nitrogen environment (N2 purging) 4A and 2.5 equivalents of CuCN were placed in DMFStir at 150 ° C.After 72 hours, slowly pour the reaction into ice water at 0 ° C, stir for 30 minutes, and then add an aqueous ammonia solution. After removing the solvent of the solvent layer and adsorbing it on the silica, ivory solid 4B is obtained after the column using the MC developing solvent.
3.6-Diphenyl-9H-carbazole The same method as employed in the preparation of Intermediate 7 but starting from 3,6-dibromo-9H-carbazole gave after flash chromatography the title compound as a white solid in a 33percent yield. M.p.: 145-147 °C. 1H NMR (CDCl3, 300 MHz) delta 8.32 (d, 2H,J= 1.2 Hz), 8.09 (s, 1H), 7.72-7.66 (m, 6H), 7.50-7.43 (m, 6H), 7.35-7.30 (m, 2H).
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In 1,2-dimethoxyethane; water; at 80℃; for 3.5h;
As an example of a material according to the invention, a synthetic method of a compound represented by formula (67) 9-{4-[3, 6-di(2-phenyl) phenyl-N-carbazolyl] phenyl} -10-phenylanthracene (hereinafter referred to as BPCzPA) below will be described. Note that 9-phenyl-10-(4-bromophenyl) anthracene is prepared in the manner shown in Example 1. [0220][0221]First, a synthetic method of 3,6-di(2-phenyl-phenyl)-carbazole will be shown below. A mixture of 3.25 g (10.0 mmol) 3,6-dibromocarbazole, 4.2 g (21.0 mmol) of o-biphenylboronic acid, 50 mg (0.21 mmol) of palladium acetate, 6.9 g (5.2 mmol) of potassium carbonate, water (25 mL), 308 mg (1.0 mmol) of tri(ortho-tolyl) phosphine, and 30 mL of dimethoxyethane is heated to reflux at 80 0C for 3.5 three hours. After the reaction, the solution is rinsed with water, aqueous layer is thereafter extracted with toluene, and it is rinsed together with the organic layer using saturated salt solution, and thereafter dried with magnesium sulfate. After natural filtration, the filtrate is condensed to obtain 2.2 g of 3, 6-di(2 -phenyl -phenyl)-carbazole as a white solid at a EPO <DP n="70"/>yield of 46 %. A synthetic scheme of 3,6-di(2-phenyl-phenyl)-carbazole is shown below.
With sodium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene;
Manufacturing Example 29 A solution was prepared by adding 6.50 g of 3,6-dibromocarbazole, 10.00 g of biphenyl-2-boronic acid, and 0.72 g of tetrakis(triphenylphosphine)palladium into 20 ml of ethanol and 100 ml of toluene. The resulting solution was blended with 40.g of a 22% aqueous solution of sodium carbonate, and the mixture was heated under reflux in a nitrogen atmosphere for 7.5 hours. After the mixture was cooled, toluene and water were added thereto, and insoluble materials were removed therefrom using a filter aid. Subsequently, the organic layer was separated from the aqueous layer, and the solvent was distilled off under a reduced pressure. After washing with water and drying were performed, so that a light brown powder was prepared. The resulting powder was subjected to a silica gel column chromatography treatment (eluant: toluene/hexane=2/1 on a volume ratio basis), followed by washing with ethanol. Thus, 5.22 g of a colorless needle crystal of 3,6-bis(2-biphenylyl)carbazole was produced. The physical properties thereof were as follows. melting point: 186.0C to 189.0 C. elemental analysis value (%) measured value/calculated value C 91.46/91.69 H 5.13/5.34 N 3.05/2.97 infrared absorption spectrum (KBr pellet-method) NH stretching vibration: 3425 cm-1
With copper(l) iodide; 18-crown-6 ether; potassium carbonate; In DMPU (1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone); at 170℃; for 20h;
Synthesis of Intermediate E; 3.25 g (10.0 mmol) of 3, 6-dibromocarbazole, 10.2 g (50.0 mmole) iodobenzene, 190 mg (1.0 mmole) of Cul, 132 mg (0.5 mmole) of 18-C-6, 2.76 g (20.0mmole) of K2CO3 were dissolved in 50 mL of DMPU, and stirred at 170°C for 20 hours. The mixture was cooled to room temperature and 50 mL of diethylether was added thereto. Then the mixture was washed with plenty of water and ammonium hydroxide solution. A collected organic layer was dried over MgSO4 to evaporate the solvent. The residue was purified using silica gel column chromatography to obtain 3.40 g of white solid Intermediate E (Yield: 85 percent). (NMR (CDCl3, 400MHz) delta (ppm) 7.92 (m, 2H), 7.55-7.47 (m, 6H), 7.36-7.16 (m, 3H); 13C NMR (CDCl3, 100MHz) delta (ppm) 142.6, 137.6, 130.2, 129.8, 127.4, 127.0, 122.8, 122.5, 115.3, 111.3).
75%
With tris-(dibenzylideneacetone)dipalladium(0); triphenylphosphine; sodium t-butanolate; In toluene; at 100℃; for 24h;
To a round bottom flask Sub 2-1-6 (6.5g, 20mmol), Sub 2-1-2-1 (4.1g, 20mmol),Pd2 (dba) 3 (0.9g, 1mmol), PPh3 (0.5g, 2mmol ), NaOt-Bu (5.8g, 60mmol), were addedto toluene (210mL), respectively, and refluxed under stirring for 24 hours at 100 ° C.The organic layer was dried and the ether was extracted with water over MgSO4 andconcentrated and to the resulting organic silicagel column and recrystallized from a Sub2-1-7-1 6.0g (yield: 75percent) was obtained.
3.27 g
With palladium diacetate; tris-(o-tolyl)phosphine; sodium t-butanolate; In toluene;
2.5 g (14.9 mmol) ofcarbazole was used (utilized) to perform an NI3S bromination to synthesize 2,5-dibromocar- bazole. Then, the 2,5-dibromocarbazole and iodinated phenyl were used (utilized) to synthesize 3.72 g (9.28 mmol) of Intermediate E through a buchwald reaction.
1
Synthesis of Compound 3:; Compound 3 was obtained by coupling compound 2 with 4-fluoronitrobenzene.This coupling was carried out by making the compound 2 (1.00 g, 3.1 mmol), previously dissolved in dimethylformamide (15 ml), react with 4-fluoronitrobenzene (699.40 mg, 4.9 mmol) in the presence of sodium hydride (236.00 mg, 9.8 mmol) in order to deprotonate the nitrogen atom of compound 2, as described by Zhu et al. in Macromolecules, 2000, 33, 801-807, [5].However, unlike the protocol proposed by Zhu et al., the solvents (CH2Cl2 and hexane) used for washing the precipitate formed were at 0° C.The yield of this reaction was quantitative.
81%
With potassium carbonate In N,N-dimethyl-formamide for 12h; Reflux;
General procedure: 3a-3c, a 100-mL two-necked flask equipped with a reflux condenser was charged with a carbazole compound with a -OCH3, H or Br substituent (15.3 mmol), K2CO3 (76.52 mmol), p-nitrofluorobenzene (61.4 mmol), and DMF (80 mL). Each reaction mixture was heated under reflux for 12 h, cooled, and then poured into water (500 mL). Each precipitate was filtered, dried, and recrystallized. 3c, bright yellow powder, yield 81%. 1H NMR (500 MHz, CDCl3): δ 8.57-8.47 (m, 2H), 8.23 (d, J = 1.9 Hz, 2H), 7.79-7.71 (m, 2H), 7.57 (d, J= 8.7, 2.0 Hz, 2H), 7.35 (d, J = 8.7 Hz, 2H).
80%
With potassium <i>tert</i>-butylate In N,N-dimethyl-formamide at 110℃; for 24h; Inert atmosphere;
5.1 (1) Synthesis of Intermediate 3,6-dibromo-9- (4-nitrophenyl) -9H-carbazole
The 3.250g (0.01mol) 3,6- dibromo-carbazole, 2.122ml (0.02mol) difluoronitrobenzene was added and 50mlDMF three 100ml flask, with magnetic stirring and argon gas, was added 1.122g (0.01mol) potassium t-butoxide, the reaction temperature was raised to 110 24h; the reaction mixture was poured into ice water, the resulting filtration cake was recrystallized from ethanol twice, the obtained cake was suction filtered and dried in vacuo 80 24h, to give the product 3.569g, the yield was 80%; the intermediate structure is as follows
78%
Stage #1: 3,6-dibromo-9H-carbazole With potassium <i>tert</i>-butylate In N,N-dimethyl-formamide at 110℃; for 0.5h;
Stage #2: 4-Fluoronitrobenzene In N,N-dimethyl-formamide at 70℃; for 12h;
Synthesis of DNB and NDTC
3,6-Dibromocarbazole (1.63 g, 5 mmol) and Potassium tert-butoxide (0.56 g, 5 mmol) were dissolved in dry DMF (50 mL) in a flask fitted with a magnetic stirrer and condenser. The mixture was heated at 110 °C for 30 min and then 4-Fluoronitrobenzene (0.71 g, 5 mmol) was added with stirring for overnight. The mixture was cooled to room temperature, poured into ice-water, filtered, and the crude residue was recrystallized from ethanol. A light yellow powder was obtained (1.74 g, 78% yield). 1H NMR (500 MHz, CDCl3) δ: 8.41 (d, J = 8.5 Hz, 2H), 8.18 (s, 2H), 7.56(d, J = 8.5 Hz, 2H), 7.48 (d, J = 8.5 Hz, 2H), 7.33 (d, J = 8.5 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 147.12, 144.96, 139.18, 122.79, 122.13, 121.83, 121.15, 117.69, 113.26, 111.25. MS (EI): calculated for C18H10Br2N2O2 m/z: 446.09, found m/z: 445.86.
76%
With potassium carbonate In N,N-dimethyl-formamide Inert atmosphere;
70%
With potassium carbonate In dimethyl sulfoxide at 100℃; for 7h;
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 110℃; for 5h;
[Step 1: Synthesis of 3-6-bis[4-(9H-carbazol-9-yl)phenyl]-9H-carbazole (CP2C)] 1.0 g (3.1 mmol) of 3,6-dibromo-9H-carbazole, 1.8 g (6.2 mmol) of 4-(9H-carbazol-9-yl)phenylboronic acid, and 457 mg (1.5 mmol) of tri(ortho-tolyl)phosphine were put into a 300 mL three-neck flask. To the mixture were added 20 mL of ethanol, 50 mL of toluene, and 20 mL (2.0 mol/L) of an aqueous solution of potassium carbonate. This mixture was stirred to be degassed while the pressure was reduced. To the mixture was added 70 mg (0.30 mmol) of palladium(II) acetate. This mixture was refluxed at 110 C for 5 hours, cooled to room temperature, and then left for 15 hours; accordingly a black solid was precipitated. The precipitated solid was subjected to suction filtration and then collected. The collected solid was dissolved in toluene which was heated, and this solution was subjected to filtration through celite (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 531-16855), alumina, and Florisil (produced by Wako Pure Chemical Industries, Ltd., Catalog No. 540-00135). The obtained filtrate was condensed to give a white solid. The obtained solid was recrystallized with toluene/hexane to give 1.1 g of a white powder, which was the object of the synthesis, at a yield of 58 %.
With potassium carbonate;tetrakis(triphenylphosphine) palladium(0); In tetrahydrofuran;Inert atmosphere; Reflux;
Example 9; a) 2.41 g (7.2 mmol) of 3,6-dibromocarbazole and 5.00 g (17.29 mmol) of 4-(Lambda/,/V- diphenylamino)-1-phenylboronic acid are stirred in 1 10 ml of THF under argon. 9.95 g of potassium carbonate dissolved in 36 ml of water are added and everything is further stirred under argon. Then 0.21 g (0.18 mmol) of palladium tetrakis(triphenylphosphine) are added and the reaction is heated at reflux over night. At room temperature, a 1 % aqueous solution of NaCN is added to the medium which is boiled for 20 min. At room temperature, ethyl acetate is added and the organic phase is washed with water, dried over magnesium sulfate, filtered and evaporated under reduced pressure. The crude material is purified by chromatography over silica gel (cyclohexane/ethyl acetate 5:1 ) to give 4.48 g of white crystals (95%). 1H NMR (CDCI3, 300 MHz) 8.30 (d, J = 1.5 Hz, 2H), 8.07 (s, 1 H), 7.66 (dd, J = 1.8 Hz, J = 8.4 Hz, 2H), 7.61-7.57 (m, 4H), 7.48 (d, J = 8.4 Hz, 2H), 7.30-7.25 (m, 8H), 7.20-7.14 (m, 12H), 7.05-7.00 (m, 4H).
68%
With palladium diacetate; potassium carbonate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; for 4h;Reflux;
(121.8 mmol) of 18.9 g (58 mmol) of 3,6-dibromo-9H-carbazole and (4- (diphenylamino) phenyl) boronic acid were dissolved in 100 ml of toluene, 20 ml of ethanol and 40 ml of water and 24.0 g of potassium carbonate was added. Saturated with nitrogen and 1.1 g (4 mmol) of tris-o-tolylphosphine and 262 mg (2 mmol) of palladium acetate were added, The mixture was stirred at reflux for 4 hours. Next, the organic layer was separated, washed with water, filtered, and the solvent was removed in vacuo. Recrystallization twice with toluene gave 25.7 g (68%) of a solid
51%
With potassium carbonate;palladium diacetate; tris-(o-tolyl)phosphine; In ethanol; water; toluene; at 80℃; for 3h;
[Step 1: Synthesis of 4,4'-(9H-carbazol-3,6-diyl)bis(N,N-diphenylaniline) (TP2C)] 1.1 g (3.5 mmol) of 3,6-dibromocarbazole, 2.0 g (7.0 mmol) of triphenylamine-4-boronic acid, and 0.24 g (1.1 mmol) of tri(ortho-tolyl)phosphine were put into a 300 mL three-neck flask. To the mixture were added 30 mL of ethanol, 50 mL of toluene, and 10 mL (2.0 mol/L) of an aqueous solution of potassium carbonate. This mixture was stirred to be degassed while the pressure was reduced. After the degassing, 47 mg (0.21 mmol) of palladium(II) acetate was added to the mixture. This mixture was stirred at 80 C for 3 hours under a stream of nitrogen. After the stirring, the aqueous layer of the mixture was extracted with toluene. The extract was combined with the organic layer and then washed with a saturated saline solution. Then, the obtained organic layer was dried with magnesium sulfate. After the drying, the mixture was subjected to gravity filtration. The obtained filtrate was condensed to give an oily light yellow substance. The obtained oily substance was purified by silica gel column chromatography (a developing solvent was a mixed solvent of toluene: hexane = 1: 1) to give an oily light yellow substance. This oily substance was recrystallized with toluene/hexane to give 1.2 g of a white powder, which was the object of the synthesis, at a yield of 51 %.
phenylboronic acid, K2CO3 (2 mol), Pd(PPh3)4[ No CAS ]
[ 56525-79-2 ]
Yield
Reaction Conditions
Operation in experiment
73%
In toluene;
Preparation of 3,6-diphenylcarbazole As depicted in Reaction 5, 15 g (46 mmol) of 3,6-dibromocarbazole, 14 g (92 mmol) of phenylboronic acid, K2CO3 (2 mol), Pd(PPh3)4 and toluene were refluxed for 10 hours. The reaction mixture was extracted with methylene chloride. The solvent was removed, and then the residue was purified by column chromatography (eluent:hexane) to give the title compound. Yield: 73percent. MS (EI) (Calcd. for C24H17N, 319.14. Found: 319).
Stage #1: 3,6-dibromo-9H-carbazole With potassium hydroxide In N,N-dimethyl-formamide for 0.5h;
Stage #2: (2-bromoethyl)oxirane In N,N-dimethyl-formamide at 20℃;
97.9%
Stage #1: 3,6-dibromo-9H-carbazole With potassium hydroxide In N,N-dimethyl-formamide for 0.5h;
Stage #2: (2-bromoethyl)oxirane In N,N-dimethyl-formamide at 20℃;
19.1 Step 1. Synthesis of 3,6-dibromo-9-(2-(oxiran-2-yl)ethyl)-9H-carbazole
Crushed KOH (0.0054 g, 0.0954 mmol, 1.2 equiv) was added to 3,6-dibromocarbazole (0.0258 g, 0.0795 mmol, 1 equiv.) in 0.5 mL DMF solution and the mixture was stirred for 30 min. 1-Bromo-3,4-epoxybutane (0.0300 g, 0.199 mmol) in 0.5 mL DMF solution was dropwise added into the mixture and it was stirred at room temperature for overnight. Reaction crude was diluted with 20 mL EtOAc and washed with water 5*10 mL. The organic layer was dried over anhydrous Na2SO4 and evaporated to afford 31.2 mg white solid as product, yield 97.9%. 1H NMR (CDCl3, 400 MHz) δ ppm 1.65-1.81 (m, 1H) 2.13-2.27 (m, 1H) 2.34 (dd, J=4.88, 2.64 Hz, 1H) 2.64 (dd, J=4.78, 4.05 Hz, 1H) 2.69-2.80 (m, 1H) 4.26-4.54 (m, 2H) 7.27 (d, J=8.69 Hz, 2H) 7.50 (dd, J=8.69, 1.90 Hz, 2H) 8.08 (d, J=1.90 Hz, 2H)
Multi-step reaction with 2 steps
1.1: potassium hydroxide / N,N-dimethyl-formamide / 0.5 h / 20 °C
1.2: 20 °C
2.1: bismuth(III) chloride / cyclohexane / Reflux
Multi-step reaction with 2 steps
1: potassium hydroxide / N,N-dimethyl-formamide / 1 h
2: lithium amide / hexane; 1,4-dioxane / 160 °C / Microwave irradiation
Multi-step reaction with 2 steps
1: potassium hydroxide / N,N-dimethyl-formamide / 20 °C
2: bismuth(III) chloride / cyclohexane / Reflux
Multi-step reaction with 2 steps
1.1: potassium hydroxide / N,N-dimethyl-formamide / 1 h / 20 °C
1.2: 3 h / 20 °C
2.1: bismuth(III) chloride / cyclohexane / 48 h / 90 °C
With copper(l) iodide; caesium carbonate; In N,N-dimethyl-formamide; at 220℃; for 0.5h;Microwave irradiation;
General procedure: 9H-Carbazole (1.0 mmol), Cs2CO3 (1.0 mmol), iodobenzene (1.1 mmol), CuI (0.1 mmol), and DMF (2 mL) were added to a 5-mL vial. The vial was sealed with a crimp cap and placed in a Biotage initiator microwave cavity. After irradiation at 220 C for the appropriate time and subsequent cooling, the reaction mixture was diluted with saturated aqueous ammonium chloride. Products were isolated by extraction into ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated. Products were purified by silica gel column chromatography using a hexane/ethyl acetate solvent. N-Phenyl-carbazole (2a)22 was obtained (96% yield) as a white solid.
With copper(l) iodide; caesium carbonate In N,N-dimethyl-formamide at 220℃; for 0.5h; Microwave irradiation;
4.2. General experimental procedure for microwave-assisted N-arylation of carbazoles
General procedure: 9H-Carbazole (1.0 mmol), Cs2CO3 (1.0 mmol), iodobenzene (1.1 mmol), CuI (0.1 mmol), and DMF (2 mL) were added to a 5-mL vial. The vial was sealed with a crimp cap and placed in a Biotage initiator microwave cavity. After irradiation at 220 °C for the appropriate time and subsequent cooling, the reaction mixture was diluted with saturated aqueous ammonium chloride. Products were isolated by extraction into ethyl acetate. The organic layer was dried over anhydrous magnesium sulfate, filtered, and concentrated. Products were purified by silica gel column chromatography using a hexane/ethyl acetate solvent. N-Phenyl-carbazole (2a)22 was obtained (96% yield) as a white solid.
74%
With potassium phosphate; copper(l) iodide; N,N`-dimethylethylenediamine In toluene at 100℃; for 48h; Inert atmosphere;
Synthesis of 2:
To a solution of 3,6-dibromo-9H-carbazole (1, 10.0 g, 30.77mmol) in anhydrous toluene (300 mL), K3PO4 (13.06 g, 61.54 mmol),N,N'-dimethyl-1,2-ethanediamine (2.71 g, 30.77 mmol), and CuI (0.29 g, 0,05 mmol)were added under nitrogen atmosphere. The reaction mixture was stirred at 100 °C for48 h. After completion of the reaction, the mixture was cooled down to roomtemperature, and filtered. The filtrate was diluted with ethyl acetate (300 mL), washedwith water (200 mL × 3) and brine (300 mL), dried over anhydrous Na2SO4, and thenfiltered. The organic solvent was removed under reduced pressure. The crude productwas purified by column chromatography on silica (hexane:ethyl acetate = 6:1) to give2 as a yellow solid (10.5 g, 74% based on 1).
61%
With copper(l) iodide; caesium carbonate; lithium chloride In N,N-dimethyl-formamide at 150℃; for 48h;
With tetrakis(triphenylphosphine) palladium(0); caesium carbonate; In ethanol; water; toluene; at 100℃; for 24h;Inert atmosphere;
To 3,6-dibromo-9H-carbazole (1.00 g, 3.09 mmol), 2,4,6-trimethylphenylboronic acid (1.523 g, 9.29 mmol), Cs2CO3 (3.023 g, 9.27 mmol), and Pd(PPh3)4 (71.4 mg, 0.0618 mmol) in a test tube under argon atmosphere was added toluene (15 mL), ethanol (5 mL), and water (5 mL). The mixture was stirred at 100 °C fro 24 h. Then reaction mixture was cooled down and water was added. The mixture was extracted by dichloromethane (10 mL × 3). The combined organic layer was washed by water (20 mL × 3), dried over Na2SO4, filtered, and evaporated under reduced pressure. The crude product was purified by silica gel chromatography (15percent ethyl acetate/hexane) to afford 5 (850 mg,68percent yield).
68%
With tetrakis(triphenylphosphine) palladium(0); caesium carbonate; In ethanol; water; toluene; at 100℃; for 24h;Inert atmosphere;
3,6-dibromo-9H-carbazole (2b) (1.00g, 3.09mmol),Mesityl boronic acid (1.52g, 9.29mmol),Cesium carbonate (Cs2CO3,3.02g, 9.27mmol) and tetrakis triphenylphosphine palladium (0) (Pd (PPh3) 4,71.4mg, 0.0618mmol) under an argon stream,Toluene (15 mL), and ethanol (5 mL), water (5 mL) was added,The mixture was heated and stirred for 24 hours at 100 .The reaction mixture was cooled to room temperature, water was added. Extracted with dichloromethane, washed the organic layer twice with water, dried over anhydrous sodium sulfate, filtered, and evaporated at room temperature under reduced pressure to give the crude product.The crude product, MPLC: purified by (solvent ethyl acetate 15percent by volume containing hexane), 3,6-dimesityl-9H-carbazole the (2c) 850mg (yield: 68percent) was obtained.
60%
With caesium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 100℃; for 8h;
[Chem. 11]; A-2; Intermediate (B); (1) Synthesis of intermediate (B); [0079] A-1 (13.2 g (41 mmol) , trade name: 3,6- Dibromocarbazole, Wako Pure Chemical Industries, Ltd.), A-2 (20 g (122 mmol), trade name: 2 , 4 , 6-Trimethylphenylboronic Acid, Wako Pure Chemical Industries, Ltd.), Pd(PPh3)4 (0.938 g (0.8 mmol)), Cs2C03 (40 g (123 mmol)), toluene (150 mL), ethanol (100 mL), and water (100 mL) were placed in a 500-mL flask, followed by stirring at 100°C for 8 hours. The reaction solution was cooled to room temperature, and the organic layer was extracted and concentrated. The resulting residue was purified by a column (developing solvent: ethyl acetate/heptane = 1/4) to obtain 9.8 g (24 mmol) of an intermediate (B). The yield was 60percent.
60%
With caesium carbonate;tetrakis(triphenylphosphine) palladium(0); In ethanol; water; toluene; at 100℃; for 8h;
Example 1; [0070]; Synthesis of Compound H-08; (1) Synthesis of Intermediate A-03; [0071][Chem. 12][0072] After 13.2 g (41 mmol) of the above A-01 (trade name: 3 , 6-Dibromocarbazole, manufactured by Wako PureChemical Industries, Ltd.), 20 g (122 mmol) of A-02 (trade name: 2 , 4 , 6-trimethylphenylboronic acid, manufactured by Wako Pure Chemical Industries, Ltd.), 0.938 g (0.8 mmol) of tetrakis( triphenylphosphine) palladium (Pd(PPh3)4), 40 g (123 mmol) of Cs2C03, 150 ml of toluene, 100 ml of ethanol, and 100 ml of water were prepared and were then added in a 500 ml flask, a reaction was carried out by stirring at 100°C for 8 hours.[0073] After a reaction mixture was cooled to room temperature, a separated organic layer was extracted and condensed, and a residue obtained thereby was purified by chromatography on a silicagel column (EtOAc/heptane=l/4 ) , so that 9.8 g (24 mmol) of an intermediate A-03 was obtained. The yield was 60percent.
With dibenzo-18-crown-6; copper; potassium carbonate; In 1,2-dichloro-benzene; at 100℃;
4.75 g of 9-(3-bromophenyly9H-carbazole, 4 g of 3,6-dibromo-9-carbazole, 6.8 g of potassium carbonate, 1.56 g ofcopper powder and 0.97 g of dibenzo-18-crown-6 were dissolved in 40 mL of o-dichlorobenzene, and the resultingsolution was refluxed at a steady 100C. After completion of the reaction, the reaction mixture was extracted withdichloromethane and distilled water, and the solvent was dried. The resulting solid was filtered and purified, yielding9-(3-(9H-carbazol-9-yl)phenyl)-3,6-dibromo-9H-carbazole as an intermediate.
With tetrakis(triphenylphosphine) palladium(0); tri-tert-butyl phosphine; potassium carbonate; In water; toluene; at 85℃; for 24h;Inert atmosphere;
4.3.1 3,6-di-n-butylcarbazole 1 A mixture of 3,6-dibromocarbazole (11.1 g, 34.3 mmol), n-<strong>[4426-47-5]butylboronic acid</strong> (8.41 g, 82.4 mmol), toluene (150 mL) and potassium carbonate (aq) (2 M, 50 mL) was deoxygenated with nitrogen for 30 min. Tetrakis(triphenylphosphine)palladium (0) (0.196 g, 0.170 mmol) and tri-tert-butylphosphine (0.208 g, 1.03 mmol) were added. The reaction mixture was stirred at 85 C for 24 h. After cooling to room temperature, water was added. The aqueous layer was extracted with toluene. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, filtered and concentrated invacuo. The crude material was purified by column chromatography on a silica gel (0:1-1:1 chloroform: hexane) to give 2 as a white solid (5.90 g, 62%). 1H-NMR (400 MHz, DMSO-d6) delta 10.90 (s, 1H), 8.85 (s, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 2.65 (t, J = 7.8 Hz, 4H), 1.60 (m, 4H), 1.30 (m, 4H), 0.85 (m, J = 7.4 Hz, 6H). C-NMR (400 MHz, CDCl3): delta 138.40, 134.05, 126.62, 123.64, 119.71, 110.43, 36.02, 34.79, 22.72, 12.38. EI-MS (M+m/z): Calcd. For C20H25N: 279.2, found: 279.
With tetrakis(triphenylphosphine) palladium(0); In ethanol; hexane; dichloromethane; water; toluene;
Example 1 Synthesis of Carbazole Derivative 1 3,6-Dibromocarbazole (14.31 g, 44.0 mol), <strong>[162607-20-7]5-methyl-2-thiophene boronic acid</strong> (25.01 g, 176.1 mmol) and tetrakis(triphenylphosphine)palladium (1.30 g) were added to a solvent mixture of toluene (180 mL) and ethanol (60 mL). Then, an aqueous solution of sodium carbonate (37.3 g) in distilled water (90 mL) was added to the mixture, followed by refluxing for 15 hours in a nitrogen atmosphere. Next, the resultant mixture was treated through hot filtration using a filtration aid to remove insoluble matter. Subsequently, the organic layer was separated, and the solvent was evaporated under reduced pressure. In addition, the residue was washed with water and dried to obtain a yellow-brown solid. Next, the obtained solid was purified through silica gel column chromatography using as an eluent a solvent mixture of methylene chloride/hexane (1/1 by volume), to thereby obtain 12.25 g of 3,6-bis(5-methylthiophen-2-yl)carbazole.
12.25 g
With tetrakis(triphenylphosphine) palladium(0); sodium carbonate; In ethanol; water; toluene; for 15h;Reflux; Inert atmosphere;
3,6-Dibromocarbazole (14.31 g, 44.0 mol), <strong>[162607-20-7]5-methyl-2-thiophene boronic acid</strong> (25.01 g, 176.1 mmol) and tetrakis(triphenylphosphine)palladium (1.30 g) were added to a solvent mixture of toluene (180 mL) and ethanol (60 mL). Then, an aqueous solution of sodium carbonate (37.3 g) in distilled water (90 mL) was added to the mixture, followed by refluxing for 15 hours in a nitrogen atmosphere. Next, the resultant mixture was treated through hot filtration using a filtration aid to remove insoluble matter. Subsequently, the organic layer was separated, and the solvent was evaporated under reduced pressure. In addition, the residue was washed with water and dried to obtain a yellow-brown solid. Next, the obtained solid was purified through silica gel column chromatography using as an eluent a solvent mixture of methylene chloride/hexane (1/1 by volume), to thereby obtain 12.25 g of 3,6-bis(5-methylthiophen-2-yl)carbazole.
With tris-(dibenzylideneacetone)dipalladium(0); tributylphosphine; sodium t-butanolate In toluene for 12h; Reflux;
1-2 1-2) Synthesis of compound 1-2
3,6-dibromo-9H-carbazole 20g (61.5 mmol), carbazole 20.6g (123.1 mmol), sodium tertiarybutoxide 11.8g (123.1 mmol), Pd2(dba)32.8g (3.1 mmmol) ), triteributylphosphine (a solution dissolved in 50% concentration in toluene) 1.5 ml (3.1 mmol) was suspended in 300 ml of toluene and stirred under reflux for 12 hours.Extracted with dichloromethane and distilled water, and the organic layer was purified with silica gel.The organic solution was removed and purified by a silica gel column to obtain compound 1-2, 21.4g (yield: 70%).
4.5%
With potassium phosphate; copper(l) iodide; N,N,N,N,-tetramethylethylenediamine In 1,4-dioxane at 135℃; for 20h; Inert atmosphere; Sealed tube;
Stage #1: 3,6-dibromo-9H-carbazole With potassium <i>tert</i>-butylate In N,N-dimethyl-formamide at 110℃; for 0.5h;
Stage #2: 4-fluorobenzoic acid ethyl ester In N,N-dimethyl-formamide at 70℃; for 12h;
Synthesis of EBCB and ETCB
3,6-Dibromocarbazole (1.63 g, 5 mmol) and Potassium tert-butoxide (0.56 g, 5 mmol) were dissolved in dry DMF (50 mL) in a flask fitted with a magnetic stirrer and condenser. The mixture was heated at 110 °C for 30 min and then Ethyl 4-fluorobenzoate (0.84 g, 5 mmol) was added with stirring for overnight. The mixture was cooled to room temperature, poured into ice-water, filtered, and the crude residue was recrystallized from ethanol. A white powder was obtained (1.96 g, 83% yield). 1H NMR (500 MHz, CDCl3) δ: 8.31 (d, J = 8.5 Hz, 2H), 8.21 (s, 2H), 7.62 (d, J = 8.5 Hz, 2H), 7.54 (d, J = 8.5 Hz, 2H), 7.32 (d, J = 8.5 Hz, 2H), 4.47 (dd, J = 7.0 Hz, 2H), 1.47 (t, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ: 167.33, 147.99, 141.05, 134.19, 130.66, 124.60, 124.18, 123.80, 117.46, 61.28, 14.70. MS (EI): calculated for C21H15Br2NO2 m/z: 473.16, found m/z: 473.06.
Stage #1: 3,6-dibromo-9H-carbazole With sodium hydride In tetrahydrofuran at 0℃;
Stage #2: 3-methoxy-N-(oxiran-2-ylmethyl)aniline In tetrahydrofuran at 20℃;
1 General procedure for the synthesis of various aromatic amine compounds (1-6)
General procedure: Epichlorohydrin (0.19 mL, 2.44 mmol, 1 equiv) was dissolved in DMF (8 mL). The reaction mixture was added ZnOTf (17.7 mg, 0.05 mmol, 0.02 equiv), aromatic amine compounds (2.44 mmol, 1 equiv) and stirred at room temperature for overnight. The reaction mixture was diluted with water and extracted with ethyl acetate (EA). The organic part was washed with water and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The crude was purified by column chromatography on silica gel to give pure compounds. To a solution of pure compounds (0.70 mmol, 1 equiv) in 1.4-dioxane (2.5 mL) was added KOH (47.5 mg, 0.85 mmol, 1.2 equiv) and stirred at room temperature for overnight. The reaction mixture was diluted with water and extracted with EA. The organic part was washed with water and brine. The organic layer was dried over MgSO4 and concentrated under reduced pressure. The residue was purified by column chromatographyon silica gel to give oxirane compounds. To a solutionof 3,6-dibromo-9H-carbazole (69.5 mg, 0.21 mmol, 1 equiv) in THF(1 mL) was added NaH (9.6 mg, 0.24 mmol, 1.1 equiv) and stirred at0 C. The oxirane compound (0.26 mmol, 1.2 equiv) was added andstirred at room temperature for overnight. Thin layer chromatography(TLC) showed complete consumption of starting material. Thereaction mixture was diluted with water and extracted with EA.The organic part was washed with water and brine. The organiclayer was dried over MgSO4 and concentrated under reduced pressure. The crude was purified by column chromatography on silicagel to afford the desired product. 4.1.2.1 1-(3,6-Dibromo-9H-carbazol-9-yl)-3-((3-methoxyphenyl)amino)propan-2-ol (1) 65% yield; white solid; mp = 140-141 °C; 1H NMR (300 MHz, CDCl3) δ 8.15 (d, 2H, J = 1.83 Hz), 7.56 (dd, 2H, J = 8.61, 1.83 Hz), 7.34 (d, 2H, J = 8.58 Hz), 7.08 (t, 1H, J = 8.25 Hz), 6.32 (d, 1H, J = 8.07 Hz), 6.23 (d, 1H, J = 7.35 Hz), 6.15 (t, 1H, J = 2.19 Hz), 4.41 (m, 3H), 3.73 (s, 3H), 3.32 (m, 1H), 3.20 (m, 1H); MS (ESI) m/z 504.90 [M+H]+.
3,6-bis(2-(9H-carbazol-9-yl)ethyl)-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
Example 1.7.1 3,6-bis(2-(9H-carbazol-9-yl)ethyl)-9H-carbazole (28) <strong>[1484-13-5]9-vinyl-9H-carbazole</strong> (27.0 g, 138.5 mmol) was dissolved in tetrahydrofuran and placed into an ice bath. 9-Borabicyclo[3.3.1]nonane (840.0 mL, 415.4 mmol) was added via a cannula to the reaction mixture. The reaction mixture was slowly brought to room temperature and stirred for about 3 hours under argon pressure. Sodium hydroxide (3M solution, 30.0 g, 750.0 mmol) was then added slowly into the reaction mixture. An additional amount of tetrahydrofuran was added to make the overall ratio of tetrahydrofuran to water 5:1. 3,6-Dibromo-9H-carbazole (15.1 g, 46.2 mmol) was then added to the reaction mixture. The reaction mixture was then degassed with argon for approximately one hour, and 1,1'-bis(diphenylphosphino)ferrocene]dichloropalladium(II) (1.0 g, 1.4 mmol) was added. The reaction mixture was then heated to about 50 C. overnight under argon pressure. The solvent was then removed, and the resulting residue was extracted with ethyl acetate and washed with water and brine. The ethyl acetate was then removed and the crude product was first purified by silica chromatography with 2:3 dichloromethane:hexanes as the eluent. The crude material was then purified by columns in both 2:3 dichloromethane:hexanes and 1:9 ethyl acetate:hexanes to remove the bottom and top spots impurities, respectively, to yield the product as a white solid 28 (64% yield). 1H NMR (400 MHz, DMSO-d): delta 11.05 (s, 1H), 8.16 (d, J=7.7 Hz, 4H), 8.10 (s, 2H), 7.68 (d, J=8.0 Hz, 4H), 7.47-7.43 (m, 4H), 7.40-7.31 (m, 4H) 7.20 (t, J=7.2 Hz, 4H), 4.70-4.66 (m, 4H), 3.19 (t, J=7.7 Hz, 4H)
5-(3,6-dibromo-9H-carbazol-9-yl)-N,N,N-trimethylpentan-1-aminium chloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.28 g
Example 1: Preparation of 5-(3,6-dibromo-9H-carbazol-9-yl)-N,N,N- trimethylpentan-l-aminium chloride (MTK-012) 1.00 g (3.0mM) 3,6-dibromocarbazole was dissolved in 100 ml dimethylformamide (DMF). 0.57 g (3.0 mM). <strong>[15008-33-0](5-bromopentyl)-trimethyl-ammonium bromide</strong> was added at once. After 10 min. of magnetic stirring 1.40 g (10 mM) potassium carbonate was added. After additional 10 min of stirring the temperature was raised to 50C and the mixture was stirred at this temperature for 4 h. After cooling to RT the solution was transferred to a separatory funnel and 200 ml of H20 and 200 ml dichloromethane were added. The solvent mixture was shaken and the lower phase was collected. The upper aqueous phase was extracted four times with 50 ml 3:1 dichloromethane methanol and the 5 lower phases were combined and washed with 100 ml saturated sodium chloride solution. Dried with MgSC , filtered and evaporated to dryness. The residue was crystallized from H20. Yield: 1.28g. Proton NMR in CD3OD: 1.31 m 2H, 1.70 m 2H, 1.93 m 2H, 3.01 s 9H, 3.16 m 2H, 4.39 t 2H, J=0.6, 7.47 d 2H, J= 2.0, 7.54 dd 2H, Jl= 2.0, 32= 0.4, 8.21 d 2H, J=0.4. MS: 451, 453, 455 M+ (symetrical 2Br triplet) 452, 454, 456 (MH)+ (symetrical 2Br triplet).
With potassium hydroxide; In N,N-dimethyl-formamide; at 20℃;Cooling with ice;
Chemistry. (S)-3,6-dibromo-9-(oxiran-2-ylmethyl)-9H-carbazole: A stirred solution of 3,6- dibromocarbazole (6.27 g, 19.3 mmol) and crushed potassium hydroxide pellets (1.30 g, 23.2 mmol) in dimethylformamide (100 ml) was cooled in ice before the slow drop wise addition of (R)-glycidyl75 3-nitrobenzenesulfonate, (20) (5.01 g, 19.3 mmcl). The ice bath was removed and the reaction was stirred overnight at ambient temperature. The reaction mixture was diluted with water and washed several times with water and then brine. The organic layer was dried over Na2SO4, filtered, and concentrated to give epoxide in quantitative yield. Crude material was carried forward. 1H NMR(CDCI3, 400 MHz) : 5 8.13 (d, J = 2.0 Hz, 2H), 7.57 (dd, J = 8.7, 1.8 Hz, 2H), 7.39 -7.31 (m, 2H), 4.65(dd, J = 16.0, 2.5 Hz, 1H), 4.28 (dd, J = 16.1, 5.0 Hz, 1H), 3.32 (ddd, J = 5.2, 2.6, 1.3 Hz, 1H), 2.82 (t, J4.3 Hz, 1H), 2.50 (dd, J = 4.7, 2.6 Hz, 1H). HPLC conditions: ChiralPak AD-H column, flowrate = 1.0mI/mm, 1 % IPOH/hexanes. Retention time of peak 1(major) is 24.3 - 34.0 minutes and retentiontime of peak 2 (minor) is 48.4- 58.6 minutes; ee = 95.9 %. [a]L,20= -1-5.737 (c = 0.244, THE)
Stage #1: 3,6-dibromo-9H-carbazole With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride In water; N,N-dimethyl-formamide for 0.75h; Inert atmosphere;
Stage #2: zinc(II) cyanide With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; zinc diacetate; zinc In water; N,N-dimethyl-formamide at 100℃; for 20h; Inert atmosphere;
9H-Carbazole-3,6-dicarbonitrile (2)
To a mixture of DMF (30 mL) and water (0.3 mL) in a 150-mLround-bottom flask, 3,6-dibromo-9H-carbazole (1; 9.75 g, 30mmol) and dppf (80 mg, 0.144 mmol, 0.48 mol%) were added. Theflask was evacuated/backfilled with argon (3 ×) and then argon was bubbled through the solution for 45 min. Subsequently, Zn(CN)2(4.21 g, 36 mmol, 1.2 equiv), Zn (78 mg, 1.2 mmol, 4 mol%),Zn(OAc)2 (0.22 g, 1.2 mmol, 4 mol%) and Pd2(dba)3 (55 mg, 0.06mmol, 0.2 mol%) were added in one portion under a positive pressureof argon. The flask was quickly evacuated/backfilled with argon(2 ×), then immediately placed in a preheated oil bath (100 °C),and the mixture was vigorously stirred for 20 h. For workup, a literatureprocedure19 was followed: the mixture was cooled, pouredinto aq NH4Cl/aq NH3/DI water (4:1:5, 100 mL) and filtered. Thefilter cake was washed again with the same volume of the abovemixture, and then thoroughly with DI water, followed by toluene(3 × 30 mL) and MeOH (3 × 30 mL), and then dried under air withsuction. A pale yellow solid was obtained; yield: 6.3 g (97%). Exclusivelyfor determination of the melting point, a sample was recrystallizedfrom DMF to provide a white solid; mp 390 °C (dec)[Lit.17b,c >360 °C (dec)]. FT-IR (neat solid): 3291, 2215, 1871, 1633, 1598, 1482, 1455,1402, 1297, 1257, 1224, 1189, 1133, 1050, 1020, 931, 899, 876,812, 801, 742 cm-1.1H NMR (600.1 MHz, DMSO-d6): δ = 12.4 (s, 1 H), 8.8 (d, J = 1.1Hz, 2 H), 7.8 (dd, J = 1.6, 8.4 Hz, 2 H), 7.7 (d, J = 8.4 Hz, 2 H).13C NMR (150.9 MHz, DMSO-d6): δ = 142.3, 129.9, 126.3, 121.8,120.1, 112.8, 101.7.HRMS (ESI): m/z [M + H]+ calcd for C14H8N3: 218.07127; found:218.07041; m/z [M - H]+ calcd for C14H6N3: 216.05562; found:216.05575.Anal. Calcd for C14H7N3: C, 77.41; H, 3.25; N, 19.34. Found: C,76.92; H, 3.45; N, 18.85.
81%
With 1,1'-bis-(diphenylphosphino)ferrocene; tris-(dibenzylideneacetone)dipalladium(0); zinc(II) acetate dihydrate; zinc In water; N,N-dimethyl-formamide at 110 - 120℃; for 48h; Inert atmosphere;
Carbazole-3,6-Dicarbonitrile (3)
3,6-Dibromocarbazole 2 (9.75 g,30.0 mmol) and dppf (100 mg, 0.18 mmol) were added to a 100 mLSchlenk flask and solved in 30 mL DMF and 0.3 mL water. The suspensionwas degassed via bubbling argon for 1 h through the mixture.Subsequently, Zn(CN)2 (4.21 g, 36 mmol), zinc powder (78 mg, 1.2mmol), Zn(OAc)2 2 H2O (0.26 g, 1.2 mmol) and Pd2(dba)3 dba (69.5mg, 0,06 mmol) were added under a positive pressure of argon. Thismixture was heated to 110 C for 2 days. The suspension was subsequentlycooled and then poured into a 100 mL mixture of H2O/NH4Cl/NH3 (5/4/1) and filtered through a suction filter. The filter cake waswashed with the same volume of the above mixture, toluene (3 x 30 mL)and MeOH (3 x 30 mL) to give a grey solid. The crude product wasrecrystallized with DMF to give a white solid (3) (5.2 g, 81%). 1H NMR(400 MHz, 298K, DMSO-d6) (ppm) = 7.72 (d, J = 8.5 Hz, 2H), 7.85 (d,J = 9.9 Hz, 2H), 8.80 (s, 2H), 12.38 (s, 1H, N-H). 13C NMR (101 MHz,298K, DMSO-d6) (ppm) = 101.74, 112.84, 120.10, 121.85, 126.8,129.93, 142.32.
5-(3,6-dibromo-9H-carbazol-9-yl)-N,N,N-trimethylpentan-1-aminium[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
1.28 g
Example 1 Preparation of 5-(3,6-dibromo-9H-carbazol-9-yl)-N,N,N-trimethylpentan-1-aminium chloride (MTK-012) 1.00 g (3.0 mM) 3,6-dibromocarbazole was dissolved in 100 ml dimethylformamide (DMF). 0.57 g (3.0 mM). <strong>[15008-33-0](5-bromopentyl)-trimethyl-ammonium bromide</strong> was added at once. After 10 min of magnetic stirring 1.40 g (10 mM) potassium carbonate was added. After additional 10 min of stirring the temperature was raised to 50 C. and the mixture was stirred at this temperature for 4 h. After cooling to RT the solution was transferred to a separatory funnel and 200 ml of H2O and 200 ml dichloromethane were added. The solvent mixture was shaken and the lower phase was collected. The upper aqueous phase was extracted four times with 50 ml 3:1 dichloromethane:methanol and the 5 lower phases were combined and washed with 100 ml saturated sodium chloride solution. Dried with MgSO4, filtered and evaporated to dryness. The residue was crystallized from H2O. Yield: 1.28 g. Proton NMR in CD3OD: 1.31 m 2H, 1.70 m 2H, 1.93 m 2H, 3.01 s 9H, 3.16 m 2H, 4.39 t 2H, J=0.6, 7.47 d 2H, J=2.0, 7.54 dd 2H, J1=2.0, J2=0.4, 8.21 d 2H, J=0.4. MS: 451, 453, 455 M+ (symetrical 2Br triplet) 452, 454, 456 (MH)+ (symetrical 2Br triplet).
With sodium carbonate; In 1,4-dioxane; dichloromethane; water;
(2a) Preparation of 3,6-diphenylcarbazole 25g 3,6-dibromocarbazole and 20.5g phenylboronic acid were mixed in 150mL water and 300mL dioxane in glove box. Stir vigorously and add 45g sodium carbonate. Finally add 3g Pd2DBA3 and 1.5g tri-t-butylphosphine. Stir and reflux overnight then cool and remove from glove box. Add water to precipitate white solid then filter and collect. Dissolve in dichloromethane and chromatograph on silica eluting with DCM:hexanes 1:2 and elute product as colorless solution which on evaporation and washing with hexanes gives the desired product in ~80percent yield as a fluffy white solid.
With caesium carbonate In N,N-dimethyl-formamide at 150℃; for 24h;
Metal-Free N-Arylation of Carbazoles; General Procedure
General procedure: A mixture of a fluorinated aryl halide (2.0 mmol), a carbazole (0.5 mmol), and a base (2.0 mmol) in solvent (2 mL) was allowed to react under air atmosphere. The reaction mixture was heated to the specified temperature for 24 h. After reaction completion, the mixture was added to brine (15 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined extract was concentrated under reduced pressure and the product was isolated by short chromatography on a silica gel (200-300 mesh) column.
With caesium carbonate In N,N-dimethyl acetamide at 150℃; for 24h;
With caesium carbonate In N,N-dimethyl-formamide at 150℃; for 24h;
With caesium carbonate; In N,N-dimethyl-formamide; at 150℃; for 24h;
General procedure: A mixture of a fluorinated aryl halide (2.0 mmol), a carbazole (0.5 mmol), and a base (2.0 mmol) in solvent (2 mL) was allowed to react under air atmosphere. The reaction mixture was heated to the specified temperature for 24 h. After reaction completion, the mixture was added to brine (15 mL) and extracted with CH2Cl2 (3 × 15 mL). The combined extract was concentrated under reduced pressure and the product was isolated by short chromatography on a silica gel (200-300 mesh) column.
3',6'-dibromo-9-phenyl-9H-3,9'-bicarbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium carbonate;
Example 4 Synthesis of 3',6'-dibromo-9-phenyl-9H-3,9'-bicarbazole A mixture of 32.5 g (100 mmole) 3,6-dibromo-9H-carbazole, 36.9 g (100 mmole) <strong>[502161-03-7]3-iodo-9-phenyl-9H-carbazole</strong>, 9.5 g (150 mmole) of copper powder, 27.6 g (200 mmole) of potassium carbonate, and 600 ml dimethylformamide were heated at 130 C. under nitrogen overnight, then cooled to room temperature, the solution was filtered. The filtrate was extracted three times with dichloromethane and water, dried with anhydrous magnesium sulfate, the solvent was evaporated in vacuum. The residue was purified by column chromatography on silica(hexane-dichloromethane) afforded a white solid (34.5 g, 61 mmol, 61%)
4.05 g (101.3 mmol) of sodium hydride (NaH) was put in a 1 L round flask, and 100 mL of dimethylformamide (DMF) was added thereto. Next, a solution prepared by dissolving 21.95 g (67.5 mmol) of 3,6-dibromocarbazole in 100 mL of DMF was slowly added in a dropwise fashion and then the re<strong>[2915-16-4]su</strong>ltant was agitated at room temperature for 40 minutes. 21.62 g (81.0 mmol) of <strong>[2915-16-4]2-chloro-4,6-diphenylpyrimidine</strong> was slowly added in a dropwise fashion, and the mixture was agitated for 6 hours. The re<strong>[2915-16-4]su</strong>lting reactant was poured into water to complete the reaction, and a solid produced therein was filtered. The solid was washed with water and methanol and then heated and dissolve in 400 mL of chlorobenzene, and hexane was added thereto for solidification. The obtained solid was filtered and dried in a vacuum oven, obtaining 34.9 g of a compound E (a yield 93%).
Potassium hydroxide (1.71g, 0.031 mol) was added in dibromo carbazole (5.0 g, 0.015 mol) dissolved in 20 ml of DMSO. The solutionwas heated for 3 hours at 50 ~ 60 oC. 1- bromo -6- chloro hexa (3.7g, 0.018 mol) was slowly added and it was heated for 2 hours at 50 ~ 60 o C.After confirming that the reaction was complete, the salt which had beenproduced during the reaction and potassium hydroxide were removed by using waterand chloroform. Then, the solvent was removed through adistillation under reduced pressure. Using methylene chloride and hexane (1:5), it was also separated by achromatography to obtain a white solid (4.8 g, 0.0108 mol, 72percent).
4-(1-bromo-9H-carbazol-9-yl)benzofuro[3,2-d]pyrimidine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
The 3,6-dibromo-9H-carbazole (1.0 g, 3.1 mmol) and sodium hydride (0.2 g, 5.2 mmol) were mixed in dry DMF 40 mL. The solution was stirred for 4 hours and the addition of <strong>[39876-88-5]4-chlorobenzofuro[3,2-d]pyrimidine</strong> (1.2 g, 5.9 mmol). The mixture was refluxed under nitrogen overnight. The reaction mixture was placed in water and the precipitate was then filtered off. After the residue was purified by column chromatography using THF as eluent. 13.5 was collected yellow product of g (99%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In toluene; for 15h;Inert atmosphere; Reflux;
Under nitrogen atmosphere, 3,6-dibromo-9H-carbazole 3.2g (10mmol) , <strong>[108847-76-3]thianthren-1-ylboronic acid</strong> 5.4g (0.5mmol) was dissolved in toluene 10ml Pd(PPh3)4 72mg (0.062mmol), 25ml 2M K2CO3 (50mmol) was added and refluxed for 15 hours. After completion of the reaction, MC 200ml, H2O 200ml was added and then the the MC layer was extracted, concentrated and separated by column chromatography using Hex : EA =5 : 1 and dried over anhydrous MgSO4 to give the intermediate Q 4.29g (72%).
tetramethyl 5,5'-(9H-carbazole-3,6-diyl)diisophthalate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 18h;Inert atmosphere;
Under a nitrogen atmosphere, to a solution of 3,6-dibromo-9Hcarbazole(325 mg, 1 mmol) in DMF solvent (30 mL) were added<strong>[944392-68-1]dimethyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)isophthalate</strong>(640 mg, 2 mmol), Pd(PPh3)4 (93 mg, 0.05 mmol), K2CO3(324 mg, 3 mmol). After the mixture was stirred at 100 C for18 h, then filtered and concentrated. The crude product was purifiedby column chromatography on silica gel with dichloromethane/methanol (100:5, V:V) to give pure tetramethyl 5,50-(9H-carbazole-3,6-diyl)diisophthalate as a gray solid. Yelid:330 mg, 60%. 1H NMR (600 MHz, DMSO-d6): delta 11.557 (s, 1H),8.858 (s, 2H), 8.559 (s, 4H), 8.439 (s, 2H), 7.832 (d, 2H), 7.645 (d,2H), 4.519(s, 12H). MS m/z 550.2 (M+H+).
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In tetrahydrofuran; water; for 48h;Inert atmosphere; Reflux;
3,6-dibromocarbazole (1.97 g, 6.05 mmol) under closed, argon-containing conditionsN,N-diphenyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)amine (5.63 g, 15.15 mmol),The catalyst tetrakistriphenylphosphine palladium (Pd(PPh3)4) (0.70 g, 0.61 mmol) was dissolved in tetrahydrofuran (80 ml).After heating to reflux, the aqueous K2CO3 solution (2 mol/L, 30.5 ml, 60.5 mmol) was added to the reaction system, and the reaction was carried out under reflux for 48 hours.The solvent was distilled off and the column chromatography was carried out. The eluent was petroleum ether/dichloromethane = 4/1 (volume ratio).It was then recrystallized from tetrahydrofuran/ethanol to give a white powder (yield: 88%).
80%
With tetrabutylammomium bromide; sodium carbonate; triphenylphosphine; In toluene; at 110℃; for 18h;Inert atmosphere;
Under argon atmosphere,3,6-dibromocarbazole (5 g, 915.38 mmol) andTriphenylamine borate (17.14 g,46.15 mmol) was added to two vials,Then add 100ml toluene to complete dissolution,Sodium carbonate (8.15 g,76.92 mmol),Tetrabutylammonium bromide (312.01 mg, 967.86 mol)withTetraphenylphenylphosphine (355.56 mg,307.69umol),The reaction was carried out at 110 C for 18 h.The reaction mixture was poured into water,Extracted with ethyl acetate,After the organic layer was completely washed with brine,Add anhydrous magnesium sulfate dry.After the solution was concentrated,Purification by silica gel column chromatography (eluent selection petroleum ether / dichloromethane = 6/1, v / v) gave a white solid in 80% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
Under argon atmosphere, 3,6-dibromocarbazole (5 g, 915.38 mmol) and triphenylamine borate(17.14 g, 46.15 mmol) was added to two vials and 100 ml of toluene was added for complete dissolution,Sodium carbonate (8.15 g, 76.92 mmol) was added,Tetrabutylammonium bromide (312.01 mg, 967.86 mol) andTetraphenylphenyl phosphatePalladium (355.56 mg, 307.69 mol)The reaction was carried out at 110 C for 18 h.The reaction mixture was poured into water, extracted with ethyl acetate, and the organic layer was completely washed with brine,Add anhydrous magnesium sulfate dry. The solution was concentrated and purified by silica gel column chromatography (eluent selection of petroleum ether / bisMethyl chloride = 6/1, v / v) to give a white solid in 80% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
Under argon atmosphere,A mixture of 3,6-dibromocarbazole (5 g, 915.38 mmol)And triphenylamine borate (17.14 g, 46.15 mmol) were added to two vials,Then add 100ml toluene to complete dissolution,Sodium carbonate (8.15 g, 76.92 mmol) was added,Tetrabutylammonium bromide (312.01 mg, 967.86 mol)And tetraphenylphenylphosphine (355.56 mg, 307.69 mol)The reaction was carried out at 110 C for 18 h.The reaction mixture was poured into water,Extracted with ethyl acetate, and the organic layer was completely washed with brine,Add anhydrous magnesium sulfate dry. After the solution was concentrated,Purification by silica gel column chromatography (eluent selection of petroleum ether / dichloromethane = 6/1, v / v)The final white solid was obtained in 80% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
Under an argon atmosphere, 3,6-dibromocarbazole (5 g, 915.38 mmol) and triphenylamine borate (17.14 g,46.15 mmol) was added to two vials and 100 ml of toluene was added for complete dissolution followed by addition of sodium carbonate (8.15 g,(35.01 mg, 967.86 mol) and tetraphenylphenylphosphine (355.56 mg, 307.69 mol) were reacted at 110 C for 18 h. The reaction mixture was poured into water,After extraction with ethyl acetate, the organic layer was washed thoroughly with brine and dried over anhydrous magnesium sulfate. The solution was concentrated and purified by silica gel column chromatography (eluent selection of petroleum ether / diMethyl chloride = 6/1, v / v) to give a white solid in 80% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
Under argon atmosphere,A mixture of 3,6-dibromocarbazole (5 g, 915.38 mmol)And triphenylamine borate (17.14 g, 46.15 mmol)Add to two bottles,Then add 100ml toluene to complete dissolution,Sodium carbonate (8.15 g, 76.92 mmol) was added,Tetrabutylammonium bromide (312.01 mg, 967.86 mol)And tetrakis triphenylphosphine palladium (355.56mg, 307.69umol),The reaction was carried out at 110 C for 18 h.The reaction mixture was poured into water,Extracted with ethyl acetate,After the organic layer was completely washed with brine,Add anhydrous magnesium sulfate dry.After the solution was concentrated,Purification by silica gel column chromatography (eluent selection petroleum ether / dichloromethane = 6/1, v / v)The final white solid was obtained in 80% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
In the argon atmosphere, will be 3, 6 - dibromo carbazole (5 g, 915.38 mmol) and triphenylamine borate (17.14 g, 46 . 15 mmol) is added to the two bottles in, add 100 ml toluene completely dissolved, add the sodium carbonate (8.15 g, 76 . 92 mmol), tetrabutyl ammonium bromide (312.01 mg, 967 . 86 umol) and four triphenyl phosphate palladium (355.56 mg, 307 . 69 umol), in 110 C reaction under 18 h; the reaction mixture is poured into water, extracted with ethyl acetate, the organic layer using salt water completely after washing, add anhydrous magnesium sulfate drying; solution after concentration, purification with silica gel column chromatography (elution agent selected petroleum ether/dichloromethane=6/1, v/v), at the end of the white solid, yield 80%.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
Under argon atmosphere, 3,6-dibromocarbazole (5 g, 915.38 mmol) and triphenylamine boronate (17.14 g, 46.15 mmol) were added to the two-necked flask, and 100 ml of toluene was further added for complete dissolution Followed by sodium carbonate (8.15 g, 76.92 mmol), tetrabutylammonium bromide (312.01 mg, 967.86 umol) and palladium tetrakistriphenylphosphine (355.56 mg, 307.69 mol) and reacted at 110 C for 18 h. The reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was washed thoroughly with brine and then dried over anhydrous magnesium sulfate. The solution was concentrated and purified by silica gel column chromatography (eluent petroleum ether / methylene chloride = 6/1, v / v) to give a white solid in a yield of 80%.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;
Under an argon atmosphere,Add 3,6-dibromocarbazole (5g, 915.38mmol) and triphenylamine boronate (17.14g, 46.15mmol) into two-necked flask and add 100ml of toluene for complete dissolution. Add sodium carbonate (8.15g, 76.92 mmol), tetrabutylammonium bromide (312.01 mg, 967.86 umol) and palladium tetrakistriphenylphosphine (355.56 mg, 307.69 umol) were added and reacted at 110 C for 18 h; the reaction mixture was poured into water and extracted with ethyl acetate The organic layer was washed thoroughly with brine and dried over anhydrous magnesium sulfate. After the solution was concentrated, the residue was purified by silica gel column chromatography (eluting with petroleum ether / methylene chloride = 6/1, v / v) A white solid was obtained in 80% yield.
80%
With tetrakis(triphenylphosphine) palladium(0); tetrabutylammomium bromide; sodium carbonate; In toluene; at 110℃; for 18h;Inert atmosphere;
Under an argon atmosphere, 3,6-dibromocarbazole (5 g, 915.38 mmol) and triphenylamine boronate (17.14 g,46.15 mmol) was added to two bottles, 100 ml of toluene was added for complete dissolution, sodium carbonate (8.15 g, 76.92 mmol), tetrabutylammonium bromide (312.01 mg, 967 · 86 μmol) and tetraphenylphosphine palladium (355.56 mg, 307.69 umol) was reacted at 110 C. for 18 h; the reaction mixture was poured into water and extracted with ethyl acetate. The organic layer was completely washed with brine and dried over anhydrous magnesium sulfate; the solution was concentrated. After purification by silica gel column chromatography (eluent selected petroleum ether/dichloromethane = 6/1, v/v), a white solid was finally obtained with a yield of 80%.
N,N'-bis(9,9-dimethyl-9H-fluoren-2-yl)-N,N'-diphenyl-9H-carbazole-3,6-diamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With tris-(dibenzylideneacetone)dipalladium(0); tributylphosphine; sodium t-butanolate; In toluene; at 120℃; for 24h;Inert atmosphere;
In a 500 ml three-opening in the bottle, through under the protection of nitrogen, adding 0.03 muM 3, 6 - dibromo carbazole, 0.07 muM raw material B3, 250mL toluene stirring and mixing, and then adding 0.09 muM tert sodium butylate, 0.0015 muM Pd2 (Dba)3 , 0.002 muM butyl phosphine tri, heating to 120 C, reflux reaction for 24 hours; the natural cooling to room temperature, filtered, and the filtrate is pressure reducing and steaming and (-0.09 mpa, 85 C), through neutral silica gel column, to obtain the intermediate P3
With copper(l) iodide; caesium carbonate; In N,N-dimethyl-formamide; at 110℃; for 12h;Inert atmosphere;
A.3,6-dibromocarbazole (200 mg, 0.62 mmol) was dissolved in DMF solvent,Then iodobenzene (188.3 mg, 0.92 mmol) was added,DL-piperidinecarboxylic acid (11.88 mg, 0.09 mmol),Cuprous oxide (8.27 mg, 0.04 mmol),Cesium carbonate (299.75 mg, 0.92 mmol),Under nitrogen protection,The reaction system was placed at 110 ° C under reflux for 12 h,After the reaction is complete,Ethyl acetate extraction,Organic layer washed three times,Dried over anhydrous sodium sulfate,The solvent is evaporated under reduced pressure;The crude product was purified by column chromatography with ethyl acetate and petroleum ether mixed solvent (20: 1)To obtain compound 3,6-dibromo-9-phenylcarbazole (white solid, 225 mg, yield 90.5percent).
9-(6-(9H-carbazol-9-yl)hexyl)-3,6-dibromo-9H-carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
Stage #1: 3,6-dibromo-9H-carbazole; N-(6-bromohexyl)carbazole With tetrabutylammomium bromide In toluene for 0.25h; Inert atmosphere;
Stage #2: With potassium carbonate In water; toluene at 90℃; for 18h; Inert atmosphere;
Synthesis of 9-(9-(6-hexyl)-carbazole)-3,6-dibromocarbazole (M1)
Under nitrogen atmosphere, 9-(6-brominehexyl)-carbazole (990 mg, 3 mmol), 3,6-dibromocarbazole (975 mg, 3 mmol) and tertbutyl ammonium bromide (TBAB) (322 mg, 1 mmol) were all added into 50 mL flask with 25 mL toluene solution and stirred for 15 min. Then 5 mL K2CO3 aqueous solution (2 M) was added and the mixture was slowly heated to 90°C for 18 h. The reaction solution was stopped by adding some water, extracted by CH2Cl2 and dried by anhydrous MgSO4, and then the organic layer was evaporated to obtain the crude product. Finally, column chromatography on silica gel with the eluent of petroleum ether/CHCl2 (5:1) was used to purify the crude product. Yield: 1.25 g, 73%. M.P. 110-115°C. 1H NMR (600 MHz, CDCl3) d(ppm) 8.11 (d, J 1.8 Hz, 2 H), 8.08 (d,J 7.8 Hz, 2 H), 7.48 (dd, J1 8.4 Hz, J2 1.8 Hz, 2 H), 7.43 (ddd,J1 8.4 Hz, J2 7.2 Hz, J3 1.2 Hz, 2 H), 7.31 (d, J 7.8 Hz, 2 H), 7.22(t, J 7.2 Hz, 2 H), 7.12 (d, J 9 Hz, 2 H), 4.24 (t, J 6.6 Hz, 2 H), 4.12(t, J 6.6 Hz, 2 H), 1.84-1.78 (m, 2 H), 1.77-1.71 (m, 2 H), 1.37-1.24(m, 4 H). 13C NMR: (600 MHz, CDCl3): d(ppm) 140.4, 139.3, 129.1,125.7, 123.5, 123.4, 122.9, 120.5, 118.9, 112.1, 110.4, 108.7, 43.1, 42.9, 28.9, 28.7, 27.1, 27.0. Element anal. calcd for C30H26Br2N2 (%): C,62.74; H, 4.56; N, 4.88. Found: C, 63.09; H, 4.77; N, 4.65. MALDI-TOF-MS (m/z): calcd for C30H26Br2N2, 574.35; found, 575.07.
With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; lithium hexamethyldisilazane; tri tert-butylphosphoniumtetrafluoroborate In tetrahydrofuran at 65℃; for 10h; Inert atmosphere;
23 Example-23
Under argon atmosphere3,6-dibromocarbazole (325 mg, 1.00 mmol),Diphenylamine (374 mg, 2.21 mmol),Tris (dibenzylideneacetone) dipalladium / chloroform adduct (11 mg, 10 μmol)Tetrahydrofuran (2 mL) was added to a mixture of tri-tert-butylphosphonium tetrafluoroborate (15 mg, 51 μmol).Lithium bis (trimethylsilyl) amide (1M tetrahydrofuran solution, 3.3 mL, 3.3 mmol) was added to the resulting mixture,Stir at 65 ° C. for 10 hours.After cooling to room temperature, the reaction solution was filtered using silica gel and washed with chloroform.The filtrate was concentrated under reduced pressure, and the resulting crude product was purified by silica gel column chromatography (hexane / ethyl acetate = 9/1),The target 3,6-bis (diphenylamino) carbazole was obtained as a pale yellow solid (482 mg, 0.962 mmol, yield 96%).
70%
With tris-(dibenzylideneacetone)dipalladium(0); tri-tert-butyl phosphine; sodium t-butanolate In toluene for 12h; Reflux;
2.2-1 2-1) Synthesis of compound 2-1
3,6-dibromo-9H-carbazole 10g (30.8 mmol), diphenylamine 5.2g (30.8 mmol), sodium tertiarybutoxide 5.9g (61.5 mmol), Pd2(dba) 31.4g (1.5 mmmol) , Tritertiary butylphosphine (50 wt% in toluene) 1.5 ml (3.1 mmol) was suspended in 50 ml of toluene and stirred under reflux for 12 hours. Extracted with dichloromethane and distilled water, and the organic layer was filtered with silica gel. The organic solution was removed and subjected to silica gel column to obtain compound 2-1, 10.8 g (yield: 70%).
With tris-(dibenzylideneacetone)dipalladium(0); tri-tert-butyl phosphine; sodium t-butanolate In toluene at 110℃; for 24h; Inert atmosphere;
1 Take intermediate M1-2 as an example:
In a 250-ml three-necked flask, 0.05 mol of raw material J-1, 0.12 mol of raw material II-1, and 100 ml of toluene were added and mixed by stirring. After deoxygenation, 0.0025 mol of Pd2(dba)3, 0.075 mol of tri-tert-butylphosphine and sodium tert-butoxide were added, and the reaction was carried out at 110° C. in an inert atmosphere for 24 hours. The progress of the reaction was continuously monitored by TLC during the reaction. After the reaction of the raw materials is completed, the mixture is cooled and filtered, the filtrate is evaporated to remove the solvent, and the crude product is passed through a silica gel column to obtain the intermediate M1-2.
With tris(dibenzylideneacetone)dipalladium(0) chloroform complex; tri-tert-butyl phosphine; sodium t-butanolate; In toluene; at 115℃; for 24h;Inert atmosphere;
1) In a 250 ml three-necked flask, 0.03 mol of 3,6-dibromo-9H-carbazole was added under nitrogen protection.0.036 mol of 9,9-dimethyl-N-phenyl-9H-indole-2-amine, 150 ml of toluene, stirred and mixed, and then added 0.045 mol of sodium t-butoxide.0.0015mol Pd2(dba)3, 0.0015mol of tri-tert-butylphosphine, heated to 115 C with stirring, reflux reaction for 24 hours,Sampling point plate, showing no 3,6-dibromo-9H-carbazole remaining, the reaction is complete; naturally cooled to room temperature, filtered, and the filtrate was steamed under reduced pressure.To a fraction free, over a neutral silica gel column to give intermediate 2-1.HPLC purity 99.3%, yield 65.9%;
With copper(l) iodide; sodium In N,N-dimethyl-formamide at 120℃; for 4h;
1.1-2 1-2: Synthesis of 3,6-bis (2-ethylhexyloxy) -9H-carbazole (b)
The obtained 3,6-dibromo-9H-carbazole (a) (8.1 g, 25.08 mmol)And 2-ethylhexan-1-ol (39.20 g, 301.02 mmol),Sodium (6.82 g, 301.02 mmol) and CuI (11.94 g, 62.71 mmol)Were dissolved in 500 ml of DMF, and the mixture was stirred under reflux at 120 ° C for 4 hours.After completion of the reaction, the solvent was removed, and the reaction product was extracted with water and ethyl acetate.Dried over MgSO4 and then purified by column chromatography (n-hexane / DCM = 3/1) to give the title compound. The yield was 70% (7 g).
With sodium hydride In N,N-dimethyl-formamide at 90℃; for 24h; Schlenk technique; Inert atmosphere;
3,6-Dibromo-9-(2-(2-(2-methoxyethoxy)ethoxy)ethyl)-9H-carbazole (3)
To a solution of di-bromo-9H-carbazole (1) (1 g, 3.07 mmol), in anhydrous DMF (15 ml), NaH (160 mg, 4 mmol) and 1-bromo-2-(2-(2-methoxyethoxy)ethoxy)ethane (840 mg, 3.7 mmol) were added. The reaction mixture was stirred at 90 °C for 24 h. After consumption of carbazole substrate (the reaction was monitored by TLC, n-hexane: ethyl acetate was used as the eluent) the reaction mixture was poured into water (200 ml), extracted with ethyl acetate (3×50 ml) and dried over anhydrous MgSO4. The solvent was removed under diminished pressure and the residue was purified by column chromatography eluting with n-hexane: ethyl acetate (1:4) to afford colourless liquid with 70 % yield (1.01 g). 1H NMR (400 MHz, CDCl3): δ 8.12 (s, 2H, carbazolyl), 7.54 (d, 2H, J=8Hz, carbazolyl), 7.34 (d, 2H, J=8Hz, carbazolyl), 4.43 (t, 2H, J=2Hz, -NCH2-), 3.83 (t, J=2Hz, -NCH2CH2-O-), 3.50-3.40 (m, 8H), 3.34 (s, 3H, -OCH3). 13C{1H} NMR (100 MHz, CDCl3): δ 139.4, 128.9, 123.3, 122.3, 112.0 (carbazolyl carbon attached to bromine), 110.7 (carbazolyl carbon attached to bromine), 71.8 (alkyl carbon attached to N), 70.9, 70.5, 70.1, 69.3, 58.9, 43.4 (-OCH3). MALDI-TOF (m/z): C19H21Br2NO3, calculated value 471.986 (M)+, found 471.521 (M)+.
With tetrakis(triphenylphosphine) palladium(0); sodium hydroxide; In tetrahydrofuran; water; at 80℃;
The starting material, 2-([1,1'-biphenyl]-4-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (34.83 g, 124.3 mmol) was dissolved in THF in a round bottom flask, 3,6-dibromo-9H-carbazole (60.6 g, 186.5 mmol), Pd(PPh3)4 (7.18 g, 6.2 mmol), NaOH (14.91 g, 372.8 mmol) and water were added and stirred at 80 C. After completion of the reaction, the reaction mixture was extracted with CH2Cl2 and water. The organic layer was dried over MgSO4 and concentrated. The resulting compound was purified by silicagel column and recrystallized to obtain 41.71 g (yield: 84%) of the product
bis(4-(3,6-dibromo-9H-carbazol-9yl)phenyl)methanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With sodium cyanide In N,N-dimethyl-formamide at 100℃; for 3.5h; Inert atmosphere;
1; 3
S1. In a dry flask, add 3,6-dibromocarbazole and 4,4'-difluorobenzophenone in an amount ratio of 2.2: 1, and then add an appropriate amount of sodium cyanide,N, N dimethylformamide, nitrogen-filled deoxidation, fully stirred for 0.5h, and then reacted at 100 ° C for 3h under anhydrous and anaerobic conditions;S2, the system is cooled to room temperature after the reaction, most of the solvent is distilled off under reduced pressure, a large amount of water is added, a small amount of sodium chloride is added at the same time, and the mixture is left to stand for 15 minutes, and then filtered to obtain a crude product. The mixed solvent of ethyl acetate and petroleum ether and the mixed solvent of tetrahydrofuran and petroleum ether with a volume ratio of 1: 8 were thoroughly washed and vacuum filtered, and the obtained solid was dried under vacuum for a period of time to obtain the final product. The final product was bis (4- (3,6-dibromo-9H-carbazol-9yl) phenyl) methanone (DBCz-BP) with a yield of 86.3%.
9,9'-(6-(3,6-dibromo-9H-carbazol-9-yl)-1,3,5-triazine-2,4-diyl)bis(9H-carbazole)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
Stage #1: 3,6-dibromo-9H-carbazole With n-butyllithium In tetrahydrofuran; hexane for 0.5h; Inert atmosphere;
Stage #2: 9,9’-(6-chloro-1,3,5-triazine-2,4-diyl)bis(9H-carbazole) In tetrahydrofuran; hexane at 20℃; for 12.1667h; Inert atmosphere;
With caesium carbonate In N,N-dimethyl-formamide at 150℃; for 12h; Inert atmosphere;
Preparation of M5:
Weigh 3,6-dibromocarbazole (1.625g, 5mmol), cesium carbonate (1.95g, 6mmol), 3-fluorobenzophenone (0.9g, 4.5mmol,) and pour into double Set up a reflux condensing device, vacuum and fill with nitrogen, measure 50mL N,N-dimethylformamide solvent into the flask, reflux and condense at 150°C for 12h, and obtain the crude product using silica gel column chromatography (eluent: two Methyl chloride/petroleum ether = 2/1, v/v), 0.243 g of light yellow solid was obtained (yield: 11%)
Stage #1: 3,6-dibromo-9H-carbazole With potassium <i>tert</i>-butylate In N,N-dimethyl-formamide at 30℃; for 0.5h; Inert atmosphere;
Stage #2: o-fluorobenzophenone In N,N-dimethyl-formamide at 80℃; for 18h; Inert atmosphere;
Preparation of M2:
Weigh 3,6-dibromocarbazole (1.625g, 5mmol), potassium tert-butoxide (0.672g, 6mmol) and pour it into a two-necked flask, set up a reflux condenser, vacuum and fill with nitrogen Measure 50mL N,N-dimethylformamide solvent into the flask, stir at 30°C for 30min, weigh 2-fluorobenzophenone (0.9g, 4.5mmol,) into the reactor, reflux and condense at 80°C At 18h, the obtained crude product was separated by silica gel column chromatography (eluent: dichloromethane/petroleum ether=2/1, v/v) to obtain 1.915 g of light yellow solid (yield 84%).
3,6-bis(3,5-di-tert-butylphenyl)carbazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; In 1,4-dioxane; water; at 100℃; for 48h;Inert atmosphere;
A mixture of the carbazole (1.062 g, 3.267 mmol, 1.00 eq), 3,5-di-t-butylphenyl boropinacolate ester (3.100 g, 9.801 mmol, 3.00 eq), Pd(PPh3)4 (0.755 g, 0.6534 mmol, 0.20 eq), and K3PO4 (6.241 g, 29.403 mmol, 9.00 eq) equipped with a reflux condenser was evacuated, then back-filled with nitrogen, this evacuation/re-fill process was repeated 3× more, freshly deoxygenated 1,4-dioxane (30 mL) and H2O (5.0 mL) were added simultaneously via syringes, the golden yellow mixture was placed in a mantle heated to 100 C., stirred vigorously (1000 rpm) for 48 hrs, removed from the mantle, allowed to cool gradually to 23 C., the golden yellow suspension was suction filtered through silica gel, rinsed with CH2Cl2 (4×20 mL), the yellow filtrate solution was concentrated onto celite, and purified via silica gel chromatography; hexanes 50% CH2Cl2 in hexanes to afford the disubstituted carbazole as a white foam (1.551 g, 2.852 mmol, 87%). NMR indicated pure product. (0375) 1H NMR (500 MHz, Chloroform-d) δ 8.34 8.29 (m, 2H), 8.10 (s, 1H), 7.68 (dd, J=8.4, 1.8 Hz, 2H), 7.54 (d, J=1.7 Hz, 4H), 7.51 (d, J=8.3 Hz, 2H), 7.45 (t, J=1.8 Hz, 2H), 1.43 (s, 36H). 13C NMR (126 MHz, Chloroform-d) δ 151.03, 141.57, 139.25, 134.55, 126.04, 123.93, 122.02, 120.74, 119.18, 110.71, 35.01, 31.60.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






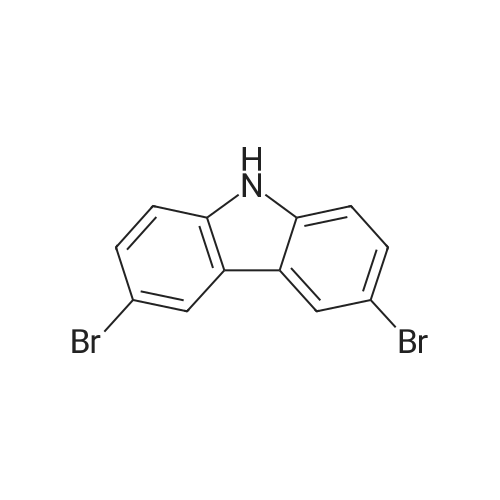

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping