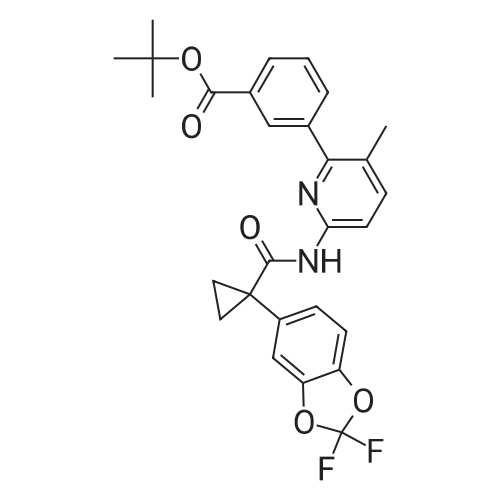
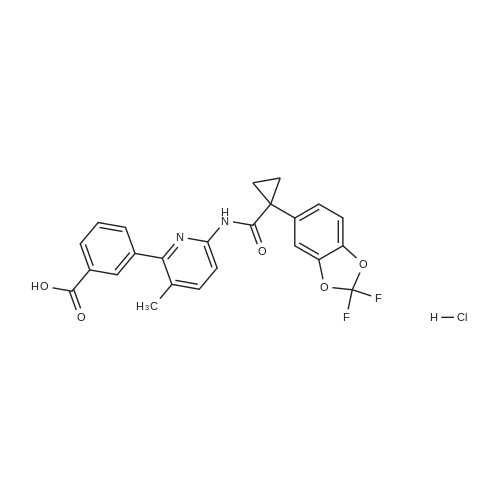
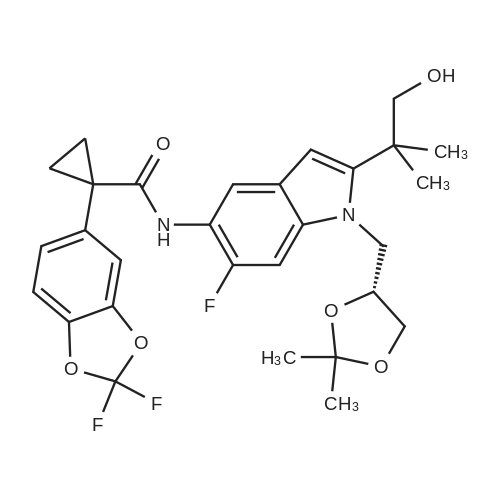
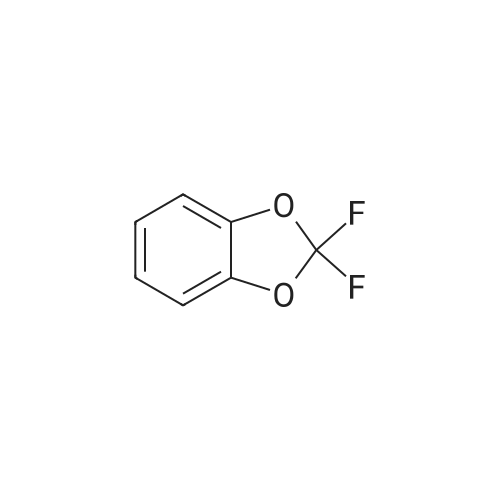
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
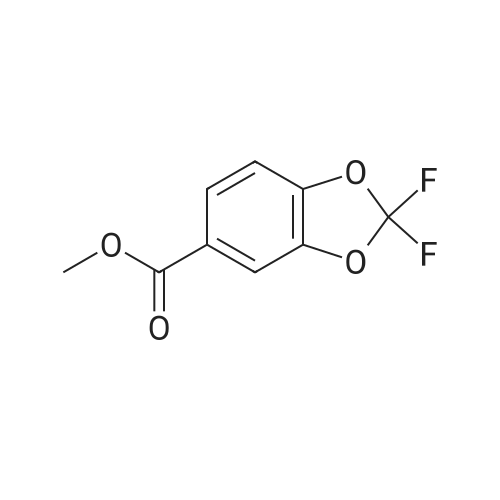
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40° C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95percent) that is used directly in the next step. The remaining steps are the same as described above for the synthesis of the acid moiety.
92%
With sodium cyanide In dimethyl sulfoxide at 20 - 40℃; for 2 h;
Step 4step 4 5[00160] 2-(2.2-difluorobenzordi ri .31dioxol-5-yl)acetonitrile: 5-(chloromethyl)-2,2- difluoro-2H-l,3-benzodioxole (12.36 g, 60 mmol, 1.00 equiv.) was dissolved in DMSO (120 mL). This was followed by the addition of NaCN (4.41 g, 1.50 equiv.) with the inert temperature below 40 °C. The resulting solution was stirred for 2 hours at room temperature. The reaction progress was monitored by GCMS. The reaction was then quenched by the addition of 300 mL of water/ice. The resulting solution was extracted with 3 x 100 mL of ethyl acetate. The organic layers combined and washed with 3 x 100 mL brine dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.84 g (92percent) of 2-(2,2- difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile as brown oil.
With hydrogenchloride; dimethyl sulfoxide In water at 110℃; for 5 h;
To the solution of Example 2D (8 g, 29.7 mmol) in dimethyl sulfoxide (80 mL) was added 3 M aqueous HC1 (80 mL) and the reaction was heated at 110 °C for 5 hours. Two additional vials were set up as described above. After completion of the reaction, all three reaction mixtures were combined. The reaction was extracted with methyl tert-butyl ether (2 x 500 mL) and the combined organics were washed with brine, dried over Na2SO4, filtered and concentrated to give a residue, which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5/1) to provide the title compound (12 g, 68.3 percent yield). ‘H NMR (400 MHz, DMSO-d6) 6 ppm 3.24 - 3.56 (m, 1 H) 4.02 (s, 2 H) 7.20 (d, J=8.38 Hz, 1 H) 7.34 - 7.43 (m, 2 H).
95 %Chromat.
With hydrogenchloride In water; dimethyl sulfoxide at 40 - 75℃;
The DMSO solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature 99percent was observed (typically after 5-6 h), the reaction was cooled to 20-25° C. and extracted with MTBE (2×525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5percent NaCl (2×375 mL). The solution was then transferred to equipment appropriate for a 1.5-2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at <60° C. to remove the solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125-130° C. (oven temperature) and 1.5-2.0 Ton. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66percent yield from 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of 91.5percent AUC (corresponds to a w/w assay of 95percent). 1H NMR (500 MHz, DMSO) δ 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (compound 20).[00242] A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 0C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 20 (95percent) that is used directly in the next step.
66%
at 25℃; for 2 h;
[00195] 2-(2,2-difluorobenzo[rf] [l,3]dioxol-5-yl)acetonitrileA mixture of 5-(chloromemyl)-2,2-difluorobenzo[
A solution of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 0C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)- acetonitrile (95percent) that is used directly in the next step.
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40° C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95percent) that is used directly in the next step. The remaining steps are the same as described above for the synthesis of the acid moiety. Amine Moiety
95%
With NaCN In water; dimethyl sulfoxide
(2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40° C. The mixture was stirred for 1 hour then water (6 vol) was added followed by MTBE (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95percent) that was used directly in the next step. 1H NMR (500 MHz, DMSO) δ 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
95%
With NaCN In water; dimethyl sulfoxide
(2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40° C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl tert-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95percent) that was used directly in the next step.
General procedure: To a solution of 2, 2-difluorobenzo [1, 3] dioxol-5-yl-methanol (930 mg, 5 mmol) in anhydrous THF (5 ml) was added NaBH4 (208 mg, 5.5 mmol) in portions at T = 0 °C in ten minutes. The mixture was then stirred at the same temperature for about 2 h (controlled with HPLC). H2O (1.5 ml) was added and the mixture was stirred for about 15 min. The mixture was then extracted with diethyl ether (3 * 3 ml) and washed with water (2 * 1 ml). The organic phase was completely dried and lyophilized to obtain 2,2-difluorobenzo [1, 3] dioxol-5-yl-methanol as colorless oil and used without further purification (865 mg, 85percent). A solution of 2,2-difluorobenzo [1, 3] dioxol-5-yl-methanol (865mg, 4.6mmol) in thionyl chloride (2ml) was stirred at room temperature upon completeness. When the reaction was complete (about 1h), the solution was concentrated under vacuum to remove the excess of thionyl chloride. Then, the residue was resuspended in a solution of dichloromethane and saturated NaHCO3 1:1 (2ml). The aqueous layer was then back-extracted with dichloromethane (2×1ml) and the combined organic layers were dried over Na2SO4 and finally concentrated to vacuum to give 5(chloromethyl)-2,2-difluorobenzo [1, 3] dioxole (823mg, 87percent) which was used directly in the next step. A mixture of 5(chloromethyl)-2,2-difluorobenzo [1, 3] dioxole (823 mg, 3.99 mmol) and KCN (519 mg, 7.98 mmol) was stirred in dimethyl sulfoxide (DMSO) (3 ml) for 3 h at room temperature. H2O (1.5 ml) was added and the mixture extracted with ethyl acetate (EtOAc) (3 * 3 ml) and the organic phases were dried over Na2SO4 and concentrated under reduced pressure. The product was purified by preparative HPLC to afford 2(2, 2-difluorbenzo [1, 3] dioxol-5-yl)-acetonitrile (432 mg, 55percent).
Reference:
[1] European Journal of Medicinal Chemistry, 2018, vol. 144, p. 179 - 200
With tetraoctyl ammonium bromide; potassium hydroxide In water at 70℃; for 1 h;
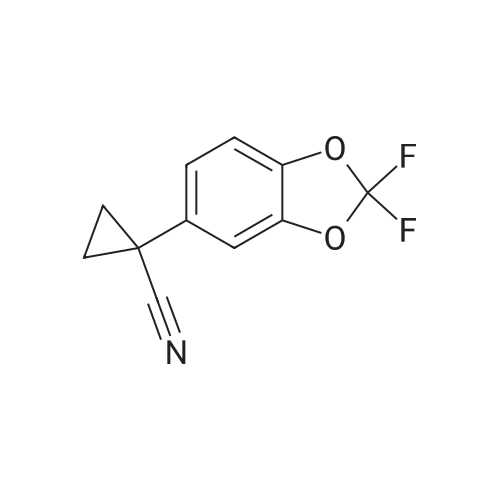
A mixture of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt percent aqueous KOH (5.0 eq) l-bromo-2-chloroethane (1.5 eq), and OCt4NBr (0.02 eq) is heated at 70 0C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford (2,2-difluoro-l,3- benzodioxol-5-yl)-cyclopropanecarbonitrile.
100%
With potassium hydroxide In water at 70℃; for 1 h;
A mixture of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt percent aqueous KOH (5.0 eq) l-bromo-2-chloroethane (1.5 eq), and OCt4NBr (0.02 eq) is heated at 70 0C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford (2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
100%
With tetraoctyl ammonium bromide; potassium hydroxide In water at 70℃; for 1 h;
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt percent aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70° C. for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
88%
With potassium hydroxide; tetraoctyl ammonium bromide In water at 70℃; for 1 h;
Synthesis of ^^-difluoro-l^-benzodioxol-S-y^-cyclopropanecarbonitrile (compound 21). l-bromo-2-chloroethane (1.5 equiv) 50percent KOH (5.0 equiv) OCt4NBr (0.02 equiv) 70 degrees C 88- 100percent yield [00244] A mixture of compound 20 (1.0 eq), 50 wt percent aqueous KOH (5.0 eq) l-bromo-2- chloroethane (1.5 eq), and OCt4NBr (0.02 eq) is heated at 70 0C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford compound 21.
10.12 g
With tetrabutylammomium bromide; sodium hydroxide In ethanol; water at 70℃; for 48 h; Inert atmosphere
Step 5[00161] l -(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l -carbonitrile: To a 100 mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, were placed 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile (10.84 g, 55 mmol, 1.00 equiv.),NaOH (50percent) in water), 1 -bromo-2-chloroethane (11.92g, 82.5 mmol, 1.50 equiv.), BmNBr(361 mg, 1.1 mmol, 0.02 equiv.). The resulting solution was stirred for 48 h at 70 °C. The reaction progress was monitored by GCMS. The reaction mixture was cooled. The resulting solution was extracted with 3 x 200 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.12g of 1 -(2,2- difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile as brown oil.
With 1,8-diazabicyclo[5.4.0]undec-7-ene In dimethyl sulfoxide at 21℃; for 12 h;
General procedure: To a 5 mL Schlenk tube were added aryl / heteroaryl acetonitriles 1 (1.0 mmol, 1.0 equiv.), vinyl diphenylsulfonium triflate (434.4 mg, 1.2 mmol, 1.2 equiv.), and DMSO (5 mL). The mixture was stirred at room temperature for 2 min and to the mixture DBU (456 mg, 3 mmol, 3.0 equiv.) was added. The mixture was stirred for 12 hours at room temperature, and then to the mixture was added saturated ammonium chloride solution (25 mL). The resulting mixture was extracted with EtOAc(3 x 150 mL). The combined organic layers were washed with H2O (2 x 30 mL), dried with anhydrous sodium sulfate. After concentration, product 2 was purified using silica gel column chromatography using an appropriate eluent.
Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (compound 20).[00242] A solution of Compound 19 (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 0C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude compound 20 (95%) that is used directly in the next step.
66%
In dimethyl sulfoxide; at 25℃; for 2h;
[00195] 2-(2,2-difluorobenzo[rf] [l,3]dioxol-5-yl)acetonitrileA mixture of 5-(chloromemyl)-2,2-difluorobenzo[<J][l,3]dioxole (117.6 g, crude from last step) and NaCN (84 g, 1.7 mmol) in DMSO (800 mL) was stirred for 2 h at 25 0C. The reaction mixture was poured into ice and extracted with EtOAc (500 mL x 3). The combined organic layers were dried with anhydrous Na2SO4 and concentrated in vacuo to give crude product which was purified by column chromatography (P.E./EtOAc 10:1) to give 2-(2,2- difluorobenzo[?,3]dioxol-5-yl)acetonitrile (77.8 g, 66%). 1H NMR (300 MHz , CDCl3) ? 7.06-7.07 (m, 3 H), 3.75 (s, 2 H).
In dimethyl sulfoxide; at 20℃;
A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyamide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 20℃;
Step d: (2,2-Difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 20℃;
2, 2-Difluoro-benzo [1, 3] dioxol-5-yl)-acetonitrile; A mixture of crude 5-chloromethyl- 2, 2-difluoro-benzo [1, 3] dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro- benzo [1, 3] dioxol-5-yl) -acetonitrile (3.3 g) which was used directly in the next step
In dimethyl sulfoxide; at 20℃;
Step d: (2,2-Difluoro-benzo[l,3]dioxol-5-yl)-acetonitrile; A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[l,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[l,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 20℃;
A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
A mixture of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt % aqueous KOH (5.0 eq) l-bromo-2-chloroethane (1.5 eq), and OCt4NBr (0.02 eq) is heated at 70 0C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford (2,2-difluoro-l,3- benzodioxol-5-yl)-cyclopropanecarbonitrile.
100%
With potassium hydroxide;tetraoctyl ammonium bromide; In water; at 70℃; for 1h;
A mixture of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt % aqueous KOH (5.0 eq) l-bromo-2-chloroethane (1.5 eq), and OCt4NBr (0.02 eq) is heated at 70 0C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford (2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
100%
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
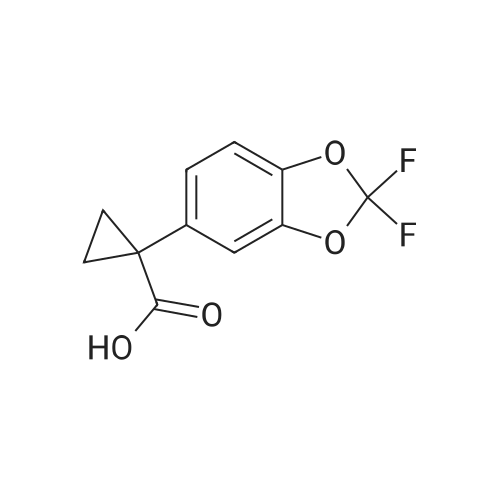
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C. for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
96.3%
With tetraoctyl ammonium bromide; triethylamine; In water; at 35℃; for 1h;
288 g of water and 67 g of triethylamine were placed in the reaction tank.Stir well, add 4g of tetraoctyl ammonium bromide and 44g of 2,2-difluoro pepper acetonitrile.80 g of 1-bromo-2-chloroethane was added dropwise at 35 C, and the reaction was kept at 35 C for 1 h, and the reaction was completed, and the solid product was directly filtered to obtain the target product, which was 48 g after drying.After HPLC purification,The content of 1-(2,2-difluorobenzo[D][1,3]dioxol-5-yl)cyclopropylcarbonitrile is 99.6%,The yield was 96.3%.
88 - 100%
With potassium hydroxide; tetraoctyl ammonium bromide; In water; at 70℃; for 1h;
Synthesis of ^^-difluoro-l^-benzodioxol-S-y^-cyclopropanecarbonitrile (compound 21). l-bromo-2-chloroethane (1.5 equiv) 50% KOH (5.0 equiv) OCt4NBr (0.02 equiv) 70 degrees C 88- 100% yield [00244] A mixture of compound 20 (1.0 eq), 50 wt % aqueous KOH (5.0 eq) l-bromo-2- chloroethane (1.5 eq), and OCt4NBr (0.02 eq) is heated at 70 0C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford compound 21.
With sodium hydroxide; N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With sodium hydroxide; N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
Step e: 1-(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
Step e: 1-(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrileSodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide; In water; at 70℃;
Step e: 1-(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With sodium hydroxide;N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With sodium hydroxide; N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
1- (2, 2-Difluoro-benzo [1, 3] dioxol-5-yl)-cyclopropanecarbonitrile; Sodium hydroxide (50 % aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro- benzo [1, 3] dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C and the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to to give crude 1- (2, 2-difluoro-benzo [1, 3] dioxol-5-yl) -cyclopropanecarbonitrile, which was used directly in the next step
With sodium hydroxide;N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[l,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and l-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 0C.[00171] The mixture was stirred overnight at 70 0C before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude l-(2,2-difluoro-benzo[l,3]dioxol-5- yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
[00203] Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile. l-bromo-2-chloroethane (1.5 equiv)50% KOH (5.0 equiv)Oct NBr 0.02 e uiv 88- 100% yield[00204] A mixture of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt % aqueous KOH (5.0 eq) l-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) is heated at 70 C for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford (2,2-difluoro-l,3- benzodioxol-5-yl)-cyclopropanecarbonitrile. 1H NMR (500 MHz, DMSO) delta 7.43 (d, J= 8.4 Hz, 1H), 7.40 (d, J= 1.9 Hz, 1H), 7.30 (dd, J= 8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H).
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) is heated at 70 C. for 1 h. The reaction mixture is cooled then worked up with MTBE and water. The organic phase is washed with water and brine then the solvent is removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile. 1H NMR (500 MHz, DMSO) delta 7.43 (d, J=8.4 Hz, 1H), 7.40 (d, J=1.9 Hz, 1H), 7.30 (dd, J=8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H).
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile [0277] [0278] A stock solution of 50% w/w NaOH was degassed via nitrogen sparge for no less than 16 h. An appropriate amount of MTBE was similarly degassed for several hours. To a reactor purged with nitrogen was charged degassed MTBE (143 mL) followed by <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (40.95 g, 207.7 mmol) and tetrabutylammonium bromide (2.25 g, 10.38 mmol). The volume of the mixture was noted and the mixture was degassed via nitrogen sparge for 30 min. Enough degassed MTBE is charged to return the mixture to the original volume prior to degassing. To the stirring mixture at 23.0 C. was charged degassed 50% w/w NaOH (143 mL) over 10 min followed by 1-bromo-2-chloroethane (44.7 g, 311.6 mmol) over 30 min. The reaction was analyzed by HPLC in 1 h intervals for % conversion. Before sampling, stirring was stopped and the phases allowed to separate. The top organic phase was sampled for analysis. When a % conversion >99% was observed (typically after 2.5-3 h), the reaction mixture was cooled to 10 C. and was charged with water (461 mL) at such a rate as to maintain a temperature <25 C. The temperature was adjusted to 20-25 C. and the phases separated. Note: sufficient time should be allowed for complete phase separation. The aqueous phase was extracted with MTBE (123 mL), and the combined organic phase was washed with 1 N HCl (163 mL) and 5% NaCl (163 mL). The solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile in MTBE was concentrated to 164 mL under vacuum at 40-50 C. The solution was charged with ethanol (256 mL) and again concentrated to 164 mL under vacuum at 50-60 C. Ethanol (256 mL) was charged and the mixture concentrated to 164 mL under vacuum at 50-60 C. The resulting mixture was cooled to 20-25 C. and diluted with ethanol to 266 mL in preparation for the next step. 1H NMR (500 MHz, DMSO) delta 7.43 (d, J=8.4 Hz, 1H), 7.40 (d, J=1.9 Hz, 1H), 7.30 (dd, J=8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H).
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C. for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
A mixture of(2,2-dit1uoro-1,3-benzodioxol-5-yl)-acetonitrile (LO eq), 50 wt%aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oc~NBr (0.02 eq) was heated at70 oc for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile. 1H Nl..1R (500 MHz, DMSO) 8 7.43(d,J=8.4Hz, 1H),7.40(d,J= 1.9Hz, 1H), 7.30(dd,J=8.4, 1.9Hz, 1H), 1.75(m,2H), 1.53(m, 2H).
With tetrabutylammomium bromide; sodium hydroxide; In tert-butyl methyl ether; water; at 23℃;Inert atmosphere;
A stock solution of 50% w/w NaOH was degassed via nitrogen sparge for no less than 16 h. An appropriate amount of MTBE was similarly degassed for several hours. To a reactor purged with nitrogen was charged degassed MTBE (143 mL) followed by <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (40.95 g, 207.7 mmol) and tetrabutylammonium bromide (2.25 g, 10.38 mmol). The volume of the mixture was noted and the mixture was degassed via nitrogen sparge for 30 min. Enough degassed MTBE is charged to return the mixture to the original volume prior to degassing. To the stirring mixture at 23.0 C was charged degassed 50% w/w NaOH (143 mL) over 10 min followed by 1-bromo-2-chloroethane (44.7 g, 311.6 mmol) over 30 min. The reaction was analyzed by HPLC in 1 h intervals for % conversion. Before sampling, stirring was stopped and the phases allowed to separate. The top organic phase was sampled for analysis. When a % conversion > 99 % was observed (typically after 2.5 - 3 h), the reaction mixture was cooled to 10 C and was charged with water (461 mL) at such a rate as to maintain a temperature < 25 C. The temperature was adjusted to 20 - 25 C and the phases separated. Note: sufficient time should be allowed for complete phase separation. The aqueous phase was extracted with MTBE (123 mL), and the combined organic phase was washed with 1 N HCl (163mL) and 5% NaCl (163 mL). The solution of (2,2-difluoro-1,3-benzodioxol-5-yl)cyclopropanecarbonitrile in MTBE was concentrated to 164 mL under vacuum at 40 - 50 C. The solution was charged with ethanol (256 mL) and again concentrated to 164 mL under vacuum at 50 - 60 C. Ethanol (256 mL) was charged and the mixture concentrated to 164 mL under vacuum at 50 - 60 C. The resulting mixture was cooled to 20 - 25 C and diluted with ethanol to 266 mL in preparation for the next step. 1H NMR (500 MHz, DMSO) delta 7.43 (d, J= 8.4 Hz, 1H), 7.40 (d, J= 1.9 Hz, 1H), 7.30 (dd, J= 8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H).
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
[00293] A mixture of<strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (1.0 eq), 50 wt%aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70oc for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organicphase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1 ,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
[00308] A mixture of (2,2-difluoro-l ,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1 -bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-l ,3- benzodioxol-5-yl)-cyclopropanecarbonitrile.
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide; at 70℃;
Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
10.12 g
With tetrabutylammomium bromide; sodium hydroxide; In ethanol; water; at 70℃; for 48h;Inert atmosphere;
Step 5[00161] l -(2.2-difluoro-2H-1.3-benzodioxol-5-yl)cvclopropane-l -carbonitrile: To a 100 mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, were placed 2-(2,2-difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile (10.84 g, 55 mmol, 1.00 equiv.),NaOH (50%) in water), 1 -bromo-2-chloroethane (11.92g, 82.5 mmol, 1.50 equiv.), BmNBr(361 mg, 1.1 mmol, 0.02 equiv.). The resulting solution was stirred for 48 h at 70 C. The reaction progress was monitored by GCMS. The reaction mixture was cooled. The resulting solution was extracted with 3 x 200 mL of ethyl acetate and the organic layers combined. The resulting mixture was washed with 1 x 200 mL of brine. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.12g of 1 -(2,2- difluoro-2H-l,3-benzodioxol-5-yl)cyclopropane-l-carbonitrile as brown oil.
With sodium hydroxide; N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
Step e: l-(2,2-Difluoro-benzo[l,3]dioxol-5-yl)-cyclopropanecarbonitrile; Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[l,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and l-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 0C. The mixture was stirred overnight at 70 0C before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude l-(2,2-difluoro-benzo[l,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With tetraoctyl ammonium bromide; potassium hydroxide; at 70℃; for 1h;
(2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile (1.0 eq.), 50 wt% aqueous KOH (5.0 eq.), 1-bromo-2-chloro A mixture of ethane (1.5 eq.) and Oct 4 NBr (0.02 eq.) was heated at 70 C for 1 h. The reaction mixture was cooled and then treated with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to provide (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
With sodium hydroxide; N-benzyl-N,N,N-triethylammonium chloride; In water; at 70℃;
Sodium hydroxide (50% aqueous solution, 10 mL) was slowly added to a mixture of crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, benzyltriethylammonium chloride (3.00 g, 15.3 mmol), and 1-bromo-2-chloroethane (4.9 g, 38 mmol) at 70 C. The mixture was stirred overnight at 70 C. before the reaction mixture was diluted with water (30 mL) and extracted with ethyl acetate. The combined organic layers were dried over sodium sulfate and evaporated to dryness to give crude 1-(2,2-difluoro-benzo[1,3]dioxol-5-yl)-cyclopropanecarbonitrile, which was used directly in the next step.
With tetraoctyl ammonium bromide; potassium hydroxide; In water; at 70℃; for 1h;
l-bromo-2-chloroethane (1.5 equiv) (0474) uu 1 / v y ici u (0475) [00135] A mixture of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt % aqueous KOH (5.0 eq) l-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-l,3-benzodioxol-5-yl)-cyclopropanecarbonitrile. 1H NMR (500 MHz, DMSO) d 7.43 (d, J = 8.4 Hz, 1H), 7.40 (d, J = 1.9 Hz, 1H), 7.30 (dd, J = 8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H).
5-bromomethyl-2,2-difluro-1,3-benzodioxolane[ No CAS ]
[ 68119-31-3 ]
Yield
Reaction Conditions
Operation in experiment
64%
With potassium bromide; In ice-water; water;
EXAMPLE 4 Preparation of 2,2-Difluoro-1,3-benzodioxole-5-acetonitrile To a solution of 2,2-difluoro-5-bromomethyl-1,3-benzodioxole (41.0 g) in absolute alcohol (160 ml) at 60-70 C a hot solution of potassium cyanide (22.1 g, 0.34 mole) in water (30 ml) is added. There is a slight exotherm and within 5 minutes potassium bromide separates out of the reaction mixture. The reaction mixture is refluxed for 1.5 hours, cooled and added to ice-water. The mixture is extracted with ether (3*100 ml), the combined extracts are washed with water (2*50 ml), dried over sodium sulfate and evaporated to afford a dark oil. Vacuum distillation yields 21.2 g of product (64% yield); b.p. 64-67 C (at 0.03 mm); i.r. (neat) 2255 cm-1; nmr (CCl4) delta 3.68 (S, 2H), 7.00 (S, 3H).
With potassium bromide; In ice-water; water;
EXAMPLE 4 Preparation of 2,2-Difluoro-1,3-benzodioxole-5-acetonitrile To a solution of 2,2-difluoro-5-bromomethyl-1,3-benzodioxole (41.0 g) in absolute alcohol (160 ml) at 60 to 70 C a hot solution of potassium cyanide (22.1 g, 0.34 mole) in water (30 ml) is added. There is a slight exotherm and within 5 minutes potassium bromide separates out of the reaction mixture. The reaction mixture is refluxed for 1.5 hours, cooled and added to ice-water. The mixture is extracted with ether (3*100 ml), the combined extracts are washed with water (2*50 ml), dried over sodium sulfate and evaporated to afford a dark oil. Vacuum distillation yields 21.2 g of title product; b.p. 64 to 67 C (at 0.03 mm); i.r. (neat) 2255 cm-1; nmr (CCl4) delta 3.68 (S, 2H), 7.00 (S, 3H).
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step. The remaining steps are the same as described above for the synthesis of the acid moiety.
92%
With sodium cyanide; In dimethyl sulfoxide; at 20 - 40℃; for 2h;
Step 4step 4 5[00160] 2-(2.2-difluorobenzordi ri .31dioxol-5-yl)acetonitrile: 5-(chloromethyl)-2,2- difluoro-2H-l,3-benzodioxole (12.36 g, 60 mmol, 1.00 equiv.) was dissolved in DMSO (120 mL). This was followed by the addition of NaCN (4.41 g, 1.50 equiv.) with the inert temperature below 40 C. The resulting solution was stirred for 2 hours at room temperature. The reaction progress was monitored by GCMS. The reaction was then quenched by the addition of 300 mL of water/ice. The resulting solution was extracted with 3 x 100 mL of ethyl acetate. The organic layers combined and washed with 3 x 100 mL brine dried over anhydrous sodium sulfate and concentrated under vacuum to afford 10.84 g (92%) of 2-(2,2- difluoro-2H-l ,3-benzodioxol-5-yl)acetonitrile as brown oil.
With sodium cyanide; In dimethyl sulfoxide;
(2,2-Difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
With sodium cyanide; In dimethyl sulfoxide;
Step d: (2,2-Difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
With sodium cyanide; In dimethyl sulfoxide;
Step d: (2,2-Difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile which was used directly in the next step.
With sodium cyanide; In dimethyl sulfoxide; at 30 - 40℃; for 1h;
A solution of 5-chloromethyl-2,2-dit1uoro-1 ,3-benzodioxole (1 eq) in DMSO (1.25vol) was added to a slurry ofNaCN ( 1.4 eq) in DMSO (3 vol), while maintaining thetemperature between 30-40 oc_ The mixture was stirred for 1 h, and then water (6 vol) wasadded, folimved by methyl tert-butyl ether (MTBE) (4 vol), After stirring for 30 min, the layerswere separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organiclayers were washed with water (L8 vol), dried (Na2S04), filtered, and concentrated to affordcrude (2,2-difluoro-1 ,3-benzodioxol-5-yl)-acetonitriie {95%) that was used directly in the nextstep. 1H N:MR (500 1ffiz, DMSO) 8 7.44 (br s, lH), 7.43 (d, J= 8.4 Hz, lH), 7.22 (dd, J= 8.2,L8 Hz, lH), 4.07 (s, 2H),
A solution of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 0C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)- acetonitrile (95%) that is used directly in the next step.
95%
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 equivalent) in DMSO (1.25 vol) was added to sodium cyanide (1.4 eq.). In a slurry in DMSO (3 vol), the temperature was maintained between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added sequentially with methyl t-butyl ether (MTBE) (4 vol.). The combined organic layers were washed with water (1 mL), dried (EtOAc m. After stirring for 30 minutes, the layers were separated. -1,3-benzodioxol-5-yl)-acetonitrile (95%) which was used directly in the next step.
In dimethyl sulfoxide; at 20℃;
Step d: (2,2-Difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 20℃;
A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
A solution of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 0C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (TS^SO4), filtered, and concentrated to afford crude (2,2- difluoro-l,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step.
In dimethyl sulfoxide; at 20℃;
A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[l,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2- difluoro-benzo[l,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
00197] Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile.95- 100% yield[00198] A solution of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)- acetonitrile (95%) that is used directly in the next step. 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step. 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl tert-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO ( 1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1,8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step.
[00287] A solution of 5-chloromethyl-2,2-difluoro-1 ,3-benzodioxole (1 eq) in DMSO (1.25vol) was added to a slurry ofNaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperaturebetween 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followedby methyl tert-buty1 ether (MTBE) ( 4 vol). After stirring for 30 min, the layers were separated.The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers werewashed with water (1.8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
A solution of 5-chloromethyl-2,2-difluoro-l ,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl terf-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2- difluoro-l,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step.
In dimethyl sulfoxide; at 20℃;
A mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl tert-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step.
In dimethyl sulfoxide; at 30 - 40℃; for 1h;
A solution of 5-chloromethyl-2,2-difluoro-l,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl /cvV-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2S04), filtered, and concentrated to afford crude (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step. NMR (500 MHz, DMSO) d 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
Step d: (2,2-Difluoro-benzo[1,3]dioxol-5-yl)-acetonitrileA mixture of crude 5-chloromethyl-2,2-difluoro-benzo[1,3]dioxole (4.4 g) and sodium cyanide (1.36 g, 27.8 mmol) in dimethylsulfoxide (50 mL) was stirred at room temperature overnight. The reaction mixture was poured into ice and extracted with ethyl acetate (300 mL). The organic layer was dried over sodium sulfate and evaporated to dryness to give crude (2,2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile (3.3 g) which was used directly in the next step.
(2,2-Difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C. for 1 h. The reaction mixture was cooled then worked up with MTBE and water. The organic phase was washed with water and brine then the solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile. 1H NMR (500 MHz, DMSO) delta 7.43 (d, J=8.4 Hz, 1H), 7.40 (d, J=1.9 Hz, 1H), 7.30 (dd, J=8.4, 1.9 Hz, 1H), 1.75 (m, 2H), 1.53 (m, 2H).
With aqueous KOH;
(2,2-Difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of <strong>[68119-31-3](2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile</strong> (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C. for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) is added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture is stirred for 1 hour then water (6 vol) is added followed by MTBE (4 vol). After stirring for 30 min, the layers are separated. The aqueous layer is extracted with MTBE (1.8 vol). The combined organic layers are washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that is used directly in the next step. The remaining steps are the same as described above for the synthesis of the acid moiety. Amine Moiety
95%
With NaCN; In water; dimethyl sulfoxide;
(2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol) maintaining the temperature between 30-40 C. The mixture was stirred for 1 hour then water (6 vol) was added followed by MTBE (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step. 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
95%
With NaCN; In water; dimethyl sulfoxide;
(2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl tert-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step.
95%
In water; dimethyl sulfoxide;
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile A solution of 5-chloromethyl-2,2-difluoro-1,3-benzodioxole (1 eq) in DMSO (1.25 vol) was added to a slurry of NaCN (1.4 eq) in DMSO (3 vol), while maintaining the temperature between 30-40 C. The mixture was stirred for 1 h, and then water (6 vol) was added, followed by methyl tert-butyl ether (MTBE) (4 vol). After stirring for 30 min, the layers were separated. The aqueous layer was extracted with MTBE (1.8 vol). The combined organic layers were washed with water (1.8 vol), dried (Na2SO4), filtered, and concentrated to afford crude (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (95%) that was used directly in the next step.
With hydrogenchloride; dimethyl sulfoxide; In water; at 110℃; for 5h;
To the solution of Example 2D (8 g, 29.7 mmol) in dimethyl sulfoxide (80 mL) was added 3 M aqueous HC1 (80 mL) and the reaction was heated at 110 C for 5 hours. Two additional vials were set up as described above. After completion of the reaction, all three reaction mixtures were combined. The reaction was extracted with methyl tert-butyl ether (2 x 500 mL) and the combined organics were washed with brine, dried over Na2SO4, filtered and concentrated to give a residue, which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate = 5/1) to provide the title compound (12 g, 68.3 % yield). ?H NMR (400 MHz, DMSO-d6) 6 ppm 3.24 - 3.56 (m, 1 H) 4.02 (s, 2 H) 7.20 (d, J=8.38 Hz, 1 H) 7.34 - 7.43 (m, 2 H).
With hydrogenchloride; In water; dimethyl sulfoxide; at 40 - 75℃;
00201] Synthesis of (2,2-difluoro-l,3-benzodioxol-5-yl)-acetonitrile.[00202] The DMSO solution of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate- acetonitrile from above was charged with 3 N HC1 (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature < 40 C. The mixture was then heated to 75C over 1 h and analyzed by HPLC every 1 - 2 h for % conversion. When a conversion of > 99% was observed (typically after 5 - 6 h), the reaction was cooled to 20 - 25 C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 - 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at < 60C to remove the solvents. (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 - 130 C (oven temperature) and 1.5 - 2.0 Torr. (2,2- Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5- bromo-2,2-difluoro-l,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J = 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
With hydrogenchloride; In water; dimethyl sulfoxide; at 40 - 75℃;
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile [0285] [0286] The DMSO solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature 99% was observed (typically after 5-6 h), the reaction was cooled to 20-25 C. and extracted with MTBE (2×525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2×375 mL). The solution was then transferred to equipment appropriate for a 1.5-2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at <60 C. to remove the solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125-130 C. (oven temperature) and 1.5-2.0 Torr. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
95%Chromat.
With hydrogenchloride; In water; dimethyl sulfoxide; at 40 - 75℃;
The DMSO solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature 99% was observed (typically after 5-6 h), the reaction was cooled to 20-25 C. and extracted with MTBE (2×525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2×375 mL). The solution was then transferred to equipment appropriate for a 1.5-2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at <60 C. to remove the solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125-130 C. (oven temperature) and 1.5-2.0 Ton. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
With hydrogenchloride; In dimethyl sulfoxide; at 40 - 75℃;
The DMSO solution of (2,2-dii1uoro-l ,3-benzodioxol-5-yl)-1-ethylacetateacetonitrilefrom above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 minutes whilemaintaining an intemal temperature less than 40 oc. The mixture was then heated to 75 oc over1 hour and analyzed by HPLC every 1 to 2 hour for percent conversion. When a conversion ofgreater than 99% was observed (typically after 5 to 6 hours), the reaction was cooled to 20 to 25oc and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phaseseparation during the extractions. The combined organic extracts were washed with 5% NaCl (2X 375 mL). The solution was then transferred to equipment appropriate for a L5 to 2.5 TorTvacuum distillation that was equipped with a cooled receiver flask The solution wasconcentrated under vacuum at less than 60C to remove the solvents. (2,2-Difluoro-1 ,3-benzodioxoi-5-yl)-acetonitrile was then distilled from the resulting oil at 125 to 130 oc (oventemperature) and 1.5 to 2.0 Torr. (2,2-Difluoro-1 ,3-benzodioxol-·5-yl)-acetonitrile was isolatedas a clear oil in 66% yield from. 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with anHPLC purity of91.5~·o AUC (corresponds to a w/w assay of95%). 1H NMR (500 ~ffiz,DMSO)o7.44(brs, lH), 7.43 (d,J=8.4 Hz, 1H), 7.22(dd,J=8.2, L8Hz, lH),4.07(s,2H).
With hydrogenchloride; In water; dimethyl sulfoxide; at 40 - 75℃;
The DMSO solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature 99% was observed (typically after 5 - 6 h), the reaction was cooled to 20 - 25 C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 - 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at < 60C to remove the solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 - 130 C (oven temperature) and 1.5 - 2.0 Torr. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2- difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
With hydrogenchloride; In water; dimethyl sulfoxide; at 40 - 75℃; for 1.33333h;
[00291] The DMSO solution of (2,2-difluoro-1 ,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrilefrom above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining aninternal temperature 99% was observed (typicallyafter 5-6 h), the reaction was cooled to 20-25 oc and extracted with MTBE (2 X 525 mL),with sufficient time to allow for complete phase separation during the extractions. Thecombined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was thentransferred to equipment appropriate for a 1.5 - 2.5 Torr vacuum distillation that was equippedwith a cooled receiver flask. The solution was concentrated under vacuum at < 60C to removethe solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from theresulting oil at 125- 130 oc (oven temperature) and 1.5-2.0 Torr. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of91.5% AUC (corresponds to aw/w assay of95%). 1H NMR (500 MHz, DMSO) 8 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H),7.22 (dd,J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
With hydrogenchloride; In dimethyl sulfoxide; at 40 - 75℃;
[00306] The DMSO solution of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature < 40 C. The mixture was then heated to 75C over 1 h and analyzed by HPLC every 1 - 2 h for % conversion. When a conversion of > 99% was observed (typically after 5 - 6 h), the reaction was cooled to 20 - 25 C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 - 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at < 60C to remove the solvents. (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 - 130 C (oven temperature) and 1.5 - 2.0 Torr. (2,2-Difluoro-l,3- benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2- difluoro-l ,3-benzodioxole (2 steps) and with an HPLC purity of 91.5%) AUC (corresponds to a w/w assay of 95%). 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
With hydrogenchloride; In dimethyl sulfoxide; at 40 - 75℃;
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile The DMSO solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature <40 C. The mixture was then heated to 75 C. over 1 h and analyzed by HPLC every 1-2 h for % conversion. When a conversion of >99% was observed (typically after 5-6 h), the reaction was cooled to 20-25 C. and extracted with MTBE (2×525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2×375 mL). The solution was then transferred to equipment appropriate for a 1.5-2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at <60 C. to remove the solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125-130 C. (oven temperature) and 1.5-2.0 Torr. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). 1H NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
With hydrogenchloride; In dimethyl sulfoxide; at 75℃;
3 N HCl was charged to the DMSO solution from (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethyl acetate-acetonitrile above for 20 min. (617.3 mL, 1.85 mol) while maintaining the internal temperature at 99% (usually after 5-6 h) was observed, the reaction was cooled to 20-25 C and extracted with MTBE (2 x 525 mL) during which time was taken to allow complete separation of the phases. The combined organic extracts were washed with 5% NaCl (2 x 375 mL). The solution was then transferred to a facility suitable for 1.5-2.5 Torr vacuum distillation equipped with a cooled receiving flask. The solution was concentrated under vacuum at <60 C to remove the solvent. Subsequent distillation of (2,2-difluoro-1,3-benzodioxole-5-yl)-acetonitrile from the obtained oil at 125-130 C (oven temperature) and 1.5-2.0 Torr. . Isolated from 5-bromo-2,2-difluoro-1,3-benzodioxol, a 66% transparent oil (2,2-difluoro-1,3-benzoic acid) Oxocyclo-5-yl)-acetonitrile (2 steps) with HPLC purity of 91.5% AUC (corresponding to 95% w/w analysis).
With hydrogenchloride; In dimethyl sulfoxide; at 40 - 75℃; for 1h;
The DMSO solution of (2,2-difluoro-l,3-benzodioxol-5-yl)-l-ethylacetate- acetonitrile from above was charged with 3 N HC1 (617.3 mL, 1.85 mol) over 20 minutes while maintaining an internal temperature less than 40 C. The mixture was then heated to 75 C over 1 hour and analyzed by HPLC every 1 to 2 hours for percent conversion. (0472) When a conversion of greater than 99% was observed (typically after 5 to 6 hours), the reaction was cooled to 20 to 25 C and extracted with MTBE (2 X 525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2 X 375 mL). The solution was then transferred to equipment appropriate for a 1.5 to 2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at less than 60 C to remove the solvents. (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125 to 130 C (oven temperature) and 1.5 to 2.0 Torn (2,2-Difluoro-l,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2-difluoro-l,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). NMR (500 MHz, DMSO) d 7.44 (br s, 1H), 7.43 (d, J= 8.4 Hz, 1H), 7.22 (dd, J= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
(2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile[ No CAS ]
[ 68119-31-3 ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; In water; dimethyl sulfoxide; at 40 - 75℃; for 1h;
Synthesis of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile The DMSO solution of (2,2-difluoro-1,3-benzodioxol-5-yl)-1-ethylacetate-acetonitrile from above was charged with 3 N HCl (617.3 mL, 1.85 mol) over 20 min while maintaining an internal temperature 99% was observed (typically after 5-6 h), the reaction was cooled to 20-25 C. and extracted with MTBE (2×525 mL), with sufficient time to allow for complete phase separation during the extractions. The combined organic extracts were washed with 5% NaCl (2×375 mL). The solution was then transferred to equipment appropriate for a 1.5-2.5 Torr vacuum distillation that was equipped with a cooled receiver flask. The solution was concentrated under vacuum at <60 C. to remove the solvents. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was then distilled from the resulting oil at 125-130 C. (oven temperature) and 1.5-2.0 Torr. (2,2-Difluoro-1,3-benzodioxol-5-yl)-acetonitrile was isolated as a clear oil in 66% yield from 5-bromo-2,2-difluoro-1,3-benzodioxole (2 steps) and with an HPLC purity of 91.5% AUC (corresponds to a w/w assay of 95%). NMR (500 MHz, DMSO) delta 7.44 (br s, 1H), 7.43 (d, J=8.4 Hz, 1H), 7.22 (dd, J=8.2, 1.8 Hz, 1H), 4.07 (s, 2H).
2-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)propanenitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
A solution of 2- (2,2-difluorobenzo [dl [1,3] dioxol-5-yl)acetonitrile (400 mg,1.99 mmol) in anhydrous THF (18 mL) was added NaH (60% in mineral oil, 95 mg, 2.39 mmol) at -70 C. After addition, the mixture was stuffed at this temperature for 30 minutes. A solution of Mel (339 mg, 2.39 mmol) in anhydrous THF (2 mL) was added and the mixture was stirred at 17 C for 18 hours. The mixture was quenched with ice water (30 mL). The aqueous layer was extracted with EtOAc (20 mL x3) and the combined organic layer was washed with brine (20 mL), dried over Na2504, filtered and concentrated to give the residue. The crude product was purified by silica gel column chromatography (PE/EtOAc = 3/1) to afford 355 mg (yield: 83%) of the desired compound as a yellow oil. ?H NMR (CDC13, 400 MHz): (51.67 (3H, d, J= 7.2 Hz), 3.92 (1H, q, J= 7.2 Hz), 7.07-7.14 (3H, m).
2-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-2-methylpropanenitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
Step 1: A solution of 2- (2,2-difluorobenzo [dl [1,3] dioxol-5-yl)acetonitrile (400 mg, 1.99 mmol) in anhydrous THF (8 mL) was added LDA (2M in THF, 2.4 mL, 4.78 mmol) at -78 C. After addition, the mixture was stirred at this temperature for 15 minutes. A solution of Mel (1.13 g, 7.96 mmol) in anhydrous THF (2 mL) was added and the mixture was stirred at 17 C for 18 hours. Followed workup and purification procedures described in Intermediate 2 to afford 230 mg (yield: 50%) of 2-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-2- methylpropanenitrile as a yellow solid. ?H NMR (CDC13, 400 MHz): (5 1.73 (6H, s), 7.07 (1H, d, J= 8.4 Hz), 7.17-7.27 (2H, m).
50%
Step 1: Synthesis of 2-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-2- methylpropanenitrile (0713) [00276] A solution of 2-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)acetonitrile (400 mg, (0714) 1.99 mmol) in anhydrous THF (8 mL) was added LDA (2M in THF, 2.4 mL, 4.78 mmol) at - 78 C. After addition, the mixture was stirred at this temperature for 15 minutes. A solution of Mel (1.13 g, 7.96 mmol) in anhydrous THF (2 mL) was added and the mixture was stirred at 17 C for 18 hours. The mixture was quenched with ice water (30 mL). The aqueous layer was extracted with EtOAc (20 mL x3) and the combined organic layer was washed with brine (30 mL), dried over Na2S04, filtered and concentrated to give the residue. The crude product was purified by silica gel column chromatography (PE/EtOAc = 4/1) to afford 230 mg (yield: 50%) of compound 2-(2,2-difluorobenzo[d][l,3]dioxol-5-yl)-2-methylpropanenitrile as a yellow solid. 1H NMR (CDC13, Bruker Avance 400 MHz): delta 1.73 (6H, s), 7.07 (1H, d, J = 8.4 Hz), 7.17-7.27 (2H, m).
With sodium hydroxide; In water; dimethyl sulfoxide; at 0 - 20℃;
Iodomethane (3.96 g, 27.9 mmol) was added dropwise to a stirred mixture of <strong>[68119-31-3]2-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)acetonitrile</strong> (1.375 g, 6.97 mmol) and sodium hydroxide (1.116 g, 27.9 mmol) in dimethyl sulfoxide (12 mL) and water (1.9 mL) at 0 C. The reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was poured into ice water (60 mL) and extracted with 300 mL of methyl tert-butyl ether. The organic extracts were washed with brine and then concentrated with heptane to provide a brownish pink solid. 1H NMR (400 MHz, dimethyl sulfoxide-d6) delta 7.61 (d, J=1.9 Hz, 1H), 7.43 (d, J=8.5 Hz, 1H), 7.36 (dd, J=8.5, 2.0 Hz, 1H), 1.68 (s, 6H).
Preparation of (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile A mixture of (2,2-difluoro-1,3-benzodioxol-5-yl)-acetonitrile (1.0 eq), 50 wt % aqueous KOH (5.0 eq) 1-bromo-2-chloroethane (1.5 eq), and Oct4NBr (0.02 eq) was heated at 70 C. for 1 h. The reaction mixture was cooled, then worked up with MTBE and water. The organic phase was washed with water and brine. The solvent was removed to afford (2,2-difluoro-1,3-benzodioxol-5-yl)-cyclopropanecarbonitrile.
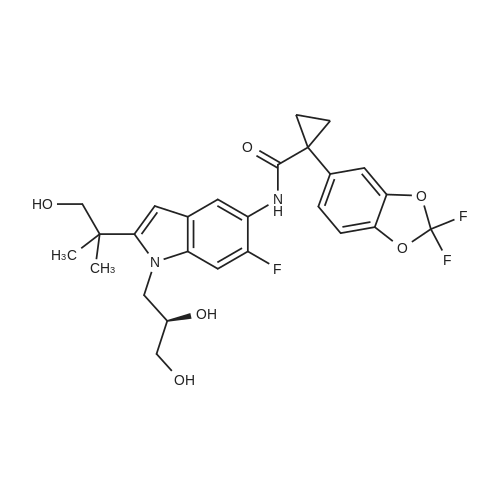
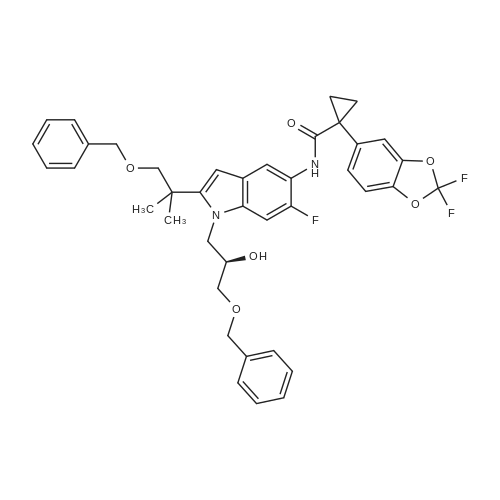
N-((R)-2-tert-butyl-1-((2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-1H-indol-5-yl)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamide[ No CAS ]
(R)-3-(2-tert-butyl-5-(1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)cyclopropanecarboxamido)-1H-indol-1-yl)-2-hydroxypropyl 4-methylbenzenesulfonate[ No CAS ]
1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)(<SUP>2</SUP>H<SUB>4</SUB>)cyclopropane-1-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
With tetrabutylammomium bromide; potassium hydroxide; In water-d2; at 70℃; for 48h;
EXAMPLE 6(R)-l-(2,2-difluorobenzo[dl [l,31dioxol-5-yl)-N-(l-(2,3-dihvdroxypropyl)-6-fluoro-2-(l-[00208] l-(2.2-difluoro-2H-1.3-benzodioxol-5-yl)(2H4)cvclopropane-l-carbonitrile: To a 25-mL round-bottom flask, was placed potassium hydroxide (4.26 g, 75.92 mmol, 4.99 equiv.), D2O (4.26 g), 2-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)acetonitrile (3 g, 15.22 mmol, 1.00 equiv.), 1 -bromo-2-chloro(2H4)ethane (4.34 g, 29.44 mmol, 1.50 equiv.), BmNBr (98 g, 305.30 mmol, 0.02 equiv.). The resulting solution was stirred for 48 h at 70 C. The resulting solution was extracted with 3 x 50 mL of ethyl acetate and the organic layers were combined, dried over anhydrous sodium sulfate and concentrated under vacuum to afford 3 g (87%) of l-(2,2-difluoro-2H-l,3-benzodioxol-5-yl)(2H4)cyclopropane-l-carbonitrile as red oil.
N-[1-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[1-(benzyloxy)-2-methyl(1.1-2H2)propan-2-yl]-5-fluoro-1H-indol-6-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
N-[2-[1-(benzyloxy)-2-methylpropan-2-yl]-1-[3-(benzyloxy)-2-oxo(3,3-2H2)propyl]-6-fluoro-1H-indol-5-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
N-[1-[3-(benzyloxy)-2-hydroxy(2,3,3-2H3)propyl]-2-[1-(benzyloxy)-2-methylpropan-2-yl]-6-fluoro-1H-indol-5-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
(R) 1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-[1-[2,3-dihydroxy(2,3,3-2H3)propyl]-6-fluoro-2-(1-hydroxy-2-methyl(1,1-2H2)propan-2-yl)-1H-indol-5-yl]cyclopropane-1-carboxamide[ No CAS ]
N-[1-[(2R)-3-(benzyloxy)-2-hydroxy(3,3-2H2)propyl]-2-[1-(benzyloxy)-2-methyl(1,1-2H2)propan-2-yl]-5-fluoro-1H-indol-6-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
N-[2-[1-(benzyloxy)-2-methyl(1,1-2H2)propan-2-yl]-1-[3-(benzyloxy)-2-oxo(3,3-2H2)propyl]-6-fluoro-1H-indol-5-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane1-carboxamide[ No CAS ]
N-[1-[3-(benzyloxy)-2-hydroxy(2,3,3-2H3)propyl]-2-[1-(benzyloxy)-2-methyl(1,2H2)propan-2-yl]-6-fluoro-1H-indol-5-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
(R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methyl(1,1-2H2)propan-2-yl)-1H-indol-5-yl)(2H4)cyclopropanecarboxamide[ No CAS ]
N-[1-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-5-fluoro-2-[1-hydroxy-2-methyl(2H2)propan-2-yl]-1H-indol-6-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)(2H4)cyclopropane-1-carboxamide[ No CAS ]
N-[1-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-5-fluoro-2-[1-hydroxy-2-methyl(2H6)propan-2-yl]-1H-indol-6-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)(2H4)cyclopropane-1-carboxamide[ No CAS ]
(R)1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-[1-[2,3-dihydroxy(3,3-2H2)propyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl]cyclopropane-1-carboxamide[ No CAS ]
(R)-1-(2,2-difluorobenzo[d][1,3]dioxol-5-yl)-N-(1-(2,3-dihydroxypropyl)-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl)cyclopropanecarboxamide[ No CAS ]
N-[1-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-2-[1-(benzyloxy)-2-methyl(1,1-2H2)propan-2-yl]-5-fluoro-1H-indol-6-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
N-[2-[1-(benzyloxy)-2-bismethyl(3,3-2H6)propan-2-yl]-1-[(2R)-3-(benzyloxy)-2-hydroxypropyl]-6-fluoro-1H-indol-5-yl]-1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)cyclopropane-1-carboxamide[ No CAS ]
1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-[1-[(2R)-2,3-dihydroxypropyl]-6-fluoro-2-[1-hydroxy-2-methyl(2H6)propan-2-yl]-1H-indol-5-yl]cyclopropane-1-carboxamide[ No CAS ]
(R)1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)-N-[1-[2,3-dihydroxy(2,3,3-2H3)propyl]-6-fluoro-2-(1-hydroxy-2-methylpropan-2-yl)-1H-indol-5-yl]cyclopropane-1-carboxamide[ No CAS ]
1-(2,2-difluorobenzo[1,3]dioxol-5-yl)cyclopropanecarboxylic acid amide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
61.3%
A mixture of 10 g of (2-2-difluoro-benzo[1,3]dioxol-5-yl)-acetonitrile, 10 gbenzyltriethylammonium chloride, and 14.8 g 1-bromo-2-chloro ethane was stined for 15mm. To the reaction mixture was added 30 ml 50% sodium hydroxide solution at room temperature, then the reaction mixture was heated to 70-75 C and maintained for 15 hours. After completion of reaction, 100 ml ethyl acetate and 100 ml water were added at room temperature and stined for 15 mm. The layers were separated, and the aqueouslayer was extracted with 25 ml ethyl acetate. The combined ethyl acetate layer waswashed with water. The solvent was completely removed under vacuum at 50-55 C. Tothe resulting residue, 50 ml dimethyl sulfoxide, 1.12 g potassium carbonate and 8.7 g30% hydrogen peroxide were added at 15-20 C. The reaction mixture was heated to 25-30 C and maintained for 3 hours. After completion of reaction, 100 ml water, 100 mltoluene were added to the reaction mixture. The layers were seapareted, and the aqueous layer was extracted with 50 ml toluene. The toluene layer was washed with 50m1 10% sodium sulfite, followed by washing with 50 ml water. The solvent was completely removed under vacuum at 50-55 C. The resulting residue was triturated with hexanes (50 mL) for 1 hour. The obtained solid was filtrered and washed with hexane (10 ml).The wet material was dried under vacuum to afford 1-(2-2-difluoro-benzo[1,3]dioxol-5- yl)-cyclopropanecarboxylic acid amide (7.5 g, 61.3% yield)
With N-benzyl-N,N,N-triethylammonium chloride; sodium hydroxide; In water; at 75℃; for 24h;
To a solution of 2, 2-difluorobenzo [1, 3] dioxol-5-yl-methanol (930 mg, 5 mmol) in anhydrous THF (5 ml) was added NaBH4 (208 mg, 5.5 mmol) in portions at T = 0 C in ten minutes. The mixture was then stirred at the same temperature for about 2 h (controlled with HPLC). H2O (1.5 ml) was added and the mixture was stirred for about 15 min. The mixture was then extracted with diethyl ether (3 * 3 ml) and washed with water (2 * 1 ml). The organic phase was completely dried and lyophilized to obtain 2,2-difluorobenzo [1, 3] dioxol-5-yl-methanol as colorless oil and used without further purification (865 mg, 85%). A solution of 2,2-difluorobenzo [1, 3] dioxol-5-yl-methanol (865mg, 4.6mmol) in thionyl chloride (2ml) was stirred at room temperature upon completeness. When the reaction was complete (about 1h), the solution was concentrated under vacuum to remove the excess of thionyl chloride. Then, the residue was resuspended in a solution of dichloromethane and saturated NaHCO3 1:1 (2ml). The aqueous layer was then back-extracted with dichloromethane (2×1ml) and the combined organic layers were dried over Na2SO4 and finally concentrated to vacuum to give 5(chloromethyl)-2,2-difluorobenzo [1, 3] dioxole (823mg, 87%) which was used directly in the next step. A mixture of 5(chloromethyl)-2,2-difluorobenzo [1, 3] dioxole (823 mg, 3.99 mmol) and KCN (519 mg, 7.98 mmol) was stirred in dimethyl sulfoxide (DMSO) (3 ml) for 3 h at room temperature. H2O (1.5 ml) was added and the mixture extracted with ethyl acetate (EtOAc) (3 * 3 ml) and the organic phases were dried over Na2SO4 and concentrated under reduced pressure. The product was purified by preparative HPLC to afford 2(2, 2-difluorbenzo [1, 3] dioxol-5-yl)-acetonitrile (432 mg, 55%). A mixture of 2(2, 2-difluorbenzo [1, 3] dioxol-5-yl)-acetonitrile (432 mg, 2.2 mmol), 1-bromo-2-chloro-ethane (615 muL, 7.5 mmol) and benzyltriethylammonium chloride (5 mg, 0.02 mmol) was heated to T = 75 C and then 50% (wt./wt.) aqueous sodium hydroxide (5 ml) was slowly added. The reaction was stirred at T = 75 C for 24 h. After this time 1-bromo-2-chloro-ethane (310 muL, 3.8 mmol) and 50% (wt./wt.) aqueous sodium hydroxide (1 mL) were added to insure the complete formation of the cyclopropyl moiety (about 12-14 h). The reaction was then heated to T = 150 C for about 60 h to insure complete conversion from the nitrile to the carboxylic acid. When the hydrolysis was complete (controlled with HPLC-MS) the dark brown mixture was diluted with water (10 mL) and extracted three times with equal volumes of dichloromethane to remove all the sub-products. The basic aqueous solution was acidified with concentrated hydrochloric acid to pH = 1. The reaction mixture was centrifuged at 4000g for 5 min and the pellet was washed with 1 M hydrochloric acid (3 * 1 ml). The solid material was dissolved in dichloromethane (5 mL) and extracted twice with equal volumes of 1 M hydrochloric acid. The organic phase was dried over Na2SO4 and concentrated under reduced pressure. The product was purified by preparative HPLC to afford the title compound (276 mg, 52%). 1H NMR (200 MHz, CDCl3) 7.17-6.96 (m, 3H), 1.82-1.65 (m, 2H), 1.39-1.22 (m, 2H). 13C NMR (50 MHz, CDCl3) delta, 179.4, 142.4, 141.9, 135.7, 133.7, 130.6, 124.6, 111.0, 107.9, 27.6 16.6. HRMS (ESI) calculated for C11H9N2O4F2: 243.0469 [M + H] + found 243.0464.
General procedure: To a solution of 2, 2-difluorobenzo [1, 3] dioxol-5-yl-methanol (930 mg, 5 mmol) in anhydrous THF (5 ml) was added NaBH4 (208 mg, 5.5 mmol) in portions at T = 0 C in ten minutes. The mixture was then stirred at the same temperature for about 2 h (controlled with HPLC). H2O (1.5 ml) was added and the mixture was stirred for about 15 min. The mixture was then extracted with diethyl ether (3 * 3 ml) and washed with water (2 * 1 ml). The organic phase was completely dried and lyophilized to obtain 2,2-difluorobenzo [1, 3] dioxol-5-yl-methanol as colorless oil and used without further purification (865 mg, 85%). A solution of 2,2-difluorobenzo [1, 3] dioxol-5-yl-methanol (865mg, 4.6mmol) in thionyl chloride (2ml) was stirred at room temperature upon completeness. When the reaction was complete (about 1h), the solution was concentrated under vacuum to remove the excess of thionyl chloride. Then, the residue was resuspended in a solution of dichloromethane and saturated NaHCO3 1:1 (2ml). The aqueous layer was then back-extracted with dichloromethane (2×1ml) and the combined organic layers were dried over Na2SO4 and finally concentrated to vacuum to give 5(chloromethyl)-2,2-difluorobenzo [1, 3] dioxole (823mg, 87%) which was used directly in the next step. A mixture of 5(chloromethyl)-2,2-difluorobenzo [1, 3] dioxole (823 mg, 3.99 mmol) and KCN (519 mg, 7.98 mmol) was stirred in dimethyl sulfoxide (DMSO) (3 ml) for 3 h at room temperature. H2O (1.5 ml) was added and the mixture extracted with ethyl acetate (EtOAc) (3 * 3 ml) and the organic phases were dried over Na2SO4 and concentrated under reduced pressure. The product was purified by preparative HPLC to afford 2(2, 2-difluorbenzo [1, 3] dioxol-5-yl)-acetonitrile (432 mg, 55%).
diphenyl(vinyl)sulfonium trifluoromethanesulfonate[ No CAS ]
[ 862574-87-6 ]
Yield
Reaction Conditions
Operation in experiment
85%
With 1,8-diazabicyclo[5.4.0]undec-7-ene; In dimethyl sulfoxide; at 21℃; for 12h;
General procedure: To a 5 mL Schlenk tube were added aryl / heteroaryl acetonitriles 1 (1.0 mmol, 1.0 equiv.), vinyl diphenylsulfonium triflate (434.4 mg, 1.2 mmol, 1.2 equiv.), and DMSO (5 mL). The mixture was stirred at room temperature for 2 min and to the mixture DBU (456 mg, 3 mmol, 3.0 equiv.) was added. The mixture was stirred for 12 hours at room temperature, and then to the mixture was added saturated ammonium chloride solution (25 mL). The resulting mixture was extracted with EtOAc (3 x 150 mL). The combined organic layers were washed with H2O (2 x 30 mL), dried with anhydrous sodium sulfate. After concentration, product 2 was purified using silica gel column chromatography using an appropriate eluent.
4-[(2R,4R)-4-([1-(2,2-difluoro-1,3 benzodioxol-5-yl)(2H4)cyclopropyl]carbonyl}amino)-7-(difluoromethoxy)-3,4-dihydro-2H-chromen-2-yl]benzoic acid[ No CAS ]
1-(2,2-difluoro-2H-1,3-benzodioxol-5-yl)(<SUP>2</SUP>H<SUB>4</SUB>)cyclopropane-1-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
43.4%
To a solution of Example 2E (4 g, 20.29 mmol) in methyl tert-butyl ether (40 mL) was added tetrabutylammonium bromide (0.327 g, 1.0 15 mmol), followed by degassed 50% aqueous NaOH (40 mL) over 20 minutes at 0 C. BrCD2CD2Br (1,2-dibromo(2H4)ethane, 6.62 g, 34.5 mmol) was added via cannula over 5 minutes. The reaction was stirred at 25 C for 12 hours. Two additional vials were set up as described above. After completion of the reaction, all three reaction mixtures were combined. The reaction was poured into ice water, maintaining a temperature of about 15 C. The mixture was extracted with methyl tert-butyl ether (2 x 200 mL) and the organic phase was dried over Na2SO4, filtered and concentrated to give a residue, which was purified by column chromatography on silica gel (eluted with petroleum/ethyl acetate = 5/1) to provide the title compound (6 g, 43.4 % yield). ?H NMR (400 MHz, DMSO-d6) oeppm 7.28 (dd,J=8.38, 1.76 Hz, 1 H) 7.36-7.48 (m, 1 H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping