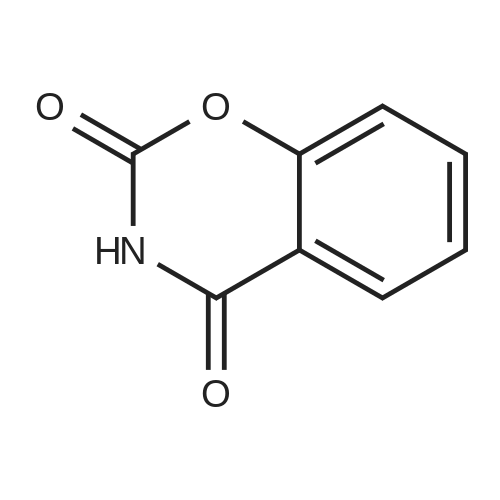
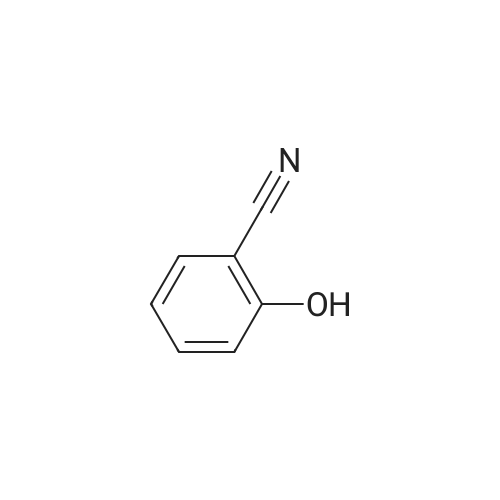
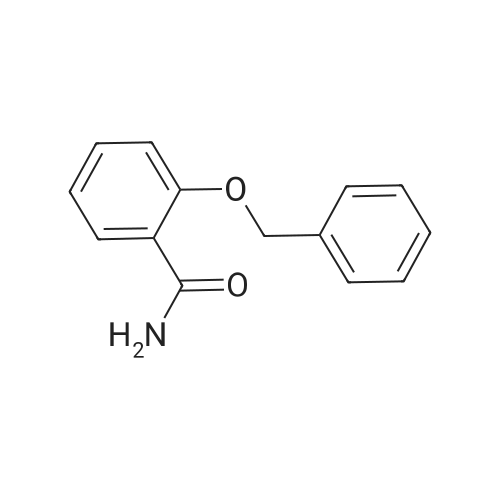
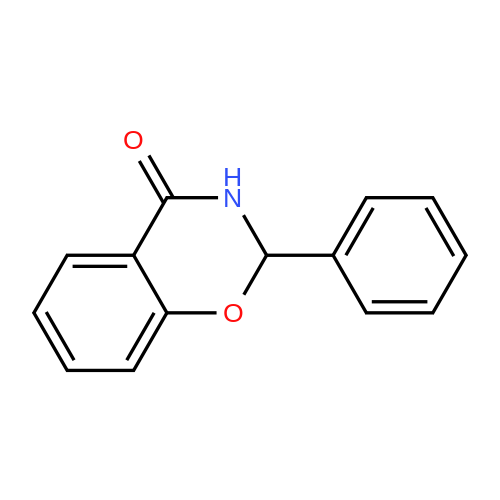
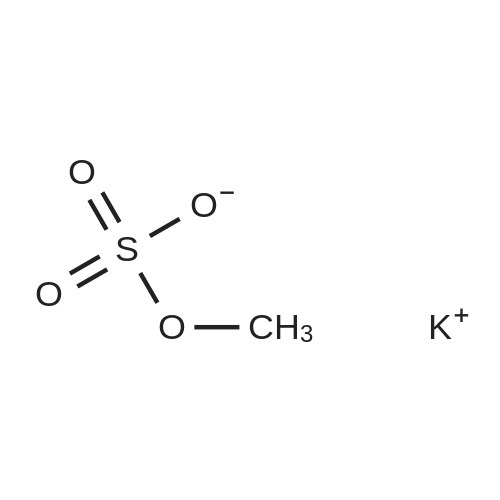
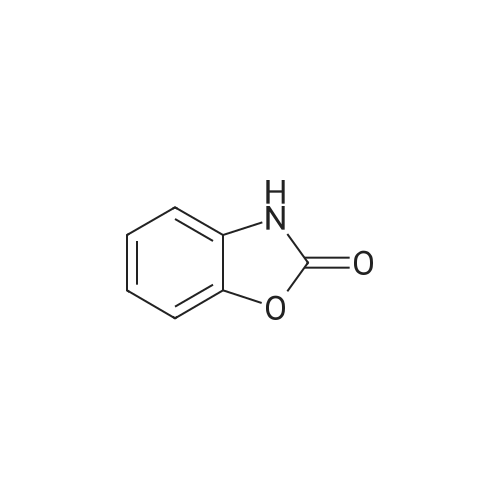
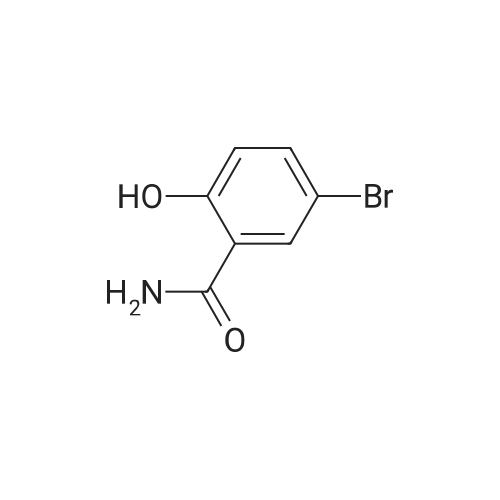
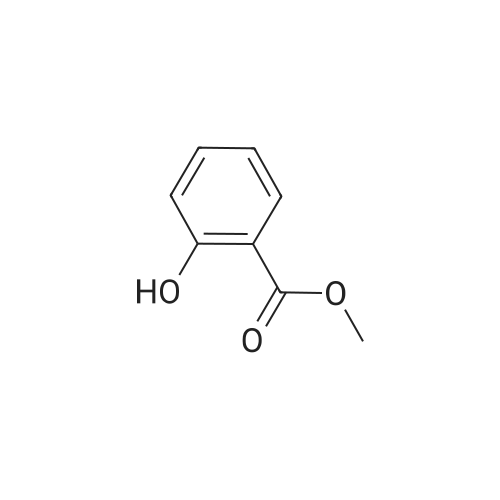
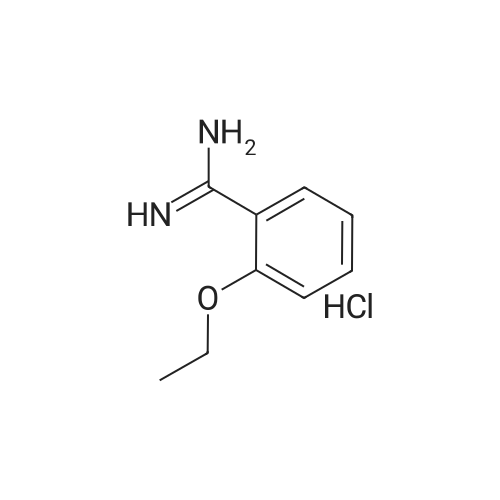
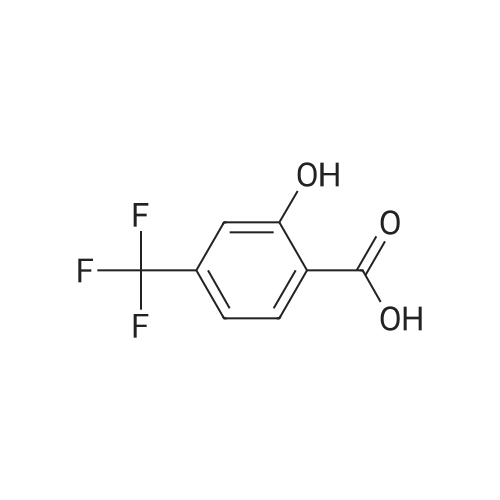
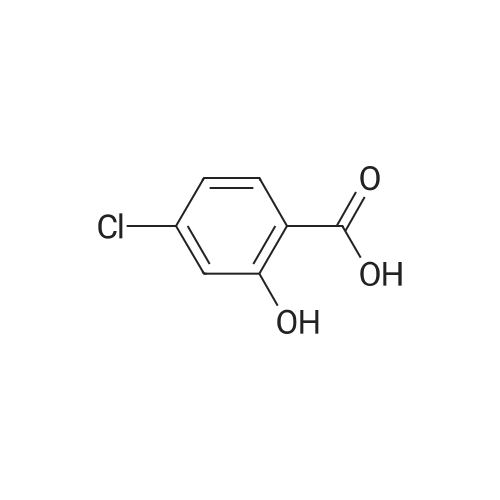
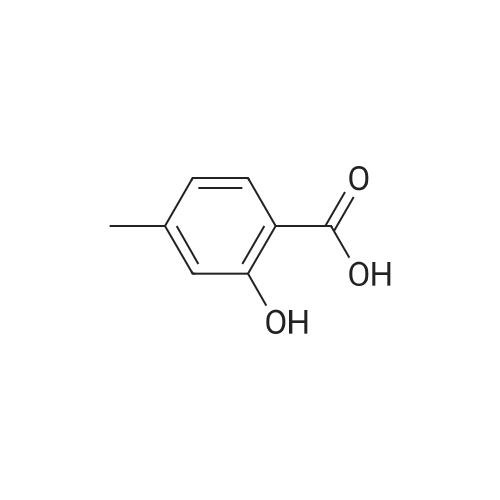
| 43% |
With pyridine; thionyl chloride; In N,N-dimethyl-formamide; for 4h;Reflux; |
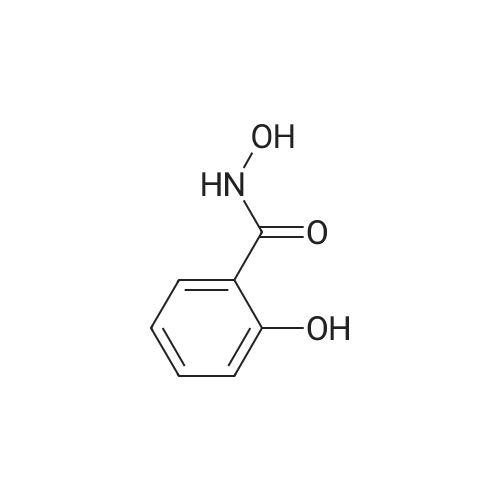
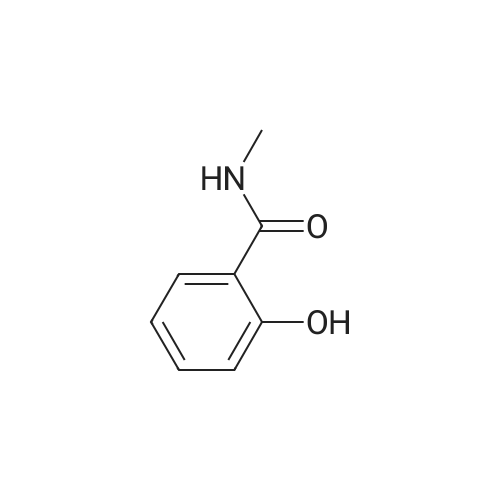
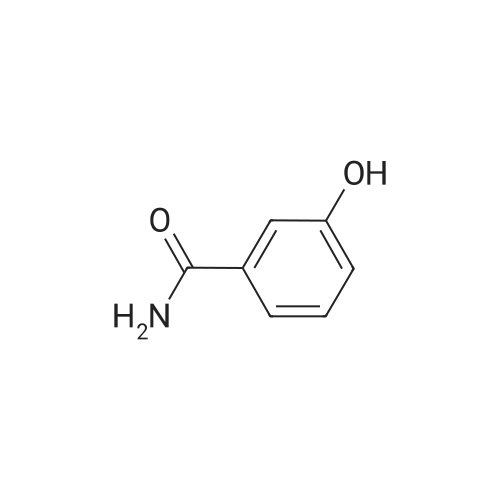
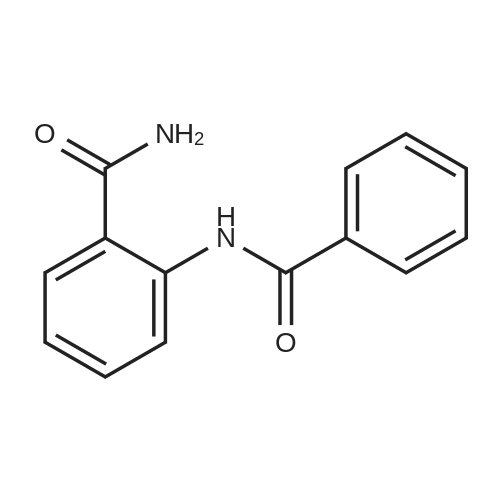
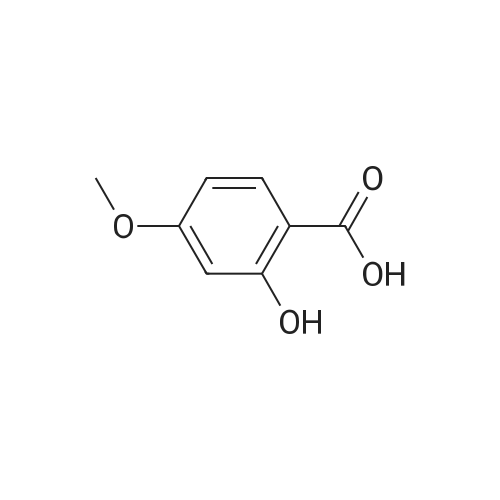
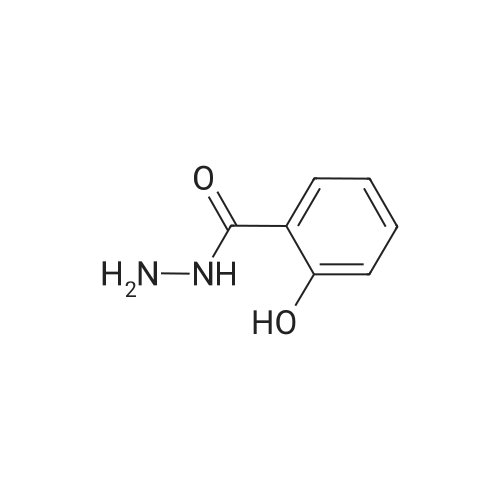
Salicylic acid (II) (2g, 14.5mmol), salicylamide (III) (1.53g, 11.1mmol) and N, N-dimethylformamide (1ml) were added to the solvent xylene (20ml), heated Reflux and mix well.After adding thionyl chloride (1.9 g, 16.0 mmol), it was stirred well for 4 h.A large amount of HCl and SO2 gas are generated during the reaction, and a tail gas absorption device is used. After a period of reaction, crystals were precipitated and stirring was continued for 30 min. After completion of the reaction, the mixture was distilled under reduced pressure, and the solvent was evaporated. The solid was washed with a mixed solution of ethanol (30 ml) and acetic acid (1 ml) and recrystallized from 2-methoxyethanol.The yellow-green solid product IV (1.14 g, 4.78 mmol, 43%) was obtained. |
| 39.3% |
|
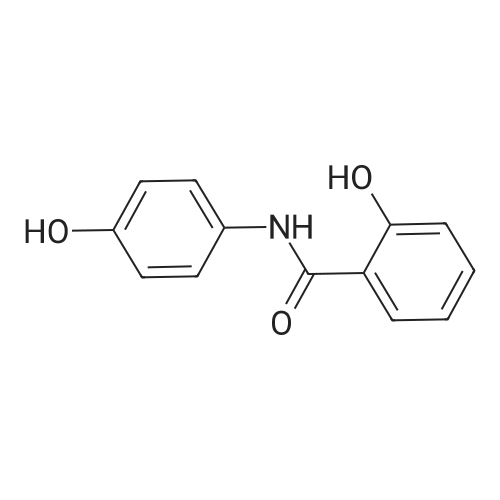
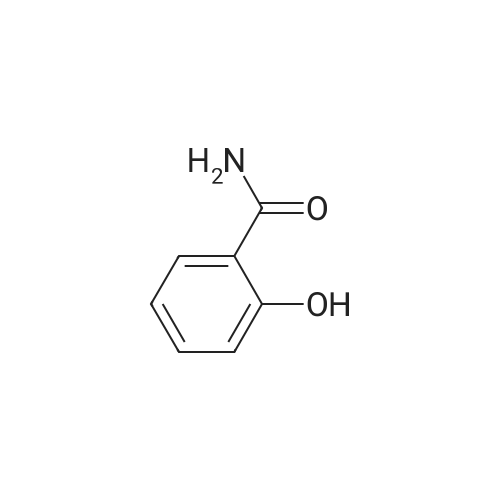
Example 1 Preparation of 2-(2-hydroxyphe41)-benz[1,3]oxazin-4-one A mixture of dichloromethane (200 ml), salicylic acid (50.0 gm) and p-toulenesulfonyl chloride (69 gm) were cooled to 10-15 C. Diisopropyl ethyl-amine (139.0 ml) was added drop-wise to the above mixture at 10-20 C. Reaction mass was stirred for 10 min at 10-20 C. and raised the temperature to 25-30 C. The reaction was maintained for 2 hours at 25-30 C. Reaction mass was cooled 0-5 C. Purified water (200 ml) was charged to the above mixture and stirred for 15 minutes. The layers were separated. Salicylamide (39.6 gm) and toluene (200.0ml) were heated to 85-90 C. and the above organic layer was added drop-wise into salicyliamide solution with simultaneous distillation of solvent at 85-90 C. and distilled the solvent upto the reaction mass temperature reaches to 110-120 C. and further reaction was maintained for 3hrs at 110-120 C. Further solvent was distilled under atmospheric pressure upto reaction mass temperature reaches to 140-160 c and further the reaction was maintained for 1-2 hrs at 140-160 C. until the starting material disappears. Reaction mass was cooled to 75-80 C. and distilled off completely toluene under vacuum. Ethanol (50 ml) was added to the above reaction mass at 75-80 C. Reaction was stirred for 15 min and distilled off the ethanol at 75-80 C. Further ethanol (50.0 ml) was added stir for 5-10 min. Ethanol was distilled off completely under vacuum at 75-80 C. Ethanol (150 ml) was charged into above contents at 75-80 C. The contents were maintained for 1 hour at 75-80 C. and slowly cooled to 0-5 C. Reaction mass was maintained for 2 hrs at 0-5 C. The reaction mass was filtered and washed with ethanol (50.0 ml). Dried the compound at 50-55 C. Yield: 39.30%. |
| 39% |
With pyridine; thionyl chloride; In 5,5-dimethyl-1,3-cyclohexadiene; ethanol; for 4h;Reflux; |
Salicylic acid (2 g, 14.5 mmol), salicylamide (1.53 g, 11.1 mmol)and pyridine (1 ml) were refluxed in xylene (20 mL). Thionylchloride (1.9 g, 16.0 mmol) was added with vigorous stirring over aperiod of 4 h. An intense evolution of SO2 and HCl was noted. At theend of the reaction, the product started to crystallize. Stirring wascontinued for an additional 30 min, and the xylenewas removed byreduced-pressure distillation. The resulting solid residue was suspendedin EtOH (30 mL) and acetic acid (1 mL). The mixture washeated gently and then allowed to cool to 20 C. The precipitatewasfiltered and recrystallized from 2-methoxyethanol (35 mL). Yield:yellow-green solid (1.04 g, 4.34 mmol, 39%). m.p. 202.2e204.6 C.1H NMR (500 MHz, DMSO-d6) d12.95 (s, 1H), 8.25e8.21 (m, 1H),8.08 (dd, J1 7.8, J2 1.5 Hz, 1H), 7.98e7.94 (m, 1H), 7.81 (dd,J1 8.4, J2 0.6 Hz, 1H), 7.68e7.60 (m, 2H), 7.13e7.08 (m, 2H). ESIMS:m/z. Calculated for C14H9NO3 239.06; found[MH] 240.0655. |
|
With pyridine; thionyl chloride; In xylene; for 11h;Reflux;Product distribution / selectivity; |
Mixture of salicylic acid (100 grams), salicylamide (89.3 grams), pyridine (15.2 ml) and xylene (600 ml) was heated to reflux temperature. Thionyl chloride (100.5 ml) was added to the above mixture at reflux temperature for 3 hours. Intense evolution of SO2 and hydrochloric acid was observed, the reaction mixture was then stirred for 8 hrs. Xylene was removed from the reaction mixture by distillation under reduced pressure. The resulting residue was suspended in methanol (200 ml) and raised the temperature to 60-65C. The reaction mixture was stirred for one hour at 60-65C and cooled to 0-5C and further stirred' for one hour. The solid obtained was filtered, washed with methanol and dried to get the title compound.Yield: 130 grams |
|
|
Example 1; Preparation of 2-(2-Hydroxyphenyl)-4H-1,3-benzoxazine-4-oneSalicylic acid (50 gm) was taken in xylene (250 ml) followed by the addition of thionyl chloride (64.5 gm) drop wise to the reaction mixture at 25-30 C. The reaction mixture was stirred for 90 minutes at 40-45 C. The excess thionyl chloride was removed by distillation. Salicylamide (49.7 gm) was added to the resulting mixture and followed by the distillation of xylene up to a reaction temperature of 170 C. The reaction mixture was further stirred for 60 minutes at 80 C. followed by the addition of ethanol (80 ml) and refluxed for 15 minutes. The resulting mass was cooled to 25 C. and stirred for 30 minutes at the same temperature. The resulting solid was filtered and dried to produce 43 gm of 2-(2-hydroxyphenyl)-4H-1,3-benzoxazine-4-one as slightly yellow crystals. (Melting point: 206-208 C.). |
|
|
Example-I [68] Preparation of 2-(2-hydroxyphenyl) benz [e] [1, 3] oxazin-4-one [69] [70] Xylene (1.5 L) and salicylic acid (1 Kg.) was added to a 5 L 4-neck round bottom flask equipped with a mechanical stirrer and thermocouple. Thionyl chloride (1.29 kg) was added at 10 oC to 15oC. After addition of thionyl chloride reaction mass is stirred at 10 0C to 15oC for 30 minutes. Reaction mass is heated at 35-40 0C for 1 hr. Salicylamide solution (0.891 kg in 2.0 lit of Xylene) was added in reaction mass at 25 0 C -30 0 C. After addition reaction mass was gradually heated at 80 0C - 126 0C and stirred for 2 hrs. After the completion of reaction excess of Xylene was distilled out. Methanol was added in the reaction mass at 70 0C -80 0C and stirred for 1 hr. gradually cooled the reaction mass at 25-30 0C. Filtered the solid and washed with Methanol and sucked it dry. Dry the obtained solid at 55-60 0C under vacuum tray drier. |
|
|
EXAMPLE-I Preparation of 2-(2-Hydroxyphenyl) Benz [e] [1, 3] Oxazin-4-One Xylene (1.5 L) and salicylic acid (1 Kg.) was added to a 5 L 4-neck round bottom flask equipped with a mechanical stirrer and thermocouple. Thionyl chloride (1.29 kg) was added at 10 C. to 15 C. After addition of thionyl chloride reaction mass is stirred at 10 C. to 15 C. for 30 minutes. Reaction mass is heated at 35-40 C. for 1 hr. Salicylamide solution (0.891 kg in 2.0 lit of Xylene) was added in reaction mass at 25 C.-30 C. After addition reaction mass was gradually heated at 80 C.-126 C. and stirred for 2 hrs. After the completion of reaction excess of Xylene was distilled out. Methanol was added in the reaction mass at 70 C. -80 C. and stirred for 1 hr. gradually cooled the reaction mass at 25-30 C. Filtered the solid and washed with Methanol and sucked it dry. Dry the obtained solid at 55-60 C. under vacuum tray drier. |
|
With pyridine; thionyl chloride; In 5,5-dimethyl-1,3-cyclohexadiene;Reflux; |
Salicylic acid (b) 24.9 g was sequentially added to a 250 mL three-necked flask. Salicylamide (c) 20.6g, 1.5 mL of pyridine and 30 mL of xylene, Slowly add 23.7 mL of thionyl chloride under stirring. After heating and refluxing, The solvent was removed under reduced pressure. The residue is dissolved by heating with a mixed solvent of ethanol and acetic acid. cool down, filter, Ethylene glycol monomethyl ether recrystallized, Yellow needle crystals, Mp 226-227 C. |
| 15 g |
With pyridine; aluminum (III) chloride; thionyl chloride; In o-xylene; at 25 - 125℃; for 1.5h; |
10 grams of salicylamide, 10.90 grams of salicylic acid, 0.20 grams aluminum chloride and 1.17 grams pyridine were added to 55 ml o-xylene at 25-30C. Reaction mass temperature was raised to 115-125C and then 16.70 grams thionyl chloride was slowly added over a period of 60 min at 1 15-125C and maintained at 1 15-125C for 30 min. The reaction mass was cooled to 25-30C and further added 33 ml absolute alcohol. The obtained slurry product was filtered and washed with absolute alcohol to give 15 grams of pale yellow crystalline 2-(2- hydroxyphenyl)-4H- l,3-benzoxazin-4-one having chromatographic purity about (0077) 95.5 %. (0078) The above product was purified by refluxing in absolute alcohol in presence of sodium methoxide followed by glacial acetic acid addition and then cooled to room temperature. The product was filtered and washed with absolute alcohol to obtain pure 2- (2-hydroxyphenyl) -4H- 1 , 3 -b enzoxazin-4-one having chromatographic purity about 99.5 %. |
|
With pyridine; thionyl chloride; In 5,5-dimethyl-1,3-cyclohexadiene; at 130 - 140℃; for 2h; |
General procedure: Substituted benzoic acid (1.2e2.0 g, 10 mmol) and o-hydroxybenzamide (1.5e2.3 g, 11 mmol) were added into 20 mL of xylene,then anhydrous pyridine (catalytic amount) was added, and thionylchloride (1.5 mL, 20 mmol) in xylene (8 mL) was added dropwiseover 30 mins at room temperature. After that, stirring was undertakenat 130e140 C. The mixturewas stirred for 2 h (TLC show acidwas consumed) and the solvent was distilled off under reducedpressure to obtain intermediate. The intermediate, 4-hydrazinobenzene-1-sulfonamide hydrochloride (1.1 g, 5 mmol)and triethylamine (1.0 g,10 mmol), were added to 15 mL of absoluteethanol, and the mixture was stirred at 80 C for 18 h. When thereaction was complete, the solution was filtered, and saturatedsodium bicarbonate was added to adjust pH to 7. The solution wasextracted with ethyl acetate, and the organic layer was washedwith 20 mL of saturated brine and dried over anhydrous sodium.The solvent was evaporated under reduced pressure to obtain thetarget compounds. The target compounds were recrystallized frommethanol. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping