* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
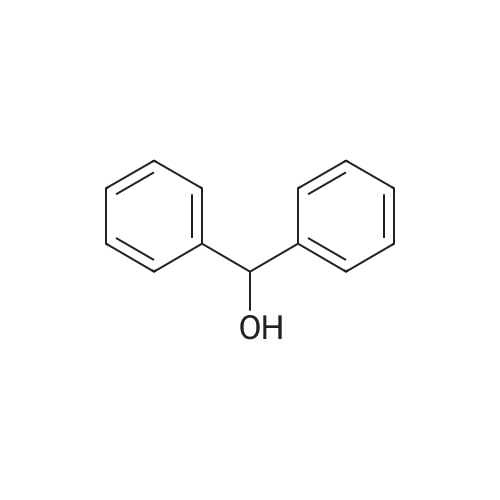
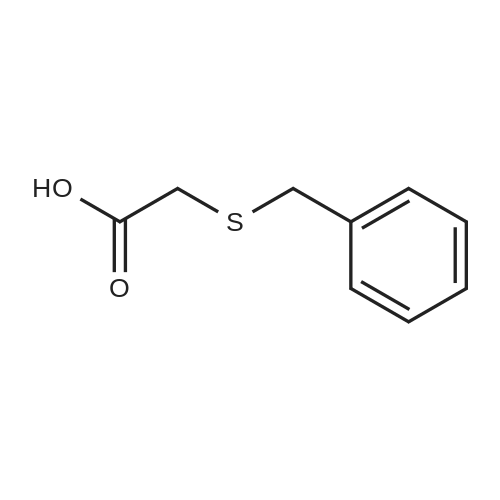
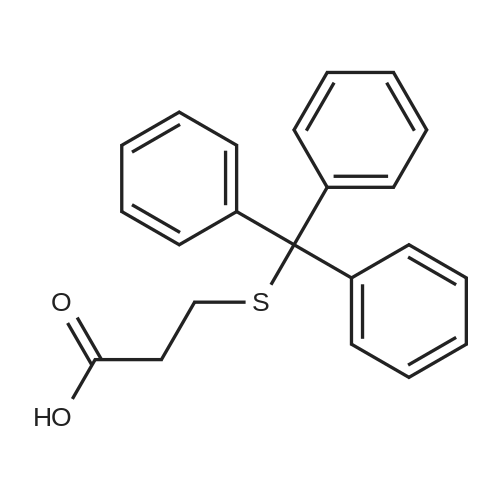
Thioglycolic acid (10.5 g, 7.92 ml (d=l .325), 0.114 mol) was added dropwise to the solution of diphenylmethanol in trifuoroacetic acid (100 ml). The formation of precipitate was observed in 30 min. The reaction mixture was stirred at room temperature for 3 h. The resulting precipitate was filtered off washed with water (3> 50 ml) and hexanes. The crude product was recrystallized from EtOAc/hexanes to get colorless precipitate of 2- (Benzhydrylthio)acetic acid. Yield: 17.93 g (64percent). M.p. 121 °C. 2-(B enzhy dryl th io)acetam ide
Reference:
[1] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2984 - 2997
[2] Tetrahedron Asymmetry, 2004, vol. 15, # 6, p. 1053 - 1058
[3] Tetrahedron Asymmetry, 2005, vol. 16, # 21, p. 3507 - 3511
[4] Patent: US7553646, 2009, B2,
[5] Molecules, 2012, vol. 17, # 9, p. 10446 - 10458
[6] Journal of Labelled Compounds and Radiopharmaceuticals, 1997, vol. 39, # 10, p. 853 - 874
[7] Journal of Organometallic Chemistry, 1996, vol. 507, # 1-2, p. 215 - 220
[8] Patent: WO2016/23997, 2016, A1, . Location in patent: Page/Page column 34
[9] Acta Chemica Scandinavica (1947-1973), 1948, vol. 2, p. 856,858
[10] Journal fuer Praktische Chemie (Leipzig), 1934, vol. <2> 141, p. 93,96[11] Arkiv foer Kemi, 1937, vol. 12 A, # 14, p. 3,4
[12] Journal fuer Praktische Chemie (Leipzig), 1934, vol. <2> 141, p. 93,96[13] Arkiv foer Kemi, 1937, vol. 12 A, # 14, p. 3,4
[14] Journal fuer Praktische Chemie (Leipzig), 1934, vol. <2> 141, p. 93,96[15] Arkiv foer Kemi, 1937, vol. 12 A, # 14, p. 3,4
[16] Patent: WO2009/90663, 2009, A1, . Location in patent: Page/Page column 8
[17] ACS Medicinal Chemistry Letters, 2011, vol. 2, # 1, p. 48 - 52
[18] Journal of Medicinal Chemistry, 2014, vol. 57, # 3, p. 1000 - 1013
3
[ 108-86-1 ]
[ 103-46-8 ]
[ 63547-22-8 ]
Yield
Reaction Conditions
Operation in experiment
99.4%
With tris-(dibenzylideneacetone)dipalladium(0); (oxan-4-yl)diphenylphosphine; silver trifluoromethanesulfonate; acetic acid; cerous nitrate In N,N-dimethyl acetamide at 100℃; for 9 h;
Example 4: To the appropriate amount of an acidic organic solvent (1: 3 mixture of acetic acid and DMA, 1: 3) was added 100 mmol of 2_ (phenylmethylthio) acetic acid, 175 mmol of bromobenzene, 7 mmol of catalyst (from 2 mmol Tri (arylene acetone) palladium and 5 mmol of Ce (N03) 3), 3.5 mmol of organic ligands L1 and 75 mmol of silver trifluoromethanesulfonate were added and the temperature was raised to 100 ° C and the reaction was carried out at that temperature for 9 hours. The reaction system obtained after the completion of the reaction was filtered while hot, the filtrate was washed with a saturated aqueous solution of sodium carbonate, and the aqueous phase and the upper organic phase were separated. The organic phase was washed with saturated brine, and the upper organic phase was taken and evaporated to evaporate to obtain a residue The eluate was purified by silica gel column chromatography (eluent was a mixed solvent of chloroform and n-butanol in a volume ratio of 10: 1). The eluted fractions were collected and the solvent was removed by evaporation to give a white The yield of the product was 99.4percent and the purity was 93.2percent.
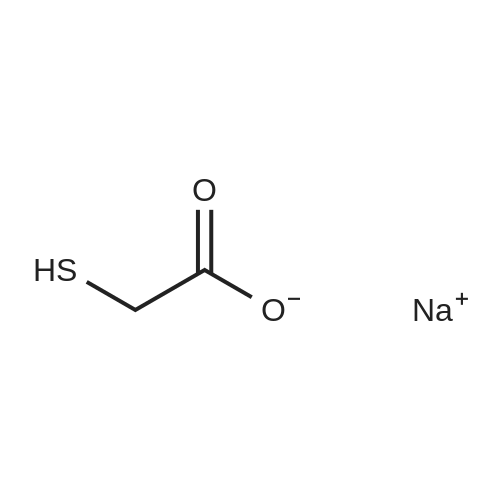
[0097] Benzhydryl bromide 1 (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H20; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60-70°C for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2C12 and saturated aqueous NaHC03. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid (3, 46percent), mp 125°C. Rf 0.2. Recrystallization from MeOH/H20 gave mp 126-128°C.
46%
at 20 - 70℃; for 3 h; Inert atmosphere
Step-1: Synthesis of Compound 3: Benzhydryl bromide 1 (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H2O; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60-70° C. for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2Cl2 and saturated aqueous NaHCO3. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid ( 3, 46percent), mp 125° C. Rf 0.2. Recrystallization from MeOH/H2O gave mp 126-128° C
Reference:
[1] Organic Process Research and Development, 2008, vol. 12, # 4, p. 614 - 617
7
[ 91-01-0 ]
[ 89619-93-2 ]
[ 63547-22-8 ]
Reference:
[1] Russian Chemical Bulletin, 2013, vol. 62, # 5, p. 1164 - 1175[2] Izv. Akad. Nauk, Ser. Khim., 2013, # 5, p. 1164 - 1175,12
8
[ 91-01-0 ]
[ 79-11-8 ]
[ 63547-22-8 ]
Reference:
[1] Journal of Organic Chemistry, 1968, vol. 33, # 5, p. 2030 - 2035
9
[ 5670-78-0 ]
[ 68-11-1 ]
[ 63547-22-8 ]
Reference:
[1] Svensk Kemisk Tidskrift, 1935, vol. 47, p. 259[2] Chem. Zentralbl., 1937, vol. 108, # I, p. 98
10
[ 68-11-1 ]
[ 776-74-9 ]
[ 63547-22-8 ]
Reference:
[1] Annales Pharmaceutiques Francaises, 1953, vol. 11, p. 509,518
11
[ 623-11-0 ]
[ 860541-61-3 ]
[ 1733-63-7 ]
[ 63547-22-8 ]
[ 65-85-0 ]
Reference:
[1] Journal of the Chemical Society, 1942, p. 90,92
[2] Chemische Berichte, 1939, vol. 72, p. 1257,1269
12
[ 63547-22-8 ]
[ 63547-24-0 ]
Yield
Reaction Conditions
Operation in experiment
94.17%
Stage #1: With sodium hydroxide In water at 25 - 30℃; for 0.25 h; Stage #2: With dihydrogen peroxide In water at 25 - 35℃; for 20 - 26 h; Stage #3: With hydrogenchloride In water; toluene at 25 - 30℃; for 1 h;
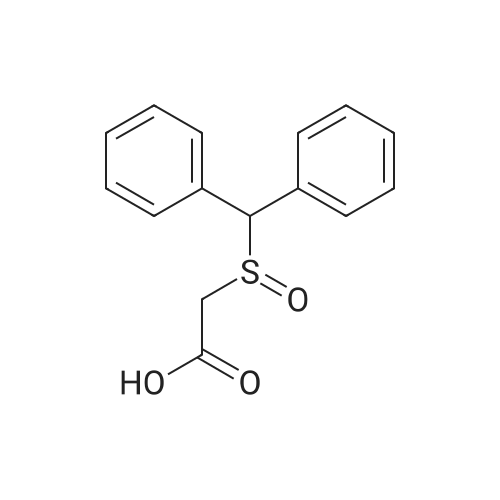
Example 1; Preparation of 2-[(Diphenylmethyl)sulfinyl] acetic acid; 2-[(Diphenylmethyl)thio] acetic acid (416 g, 1.61 mol) was suspended in purified water(4160 ml) at 25 to 300C. This was followed by the addition of 30percent sodium hydroxide solution [prepared by dissolving of 70.90 g (1.77 mol) of sodium hydroxide flakes/pellets in 235 ml of purified water] over a period of 15 minutes at 25 to 3O0C. Next, 50percent of hydrogen peroxide (142.5 g, 2.095 mol) was added in 5 to 6 hours maintaining <n="16"/>temperature 25 to 35°C under agitation. The reaction mixture was further stirred for 15 to 20 hours at 25 to 350C. After completion of the reaction (starting material should be less than 0.5percent by area) toluene (1664 ml) was added and the reaction mixture was stirred for 10 minutes. The resulting biphasic mixture was acidified with concentrated hydrochloric acid (250 ml) under stirring while maintaining the temperature between 25 to 3O0C. The resulted mass further stirred at 25 to 3O0C for 1 hour. The precipitated product was collected by filtration, washed with water (3 x 832 ml) followed by toluene (416 ml) and dried to produce 416 g of 2-[(diphenylmethyl)sulfmyl] acetic acid as a white crystalline powder (Yield: 94.17percent; HPLC Purity: 99.3percent by area).
88.15%
With tert.-butylhydroperoxide In methanol; toluene at 0 - 30℃; for 6.58333 - 7.75 h;
Example 5; Preparation of 2-[(DiphenyImethyI)sulfinyl] acetic acid; 2-[(Diphenylmethyl) thio] acetic acid (5 g) was dissolved in toluene: methanol (10:1, 55 ml) at 25 to 300C and the resulting mass was cooled at 0 to 50C. This was followed by the addition of vanadium acetyl acetonoate (0.04 g) and stirred the reaction <n="18"/>mass for 15 minutes at 0 to 50C. The tert-butylhydroperoxide (70percent solution, 3.54 g) was added at 0 to 50C under stirring over period of 20 to 30 minutes. The reaction mixture was further stirred for 6 to 7 hours at 0 to 5°C. Next, water (100 ml) was added to the reaction mixture and stirring continued for 1 hour at 10 to 15°C. The precipitated product was collected by filtration, washed with water (3 x 100 ml) followed by toluene (50 ml) and dried to give the 2-[(diphenylmethyl) sulfinyl] acetic acid as a crystalline powder (Yield = 88.15percent; HPLC Purity: 98.16percent by area).
87.15%
Stage #1: With sodium percarbonate In methanol at 10 - 30℃; for 12.25 - 14.25 h; Stage #2: With hydrogenchloride In methanol; water; toluene at 25 - 30℃; for 1 h;
Example 4; Preparation of 2-[(DiphenylmethyI)sulfinyl] acetic acid; 2-[(Diphenylmethyl) thio] acetic acid (10 g) was dissolved in methanol (50 ml) at 25 to 3O0C. This was followed by the addition of ammonium molybdate (0.40 g) and stirring the reaction mass for 15 minutes at 25 to 300C. The reaction mixture was cooled at 10 to 150C followed by the addition of sodium per carbonate (5.0 g) at 10 to 150C under stirring. The reaction mixture was further stirred for 12 to 14 hours at 20 to 25°C.This was followed by the addition of purified water (200 ml) and toluene (50 ml) under stirring for 10 minutes. The resulting biphasic mixture was acidified with cone. hydrochloric acid at pH less than 2 under stirring maintaining temperature at 25 to 300C.The resulted mass further stirred at 25 to 3O0C for 1 hour. The precipitated product was collected by filtration, washed with water (3 x 100 ml) followed by toluene (100 ml) and dried to give the 2-[(diphenylmethyl) sulfinyl] acetic acid as crystalline powder (Yield: 87.15percent; HPLC Purity: 95.70percent by area).
78.58%
Stage #1: With (-)-α-methylbenzylamine In water at 25 - 30℃; for 0.25 h; Stage #2: With sodium hypochlorite In water at 40 - 45℃; for 2.5 - 3 h; Stage #3: With hydrogenchloride In water; toluene at 25 - 30℃; for 1.5 h;
Example 3; Preparation of 2-[(Diphenylmethyl)sulfmyl] acetic acid; 2-[(Diphenylmethyl) thio] acetic acid (5 g) was suspended in purified water (50 ml) at 25 to 3O0C. This was followed by the addition of (-)-(α)-methyl benzyl amine <n="17"/>(2.345 g) and stirred the reaction mass for 15 minutes at 25 to 3O0C. The reaction mixture was further heated at 40 to 45°C followed by addition of sodium hypochlorite (5.28percent, 33 ml) over period of 30 minutes. The reaction mixture was further stirred for 2 to 2.5 hours at 40 to 450C. The reaction mass was cooled to 25 to 3O0C which is then followed by the addition of toluene (20 ml) and stirred the reaction mixture for 10 minutes. The resulting biphasic mixture was acidified with cone, hydrochloric acid under stirring at 25 to 300C. The resulted mass further stirred at 25 to 3O0C for 1.5 hour. The precipitated product was collected by filtration, washed with water (3 x 50 ml) followed by toluene (50 n) and dried to give the 2-[(diphenylmethyi) sulfmyl] acetic acid as a crystalline powder (Yield: 78.58percent; HPLC Purity: 99.4percent by area).
60.28%
Stage #1: With sodium hydroxide In water at 25 - 30℃; for 0.25 h; Stage #2: at 40 - 45℃; for 11 - 13.5 h; Stage #3: With sulfuric acid In water; toluene at 25 - 30℃; for 1 h;
Example 2; Preparation of 2-[(Diphenylmethyl)sulfinyl] acetic acid; 2-[(Diphenylmethyl) thio] acetic acid (25 g) was suspended in purified water (100 ml) at 25 to 300C. This was followed by the addition of 30percent sodium hydroxide solution (prepared by dissolving of 16.0 g of sodium hydroxide flakes/pellets in 50.0 ml purified water) over a period of 15 minutes at 25 to 3O0C. The reaction mass was heated to 40 to 450C. Further N-chlorosuccinimide (20.0 g) was added in 1 to 1.5 hours maintaining temperature 40 to 45°C under agitation. The reaction mixture was further stirred for 10 to 12 hours at 40 to 450C. The reaction mass was cooled to 25 to 3O0C after the completion of reaction followed by the addition of toluene (100 ml) and reaction mixture further stirred for 10 minutes. The resulting biphasic mixture was acidified with 50percent aqueous sulfuric acid under stirring maintaining temperature at 25 to 300C. The resulted mass was further stirred at 25 to 3O0C for 1 hour. The precipitated product was collected by filtration, washed with water (3 x 100 ml) followed by toluene (100 ml) and dried to give the 2-[(diphenylmethyl) sulfinyl] acetic acid as a white crystalline powder (Yield: 60.28percent; HPLC Purity: 98.50percent by area).
51.81%
Stage #1: With sodium perborate; water; acetic anhydride In methanol; toluene at -25 - 30℃; for 6.75 h; Stage #2: With hydrogenchloride In methanol; water; toluene for 0.25 h;
Example 6; Preparation of 2-[(Diphenylmethyl)sulfinyl] acetic acid; Sodium perborate (15.30 g) was suspended in purified water (23 ml) at 25 to 300C. The resulting mass was cooled to 10 to 15°C. This was followed by addition of acetic anhydride: methanol solution (1:1, 10.2 g dissolved in 10.2 g of methanol) at 10 to 150C and continued stirring for 15 minutes at 10 to 150C. The reaction mass was further cooled to -20 to -250C followed by the addition of 2-[(diphenylmethyl) thio] acetic acid solution (20 gm dissolved in 220 ml toluene: methanol, 10:1) at -10 to -150C under stirring over period of 2.5 hours. The reaction mixture was further stirred for 2 hours at -10 to -15°C. The reaction temperature was increased to 0 to 5°C followed by stirring for 2 hours. Then reaction mass was added to purified water (600 ml) and acidified with cone, hydrochloric acid to below 2.0 pH. The resulting precipitate was stirred for 15 minutes. The precipitated product was collected by filtration and washed with water (3 x 100 ml) and toluene (100 ml) and dried to give the 2-[(diphenylmethyl) sulfinyl] acetic acid as a crystalline powder (Yield = 51.81percent, HPLC Purity: 97.0percent by area).
91 %Chromat.
With dihydrogen peroxide In 1,2-dichloro-ethane at 20℃; for 1.5 h;
General procedure: To examine the catalytic activity of the heterogeneous catalyst, 1 mmol of sulde,1.5 ml of hydrogen peroxide 30 percent as oxidant and 5 percent mol of catalyst weredissolved in 3 ml solvent and reacted at room temperature for different times(Scheme 4; Table 1; Fig. 7). The monitoring of the sulfoxide formation was carried out by TLC (n-hexanes:EtOAc, 1:1 or CHCl3:MeOH, 9:1 as eluent). Aftercompletion of the reaction, the solvent was evaporated and the crude product waspuried by a recrystallization method (using EtOAC/n-hexane) (Table 2). Thecatalyst was recovered and reused for further runs.
Reference:
[1] Organic and Biomolecular Chemistry, 2017, vol. 15, # 12, p. 2647 - 2654
[2] Patent: WO2009/24863, 2009, A2, . Location in patent: Page/Page column 14-15
[3] Patent: WO2009/24863, 2009, A2, . Location in patent: Page/Page column 16-17
[4] Patent: WO2009/24863, 2009, A2, . Location in patent: Page/Page column 16
[5] Patent: WO2009/24863, 2009, A2, . Location in patent: Page/Page column 15-16
[6] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2984 - 2997
[7] Patent: WO2009/24863, 2009, A2, . Location in patent: Page/Page column 14-15
[8] Patent: WO2009/24863, 2009, A2, . Location in patent: Page/Page column 17
[9] Tetrahedron Asymmetry, 2004, vol. 15, # 6, p. 1053 - 1058
[10] Patent: WO2007/71035, 2007, A1, . Location in patent: Page/Page column 24
[11] Tetrahedron Asymmetry, 2007, vol. 18, # 24, p. 2959 - 2964
[12] Patent: WO2009/90663, 2009, A1, . Location in patent: Page/Page column 8-9
[13] Molecules, 2012, vol. 17, # 9, p. 10446 - 10458
[14] Molecules, 2012, vol. 17, # 9, p. 10446 - 10458
[15] Research on Chemical Intermediates, 2016, vol. 42, # 12, p. 8201 - 8215
Example 5 Benzhydrylsulfanyl acetic acid (1) This compound was prepared according to the procedure of Prisinzano. (Prisinzano, T.; Podobinski, J.; Tidgewell, K.; Luo, M.; Swenson, D. Tetrahedron: Asymmetry 2004, 15, 1053-1058). The synthesis employed benzhydrol (50.0 g, 271.4 mmol) and thioglycolic acid (25.0 g, 271.4 mmol) to give the title compound as a white solid: 69.2 g (99% yield); mp 126-129 C. Spectroscopic data was identical to lit. (Prisinzano, T.; Podobinski, J.; Tidgewell, K.; Luo, M.; Swenson, D. Tetrahedron: Asymmetry 2004, 15, 1053-1058).
64%
With trifluoroacetic acid; at 20℃; for 3h;
Thioglycolic acid (10.5 g, 7.92 ml (d=l .325), 0.114 mol) was added dropwise to the solution of diphenylmethanol in trifuoroacetic acid (100 ml). The formation of precipitate was observed in 30 min. The reaction mixture was stirred at room temperature for 3 h. The resulting precipitate was filtered off washed with water (3> 50 ml) and hexanes. The crude product was recrystallized from EtOAc/hexanes to get colorless precipitate of 2- (Benzhydrylthio)acetic acid. Yield: 17.93 g (64%). M.p. 121 C. 2-(B enzhy dryl th io)acetam ide
With toluene-4-sulfonic acid; In dichloromethane; at 20℃;Reflux;
Step I: Preparation of the benzhvdryl thio acetic acid from benzhvdrol: Benzhydrol (10 g), Thioglycolic acid (7.8 g) and pTSA (0.2g) were charged in 100 mL of dichloromethane at room temperature. The reaction mixture was refluxed azeotropically till benzhydrol is not more than 1 %. 50 mL of DM water was added and the organic layers separated. The solvent was distilled out under vacuum till slurry is formed. 100 mL of cyclohexane was added to the slurry to get a solid, which was filtered and dried, under vacuum at 40-45 0C. Yield: 8.5 g Purity: 98-99%
General procedure: Procedure B. Thioacetamides 2b-2f were synthesized in three steps. Step 1: Thioglycolic acid (1 equiv.) was reacted with diphenylmethanol or the appropriate substituted diphenylmethanol (1 equiv.) in TFA (14 equiv.) overnight at room temperature. After solvent removal in vacuo, the residue obtained was washed with water (5 mL) and hexanes (15 mL) to give the carboxylic acid product, which was carried to the next step without further purification. Step 2: The acid product from step 1 was reacted with K2C03 (1.5 equiv.) and iodomethane (CH3I; 1.5 equiv.) in acetone (50 mL) overnight under reflux conditions. After solvent removal in vacuo, the residue was suspended in water (20 mL) and extracted with CH2C12 (3 x 20 mL). The combined organic layer was dried over MgS04 and concentrated to give the methyl ester, which was carried to the next step without further purification. Step 3: A mixture of the ester (1 equiv.), NH4C1 (1.4 equiv.), concentrated NH4OH (28.0-30.0%; 20 mL) and MeOH (5.7 mL) was stirred at 50 C for 72 hours. MeOH was removed in vacuo and the reaction mixture was diluted with water (50 mL), extracted with ethyl acetate (3 x 50 mL), and dried over Na2S04. The solvent was evaporated and the recovered crude product was purified by flash column chromatography using 1 : 1 ethyl acetate/hexanes to afford the pure product. 2-((Di-p-tolylmethyl)thio)acetamide (2b). Compound 2b was synthesized according to general procedure B to give 2b (450 mg, 52% yield) as a yellow oil. 1H NMR (CDC13): 5 7.26 -7.30 (m, 4H), 7.12 (d, J = 1.6 Hz, 4H), 6.54 (brs, 1H), 5.53 (brs, 1H), 5.11 (s, 1H), 3.07 (s, 2H), 2.31 (s, 6H); GC/MS (EI): m/z 285 (M+).
16
[ 63547-22-8 ]
[ 593-56-6 ]
C16H17NO2S[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 4-methyl-morpholine; O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; In N,N-dimethyl-formamide; at 20℃;
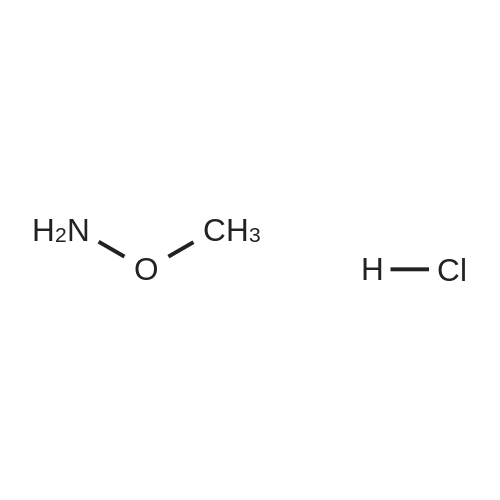
A mixture of compound 37a (7.45 g, 27.79 mmol), O-methyl hydroxylamine hydrochloride (2.75 g, 32.93 mmol), TBTU (11.4 g, 35.5 mmol) and NMM (10 mL) in dry DMF (20 mL) was stirred at room temperature overnight. Excess solvent was removed and the mixture was diluted with EtOAc that was washed successively with water, 2% citric acid, water, 2% NaHCO3, water and brine. Drying (MgSO4) and solvent evaporation gave a crude product that was purified by flash chromatography (silica gel, hexane:EtOAc 2:3) to generate 7.71 g of compound IV-1; 1H-NMR (DMSO-d6): delta 11.1 (s, 1H), 7.43-7.22 (series of m, 10H), 5.43 (s, 1H), 3.55 (s, 3H), 2.86 (s, 2H).
With NADP; sodium hydroxide; In isopropyl alcohol; at 35℃;pH 8.54;Alkaline conditions;Catalytic behavior;
Example 3 Preparation of (R)-2-(Benzhydrylsulfinyl)acetic Acid (compound (2b)) at a 15 g Scale Using a CHMO Variant (0228) This example illustrates a process for preparing the armodafinil intermediate compound, (R)-2-(Benzhydrylsulfinyl)acetic acid (compound (2b)) in enantiomeric excess at a 15 g scale via a biocatalytic conversion using an engineered CHMO polypeptide of the disclosure (e.g., a polypeptide of SEQ ID NO: 4, 6, 8, 10, 12, 14, 16, 18, 20, 22, 24, 26, 28, 30, 32, 34, 36, 38, 40, 42, 44, 46, 48, 50, 52, 54, 56, 58, 60, 62, 64, 66, 68, 70, 72, 74, 76, 78, 80, 82, 84, 86, 88, 90, 92, 94, 96, 98, 100, 102, 104, 106, 108, 110, 112, 114, 116, 118, 120, 122, 124, 126, 128, 130, 132, 134, 136, 138, 140, or 142.) The procedure described below resulted in 15.9 g (100% yield) of compound (2b) in a single crop as a white solid, and a chemical purity of 99.9% as determined by HPLC. (0229) A. Biocatalytic Reaction Protocol: (0230) A 100 mL beaker equipped with a cross shape stir bar was charged sequentially with: 15 g of benzhydrylthioacetic acid (BHTA) substrate (>98%; US patent publication 2004/0106829A1 and references therein), 77 mL of 100 mM TEA buffer solution (pH 10.3), 5.56 mL of 10 M NaOH, and 15 mL of PEG 200 (Sigma Reagent Grade). This substrate mixture was stirred at 35 C. for 20 min until all of the solid dissolved, resulting in a pH of about 8.3. A 300 mL Parr reactor vessel was fitted with a turbine impeller, an oxygen gas inlet/outlet and a dosing needle inlet. The reaction vessel at 35 C. was charged sequentially with: 30 mL of 100 mM TEA buffer solution (pH 10.3), 0.03 g of NADP, 0.15 g of KRED polypeptide of SEQ ID NO: 144, and 0.3 g of engineered CHMO polypeptide of SEQ ID NO: 136. This enzyme mixture was stirred gently at 150 rpm until all the solid powder dissolved, affording a homogenous yellow solution. The substrate solution mixture was charged into the Parr reactor vessel containing the enzyme solution. The pH of the resultant mixture was 8.54. The mixture was stirred for 1 minute at 350 rpm at 35 C. to obtain homogeneity. 7.5 mL of IPA (Sigma; >99.9% HPLC Grade) was added to initiate the KRED cofactor recycling reaction and thereby start the CHMO enzymatic reaction. The final pH was found to be 8.50. The reaction course was followed periodically by taking samples from the reaction mixture, quenching, and analyzing as described in Method 1. For the purposes of tracking the process, t=0 was set at the time at which IPA was added. The in-process reaction profile was determined using achiral HPLC analysis as described above in Example 1. The in-process sample analyses are summarized in Table 9 below. [table-us-00011-en] TABLE 9 Reaction Profile Time (h) % Conversion 0 0 3 20.5 6 36.5 26 94.5 30 97.6 33 98.8 36 99.4 48 99.9 (0232) A % conversion of >99% within 36 hours can be estimated from the kinetic profile of the reaction. The reaction mixture 48 hours after start was taken for product work-up and isolation as described below. (0233) B. Reaction Work-Up Protocol: (0234) The reaction vessel was cooled to 15 C. (internal temperature) and the pH of the reaction mixture was adjusted from pH 8.25 to 3.0 by adding 11.1 mL of 6 M HCl solution with continuous stirring at 250 rpm to precipitate out the (R)-2-(benzhydrylsulfinyl)acetic acid product as a free solid. The white slurry mixture was filtered though a standard G4 sintered glass funnel under vacuum and the reaction vessel was twice rinsed with 15 mL of cold deionized water at 5 C. (acidified with HCl to pH 3) and the filter cake was then washed with the deionized water rinse. HPLC analysis of the mother liquor indicated that 0.5% of (R)-2-(benzhydrylsulfinyl)acetic acid product was still present. The product was dried under vacuum to afford 15.9 g (100% isolated yield, 99.85% e.e.) of (R)-2-(benzhydrylsulfinyl)acetic acid as a white solid.
General Procedure for Comparative Examples 1 to 3 : Oxidation of sulphide in accordance with the method described by Kagan et al. Organic Syntheses, John Wiley and Sons INC. ed., 1993 ; vol. VIII,. 464-467. Water (0.27 mL, 0.015 mol, 1.0 eq) was added dropwise at room temperature (20C) to a solution of diethyltartrate (DET) (6.19 g, 0.03 mol, 2.0 eq) and titanium (IV) isopropoxide (4.26 g, 4.43 mL, 0.015 mol, 1.0 eq) in 125 mL of anhydrous methylene chloride, under nitrogen. Stirring was maintained until the yellow solution became homogeneous (30 min) and the sulphide (0.03 mol, 2.0 eq) was added. The solution was cooled to -30C and left in contact for 50 minutes at -30C. Then, cumene hydroperoxide (4.57 g, 5.0 mL, 0.03 mol, 2.0 eq) was added and the mixture was kept at -25C for 15 hours. After this time, 5 mL of water were added, and the solution was stirred during 1 h 30. The medium was filtered on clarcel and the filtrate worked up depending on the suphoxide obtained. As an example, when the sulfoxide of diphenylmethylthioacetic acid was generated, the compound was extracted with 3 x 100 mL of an aqueous solution of K2CO3 (0.6 M). The aqueous phases were collected, filtered on clarcel, acidified by addition of 150 mL of an aqueous solution of chlorhydric acid 4N (pH ? 1). The precipitate formed is filtered on a fritted glass, rinsed with water and then dried in vacuo at 35C.Comparative Example 1 : Enantioselectivity of asymmetric oxidation of sulphides of formula (II) with n = 1 according to X = -NH2, -OCH3, -OH The above general procedure for comparative examples was applied to diphenylmethylthioacetamide, methyldiphenylmethylthioacetate or diphenylmethylthioacetic acid as sulphide, and by using either (R,R)-DET or (S,S)-DET. [Table 11] PrecursorDETEe %Conversion rate (%)Diphenylmethylthioacetamide(R,R)-(+)-DET4290Methyldiphenylmethylthioacetate(R,R)-(+)-DET1040Diphenylmethylthioacetic acid(R,R)-(+)-DET5070Diphenylmethylthioacetic acid(S, S)-(-)-DET5083
50%
General Procedure for Comparative Examples 1 to 3 : Oxidation of sulphide in accordance with the method described by Kagan et al. Organic Syntheses, John Wiley and Sons INC. ed., 1993 ; vol. VIII,. 464-467. Water (0.27 mL, 0.015 mol, 1.0 eq) was added dropwise at room temperature (20C) to a solution of diethyltartrate (DET) (6.19 g, 0.03 mol, 2.0 eq) and titanium (IV) isopropoxide (4.26 g, 4.43 mL, 0.015 mol, 1.0 eq) in 125 mL of anhydrous methylene chloride, under nitrogen. Stirring was maintained until the yellow solution became homogeneous (30 min) and the sulphide (0.03 mol, 2.0 eq) was added. The solution was cooled to -30C and left in contact for 50 minutes at -30C. Then, cumene hydroperoxide (4.57 g, 5.0 mL, 0.03 mol, 2.0 eq) was added and the mixture was kept at -25C for 15 hours. After this time, 5 mL of water were added, and the solution was stirred during 1 h 30. The medium was filtered on clarcel and the filtrate worked up depending on the suphoxide obtained. As an example, when the sulfoxide of diphenylmethylthioacetic acid was generated, the compound was extracted with 3 x 100 mL of an aqueous solution of K2CO3 (0.6 M). The aqueous phases were collected, filtered on clarcel, acidified by addition of 150 mL of an aqueous solution of chlorhydric acid 4N (pH ? 1). The precipitate formed is filtered on a fritted glass, rinsed with water and then dried in vacuo at 35C.Comparative Example 1 : Enantioselectivity of asymmetric oxidation of sulphides of formula (II) with n = 1 according to X = -NH2, -OCH3, -OH The above general procedure for comparative examples was applied to diphenylmethylthioacetamide, methyldiphenylmethylthioacetate or diphenylmethylthioacetic acid as sulphide, and by using either (R,R)-DET or (S,S)-DET. [Table 11] PrecursorDETEe %Conversion rate (%)Diphenylmethylthioacetamide(R,R)-(+)-DET4290Methyldiphenylmethylthioacetate(R,R)-(+)-DET1040Diphenylmethylthioacetic acid(R,R)-(+)-DET5070Diphenylmethylthioacetic acid(S, S)-(-)-DET5083Comparative Example 2 :Influence of the amount of oxidizing agent on the enantioselectivity of oxidation of diphenylmethylthioacetic acid The above general procedure for comparative examples was applied to diphenylmethylthioacetic acid by varying the amount of cumene hydroperoxide from 1 to 4 equivalents. [Table 12] Cumene Hydroperoxide (eq)Ee (%)Conversion rate (%)150832509245097 The increase of the amount of the oxidizing agent allows to enhance the conversion rate of sulphide into sulphoxide but does not improve the enantioselectivity of the reaction, according to the Kagan's procedure.Comparative Example 3 :Influence of the stoichiometry of the titanium chiral complex on the enantioselectivity of oxidation of diphenylmethylthioacetic acid The above general procedure for comparative examples was applied to diphenylmethylthioacetic acid by varying the stoichiometry of the chiral titanium complex (S,S)-(-)-DET/Ti/H2O. [Table 13] (S,S)-(-)-DET / Ti / H2OEe (%)Conversion rate (%)2 / 1 /150922/1 /00974 / 1 / 0097 The water is necessary to obtain an enantioselectivity, according to the Kagan's procedure.
General Procedure for Comparative Examples 1 to 3 : Oxidation of sulphide in accordance with the method described by Kagan et al. Organic Syntheses, John Wiley and Sons INC. ed., 1993 ; vol. VIII,. 464-467. Water (0.27 mL, 0.015 mol, 1.0 eq) was added dropwise at room temperature (20C) to a solution of diethyltartrate (DET) (6.19 g, 0.03 mol, 2.0 eq) and titanium (IV) isopropoxide (4.26 g, 4.43 mL, 0.015 mol, 1.0 eq) in 125 mL of anhydrous methylene chloride, under nitrogen. Stirring was maintained until the yellow solution became homogeneous (30 min) and the sulphide (0.03 mol, 2.0 eq) was added. The solution was cooled to -30C and left in contact for 50 minutes at -30C. Then, cumene hydroperoxide (4.57 g, 5.0 mL, 0.03 mol, 2.0 eq) was added and the mixture was kept at -25C for 15 hours. After this time, 5 mL of water were added, and the solution was stirred during 1 h 30. The medium was filtered on clarcel and the filtrate worked up depending on the suphoxide obtained. As an example, when the sulfoxide of diphenylmethylthioacetic acid was generated, the compound was extracted with 3 x 100 mL of an aqueous solution of K2CO3 (0.6 M). The aqueous phases were collected, filtered on clarcel, acidified by addition of 150 mL of an aqueous solution of chlorhydric acid 4N (pH ? 1). The precipitate formed is filtered on a fritted glass, rinsed with water and then dried in vacuo at 35C.Comparative Example 3 :Influence of the stoichiometry of the titanium chiral complex on the enantioselectivity of oxidation of diphenylmethylthioacetic acid The above general procedure for comparative examples was applied to diphenylmethylthioacetic acid by varying the stoichiometry of the chiral titanium complex (S,S)-(-)-DET/Ti/H2O. [Table 13] (S,S)-(-)-DET / Ti / H2OEe (%)Conversion rate (%)2 / 1 /150922/1 /00974 / 1 / 0097 The water is necessary to obtain an enantioselectivity, according to the Kagan's procedure.
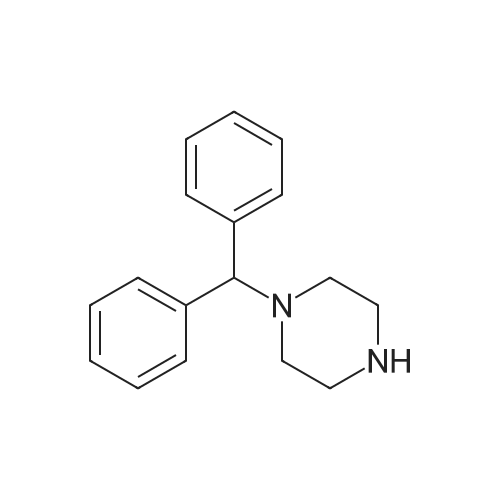
1-(4-benzhydrylpiperazin-1-yl)-2-(benzhydrylthio)ethanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride;dmap; In dichloromethane; at 20℃;
Example 14; Synthesis of l-(4-benzhvdrylpiperazin-l-yl)-2-(4-benzhvdrylthio)ethanone (compound no.44}; [00118] To a solution of 1-diphenylmethylpiperazine 0.125 g (0.5 mmol) dissolved in methylene chloride (5 ml) was added 2-<strong>[63547-22-8](benzhydrylthio)acetic acid</strong> 0.13 g (0.5 mmol) (synthesized according to Example l(d)), EDC 0.191 g (1 mmole) and trace of DMAP, and the reaction mixture was stirred at room temperature for overnight. The reaction mixture was concentrated and dissolved in ethyl acetate (10 ml) and washed with saturated sodium bicarbonate solution and brine, dried over sodium sulfate and concentrated. The residue was applied to flash column chromatography using methylene chloride and methanol (100:10) as eluents to give 0.10 g of desired product.
D. Synthesis of 2-(benzhydrylthio)acetic acid; [0080] 10 g of thiourea was dissolved in 57 ml of 48% HBr and 10 ml of water. The reaction mixture was heated to 60 C, and 20.2 g of benzhydrol was added. The temperature was increased to 900C, cooled to room temperature. The crystals were filtered off and washed with water. The above crystals were added to 35ml 30% sodium hydroxide. The mixture was heated to 70 0C, and chloroacetic acid 11.44 g in 22 ml of water was added slowly. The mixture was refluxed for half an hour after the addition. The reaction mixture was then cooled to room temperature to give 25 g of desired product. E. Synthesis of 2-(benzhvdrylsulfinyl)acetic acid[0081] 10 g of thiourea was dissolved in 57 ml of 48% HBr and 10 ml of water. The reaction mixture was heated to 60 0C, and 20.2 g of benzhydrol was added. The temperature was increased to 9O0C and then cooled to room temperature. The crystals were filtered off and washed with water. The crystals were added to 35ml 30% sodium hydroxide and the mixture was heated to 70 0C, before 11.44 g chloroacetic acid in 22 ml of water was added slowly. The mixture was refluxed for half an hour after the addition. 14.3 ml hydrogen peroxide (30%) was added to the above solution within 3 hours at room temperature. 22 ml of water was then added and the reaction mixture was filtered. The filtrate was acidified with concentrated HCl (d=1.18). The resulting solid was filtered off and dried to give 13 g of the desired product.
D. Synthesis of 2-(benzhydrylthio)acetic acid; [0063] 10 g of thiourea was dissolved in 57 ml of 48% HBr and 10 ml of water. The reaction mixture was heated to 60 C, and 20.2 g of benzhydrol was added. The temperature was increased to 900C and then cooled to room temperature. The crystals were filtered off and washed with water. The crystals were added to 35 ml of 30% sodium hydroxide. The mixture was heated to 70 0C, and chloroacetic acid (11.44 g in 22ml of water) was added slowly. The mixture was refluxed for half an hour after the addition. The reaction mixture was cooled to room temperature to give 25g of desired product.
Example 7; Synthesis of 2-(benzhydrylthio)acetic acid; [0095] 1Og of thiourea was dissolved in 57ml of 48% HBr and 10ml of water. The reaction mixture was heated to 60C, and 20.2g of benzhydrol was added. The temperature was increased to 900C and then cooled to room temperature. Crystals were filtered off and washed with water. The above crystals were then added to 30% sodium hydroxide (35ml). The mixture was heated to 70 0C, and then chloroacetic acid (1 1.44g in 22ml of water) was added slowly. The mixture was refluxed for half an hour after the addition. The reaction mixture was then cooled to room temperature to give desired product (25g).
Example 2 (Armodafinil)In 250 ml. flask, 10 gm of benzhydryl thioacetic acid was added in 100 ml toluene. The reaction mass was cooled to 0-50C. To it 5.9 gm 1,8-diazabicyclo [5.4.0] undec-7-ene was added and cooled at 0-5 0C. The reaction mass was stirred for 30 minutes. Further, 8.8 gm(+)-(2R, 8aS)-10-(Camphorylsulfonyl) oxaziridine was added and stirred till at 25 to 300C till the reaction goes to completion. The reaction mixture was filtered and solid was washed with water to get chirally pure 8 gm Armodafinic acid.Yield: 75.25% Optical purity: R isomer : 82.17 %S isomer : 17.83 %
With thionyl chloride; In dichloromethane; water; tert-butyl alcohol; benzene;
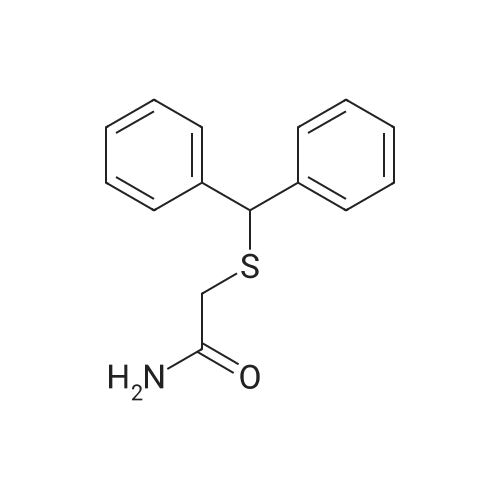
Example 6 2-Benzhydrylsulfanyl acetamide (3) Chemical method: This compound was obtained from the corresponding acid chloride. To a solution of acid 1 (777 mg, 3 mmol) in benzene was added SOCl2 (833 mg, 7 mmol). The solution was heated to reflux for 1 hour. The solvent was evaporated to give a yellow oil: 832.1 mg (99.9%). A solution of benzhydrylsulfanyl acetyl chloride (1.089 g, 4.21 mmol) in CH2Cl2 (10 mL) was added to a solution of NH4OH-THF (3:2, 30 mL) at 0 C. The reaction was stirred for 1 hour. The reaction mixture was then treated with H2O (20 mL) and extracted with CH2Cl2 (2*30 mL). The organic layer was washed with saturated NaHCO3 (20 mL) and H2O (20 mL). The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on a silica gel column (3*10 cm). Elution with 9:1 CHCl3-MeOH gave a light yellow solid: 1.044 g (96%). Chemo-enzymatic method: To a solution of Benzhydrylsulfanyl acetic acid (259 mg, 1.0 mmol) in tert-butanol (28 mL) was added ammonium carbamate (79 mg, 1.0 mmol) and Novozyme-435 (100 mg). The reaction flask was closed tightly and the reaction was stirred at 60 C. for 7 days. The reaction mixture was filtered through cotton plug and concentrated under vacuum to give a colourless turbid oil. The crude oil was purified by flash column chromatography on silicagel (2*9 cm). Elution with hexanes-ethyl acetate (3:2) furnished the sulfanyl amide as a light yellow solid: 232 mg (90% yield).
General procedure: Procedure C. Thioacetamides 21, 2p, 2s, 2v, and 2w were synthesized in two steps, while compounds 2j, 2k, 2m-2o, 2q, 2r, 2t, 2u, and 2x-2z were synthesized in two steps with slight modifications to the second step. Step 1: The same as step 1 for Procedure B. Step 2: CDI (1.1 equiv.) was added to a solution of the carboxylic acid product (1 equiv.) from step 1 in anhydrous THF (20 mL). The reaction mixture was stirred at room temperature for 2 hours, and then cooled to 0 C. Water (a few drops) was added to the reaction mixture (to quench excess CDI), followed by the dropwise addition of the appropriate amine (1 equiv.; dissolved in THF). The reaction mixture was left to warm to room temperature and stir overnight. The solvent was removed under vacuum to give a crude residue, which was dissolved in diethyl ether or ethyl acetate. The organic solution was washed with aqueous 1.0 M HC1 solution (55 mL), water (80 mL), dilute aqueous NaHC03 solution (36 mL; 1:6 dilution of saturated NaHC03 solution), and water (2 x 30 mL). The organic layer was dried over MgS04, and concentrated in vacuo to give the pure product. The bromo-substituted analogs 2q, 2t, 2x, and 2z required further purification by flash column chromatography as indicated. N-Allyl-2-(benzhydrylthio)acetamide (2j). Compound 2j was synthesized from 2-<strong>[63547-22-8](benzhydrylthio)acetic acid</strong> and allylamine according to the modified general procedure C. The product, 2j (1.97 g, 86% yield), was obtained as a viscous yellow oil that solidified over time. Mp 45-47 C; 1H NMR (CDC13): delta 7.40 (d, / = 7.2 Hz, 4H), 7.32 (t, / = 7.4 Hz, 4H), 7.25 (t, / = 8.0 Hz, 2H), 6.67 (brs, IH), 5.77-5.86 (m, IH), 5.19 (d, Jtrans = 17.6 Hz, IH), 5.15 (d, Jcis = 10.6 Hz, IH), 5.13 (s, IH), 3.84 (tt, / = 5.6, 1.6 Hz), 3.14 (s, 2H); 13C NMR (CDCI3): delta 168.0, 140.3, 133.8, 128.8, 128.2, 127.6, 116.8, 55.1, 42.1, 36.1. Anal. (CigHigNOS) C, H, N.
General procedure: The General Synthetic Example of Modafinil Derivatives [0117] Dissolve the carboxylic acid compound (0.77 mmol) represented by the following Chemical Formula 6 or 7 in dimethylformamide (DMF, 40 ml) and add each of HOBt[208 mg, 1.54 mmol, (1-hydroxybenzotriazole)] and EDC[295 mg, 1.54 mmol, (ethyl(dimethylaminopropyl)carbodiimide and stir at room temperature for 30 minutes. Add amine compound (0.77 mmol) in the reaction solution and conduct the reaction at room temperature for 1 hour, cleanse with ethylacetate (100 ml) and brine (100 ml), separate and dry the organic layer, remove the solvent under the reduced pressure and purify the residue by the chromatography to obtain the target compound of modafinil derivative. compound and the characteristics are as follows. [0119] White solid. mp 117.8; Rf=0.3 (ethyl acetate/n-hexane=1:1, v/v); IRmax (CHCl3, KBr) 3304, 3060, 3027, 2926, 2870, 1651, 1531, 1494, 1450, 1384, 1304, 1078, 1030, 922, 750, 702, 629, 587, 507 cm-1; 1H NMR (250 MHz, CDCl3) delta=7.43-7.20 (m, 10H), 6.99 (s, 1H), 5.20 (s, 1H), 3.99-3.75 (m, 3H), 3.58-3.52 (m, 1H), 3.19-3.09 (m, 3H), 2.02-1.87 (m, 4H); 13C-NMR (63 MHz, CDCl3) delta=168.5, 140.3, 128.8, 128.4, 127.6, 68.2, 54.8, 43.5, 36.1, 29.8, 25.9; LC-MS (ESI+) m/z 364-[M+Na].
In water; acetone; at 20 - 70℃; for 3h;Inert atmosphere;
[0097] Benzhydryl bromide 1 (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H20; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60-70C for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2C12 and saturated aqueous NaHC03. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid (3, 46%), mp 125C. Rf 0.2. Recrystallization from MeOH/H20 gave mp 126-128C.
46%
In water; acetone; at 20 - 70℃; for 3h;Inert atmosphere;
Step-1: Synthesis of Compound 3: Benzhydryl bromide 1 (14.78 gm, 0.059 mole) was dissolved in 75 ml of acetone in a 250-ml round-bottomed flask. To this solution was added dropwise sodium mercaptoacetate (6.59 g, 0.058 mole) in about 60 ml of H2O; the mixture was stirred under N2 for 2 h at room temperature and was thereafter warmed at about 60-70 C. for 1 h. The reaction mixture was evaporated to dryness and taken up in CH2Cl2 and saturated aqueous NaHCO3. The organic extract was rejected, and the aqueous phase was treated with acid to pH 2 and chilled. Suction filtration gave the 6.9 g of the acid ( 3, 46%), mp 125 C. Rf 0.2. Recrystallization from MeOH/H2O gave mp 126-128 C
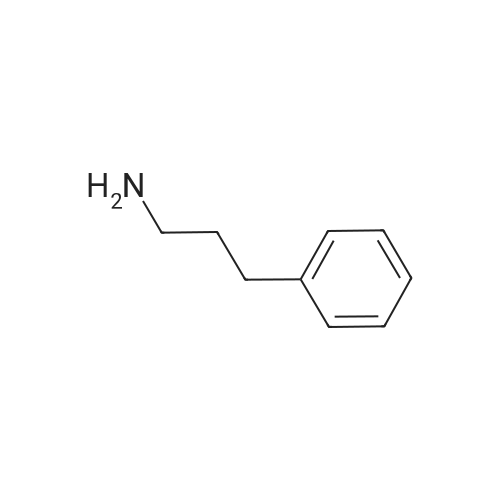
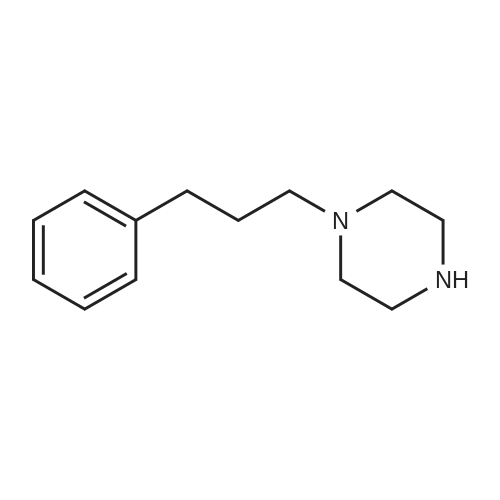
2-(benzhydrylthio)-1-(4-(3-phenylpropyl)piperazin-1-yl)ethan-1-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
2-(Benzhydrylthio)-l-(4-(3-phenylpropyl)piperazin-l-yl)ethan-l-one (11a). A mixture of CDI (583mg, 3.60 mmol) and 2-<strong>[63547-22-8](benzhydrylthio)acetic acid</strong> (775 mg, 3.00 mmol) in THF (24 mL) was stirred at r.t. under argon. After 2 hours of reaction time, l-(3- phenylpropyl)piperazine (613mg, 3.00 mmol) in THF (15 mL) was added and the reaction mixture was stirred overnight. Solvent was removed and the reaction residue was purified by flash column chromatography (ethyl acetate/triethylamine (TEA) = 95:5) to give 11a (1.2 g, 90% yield) as a yellow oil. The free base was converted to the oxalate salt and recrystallized from hot 2-propanol to give a white solid. Mp 92-95 C; 1H NMR (CDC13): delta 7.42-7.45 (m, 4H), 7.17-7.33(m, 11H), 5.34 (s, IH), 3.57-3.60 (t, 2H, J=14.1 Hz), 3.37-3.40 (t, 2H, J=5.2 Hz), 3.18 (s, 2H), 2.62-2.66 (m, 2H), 2.34-2.39 (m, 6H), 1.78-1.84 (m, 2H); 13C NMR (CDCI3): 167.2, 141.9, 140.7, 128.6, 128.5, 128.4, 128.3, 127.3, 125.8, 57.7, 54.1, 53.2, 52.7, 46.3, 41.9, 33.5, 28.4; Anal. (C28H32N2OS · C2H204 · 0.5H2O) C, H, N.
With tris-(dibenzylideneacetone)dipalladium(0); (oxan-4-yl)diphenylphosphine; silver trifluoromethanesulfonate; acetic acid; cerous nitrate; In N,N-dimethyl acetamide; at 100℃; for 9h;
Example 4: To the appropriate amount of an acidic organic solvent (1: 3 mixture of acetic acid and DMA, 1: 3) was added 100 mmol of 2_ (phenylmethylthio) acetic acid, 175 mmol of bromobenzene, 7 mmol of catalyst (from 2 mmol Tri (arylene acetone) palladium and 5 mmol of Ce (N03) 3), 3.5 mmol of organic ligands L1 and 75 mmol of silver trifluoromethanesulfonate were added and the temperature was raised to 100 C and the reaction was carried out at that temperature for 9 hours. The reaction system obtained after the completion of the reaction was filtered while hot, the filtrate was washed with a saturated aqueous solution of sodium carbonate, and the aqueous phase and the upper organic phase were separated. The organic phase was washed with saturated brine, and the upper organic phase was taken and evaporated to evaporate to obtain a residue The eluate was purified by silica gel column chromatography (eluent was a mixed solvent of chloroform and n-butanol in a volume ratio of 10: 1). The eluted fractions were collected and the solvent was removed by evaporation to give a white The yield of the product was 99.4% and the purity was 93.2%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping