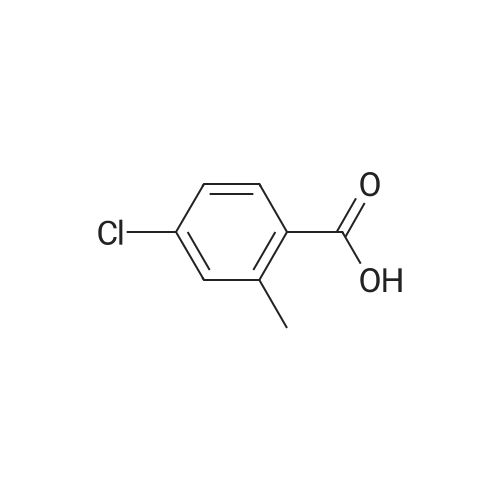
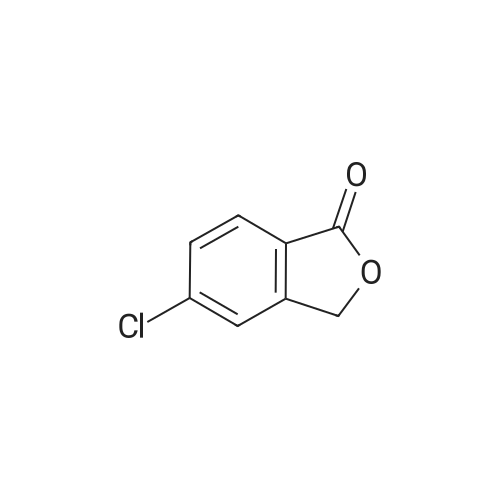
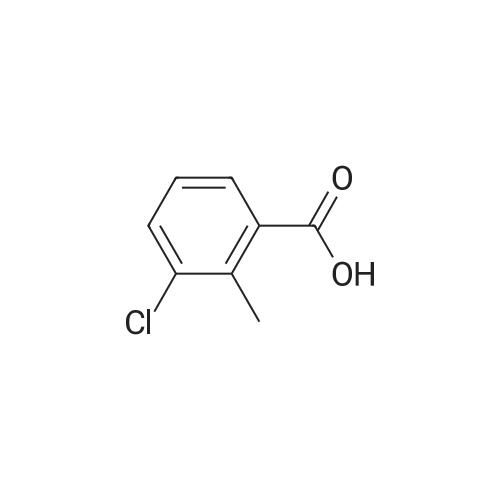
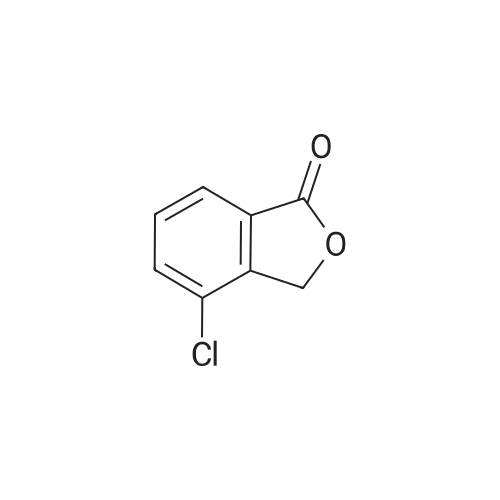
|
With thionyl chloride; for 0.5h;Heating / reflux; |
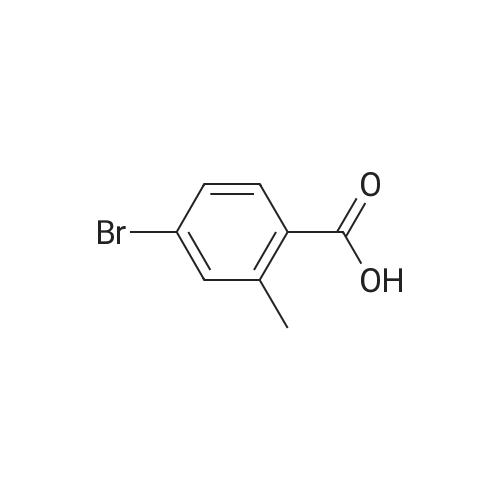
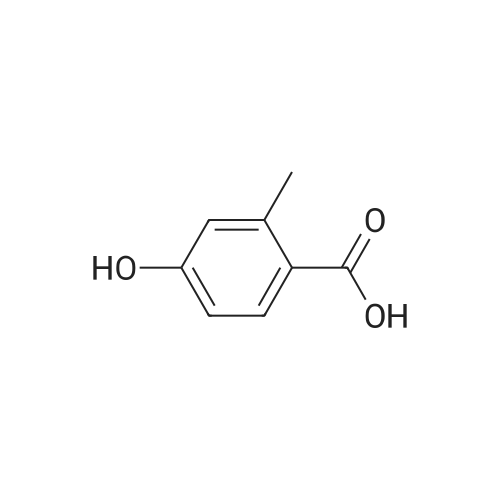
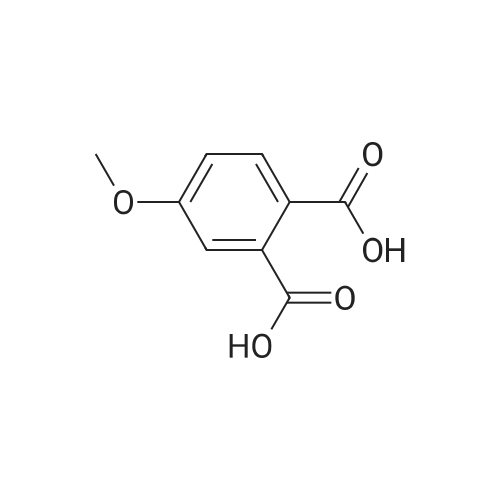
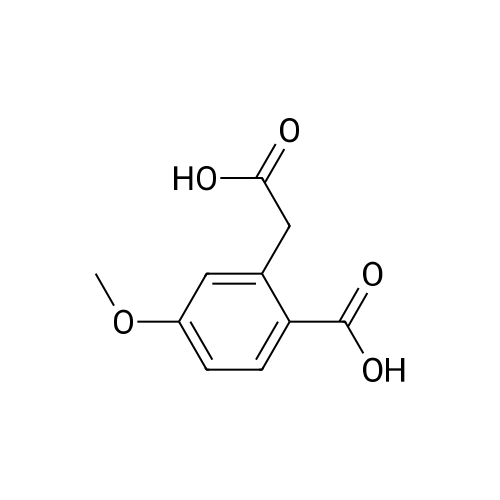
A mixture of <strong>[6245-57-4]4-methoxy-2-methyl-benzoic acid</strong> (5.00 g, 30.1 mmol) and thionyl chloride (20.0 g, 0.17 mol) was heated to reflux for 30 min. Removed the volatile in vacuo. After pumping overnight, the viscous oily acid chloride was used as crude for the next reaction without any purification. To a solution of 4-methoxy-2-methyl-benzoyl chloride inCH2Cl2 (60 mL) at0 C was added diethylamine dropwise. The formed mixture was allowed to warm up to the ambient temperature for 2 h with stirring. Removed the volatiles in vacuo. The residue was triturated with EtOAc (100 mL) and filtered. The filtrate was washed with 1MHCI, 1M NaOH and brine, dried over MgS04. Evaporation of the solvent yielded 6.51 g (98%) of the desired product as a viscous oil. LC-MS (retention time: 1.20 min, method B), MSnilz 222(M++H). |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 3h; |
Part B To a solution of 2-methyl-4-methoxybenzonic acid (166 mg, 1 mmol) in DCM (5 mL) at RT, oxalyl chloride (0.15 mL) was added followed by the DMF (1 drop). The resulting mixture was stirred at RT for 3 hours and the solvent was evaporated under vacuum. To the flask with the residue of acetyl chloride, DCM (15 mL) was added, followed by 2 (100 mg, 0.64 mmol), pyridine (0.2 mL) and catalytical amount DMAP. The mixture was heated up to 40 C. and was stirred at this temperature for overnight. After cooling down to room temperature, EtOAc (80 mL) was added and the organic was washed with 1 N HCl, brine and dried over Na2SO4. After evaporation of solvent, the crude product 3 was used in the next step directly. LC-MS m/z 306.1 (M+H). |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 2 - 12h;Product distribution / selectivity; |
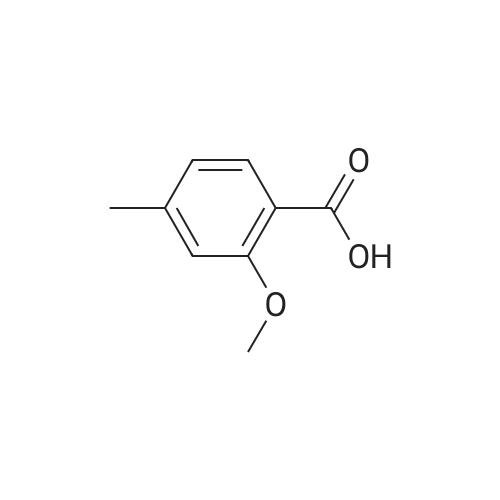
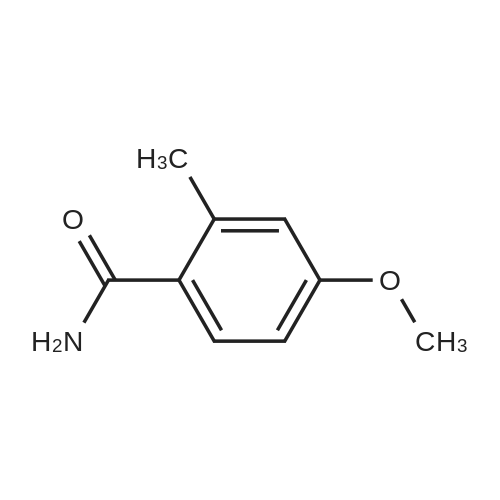
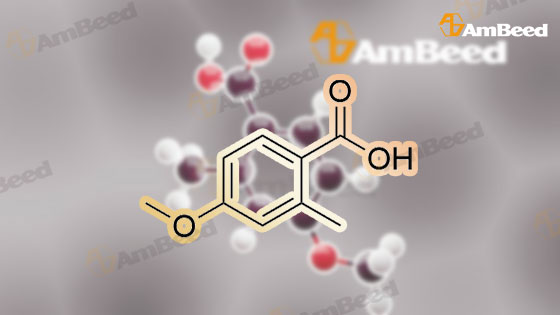
Example 1B 4-methoxy-2-methylbenzamide A mixture of Example 1A (386.05 g) in dichloromethane (3 L) was combined in a flask equipped with a mechanical stirrer, reflux condenser, and addition funnel to provide a very thick slurry. DMF (1 mL) was added as catalyst, followed by oxalyl chloride (330 g) dropwise over about 2 hours. The acidic effluent gases were scrubbed through a K2CO3 scrubber. The slurry slowly dissolved during the addition to provide a red solution. Dichloromethane (1.3 L) was distilled off at 30 C. with slight vacuum, and the resulting concentrated solution of acid chloride was polish filtered through a course sintered glass funnel to remove some insoluble matter. This filtered solution was concentrated to a crystalline residue and concentrated under high vacuum for 30 minutes to remove any excess oxalyl chloride. The crystalline residue was dissolved in THF (550 mL) and titrated into a large flask containing ice cold concentrated ammonium hydroxide (IL) over ~15 minutes. The temperature quickly rose to ~30 C. with the formation of a thick slurry of product. To this oily slurry of product, water (3 L) was added over ~15 minutes to provide a thick white slurry of product. This product was filtered over course sintered glass and washed with water (1.5 L) and dried under nitrogen/vacuum for 36 hours. The desired product was isolated (367.8 g) as an off-white solid. HPLC showed a peak at the expected retention time of 11.85 min, with a purity of 95.15% at 220 nm, and 97.29% at 254 nm. Example 3AN-(4-methoxy-2-methylbenzoyl)-N,N N',N'-tetramethylguanidineA solution of <strong>[6245-57-4]4-methoxy-2-methylbenzoic acid</strong> (10.2 g, 61.4 mmol) in dichloromethane (200 mL) was treated slowly with oxalyl chloride. A drop of DMF was added and the reaction was stirred at room temperature for 12 hours. The solution was treated with 1,1,3,3-tetramethylguanidine (14.9 g, 128.9 mmol), stirred at room temperature for 4 hours, and washed with 5% aqueous citric acid (2×100 mL). The aqueous layer was extracted with dichloromethane (2×100 mL) and the combined organic phases were dried (MgSO4), filtered, and concentrated to provide a minimal amount of the desired product. The aqueous layer was basified with ION NaOH to pH 12-13 and extracted with dichloromethane (4×200 mL). The combined organic phases were dried (MgSO4), filtered, and concentrated. The resulting oil was combined with the earlier obtained product and treated with diethyl ether to form a white precipitate that was removed by filtration. The filtrate was concentrated to provide 11.32 g (70%) of the desired product as a yellow viscous oil. 1H NMR (CD3OD) delta 2.54 (s, 3H), 3.97 (s, 6H), 3.79 (s, 3H), 6.60 (d, J=9.2 Hz, 1H), 6.75 (s, 1H), 7.65 (d, J=8.9 Hz, 1H). LC-MS (retention time: 1.00 min), MS m/z 264 (M+1). |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In chloroform; at 20℃; for 1h; |
Preparation Example 32 Synthesis of 1-bromo-2-methoxy-4-methyl-5-(4-methylbenzyl)benzene; Oxalyl chloride (3.43 mL, 0.0400 mmol) and N,N-dimethylformamide (2 drops) were added to chloroform (60 mL) solution of <strong>[6245-57-4]4-methoxy-2-methylbenzoic acid</strong> (5.0 g, 0.0300 mol). After the reaction mixture was stirred at room temperature for one hour, the reaction solvent was evaporated under reduced pressure. The obtained yellow oily substance was dissolved in chloroform (60 mL). Toluene (3.52 mL, 0.0330 mol) and aluminum chloride (8.02 g, 0.0601 mol) were added to this solution while cooled on ice, and the reaction mixture was stirred for three and a half hours while keeping the reaction mixture cooled ice. After 5% hydrochloric acid was added to the reaction mixture and extracted with chloroform, the organic phase was washed with 10% hydrochloric acid, water, a saturated sodium bicarbonate aqueous solution and brine, and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate=15:1) to obtain yellow oily (4-methoxy-2-methylphenyl) (4-methylphenyl)methanone (4.26 g, 58.9%). 1H NMR (300 MHz, CHLOROFORM-d) delta ppm 2.39 (s, 3 H) 2.42 (s, 3 H) 3.86 (s, 3 H) 6.74 (dd, J=8.5, 2.56 Hz, 1 H) 6.81 (d, J=2.6 Hz, 1 H) 7.21 - 7.27 (m, 2 H) 7.31 (d, J=8.4 Hz, 1 H) 7.64 - 7.71 (m, 2 H) ESI m/z = 263 (M+Na) Et3SiH (8.5 mL, 0.0531 mol) was added to a mixed solution of chloroform (8 mL) and acetonitrile (32 mL) of (4-methoxy-2-methyl phenyl)(4-methylphenyl)methanone and BF3·Et2O (4.5 mL, 0.0354 mol) was added dropwise while cooled on ice. The reaction mixture was warmed to room temperature and stirred at 50C for one hour. After the reaction mixture was added with a saturated sodium bicarbonate aqueous solution and extracted with ethyl acetate while cooled on ice, the organic phase was washed with brine and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography-(hexane:ethyl acetate=15:1) to obtain colorless oily 4-methoxy-2-methyl-1-(4-methylbenzyl)benzene (3.89 g, 97%). 1H NMR (300 MHz, CHLOROFORM-d) delta ppm 2.21 (s, 3 H) 2.31 (s, 3 H) 3.78 (s, 3 H) 3.88 (s, 2 H) 6.65 - 6.74 (m, 2 H) 6.97 - 7.03 (m, 3H) 7.03 - 7.11 (m, 2 H) EI 226 (M+) Br2 was added dropwise to an acetic acid (35 mL) solution of 4-methoxy-2-methyl-1-(4-methylbenzyl)benzene while cooled on ice. The reaction mixture was stirred at 110C for two hours. After the reaction mixture was added with water while cooled on ice and extracted with ethyl acetate, the organic phase was washed with a saturated sodium bicarbonate aqueous solution and brine and dried with anhydrous magnesium sulfate. After the desiccant was filtered off, the residue obtained by evaporating the solvent under reduced pressure was purified by silica gel column chromatography (hexane:ethyl acetate=15:1) to obtain a yellow oily title compound (4.21 g, 80%). 1H NMR (300 MHz, CHLOROFORM-d) delta ppm 2.20 (s, 3H) 2.31 (s, 3 H) 3.85 (s, 2 H) 3.87 (s, 3 H) 6.71 (s, 1 H) 6.94 - 7.11 (m, 4 H) 7.26 (s, 1H). EI 304 (M+), 306 (M+2). |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; for 2h; |
IG: 4-Methoxy-2-methylbenzamide; [00130] A mixture of IF (386.05 g) in CH2Cl2 (3 L) was combined in a flask equipped with a mechanical stirrer, reflux condenser and addition funnel to provide a <n="61"/>very thick slurry. DMF (1 mL) was added as catalyst, followed by oxalyl chloride (330 g) dropwise over about 2 h. The acidic effluent gasses were scrubbed through a K2CO3 scrubber. The slurry slowly dissolved during the addition to provide a red solution of acid chloride. CH2CI2 (1.3 L) was removed at 300C with slight vacuum, and the resulting concentrated solution of acid chloride was polish filtered through a course sintered glass funnel to remove some insoluble matter. This filtered solution was concentrated to a crystalline residue and pumped down under high vacuum for 30 min to remove any excess oxalyl chloride. The crystalline residue was dissolved in THF (550 mL) and titrated into a large flask containing ice cold concentrated ammonium hydroxide (1 L) over -15 min. The temperature quickly rose to -30 0C with the formation of a thick slurry of product. To this oily slurry of product, water (3 L) was added over -15 min to provide a thick white slurry of product. This product was filtered over course sintered glass and washed with water (1.5 L) and dried under nitrogen/vacuum for 36 h. 367.8g of IG was isolated as an off-white solid. HPLC showed a peak at the expected retention time of 11.85 min, with a purity of 95.15% at 220 nm, and 97.29% at 254 nm. |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 12h;Product distribution / selectivity; |
Example 17; Preparation of Compound 17: N-(4,6-dimethyl-2-pyridinyl)valyl-(4R)-N-((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-4-((3-(dimethylamino)-6-methoxy-1-isoquinolinyl)oxy)-L-prolinamide; Step 1:; A solution of <strong>[6245-57-4]4-methoxy-2-methylbenzoic acid</strong> (10.2 g, 61.4 mmol) in dichloromethane (200 mL) was treated slowly with oxalyl chloride. A drop of DMF was added and the reaction was stirred at room temperature for 12 hours. The mixture was then treated with 1,1,3,3-tetramethylguanidine (14.9 g, 128.9 mmol) and stirred at room temperature for 4 hours. The mixture was washed with 5% aqueous citric acid (2×100 mL). The aqueous phase was back-extracted with dichloromethane (2×100 mL), and the organic phases were combined, dried over anhydrous MgSO4, filtered, and concentrated to provide a minimal amount of desired product. The aqueous phase was made basic by addition of 10.0M aqueous NaOH until pH=13 was achieved and was then extracted with dichloromethane (4×200 mL). The organic phases were combined, dried over anhydrous MgSO4, filtered and concentrated. The resulting oil was combined with the earlier obtained product and the mixture was treated with diethyl ether to give a white precipitate which was removed by filtration. The filtrate was concentrated to give the desired product (11.3 g, 70% yield) as a viscous yellow oil. 1H NMR (CD3OD) delta 2.54 (s, 3H), 3.97 (s, 6H), 3.79 (s, 3H), 6.60 (d, J=9.2 Hz, 1H), 6.75 (s, 1H), 7.65 (d, J=8.9 Hz, 1H). LC-MS, MS m/z 264 (M++H). |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; 1,2-dichloro-ethane; for 1h;Product distribution / selectivity; |
Example 37; Preparation of Compound 37: N-(5,6-dihydro-4H-1,3-thiazin-2-yl)-3-methyl-L-valyl-(4R)-N-((1R,2S)-1-((cyclopropylsulfonyl)carbamoyl)-2-vinylcyclopropyl)-4-((6-methoxy-3-(4-(trifluoromethoxy)phenyl)-1-isoquinolinyl)oxy)-L-prolinamide; Step 1:; A mixture of <strong>[6245-57-4]4-methoxy-2-methyl-benzoic acid</strong> (25.3 g, 152 mmol) in 1:1 DCM:DCE was treated with oxalyl chloride (38.6 g, 304 mmol), followed by addition of DMF (0.111 g, 1.50 mmol). Vigorous gas evolution ensued, and the reaction eventually became clear after 1 h. The volatiles were removed in vacuo and the white solid residue was placed under high vacuum for 2 h. The crude material was redissolved in DCM (200 mL) and the mixture was cooled to 0 C. Diethylamine (22.8 g, 312 mmol) was added dropwise with stirring. The mixture was allowed to warm to rt for 1 h with stirring. The mixture was concentrated in vacuo to approximately volume, and Et2O (300 mL) was added and the mixture was chilled. The solid precipitate which had formed was removed by filtration. The filtrate was concentrated to give a viscous brown oil which was redissolved in DCM (200 mL) and washed with 0.1M HCl (2×50 mL). The organic phase was dried over MgSO4, filtered and concentrated in vacuo to give the desired product (33.1 g, 98% yield). LC-MS, MS m/z 222 (M++H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping