* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Agricultural and Food Chemistry, 2013, vol. 61, # 3, p. 517 - 522
[2] Organic and Biomolecular Chemistry, 2014, vol. 12, # 28, p. 5082 - 5088
[3] Patent: WO2014/125410, 2014, A1,
[4] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 9, p. 1615 - 1620
13
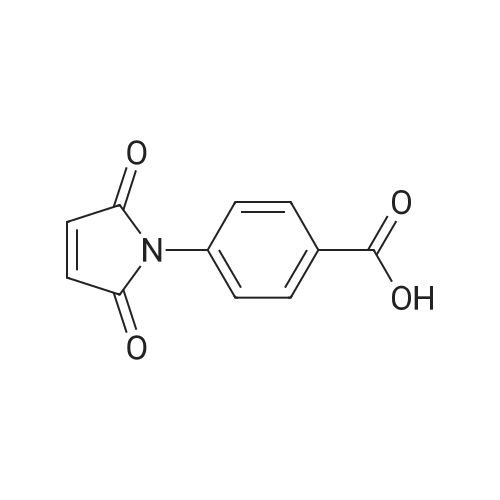
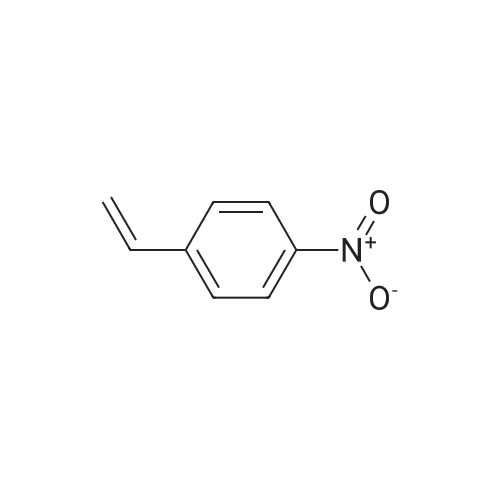
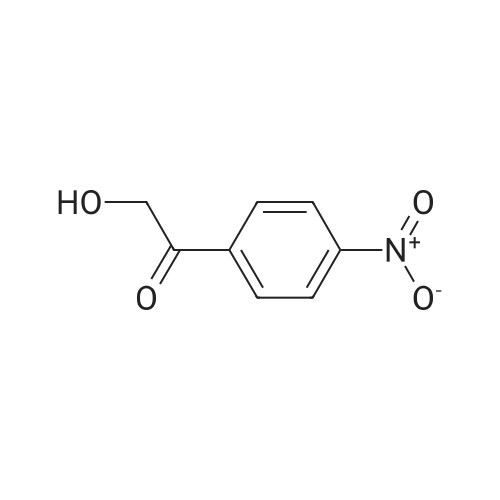
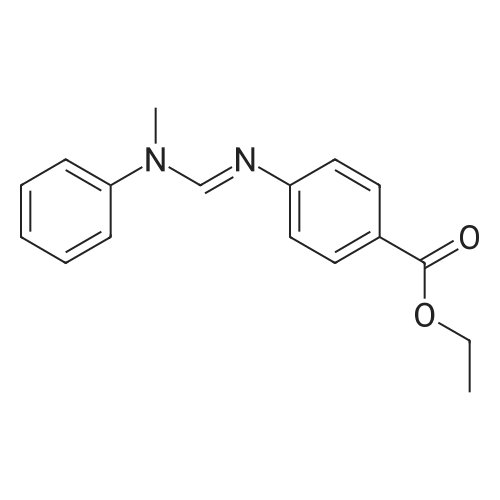
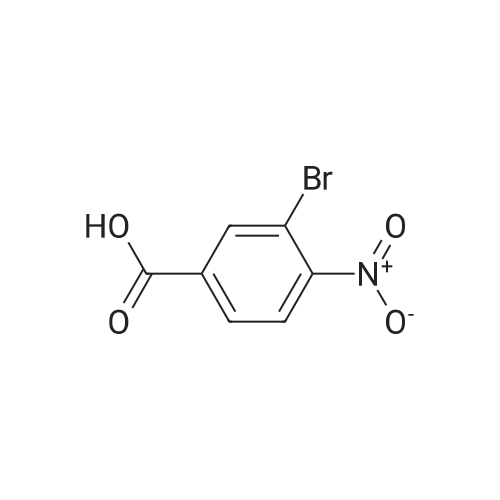
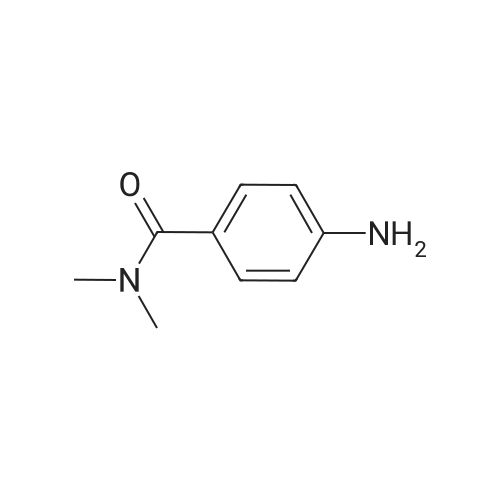
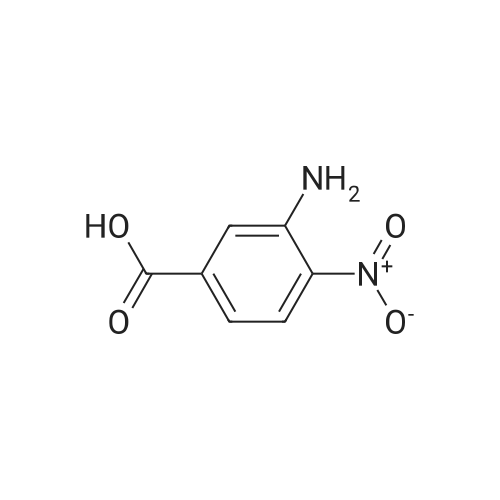
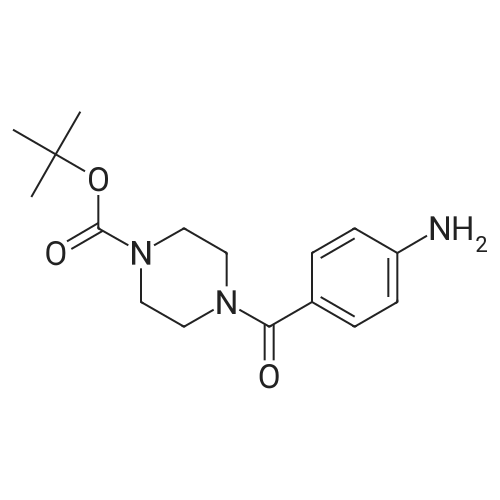
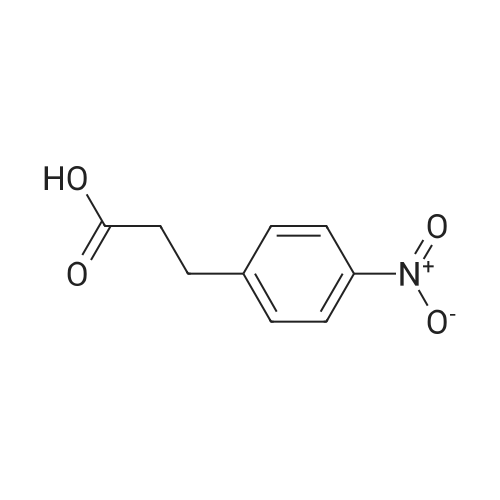
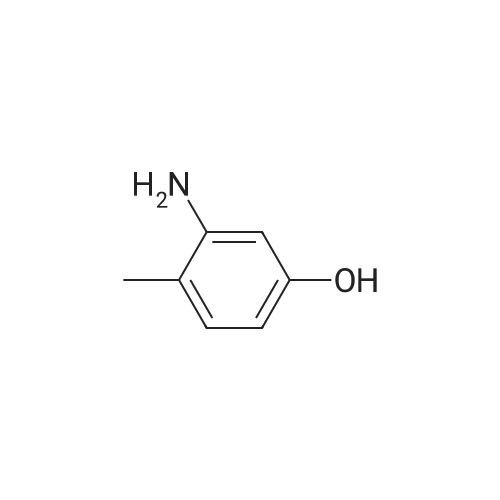
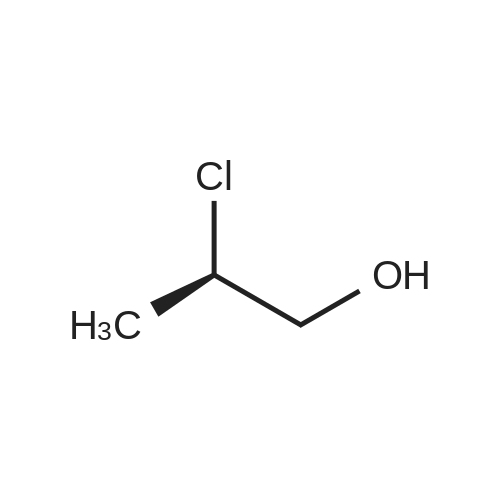
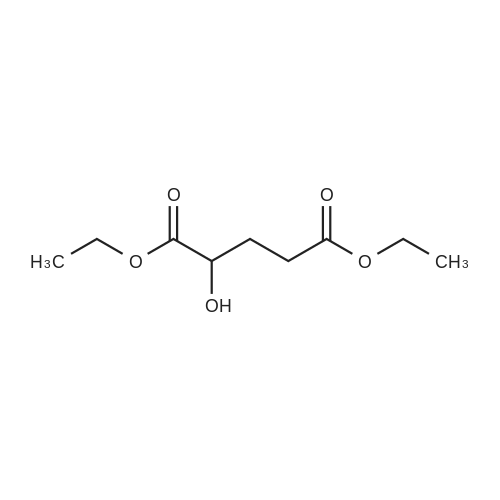
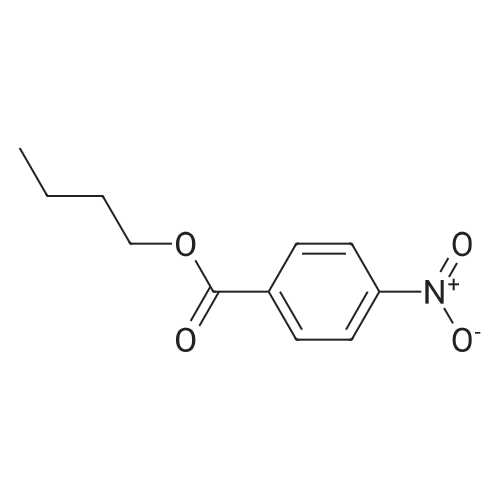
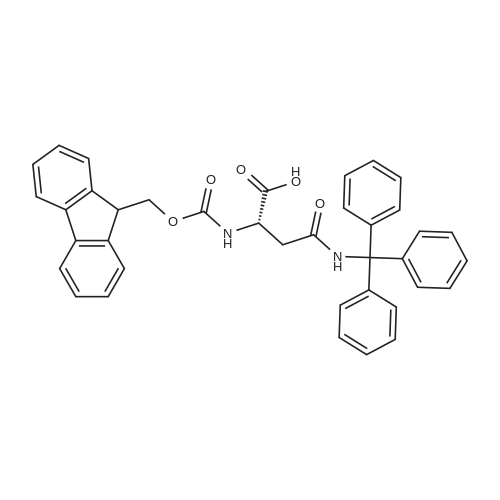
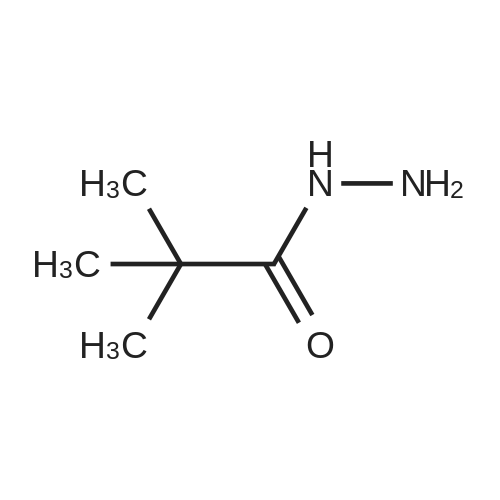
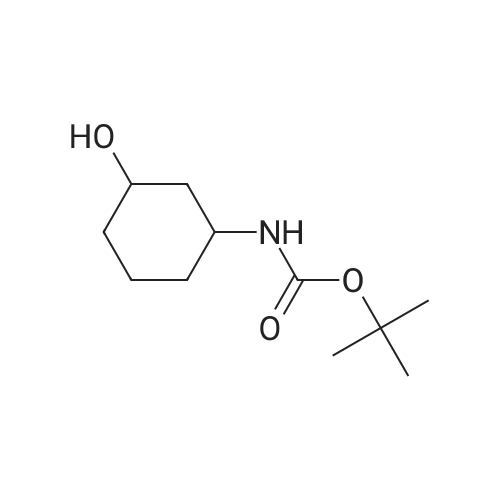
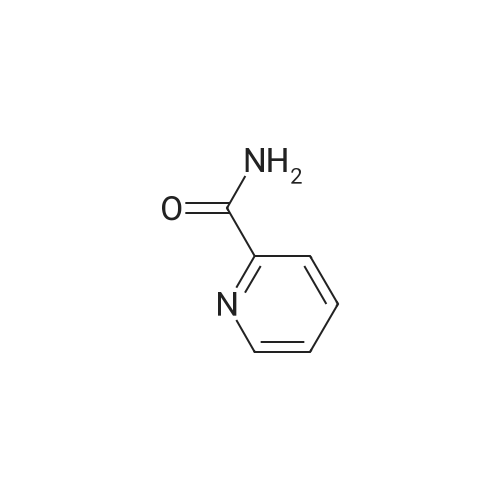
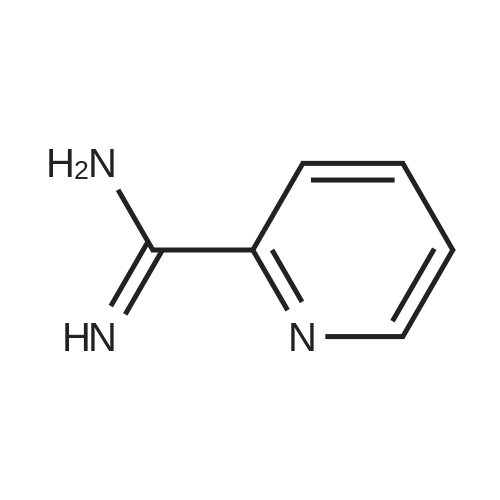
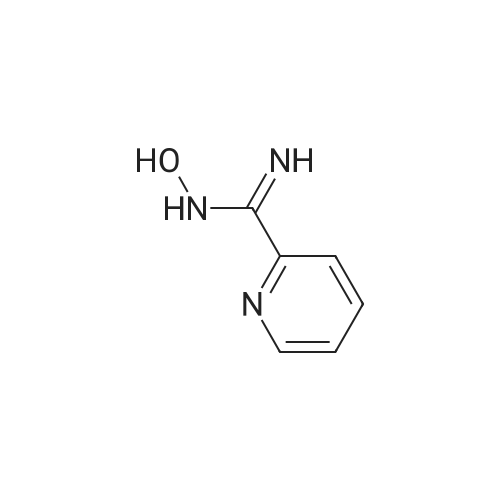
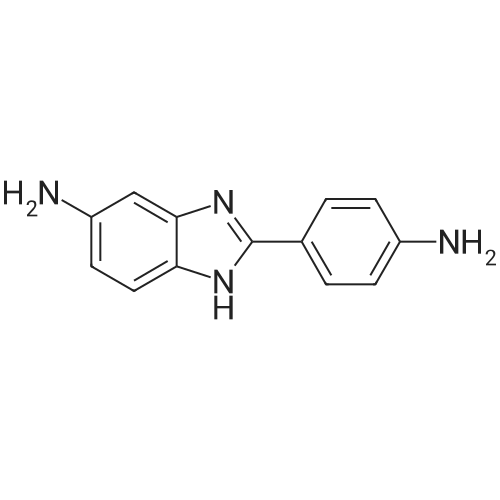
[ 109-01-3 ]
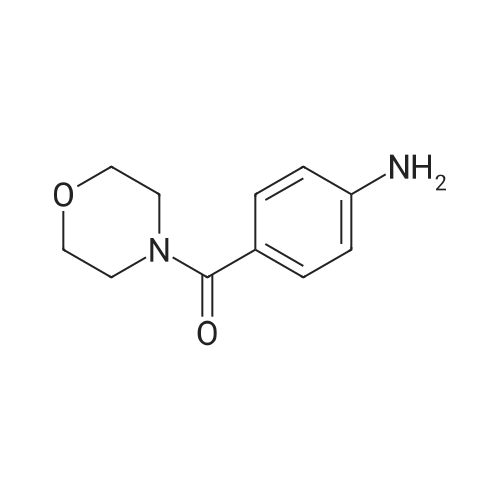
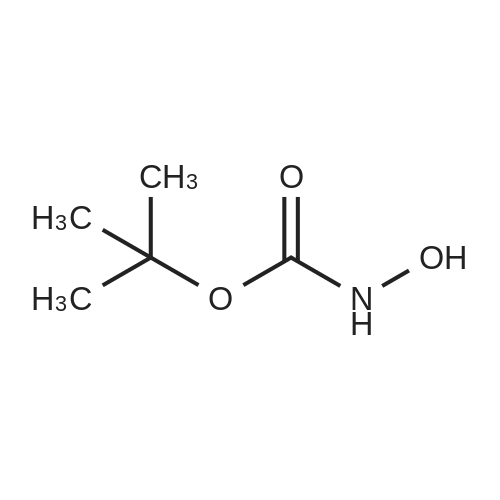
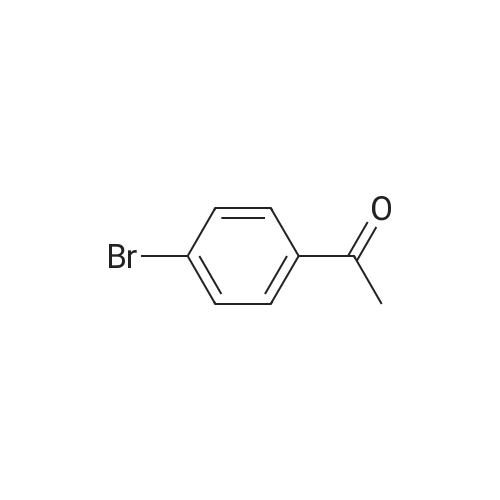
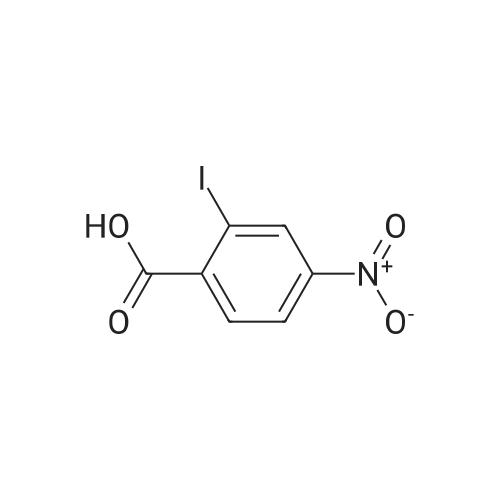
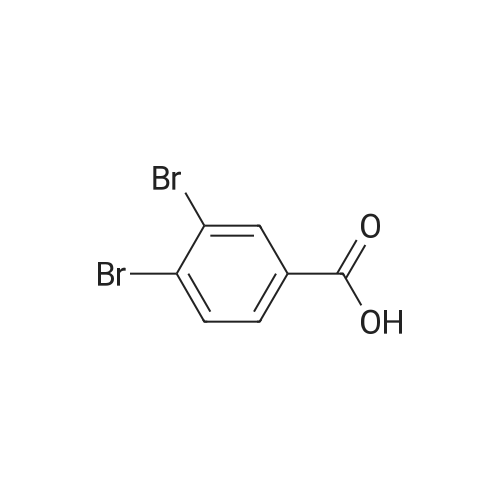
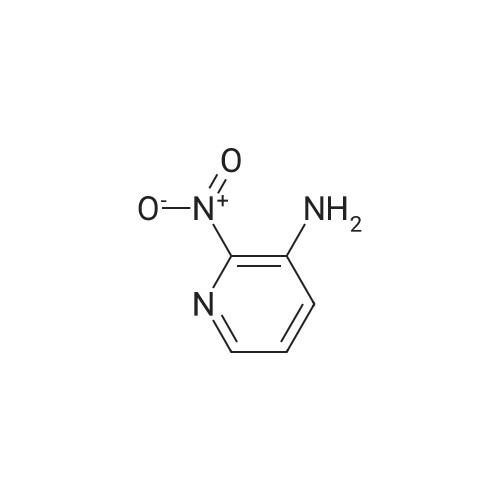
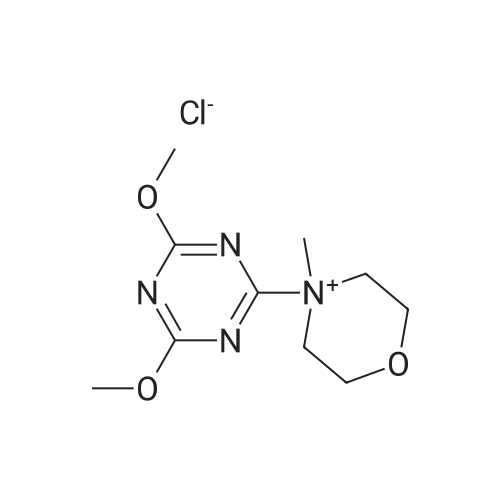
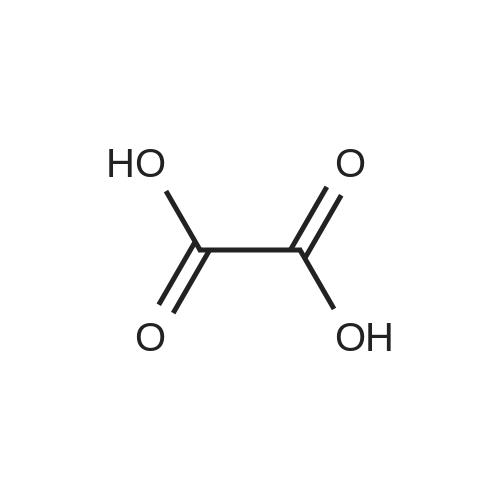
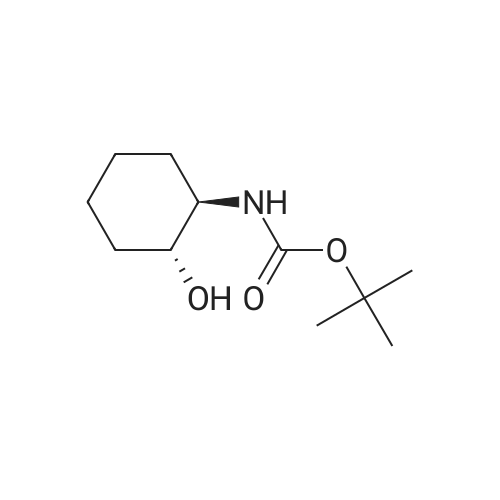
[ 62-23-7 ]
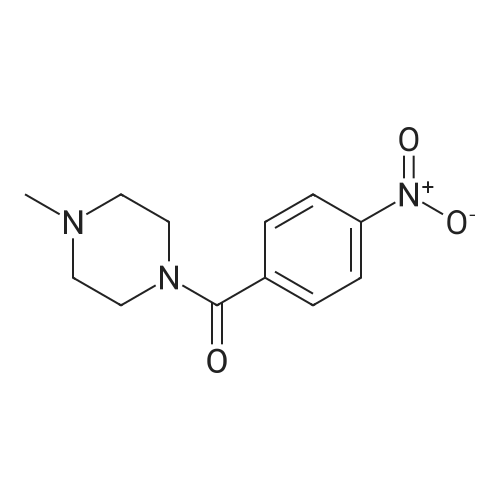
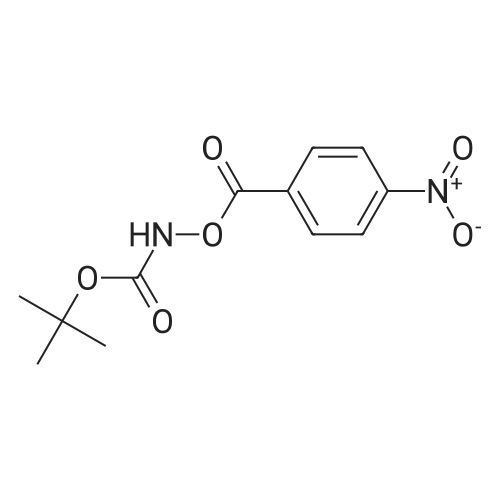
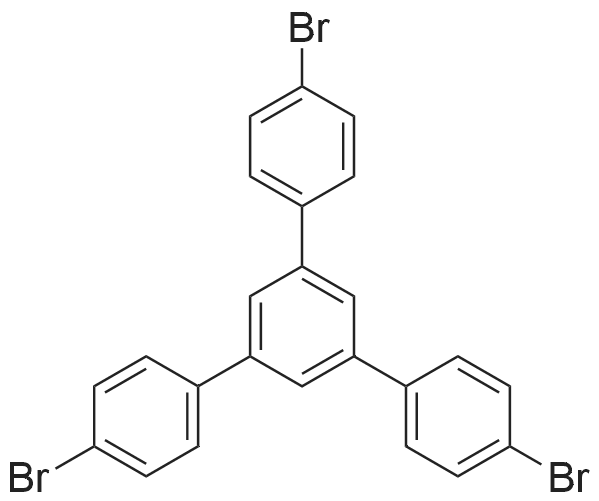
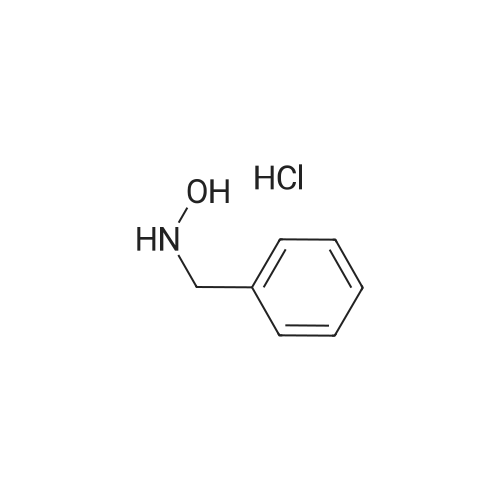
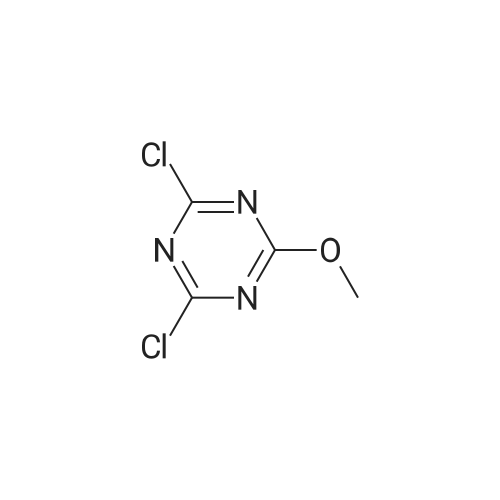
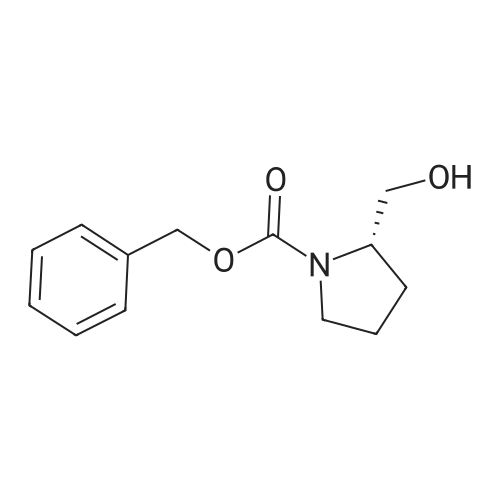
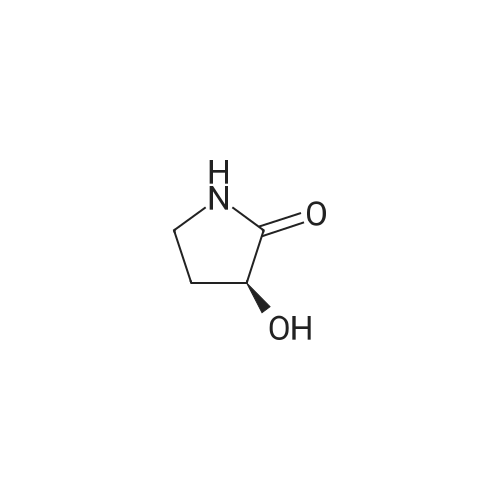
[ 21091-98-5 ]
Yield
Reaction Conditions
Operation in experiment
95%
With ethylene dichloride hydrochloride; benzotriazol-1-ol; triethylamine In N,N-dimethyl-formamide at 20℃; for 12 h;
Step 1: (4-Methyl-piperazin-l-yl)-( -nitro-phenyl)-methanoneTo a solution of 4-nitro-benzoic acid (10 g, 0.0598 mol) in DMF (300 mL) 1- methyl-piperazine (7.19 g, 0.0718 mol), HOBt (10.5 g, 0.0777 mol), EDC-HCl (17.12 g, 0.0897 mol) and TEA (12.1 g, 0.1 196 mol) were added. The reaction mixture was stirred at room temperature for 12 h. The reaction mixture was diluted with water and extracted with ethyl acetate (3 x 200 mL). The organic layer was washed with water, brine and dried over sodium sulfate. The solvent was evaporated under reduced pressure to afford the title compound [14 g, 95percent]; LC-MS (ESI): Calculated mass: 249.1; Observed mass: 250.0 [M+H]+ (RT: 0.10 min).
80%
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate In N,N-dimethyl-formamide at 20℃;
To a solution of 4-nitrobenzoicacid (1.0 g, 5.98 mmol), 1-methylpiperazine (0.599 g, 5.98 mmol) and DIPEA (2.320 g,17.95 mmol) in DMF (10 mL) was added HATU (4.55 g, 11.97 mmol). The reaction wasstirred overnight at room temperature. The mixture was quenched with water andextracted with a mix solvent of DCM and MeOH (10:1, 20 mL×3). The combinedextracts were dried over Na2SO4, filtered and concentrated. The crude was purified bycolumn chromatography on silica gel (DCM: MeOH= 20:1) to give the title compound 3a(1.2 g, 80 percent yield). LCMS: 250.1 [M+H]+.
59%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine In tetrahydrofuran at 0℃;
Step: 3A-lSynthesis of (4-Methyl-piperazin-l-yl)-(4-nitro-phenyl)-methanone. Procedure:To a solution of 4-Nitro-benzoic acid (500mg, 2.9918mmol) in THF was added EDCI (856mg, 4.4865mmol), followed by HOBt (200mg, 2.9918mmol), DIPEA (1.5ml,8.9754mmol) and 1 -Methyl -piperazine (329mg, 3.291mmol) at 0°C. The resultant was stirred overnight. The reaction was monitored by the TLC (10percent methanol: chloroform).The reaction mixture was extracted with ethyl acetate and washed with water. The organic layer was concentrated and dried over Na2S04 to afford 440mg (59percent yield) of (4- Methy 1-piperazin- 1 -yl)-(4-nitro-phenyl)-methanone.
4-nitro-benzoic acid 1-[2-(2-acetylamino-3-hydroxy-butyrylamino)-3-(4-hydroxy-phenyl)-propionyl]-5-(1,2-dicarbamoyl-ethylcarbamoyl)-pyrrolidin-3-yl ester[ No CAS ]
4-Morpholin-4-ylmethyl-phenylamine is prepared from 4-Nitro-benzoic acid by the following method: A room temperature solution of 2.49 gms. 4-nitro-benzoic acid in tetrahydrofuran (15 mL) is treated with 2.44 gms. 1',1'-carbonyl-diimidizole, and immediately immersed in an ice bath. The reaction mixture is stirred in the ice bath for 30 minutes then it is allowed to warm to room temperature. Once at room temperature the reaction mixture is treated with 1.5 mL piperidine. The reaction mixture is allowed to stir at room temperature overnight. The reaction is then made basic with the addition of saturated aqueous sodium bicarbonate solution, and the resulting mixture is extracted with ethyl acetate. The organics are separated, dried over anhydrous sodium sulfate and subsequently evaporated to dryness in vacuo. The crude product residue is then chromatographed by flash silica gel chromatography using 50% ethyl acetate in hexanes as the eluant.
EXAMPLE 5 Preparation of (S)-3-Chloro-1,2-propanediol with (S,S)-Co(salen): To a 3-necked, 250-mL, round-bottomed flask fitted with a vacuum adapter, an air inlet, and a temperature probe was added (S,S)-Co(salen) (1.51 g, 0.0023 mol, 0.23 mol %), propylene glycol monomethyl ether (20 mL), and p-nitrobenzoic acid (0.835 g, 0.0049 mol, 0.49 mol %). A slight vacuum was applied to pull air into the reaction medium, and the reaction was allowed to stir for 1 h. The air inlet was removed, and epichlorohydrin (92.52 g, 78.40 mL, 1.00 mol) was charged after the reaction vessel was purged with nitrogen. The reaction was cooled to 0 C., and water (8.1 g, 8.1 mL, 0.45 mol, 45 mol %) was slowly added, maintaining the reaction temperature below 5 C. The reaction was allowed to stir at room temperature for 16 h. When determined to be complete, the reaction was partitioned between toluene (80 mL) and water (80 mL). The aqueous layer was collected and washed with toluene (80 mL) to remove unreacted epichlorohydrin (isopropyl alcohol (20%) was added to facilitate the phase separation). After concentration of aqueous layer, the residue was vacuum distilled to give (S)-3-chloro-1,2-propanediol (37.4 g, 76% yield, >99% purity, 97% ee; b.p. 115-120 C. a 10 mm Hg).
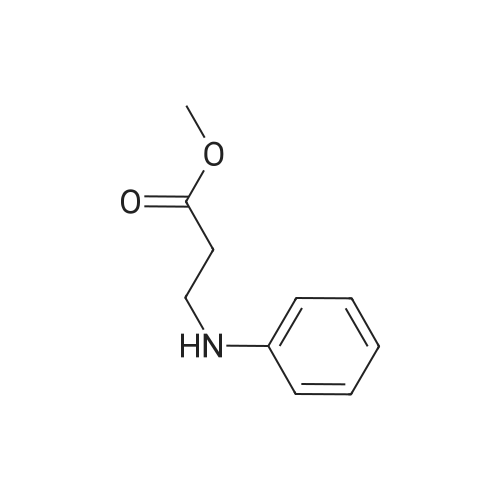
16.7 g (0.1 mol) of 4-nitrobenzoic acid are heated to reflux in 50 ml of thionyl chloride and 3 drops of dimethylformamide for 1 hour. After removing the solvent by distillation in vacuo, it was dissolved in 150 ml of tetrahydrofuran and added dropwise to a mixture of 18 g (0.1 mol) of N- (2-methoxycarbonylethyl) -aniline, 250 ml of tetrahydrofuran and 42 ml Solution. After stirring for 1 hour at room temperature, the reaction mixture was diluted with 250 ml of ethyl acetate and washed twice with 200 ml of 14% saline solution. The solvent was distilled off and purified by silica gel column chromatography to give 4-nitro-benzoic acid N-phenyl-N- (2-methoxycarbonylethyl) amide (27.68, yield 85% And the value: 0.37 (silica gel, dichloromethane / methanol = 50: 1). Mass spectrometry 31 -] ^): (] 1 + 10 + = 329.1, (M + H + Na) ++ = 352.1, Ci7Hi6N25 (328)
In tetrahydrofuran; N-methyl-acetamide; thionyl chloride; ethyl acetate;
a 4-Nitro-benzoic acid-N-phenyl-N-(2-methoxycarbonylethyl)amide 16.7 g (0.1 mol) of 4-nitrobenzoic acid were refluxed in 50 ml of thionyl chloride and 3 drops of dimethylformamide for 1 hour. After the solvent had been distilled off in vacuo the crude product was dissolved in 150 ml of tetrahydrofuran and added dropwise to a solution of 18 g (0.1 mol) of N-(2-methoxycarbonylethyl)aniline in 250 ml of tetrahydrofuran and 42 ml 0.3 mol) of triethylamine. After being stirred for one hour at ambient temperature the reaction mixture was diluted with 250 ml of ethyl acetate and washed 2* with 200 ml of 14% saline solution. After the solvent had been distilled off and the residue chromatographed (silica gel; methylene chloride) a yellow oil was obtained which slowly solidified. Yield: 32.6 g (100% of theory), Rf value: 0.37 (silica gel; methylene chloride/methanol=50:1)
With titanium(IV) oxide; sodium hydroxide; at 70℃;
General procedure: In a 10 ml three-necked flask equipped with an electric stirrer, a thermometer, a condenser and a dropping funnel, 5 mL of deionized water and 3 g of chromic anhydride were mixed and homogenized. The flask was then immersed in an ice-water bath and cooled to 25 C, The pH was adjusted to 6.0 with a hydrochloric acid solution having a mass fraction of 30%. Then, 3 g of p-nitrotoluene and 10 mL of glacial acetic acid were added thereto, and the liquid was heated to the condenser by a beaker to keep the temperature for 30 minutes. After the above- Add 80 mL of deionized water to the beaker using a dropping funnel and adjust the dropping rate to drop it in 5 min and allow to stand until no yellowish precipitate is produced. Then, the filtrate is filtered under reduced pressure. The resulting extract was placed in a round bottom beaker and 40 mL of a 40% sodium hydroxide solution with a mass fraction of 40% was added and stirred until the filter was completely dissolved, and then the resulting filtrate was air- And then 70kg of titanium dioxide, mixed evenly, in the beaker with a round bottom beaker and add the rotor, then round bottom beaker moved to the magnetic stirring heater, the temperature is set to 70 C, the speed is set to 200r / min; After the solution in the beaker has been clarified, the heating is stopped and the mixture is naturally cooled to room temperature. The liquid in the beaker is poured into a conical flask to remove the titanium dioxide. The aqueous solution is then adjusted to a pH of 7.5 with a mass fraction of 20% The conical flask is then heated until it has a pale yellow precipitateThe resulting filter material was placed in a beaker, and 1.5 g of tin dioxide was added thereto, and the resulting filtrate was added to a beaker, and the mixture was filtered and the filtrate was collected under reduced pressure. Uniform, and then use the mass fraction of 30% sulfuric acid solution soak the mixture, and then the beaker heating, heated to a temperature of 85 C, keep the temperature 2h, the hot filter, collecting the filtrate, 4-aminobenzoate; The resulting filtrate was mixed with 10 g of sodium carbonate and 30 mL of absolute ethanol, poured into a round bottom flask, and then 15 mL of hydrogen bromide was poured into it, and the vessel was transferred to a water bath. After the temperature was set to 60 C and the temperature was maintained for 70 min, the round bottom flask was charged with a distillation unit, and the round bottom flask was heated to collect 135 C fraction and cooled to room temperature to give (2 - ((4-aminobenzene Formyl) oxy) ethyl) in a yield of 47.8%.
To a solution of Compound 10 (0.70 g, 3.48 mmol) and PPh3 (1.37 g, 5.22 mmol) in CH2Cl2 (7 mL) was added DEAD (0.91 mL, 5.22 mmol) at 4 C. [Zhao et al., Eur. J. Med. Chem. 2005, 40, 231]. The resulting mixture was stirred for 10 min and then 4-nitrobenzoic acid (1.62 g, 5.22 mmol) was added. This mixture was allowed to warm up to room temperature and stirred for 16 hours. The reaction mixture was quenched with 2 N NaOH solution and extracted with AcOEt. The organic layers were combined, washed with brine, dried over Na2SO4, and concentrated. The residue was purified by flash column chromatography to give Compound 15 (0.885 g, 73%) as a pale yellow solid. Compound 15: 1H NMR (400 MHz, CDCl3): delta 1.38 (d, J=0.4 Hz, 3H), 1.48 (s, 9H), 1.96 (d, J=14.4 Hz, 1H), 2.47 (m, 1H), 3.64-3.83 (m, 2H), 4.11 (m, 1H), 5.55 (m, 1H), 8.21 (d, J=8.0 Hz, 2H), 8.31 (d, J=8.0 Hz, 2H).
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 20℃;
To a solution of tert-butyl (lS,2S)-2-hydroxycyclohexylcarbamate(1.30 g, 6.04 mmol), 4-nitrobenzoic acid (1.11 g, 6.64 mmol), triphenylphosphine (1.74 g, 6.64 mmol) in THF (30 mL) was added diethylazo dicarboxylate (1.05 ml, 6.64 mmol) and the resulting solution was stirred at ambient temperature overnight. The reaction mixture was diluted with ethyl acetate and washed with brine. The organic layer was dried and concentrated to give an orange oil that was purified by column chromatography (hexanes/ethyl acetate, 8: 1) to give (lR,2S)-2-(tert-butoxycarbonylamino)cyclohexyl 4- nitrobenzoate (1.95 g, 89%).
With di-isopropyl azodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 20℃;
(R)-(+)-3-Hydroxy-2-pyrrolidinoneTo a stirring mixture of 4-Nitrobenzoic acid (21.5 g) and (S)-(-)-3-hydroxy-2-pyrrolidinone (11.8 g) (Intermediate 17) in dry THF (360 mL) taken in a round bottomed flask fitted with anhydrous CaCl2 guard tube, triphenyl phosphine (61.2 g) was added.To this reaction mixture, diisopropyl diazodicarboxylate (DIAD) (34 mL) was added drop wise in three portions at room temperature.The reaction was stirred at room temperature.The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4).After completion, reaction mixture was concentrated under vacuum to obtain residue.Methanol (360 mL) was added to the residue followed by potassium carbonate (10 g) at room temperature.The reaction was stirred at room temperature.The progress of the reaction was monitored by TLC (developing agents: UV, I2, as well as aqueous acidic KMnO4).After completion, reaction mixture was diluted with CHCl3 and filtered through celite.Celite bed was successively washed with 1percent MeOH:CHCl3.The filtrates were combined and concentrated to dryness to remove solvents.The residues were partitioned between EtOAc: dil. HCl (200 mL, 9:1) and stirred for 15 min.Layers were separated, aq.layer was washed with EtOAc thrice until all organic impurities were washed out.The aq.Layer was concentrated to dryness to remove the water and solid residues were obtained.The residues obtained were washed with 1-2percent MeOH: CHCl3 (3*100 mL), dried over sodium sulfate, filtered trough cotton, concentrated to get brown thick liquid product.1H NMR (CDCl3, 400 MHz) delta ppm: 2.03-2.13 (m, 1H), 2.46-2.54 (m, 1H), 3.28-3.35 (m, 1H), 3.38-3.48 (m, 1H), 4.50 (t, J=8.4 Hz, 1H), 4.55 (bs, 1H), 7.02 (bs, 1H); [alpha]D25: +68, c=1, CHCl3
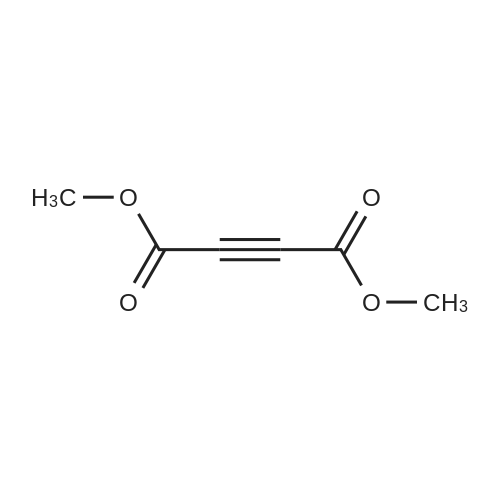
General procedure: At room temperature, organic carbonyl acid 3 (R-COOH, 0.5 mmol) was added into a reaction tube equipped with a small magnet. Then a solution of tertiary amine 1 (R1CH2-NR2R3, 0.5 mmol ) in DCM (2.5 mL) was added dropwise in 2 min. After the mixture was stirred at room temperature for a few minutes, 1 equivalents of dimethyl acetylenedicarboxylate (DMAD, 2) was added. The reaction was stirred overnight at room temperature, and then monitored by TLC with silica gel coated plates. After being stirred for 14 h, the solvent was removed and the residue was purified by a flash column chromatography with silica gel with ethyl acetate/hexane (1:25-30) as eluent to give the desired products 4, 5, and 7. Most of compounds are known and confirmed by NMR, ESI-MS, IR.
With Oxone; In water; acetonitrile; at 20℃; for 5.5h;
General procedure: To 16 mL of acetonitrile/water (1:1 v/v) mixture was added 0.5-1.2 mmol of the starting compound. The contents were heated at reflux with introduction of oxone (cf. entries for each case) incrementally over the entire duration of the reaction. For secondary benzyl halides, the reactions were run at room temperature. The progress of the reaction in each case was monitored by TLC analysis. After completion of the reaction, the reaction mixture was cooled to room temperature and the organic matter was extracted with ethyl acetate. The combined organic extract was dried over anhyd Na2SO4 and concentrated in vacuo. Short pad silica gel column chromatography of the residue led to isolation of the pure product.
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at 0 - 25℃; for 16h;
Step B - Synthesis of Intermediate Compound 6c To a solution of compound 6b (4.1 mg, 19.04 mmol), 4-nitrobenzoic acid (3.82 g, 22.85 mmol), and PPh3 (9.9g, 38.1 mmol) in THF (60 ml) was added DEAD (6.03 mL, 38.1 mmol) at 0C. The reaction mixture was then stirred at 25C for 16 h, quenched with water (100 mL) and extracted with EtOAc (100 mL x 3). The combined organic phase was washed with water (100 mL x 2), dried over Na2S04, and concentrated in vacuo. The residue was purified using column chromatography (Si02, Petroleum ether: EtOAc = 10: 1 to 5: 1) to provide compound 6c (1.3 g, 81% yield) as a white solid.
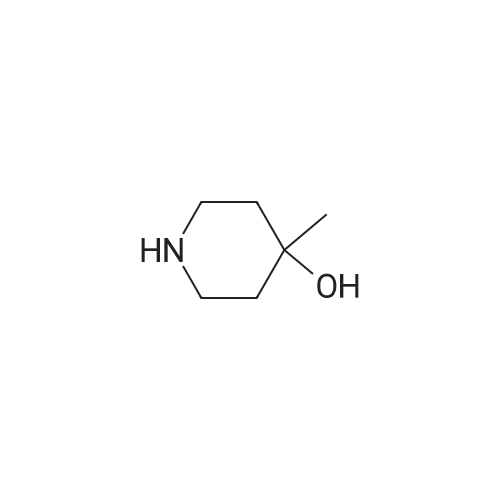
Synthesis of compound 221.4. To a solution of p-nitro benzoic acid (0.500g, 3.62mmol, l .Oeq) in DMF (10 mL) was added HATU (2.06g, 5.43 mmol, 1.5eq.). The reaction was allowed to stir at room temperature for 15 minutes. Further <strong>[3970-68-1]4-methylpiperidin-4-ol</strong> (0.500g, 4.34mmol, 1.2eq.) and DIPEA (1.402g, 10.86mmol, 3.0eq.) were added to the reaction mixture at room temperature and the reaction was allowed to stir for 2h. After completion of the reaction, reaction mixture was poured in water and product was extracted with EtOAc. Organic layer was combined, dried over sodium sulphate and concentrated under reduced pressure to obtain crude which was purified by flash chromatography to furnish 221.4 (0.270g, 34.15%). MS (ES): m/z 264.28 [M+H]+.
General procedure: To a solution of 2-Methylbenzoic acid (1.36 g, 10 mmol) in DCM (50 mL), Et3N (1.12 g, 11 mmol) and TBTU (3.60 g,11 mmol) were added in turn. After 20 min, compound b (n =4, 1.17 g, 5 mmol) and Et3N (0.50 g, 5 mmol) were added. The reaction solution was stirred at room temperature for 8 h.Then, the solvent was evaporated with the residue being takenup in EtOAc (50 mL). the EtOAc solution was washed with saturated citric acid (3 × 20 mL), NaHCO3 (3 × 20 mL), and brine (3 × 20 mL), dried over MgSO4, and evaporated under vacuum. The desired compound c (1.50 g, 76 percent yield) was derived by crystallization in EtOAc/petroleum ether (1/4) as white powder. ESI-MS: m/z: 397.8 [M+H]+.
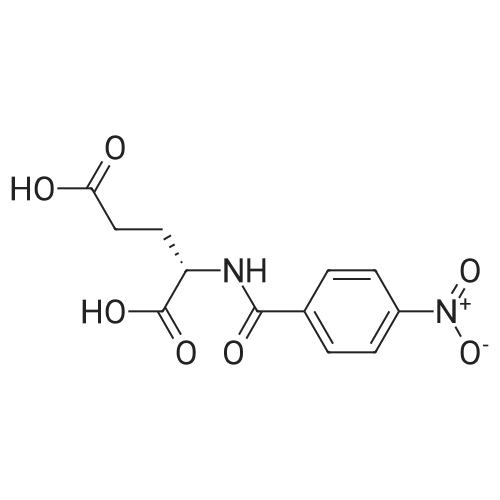
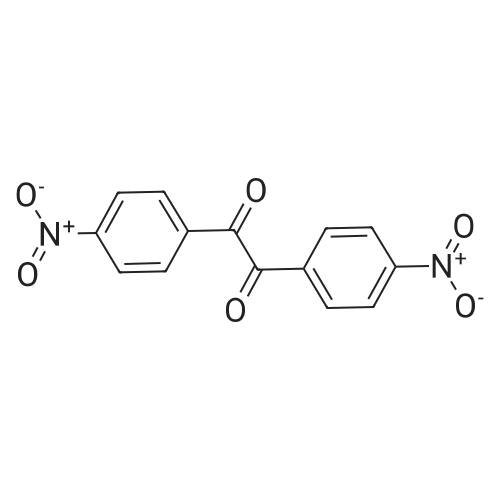
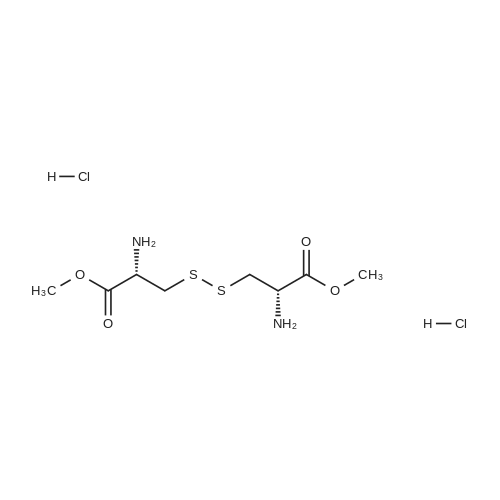
dimethyl 3,3'-disulfanediylbis(2-(4-nitrobenzamido)propanoate)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In dichloromethane; at 0 - 20℃;
General procedure: A suspension of the benzoic acid (1.17 mmol) and L-cystinedimethyl ester dihydrochloride (341 mg, 0.586 mmol) in anhydrous DCM (5mL) was cooled to 0 C to which was added Et3N (179 mL, 1.28 mmol), HOBt (17 mg, 0.117 mmol), and EDCI( 181 mg, 1.17 mmol). The resulting suspension was warmed to room temperature and stirred overnight, eventually turning into a clear solution. The mixture was diluted with EtOAc (10 mL)and washed successively with HCl (1M, 10 mL), saturated NaHCO3(10 mL), and brine (10 mL). The organic phases were dried over MgSO4, filtered, and concentrated under vacuum to afford the corresponding amide.
To a 100 ml three-mouth bottle added (3R, 3aS, 6aR)-hexahydrofuro [2,3-b] furan-3-ol (2.0g, 15 . 4mmol), pyridine (10 ml), triethylamine (2.2g, 21 . 3mmol), the ice-bath is cooled to 0 C, the 10 ml pyridine and P-nitro benzoic acid (2.7g, 16 . 2mmol) was added to the mixture in three-mouth bottle, maintained the temperature in the course of dropping 0-20C, after adding, TLC detection until the raw material the reaction is complete. After the reaction, filtration, filtrate are respectively with dilute hydrochloric acid (3×20 ml), saturated sodium carbonate (20 ml), saturated salt water (20 ml) washing, the organic phase is dried with anhydrous sodium sulfate, the filtrate concentrated to dry, the white solid obtained 4.1g, column chromatography, to obtain refined product 3.96g, yield 92%.
3α-(p-nitrophenylcarbonyloxy)-17-(1H-benzimidazol-1-yl)androsta-5,16-diene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
18.1%
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; toluene; at 4 - 22℃; for 12h;
To a two neck flask was added glaterone (1, 2 g, 5.15 mmol), 0.95 g (5.66 mmol) of pnitrobenzoicacid, 1.5 g (5.66 mmol) of PPh3 and 30 mL anhydrous THF. The mixture was stirred until all solids dissolved and then cooled to 4°C in an ice-water bath. A solution of 40percent> DEAD 2.46 rnL (5.66 mmol) in anhydrous toluene was added drop wise, allowed to attain room temperature, stirred for 12 hours. The reaction mixture concentrated in vacuo and the resulting sticky residue was suspended in 5 rnL of ethyl acetate, stirred, cooled and filtered. Mother liquor collected, stirred vigorously and 30 mL of petroleum ether added slowly. Resulting sticky suspension filtered, washed with 10percent ethyl acetate in petroleum ether (25 rnL) and dried under vacuo. The sticky solid made slurry on Buckner funnel with ether (7.5 mL x 2), filtered and dried under vacuo. Free solids (0.8 g) obtained was a mixture of product 11, traces of tripheylphosphinoxide and diethyl 1,2-hydrazinedicarboxylate. The crude product dissolved in hot IPA, allowed to cool to room temperature, resulting precipitate filtered and dried under vacuo to obtain a white solid (0.5 g, 18.1percent) of pure 11, mp 193- 194°C; Rf= 0.4 (3percent acetone in DCM); FontWeight="Bold" FontSize="10" H NMR (400 MHz, CDC13) delta 1.05 (s, 3 H, 18-CH3),
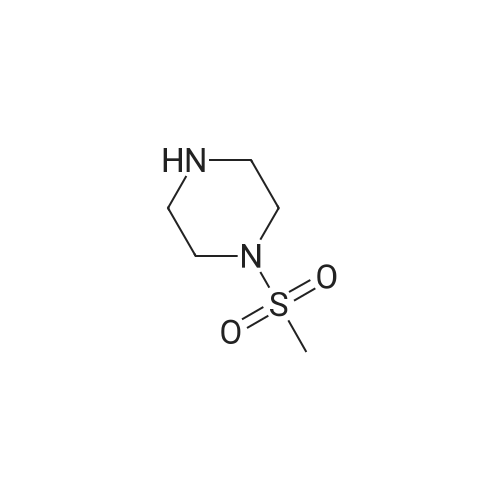
(4-(methylsulfonyl)piperazin-1-yl)(4-nitrophenyl)methanone[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50.2%
With N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In N,N-dimethyl-formamide; at 20℃; for 48h;
To a solution of 4-nitrobenzoic acid (0.43 g, 2.56 mmol) in DMF (10 mL), methanesulfonylpiperazine (0.42 g, 2.56 mmol) was added followed by HATU (0.97 g, 2.56 mmol) , And stirred at room temperature for 48 h.The organic layer was separated and the aqueous layer was extracted with ethyl acetate (30 mL x 2). The organic layers were combined, washed with water (30 mL x 3), and then washed with saturated NaCl (50 mL), washed with water (50 mL) ) Once, anhydrous Na2SO4dry.The solvent was distilled off under reduced pressure, and the residue was subjected to direct column chromatography, eluting with EA / PE = 1/1 - 3/1 to give 0.40 g of the desired product in a yield of 50.20%.
The <strong>[202409-33-4]etoricoxib</strong> 359mg was put into a 50ml methanol,2ml purified water in a single-mouth flaskPlaced in a 50C oil bathThen add 217mg of p-nitrobenzoic acid,Stir and dissolve,Pour the reaction solution into a small high pressure reactor.Then place it in an oven at 130 degrees Celsius.Stay for 2 hours,So <strong>[202409-33-4]etoricoxib</strong> and after sufficient reaction of p-nitrobenzoic acid,Let cool at room temperature.Pour the reaction solution into a 100ml single-mouth bottle.Under ice bath conditions,Vacuum evaporation (vacuum -0.03MPa),After 4h,A lot of transparent crystals,Precipitation,filter,The filter cake is washed with cold methanol.The resulting crystals are the target salt crystals.
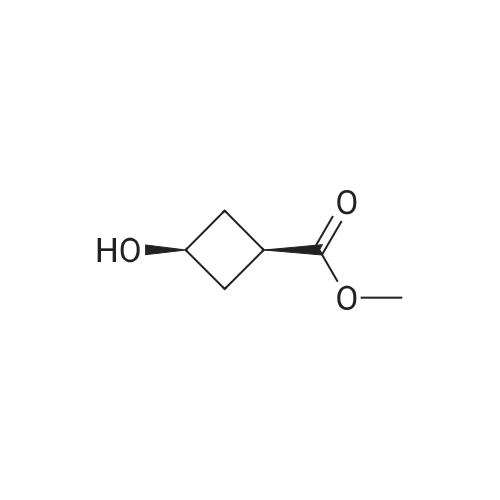
trans-p-nitrobenzoic acid (3-methoxycarbonylcyclobutyl) ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76%
With triphenylphosphine; diethylazodicarboxylate; In tetrahydrofuran; at -10 - 20℃; for 16h;Large scale;
<strong>[63485-50-7]cis-3-hydroxycyclobutylcarboxylic acid methyl ester</strong> (2730 g, 21.0 mol, 1.0 eq) prepared in Example 1 was dissolved in tetrahydrofuran (30 L) and cooled to -10 C.p-Nitrobenzoic acid (4178 g, 25.0 mol, 1.2 eq.), diethyl azodicarboxylate (4354 g, 25.0 mol, 1.2 eq.), triphenylphosphine (6557 g, 25.0 mol, 1.2 eq.).It was then stirred at room temperature for 16 h under nitrogen.The reaction was complete by TLC, the tetrahydrofuran was removed, and methyl t-butyl ether (20 L) was added and stirred for 0.5-1 h.Filter and filter cake was washed twice with methyl tert-butyl ether.All filtrates were collected and the filtrate was washed with saturated aqueous sodium bicarbonate.Dispense, dry and concentrate.The crude product was then beaten with a mixed solvent of ethyl acetate/petroleum ether, filtered and concentrated to give a pale yellow powder.Trans-III compound trans-p-nitrobenzoic acid (3-methoxycarbonylcyclobutyl) ester (4468 g, 16.0 mol), yield 76%, purity 95%.
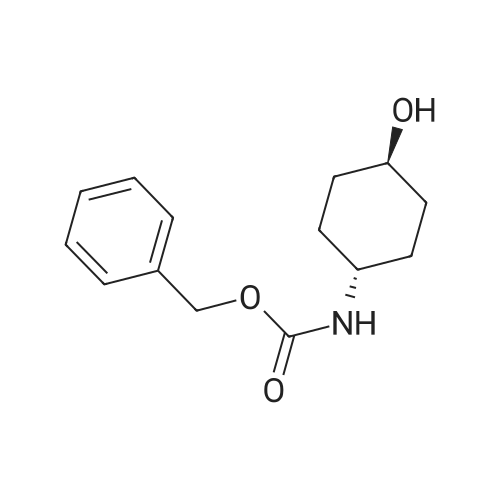
cis-4-[(benzyloxy)carbonyl]amino}cyclohexyl 4-nitrobenzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
161 mg
With di-tert-butyl-diazodicarboxylate; triphenylphosphine; In tetrahydrofuran; at 60℃; for 2h;
(1) Triphenylphosphine (421 mg) and di-tert-butyl azodicarboxylate (370 mg) were added to a solution of the compound (200 mg) obtained in Reference Example 19-7 and 4-nitrobenzoic acid (268 mg) in tetrahydrofuran (3 mL), and the mixture was stirred at 60C for 2 hours. To the above mixture, 6 mol/L hydrochloric acid was added, and the resultant mixture was stirred for 2 hours and then was extracted with ethyl acetate. The organic layer was washed with an aqueous solution of saturated sodium hydrogen carbonate and brine, and then was dried over anhydrous magnesium sulfate. After the drying agent was filtered off, the filtrate was concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (n-hexane only, to n-hexane/ethyl acetate=1:1) to give cis-4-[(benzyloxy)carbonyl]amino}cyclohexyl 4-nitrobenzoate (161 mg) as a colorless solid.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; for 8h;
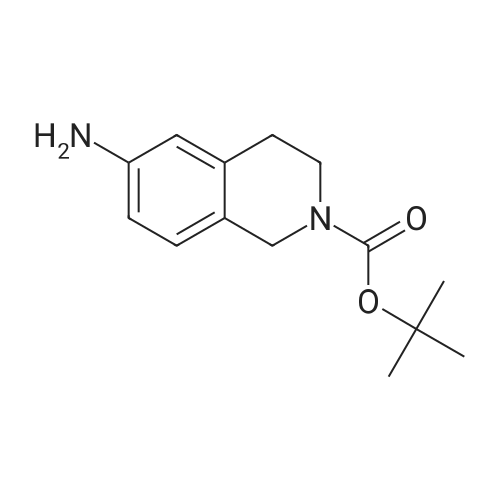
Tert-butyl 6-amino-3,4-dihydroisoquinoline-2(1H)-carboxylate (248mg, 1mmol), 4-nitrobenzoic acid (167mg, 1mmol), EDCI (212mg, 1.1mmol), and catalytic quantity of 4-DAMP are added into DCM (10mL) and the mixture were stirred for 8h. The reaction mixture was diluted with DCM and filtered using Celite, then evaporated. Starting with the residue which was not purified, Tert-butyl 6-(4-(vinylsulfonamido)benzamido)-3,4-dihydroisoquinoline-2(1H)-carboxylate (DC-TEADin07-1) was obtained as a white solid by using general synthesis procedure 1b and 2b (yield 45%). 1H NMR (400MHz, Methanol-d4) delta 7.87 (d, J=8.9Hz, 2H), 7.55-7.41 (m, 2H), 7.28 (d, J=8.9Hz, 2H), 7.11 (d, J=8.5Hz, 1H), 6.71 (dd, J=16.5, 10.0Hz, 1H), 6.24 (d, J=16.5Hz, 1H), 6.01 (d, J=9.9Hz, 1H), 5.48 (s, 2H), 4.53 (s, 2H), 3.63 (t, J=5.9Hz, 2H), 2.83 (t, J=5.9Hz, 2H), 1.49 (s, 9H).
With dmap; dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;
General procedure: A mixture of 2 or 3 (0.50 mmol), the corresponding acids RCOOH (0.60 mmol),DCC (0.60 mmol), DMAP (0.1 mmol) in dry dichloromethane (15 mL) was stirred atroom temperature. When the reaction was completed, and checked by TLC, the mixturewas filtered to remove urea from the reaction, and the filtrate was diluted bydichloromethane (45 mL). Subsequently, the diluted organic phase was washed bysaturated aqueous NaHCO3 (30 mL), and brine (30 mL), dried over anhydrousNa2SO4, concentrated in vacuo, and purified by CC to give the pure 9R/S-acyloxyderivatives of cinchonidine and cinchonine 5a-j,l-o and 6a,c,e-o [17-19]. The dataof target compounds are shown as follows.
With ferrous(II) sulfate heptahydrate; sulfuric acid; boric acid; In glycerol; at 140℃; for 42h;Inert atmosphere;
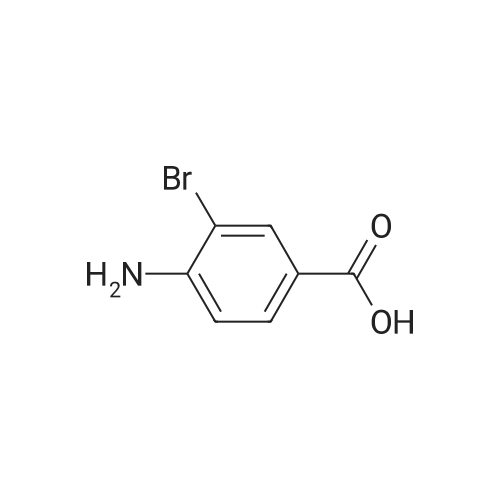
To a mixture of <strong>[6311-37-1]4-amino-3-bromobenzoic acid</strong> (500 mg, 2.31 mmol) in concentrated sulfuric acid (650 muL) was added glycerol (1.75 g, 19.0 mmol), 4-nitrobenzoic acid (391 mg, 2.34 mmol), boric acid (215 mg, 3.48 mmol) and iron(II) sulfate heptahydrate (234 mg, 0.842 mmol), and the reaction was stirred at 140 C. for 42 h. The reaction mixture was cooled to room temperature and the pH was adjusted to 10 with 10% sodium hydroxide. The aqueous layer was extracted with ethyl acetate (2*100 mL) and the combined organic layers were dried over sodium sulfate, filtered and evaporated to give the title compound (150 mg, 25% yield) as a 10 yellow powder. MS (ESI): mass calcd. for C10H6 BrNO2, 251.0; m/z found, 252.0 [M+H]+.
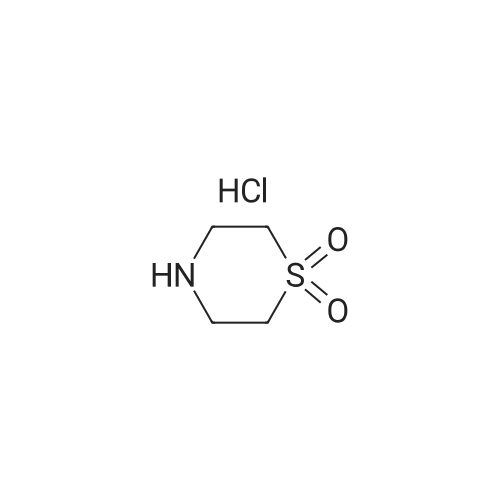
4-(4-nitrobenzoyl)-1,1-dioxo-1,4-thiazinane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; triethylamine; In N,N-dimethyl-formamide; at 15℃; for 14h;Inert atmosphere;
To a mixture of 4-nitrobenzoic acid (5 g, 29.92 mmol) and 1 ,4-thiazinane 1 ,1-dioxide hydrochloride (5.1 g, 29.92 mmol) in DMF (50 ml.) was added HOBt (6.1 g, 44.88 mmol), EDCI (8.6 g, 44.88 mmol), EbN (6.1 g, 59.84 mmol) in one portion at 15C under N2. The mixture was stirred at 15C for 14 hours. The reaction mixture was diluted with saturated Na2CC>3 (300 ml.) and extracted with EtOAc (150 ml. x 3). The combined organic layers were dried over Na2S04, filtered and concentrated under reduced pressure to give the title compound (6.5 g, crude) as a white solid. 1H NMR (400 MHz, CDCIs): d : 8.27 (d, J=8.8 Hz, 2H), 7.55 (d, J=8.8 Hz, 2H), 4.33 - 3.75 (m, 4H), 3.22 - 2.75 (m, 4H).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping