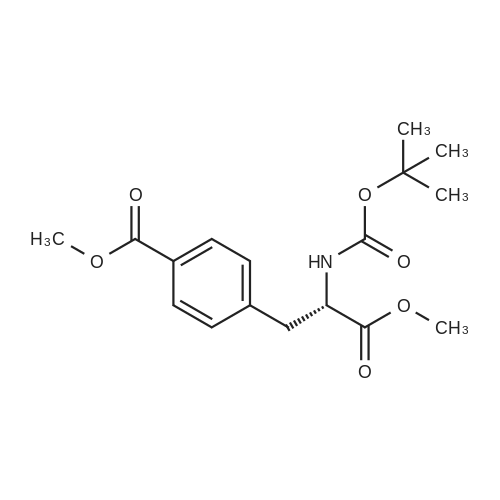
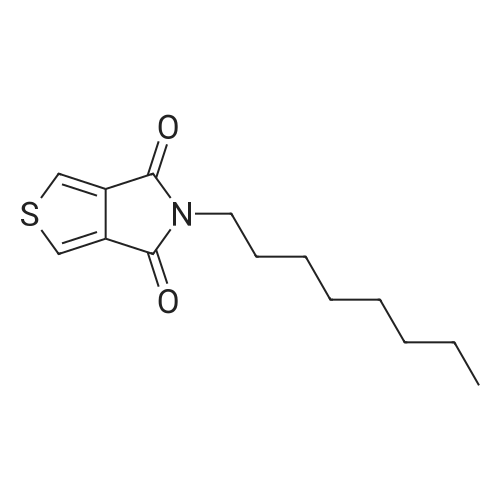
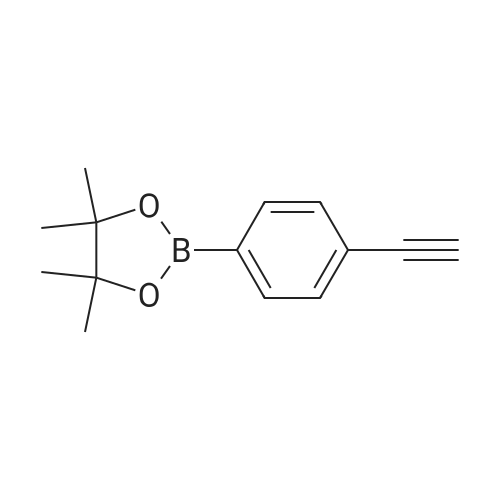
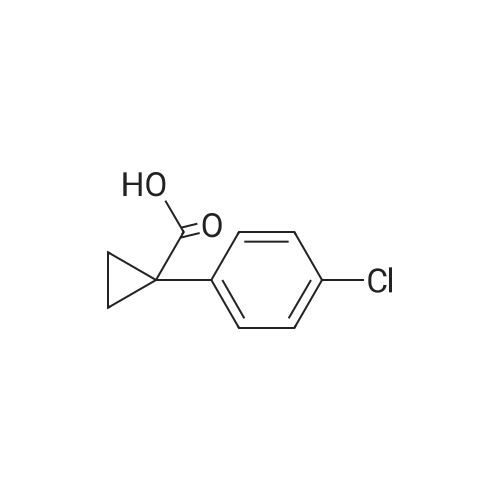
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate In 1,4-dioxane; water at 100℃; Inert atmosphere
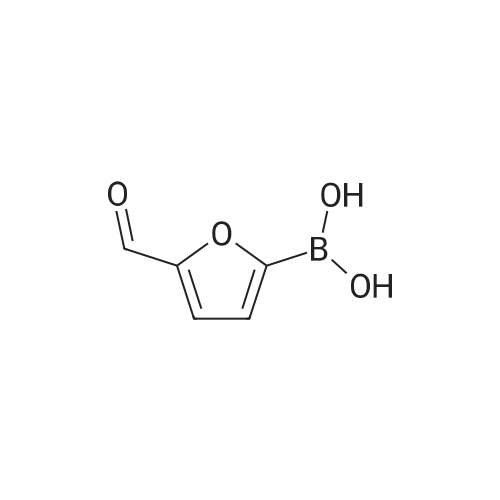
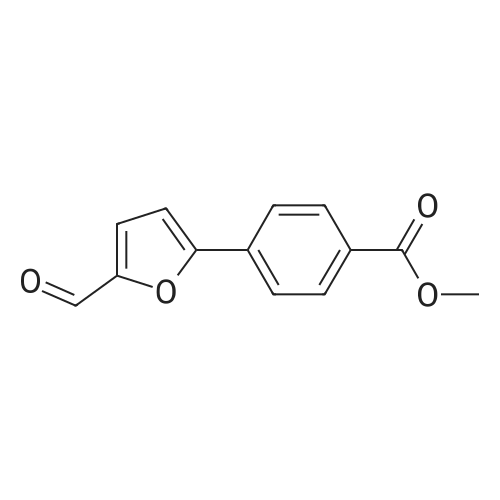
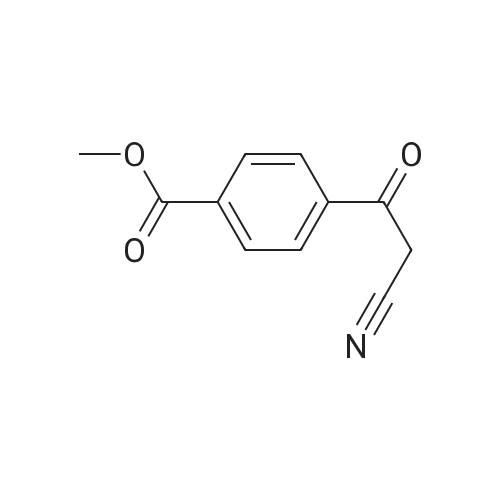
General procedure: A mixture of substituted iodobenzene (2 mmol), (5-formylfuran-2-yl)boronic acid (420 mg, 3mmol, 1.5 equiv), Pd(Ph3P)2Cl2 (0.1 mmol, 0.05 equiv, 70 mg) and potassium carbonate (6 mmol,3 equiv, 828 mg) in dioxone/H2O (6 mL/2 mL) was stirred at 100 °C under argon atmosphereuntil the starting material was consumed (typically 20 h). The reaction mixture was then diluted with 25 mL of saturated brine. The mixture was then extracted with EtOAc (25 mL × 2), and the organic layers were combined, dried over Na2SO4. The concentrated crude product was purifie dby column chromatography to afford c2a-e. The second step is the same as procedure A.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2018, vol. 28, # 6, p. 1024 - 1029
[2] Patent: CN106977474, 2017, A, . Location in patent: Paragraph 0034; 0037; 0038
[3] Chemical Biology and Drug Design, 2018, vol. 91, # 1, p. 257 - 268
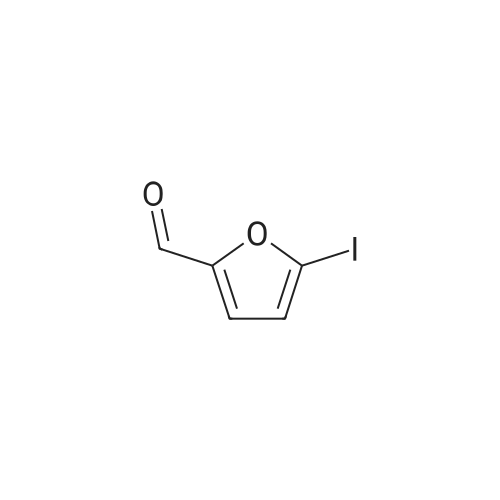
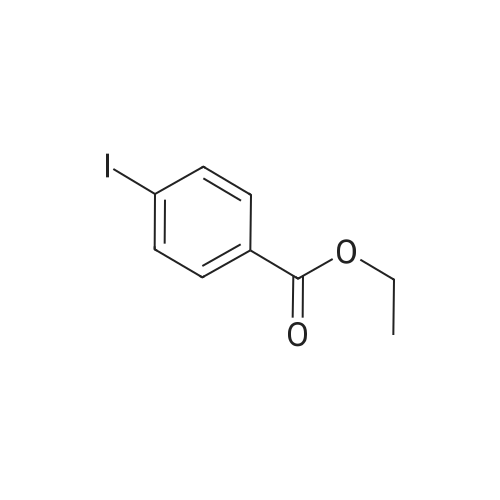
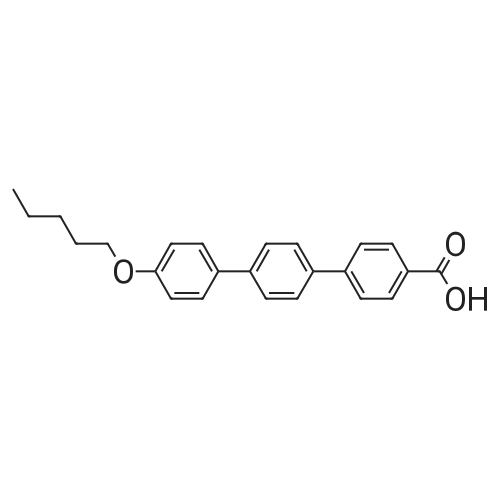
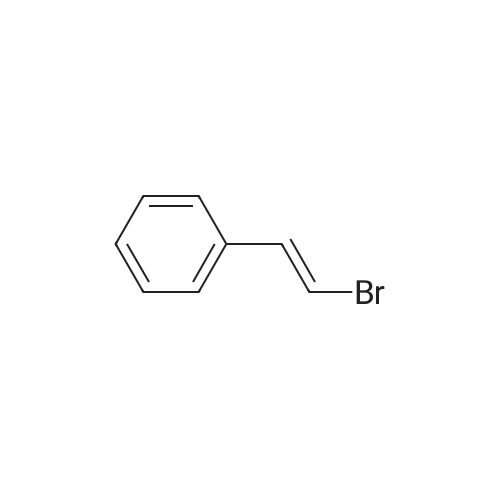
2
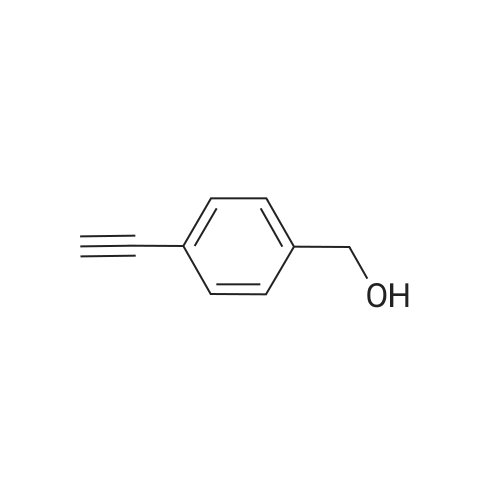
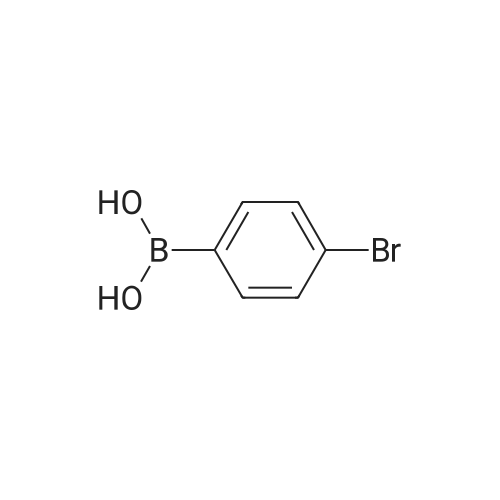
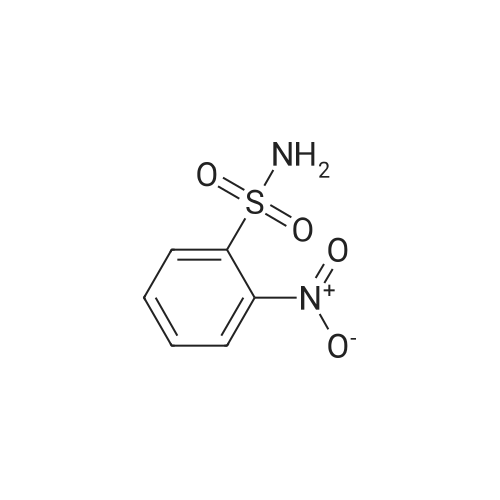
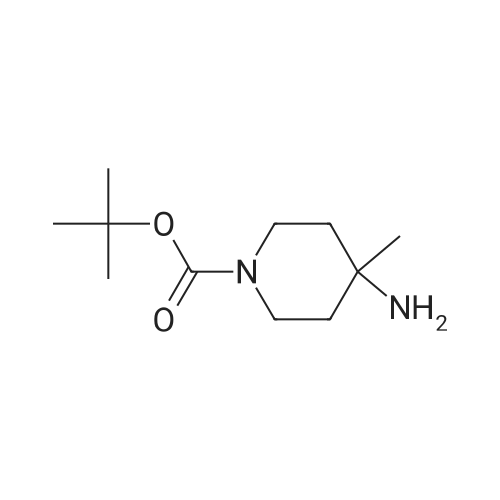
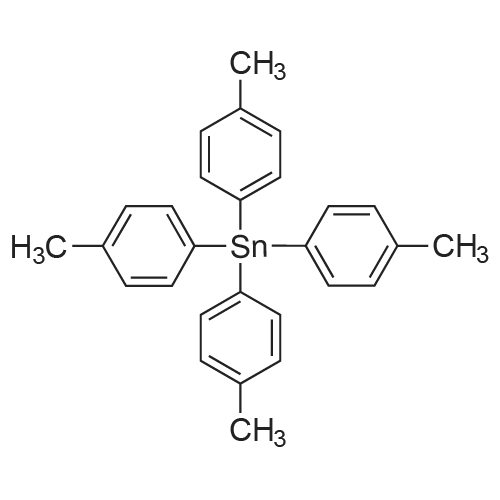
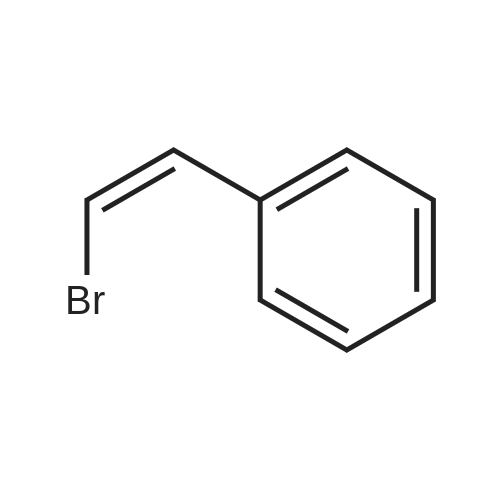
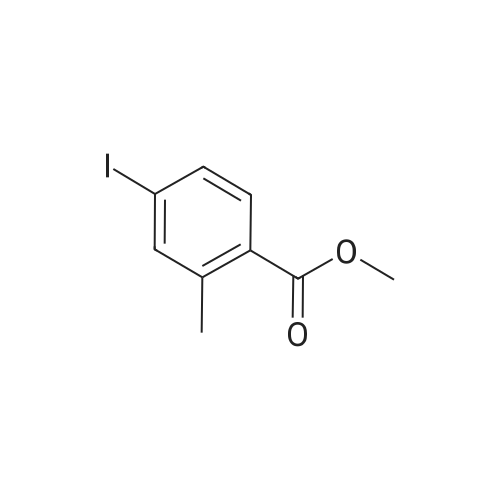
[ 1899-24-7 ]
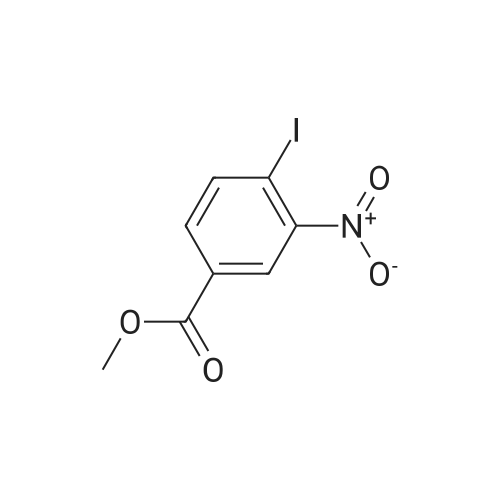
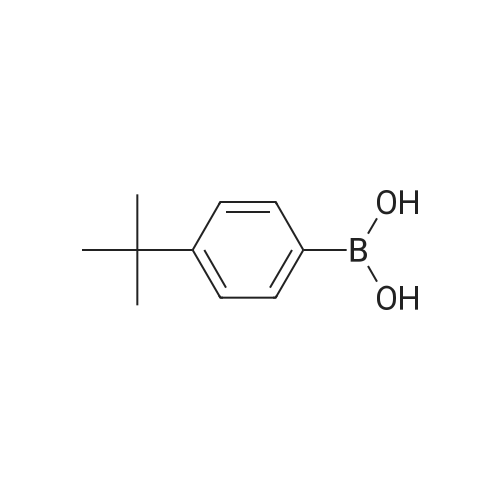
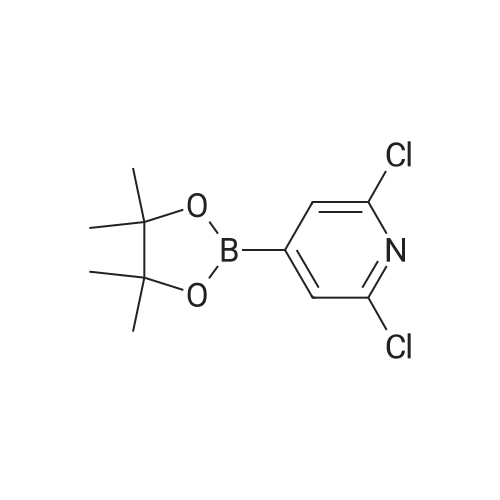
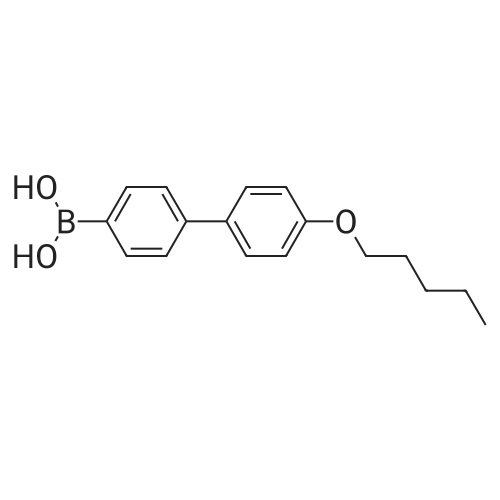
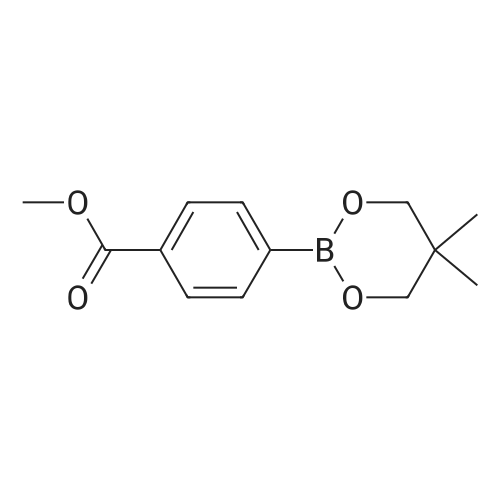
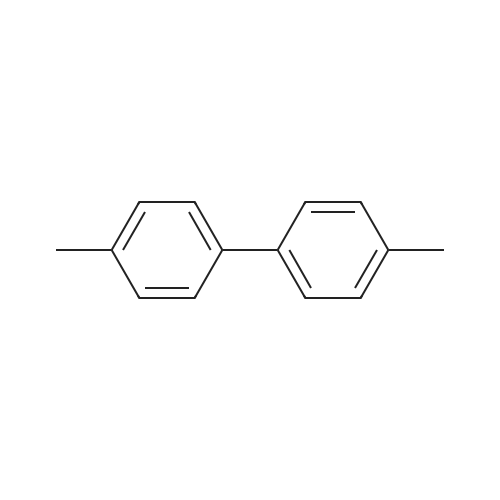
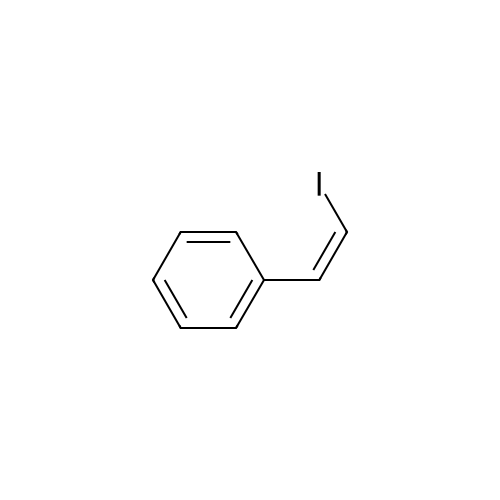
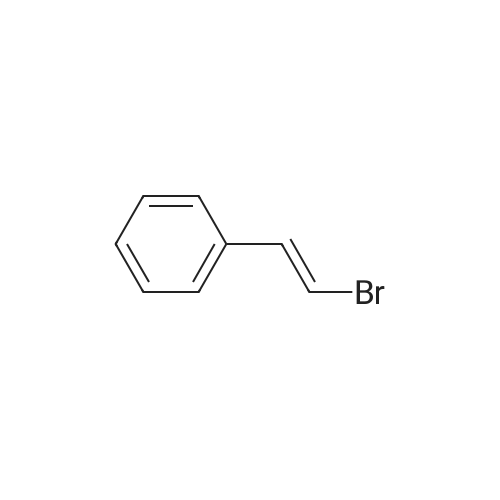
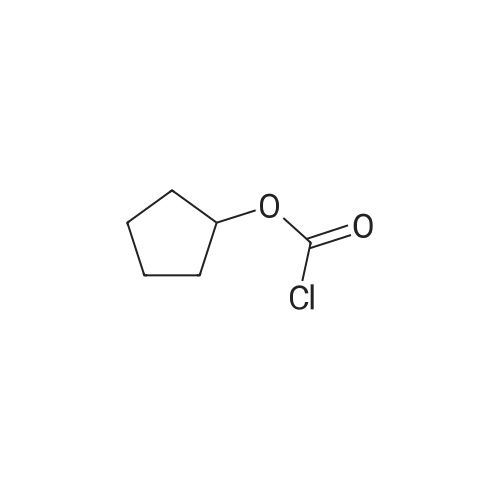
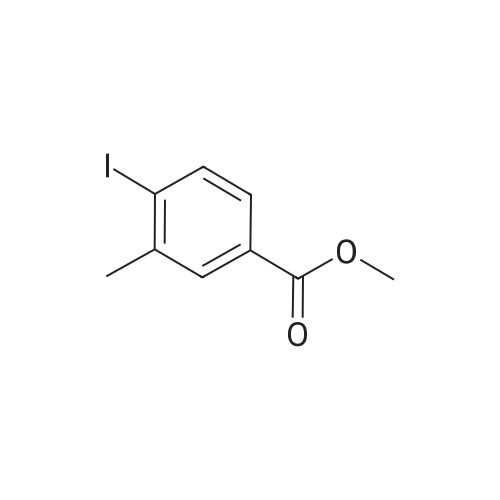
[ 619-44-3 ]
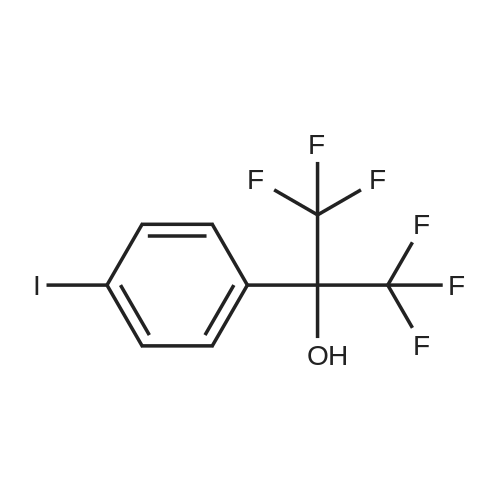
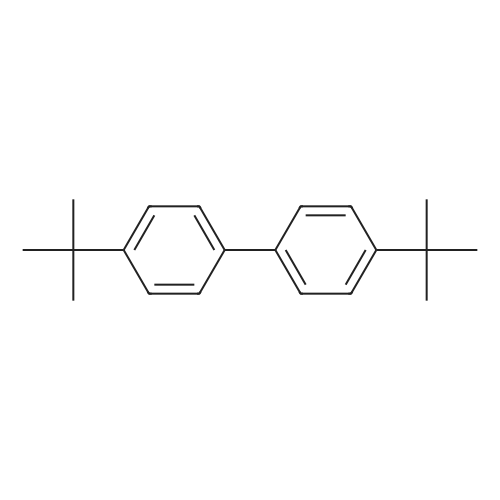
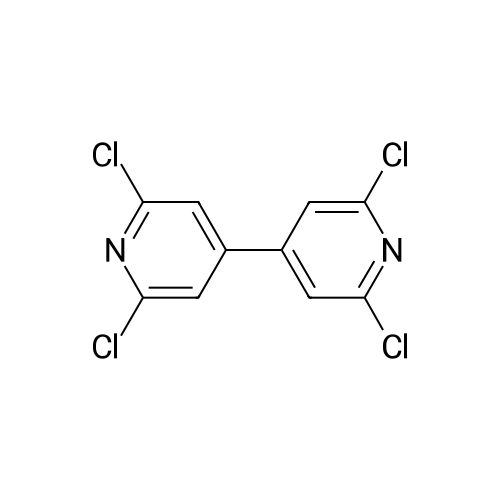
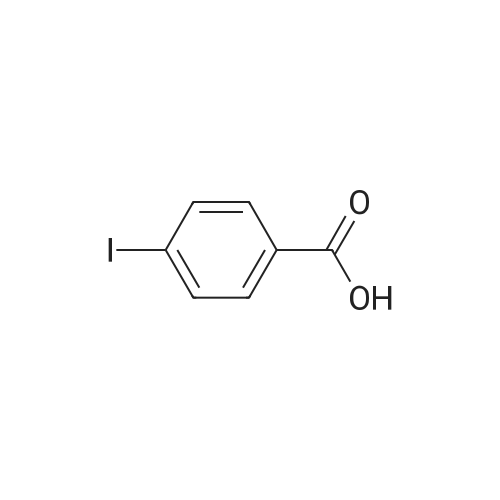
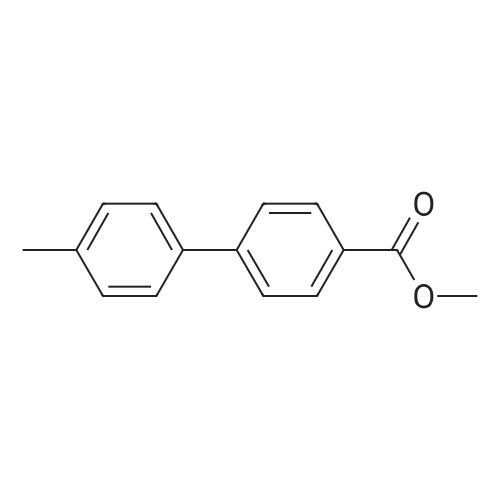
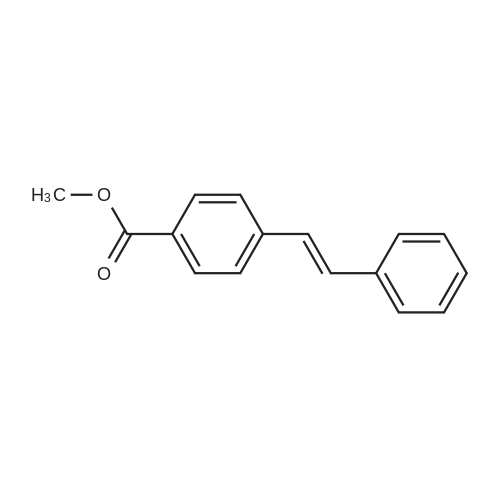
[ 53355-29-6 ]
Yield
Reaction Conditions
Operation in experiment
78%
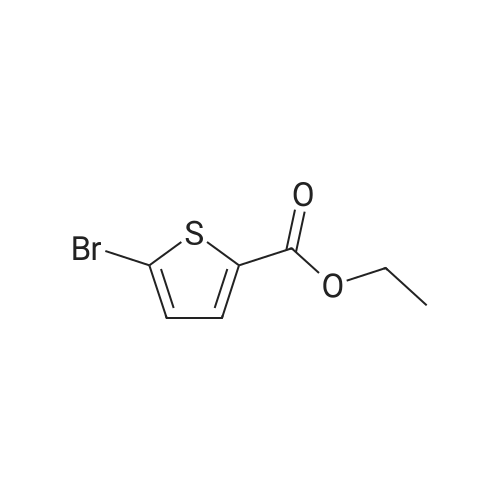
With potassium phosphate In N,N-dimethyl acetamide at 60℃; for 24 h; Irradiation; Green chemistry
General procedure: To a solution of 1 (0.3 mmol) and 2 (0.45mmol) in DMA (5 mL) was added 3wtpercent Pd/CeO2 (80 mg). The reaction mixture was stirred under incandescent light (0.79 Wcm-2) irradiation at 60°C for 24h. After reaction (monitored by TLC), when the reaction is complete, in test tube extraction and centrifugal separation allows for isolation of the desired product, while the catalyst can be washed with ethanol and water, dried under vacuum, and reused for the next run. After completion of the reaction (monitored by TLC), water (5 mL) was added and the mixture was extracted with EtOAc (3 × 5 mL). The combined organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel (10:1 petroleum ether/EtOAc) to give the target product.
Reference:
[1] Chinese Chemical Letters, 2018, vol. 29, # 6, p. 903 - 906
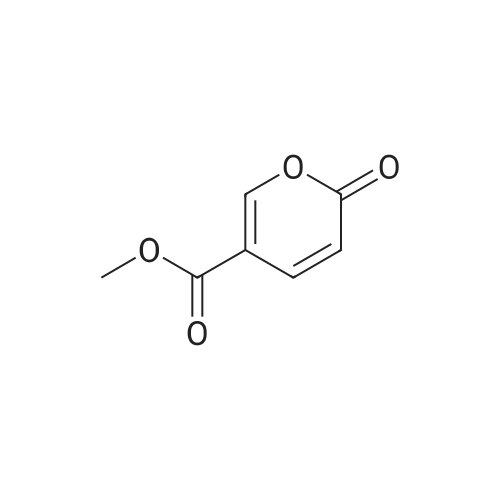
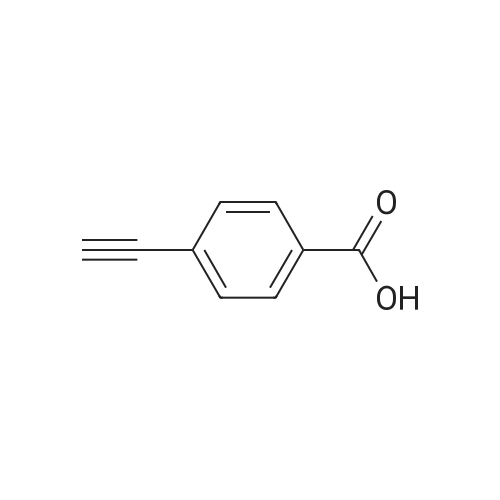
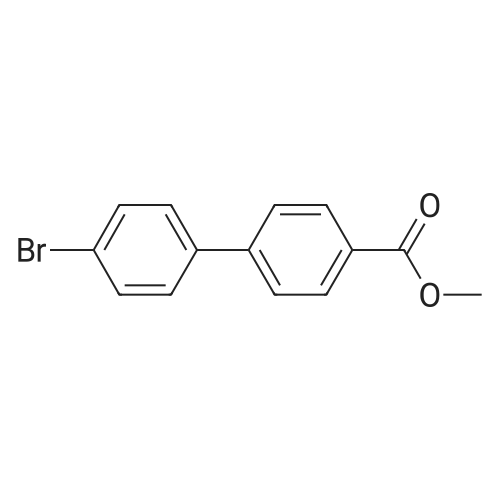
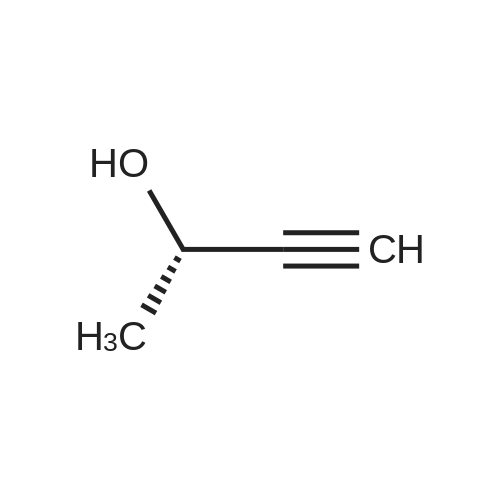
3
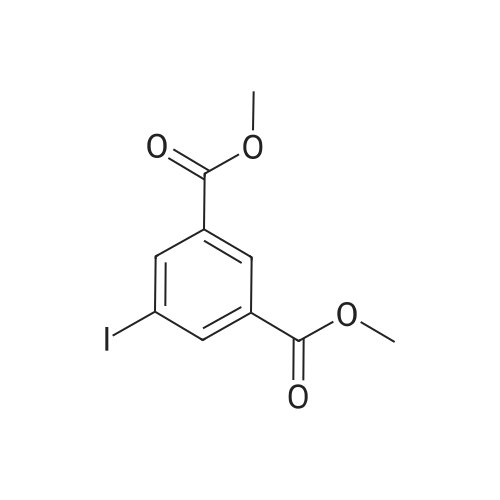
[ 619-44-3 ]
[ 378185-02-5 ]
[ 53355-29-6 ]
Reference:
[1] Synthesis, 2001, # 11, p. 1681 - 1685
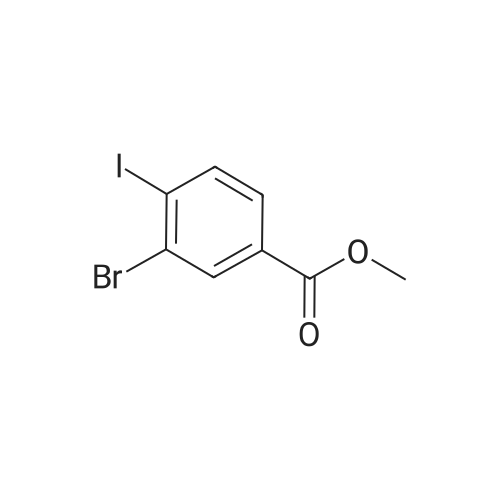
Methyl 4-iodo-3-nitrobenzoate To an ice-cooled solution of methyl 4-iodobenzoate (8.0 g, 30.5 mmol) in conc. H2SO4 (57 mL), a solution of conc. HNO3 (45 mL) and conc. H2SO4 (38.0 mL) was added drop wise. The reaction mixture was stirred at RT for 5 h, then quenched with ice, and extracted with ethyl acetate. The organic layer was washed with saturated NaHCO3 and brine (50 mL), dried over anhydrous Na2SO4 and evaporated under reduced pressure. The residue was recrystallized from DCM/n-hexane to yield the title compound as yellow solid (7.0 g, yield: 74.71percent).
33%
at 0 - 40℃; for 6 h;
A solution of 4-iodobenzoic acid methyl ester (5.24 g, 20.0 mmol) in sulfuric acid was treated with 1.43 mL of concentrated nitric acid in a dropwise fashion at 0 C. After 5 hours at room temperature, the reaction mixture was heated to 40 C for 1 hour. The resulting orange solution was added to 100 g of ice, treated with 200 mL of ethyl acetate, shaken for 30 minutes, and filtered. The phases were separated and the aqueous layer was extracted with 200 mL of ethyl acetate. The combined organic layers were washed with saturated sodium bicarbonate solution and brine, dried over magnesium sulfate, filtered, and concentrated in vacuo. The residue was passed through a column of silica gel (100percent ethyl acetate) to give 2.0 g of 4-iodo-3- nitrobenzoic acid methyl ester as a yellow solid (33percent yield). A solution of 4-iodo-3-nitrobenzoic acid methyl ester (2.0 g, 6.50 mmol) in 25 mL of absolute alcohol and 15 mL of glacial acetic acid was treated with iron powder (3.6 g, 65.0 mmol) and the mixture was heated at 80 C. After 1 hour, the reaction mixture was filtered through a pad of silica, washed with ethanol, and concentrated in vacuo. The residue was diluted with a solution of potassium carbonate and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo to give 1.6 g of 3-amino-4-iodobenzoic acid methyl ester as a white solid (88percent yield). A solution of 3-amino-4-iodobenzoic acid methyl ester (1.59 g, 5.74 mmols) in 40 ML of dichloromethane was reacted with trifluoroacetic anhydride (3 mL, 21 mmol) at room temperature. After 30 minutes, the reaction mixture was concentrated in vacuo and the residue was taken up in cold water, filtered, and dried to give 2.1 g of the title product as an off-white solid (quantitative yield).
Reference:
[1] ACS Medicinal Chemistry Letters, 2012, vol. 3, # 5, p. 427 - 432
[2] Patent: US2016/333004, 2016, A1, . Location in patent: Paragraph 0420
[3] Patent: WO2005/30213, 2005, A1, . Location in patent: Page/Page column 189
[4] Journal of the Chemical Society, 1964, p. 1825 - 1835
14
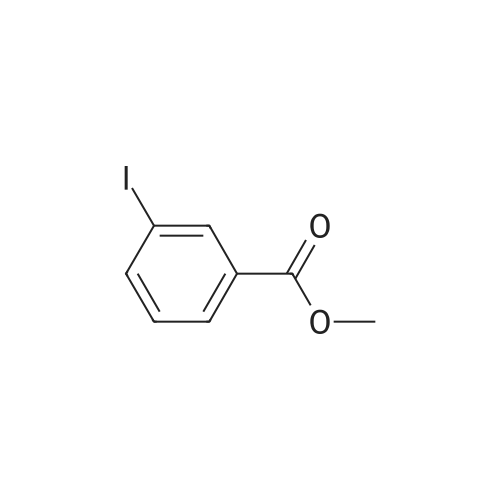
[ 610-97-9 ]
[ 619-44-3 ]
[ 55676-77-2 ]
Reference:
[1] Journal of Organic Chemistry, 2008, vol. 73, # 18, p. 7376 - 7379
[2] Chemistry Letters, 1993, # 3, p. 469 - 472
15
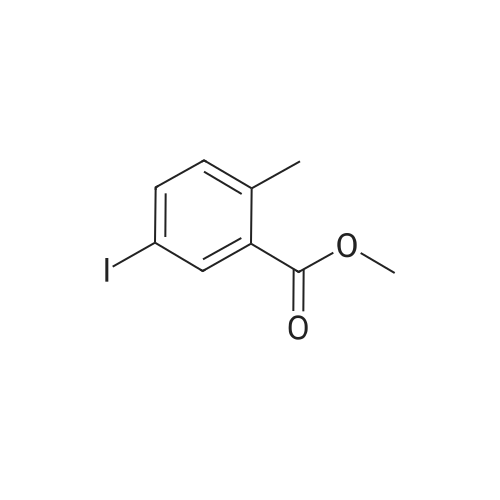
[ 619-44-3 ]
[ 81290-20-2 ]
[ 1214788-34-7 ]
[ 23516-84-9 ]
Yield
Reaction Conditions
Operation in experiment
31%
Stage #1: With cesium fluoride In tetrahydrofuran at 0 - 20℃; Stage #2: With hydrogenchloride; water In tetrahydrofuran for 5 h;
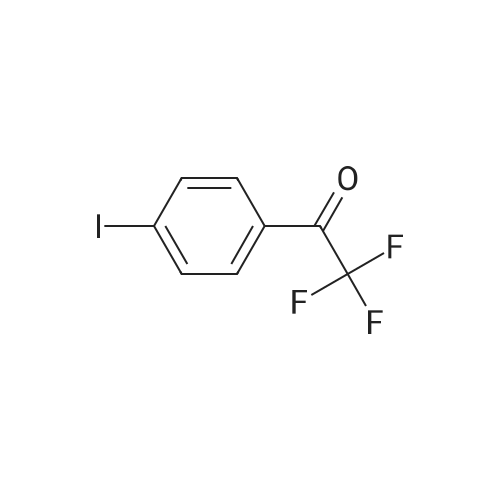
Preparation of Intermediate tert-butyld, 1, 1,3,3,3-hexafluoro-2-(4-iodψDhenyl)DroDan-2- yloxy)dimethylsilane (1 V-1); 4-lodobenzoic acid methyl ester (5g, 19.08mmol) dissolved in tetrahydrofuran (8OmL) and cooled to 00C. (Trifluoromethyl)trimethylsilane (5.43g, 38.2mmol) and cesium fluoride (145mg, 0.954mmol) added. Once addition was complete, reaction was warmed up to room temperature and stirred for 3 hours. Additional (trifluoromethyl)trimethylsilane (2.715g, 19.08mmol) was added and reaction stirred at room temperature for 4 hours. 4 M aqueous solution of hydrochloric acid (2OmL) added and stirred for 5 hours. The reaction mixture was diluted with ethyl acetate (500ml), washed with water (2x250ml), dried over sodium sulfate, filtered and concentrated. Crude purified on silica gel eluting with a gradient from 0percent to 10percent ethyl acetate in heptane to afford 2,2,2-trifluoro-1-(4-iodophenyl)ethanone (1.8g, 31 percent) GCMS= 300 at 1.47min and 1 ,1 ,1 ,3,3,3-hexafluoro-2-(4-iodophenyl)propan-2-ol (1.6g, 22percent); GCMS=370 at 1.60min.
31%
Stage #1: With cesium fluoride In tetrahydrofuran at 0 - 20℃; Stage #2: With hydrogenchloride In tetrahydrofuran; water for 5 h;
4-Iodobenzoic acid methyl ester (5 g, 19.08 mmol) dissolved in tetrahydrofuran (80 mL) and cooled to 0° C. (Trifluoromethyl)trimethylsilane (5.43 g, 38.2 mmol) and cesium fluoride (145 mg, 0.954 mmol) added. Once addition was complete, reaction was warmed up to room temperature and stirred for 3 hours. Additional (trifluoromethyl)trimethylsilane (2.715 g, 19.08 mmol) was added and reaction stirred at room temperature for 4 hours. 4 M aqueous solution of hydrochloric acid (20 mL) added and stirred for 5 hours. The reaction mixture was diluted with ethyl acetate (500 ml), washed with water (2x250 ml), dried over sodium sulfate, filtered and concentrated. Crude purified on silica gel eluting with a gradient from 0percent to 10percent ethyl acetate in heptane to afford 2,2,2-trifluoro-1-(4-iodophenyl)ethanone (1.8 g, 31percent) GCMS=300 at 1.47 min and 1,1,1,3,3,3-hexafluoro-2-(4-iodophenyl)propan-2-ol (1.6 g, 22percent); GCMS=370 at 1.60 min.
With sodium carbonate In ethanol; water; toluene at 80℃; for 4 h; Heating / reflux
A mixture of methyl 4-iodobenzoate, 9.38g (35.8 mmol), 4- bromophenylboronic acid 7.18g (35.8 mmol), Pd (PPh3) 4,2. 07g (1.79 mmol), in 180ML of toluene and 100mL of ethanol was heated to obtain a clear solution. To the solution was added 30mL of 4. OM aq. NA2CO3. The reaction mixture refluxed for 4h at 80 °C. The mixture was cooled to room temperature and diluted with 300mL ethyl acetate. The organic layer was washed with 2X300ML portions of water, 2X300ML portions of sat. aq. NACL, and dried (MGSO4). After the solution was concentrated, the residue was purified by column chromatography (eluted with 7percent ETOAC-HEPTANE) to afford the desired product in 7. 8G (78percent) as a white SOLID. 1H NMR (CDC13) 8.10 (d, 2H, J = 9. 0HZ), 7.62 (d, 2H, J = 9. 0HZ), 7.59 (d, 2H, J = 9. 3HZ), 7.48 (d, 2H, J = 9.3 Hz), 3.95 (s, 3H).
78%
With sodium carbonate In ethanol; water; toluene at 80℃; for 4 h; Heating / reflux
Example 14. 4' -Bromo-biphenyl-4-carboxylic acid methyl ester.; A mixture of methyl 4-iodobenzoate, 9.38g (35.8 mmol), 4- bromophenylboronic acid 7.18g (35.8 mmol), Pd(PPh3)4, 2.07g (1.79 mmol), in 18OmL of toluene and 10OmL of ethanol was heated to obtain a clear solution. To the solution was added 3OmL of 4.0M aq. Na2CC>3. The reaction mixture refluxed for 4h at 80 °C. The mixture was cooled to room temperature and diluted with 30OmL ethyl acetate. The organic layer was washed with 2x300mL portions of water, 2x300mL portions of sat. aq. NaCl, and dried (MgSC>4) . After the solution was concentrated, EPO <DP n="96"/>the residue was purified by column chromatography (eluted with 7percent EtOAc-Heptane) to afford the desired product in 7.8g (78percent) as a white solid. 1H NMR (CDCl3) 8.10 (d, 2H, J = 9.0Hz)Λ 7.62 (d, 2H, J = 9.0Hz), 7.59 (d, 2H, J = 9.3Hz), 7.48 (d, 2H, J = 9.3 Hz) , 3.95 (s, 3H) .
78%
With sodium carbonate In ethanol; water; toluene at 80℃; for 4 h;
Example 96; 4' -Bromo-biphenyl-4-carboxylic acid methyl ester; A mixture of methyl 4-iodobenzoate, 9.38g (35.8 mmol), 4- bromophenylboronic acid 7.18g (35.8 mmol), Pd(PPh3) 4, 2.07g (1.79 mmol), in 18OmL of toluene and 10OmL of ethanol was heated to obtain a clear solution. To the solution was added 3OmL of 4.0M aq. Na2CO3. The reaction mixture refluxed for 4h at 80 °C. The mixture was cooled to room temperature and diluted with 30OmL ethyl acetate. The organic layer was washed with 2x300mL portions of water, 2x300mL portions of sat. aq. NaCl, and dried (MgSO4) . After the solution was concentrated, the residue was purified by column chromatography (eluted with 7percent EtOAc-Heptane) to afford the desired product in 7.8g (78percent) as a white solid.
78%
With sodium carbonate In ethanol; water; toluene at 80℃; for 4 h;
A mixture of methyl 4-iodobenzoate, 9.38g (35.8 mmol), 4- BROMOPHENYLBORONIC acid 7.18g (35. 8 mmol), Pd (PPh3) 4, 2. 07G (1.79 mmol), in 180ML of toluene and LOOML of ethanol was heated to obtain a clear solution. To the solution was added 30mL of 4. OM aq. NA2CO3. The reaction mixture refluxed for 4h at 80 °C. The mixture was cooled to room temperature and diluted with 300mL ethyl acetate. The organic layer was washed with 2X300ML portions of water, 2X300ML portions of sat. aq. NaCl, and dried (MGS04). After the solution was concentrated, the residue was purified by column chromatography (eluted with 7percent ETOAC-HEPTANE) to afford the desired product in 7.8g (78percent) as a white SOLID. 1H NMR (CDC13) 8.10 (d, 2H, J = 9. 0HZ), 7. 62 (d, 2H, J = 9. 0HZ), 7. 59 (d, 2H, J = 9.3Hz), 7.48 (d, 2H, J = 9.3 Hz), 3.95 (s, 3H).
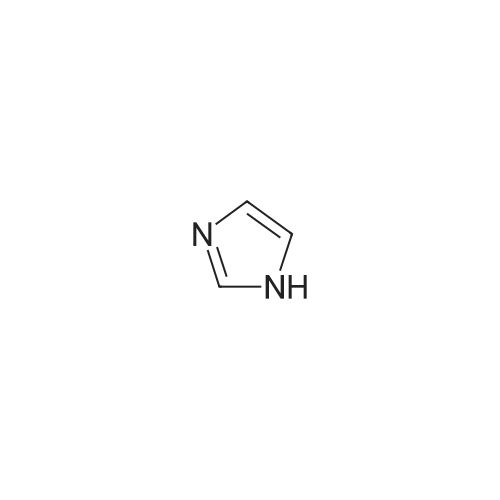
With (R,R)-N,N'-dimethyl-1,2-diaminocyclohexane; potassium carbonate; copper(I) bromide In N,N-dimethyl-formamide at 100℃; for 48 h; Inert atmosphere; Sealed tube
For the synthesis of RK464 (Scheme 2), a round bottom flask with a magnetic stir bar was usedas a reaction vessel. To this vessel, CuBr (7.2 mg, 0.025 mmol), (1R,2R)-N1,N2-dimethyl-cyclohexane-1,2-diamine (16 L, 0.05 mmol), and imidazole (82 mg, 0.6 mmol), were added with DMF (20 mL) as asolvent. The vessel was sealed with a septum and nitrogen gas was passed through it.After reacting the initial mixture of compounds, methyl 4-iodobenzoate (262 mg, 0.5 mmol). K2CO3 (357 mg, 2.6 mmol) were added. Again, nitrogen gas was back-filled and the reaction mixturewas heated in a preheated oil bath for 48 h at constant temperature of 100 °C. Reaction progress wasregularly monitored using TLC. As the reaction progressed, the color of the mixture changed from whiteto yellow. The product was extracted using ethyl acetate and purified using flash chromatography.A pure yellow viscous product (about 40 mg) was obtained and dried under vacuum. Yield: 19percent.1H-NMR (750 MHz, CD3OD) δ: 6.75 (s, 1H, Ar-H), 6.63–6.64 (d, 2H, Ar-H), 6.19-6.20 (d, 2H, Ar-H),6.16 (s, 1H, Ar-H), 2.40 (s, 3H, CH3) (Supplementary Figure S9); HRMS (ESI) calcd C11H10N2O2 for203.0821, found 203.0808 [M+H]+, (Supplementary Figure S14). FT IR νmax cm−1: 1710.61 (carboxyl, for 203.0821, found 203.0808 [M+H]+, (Supplementary Figure S14). FT IR max cm1: 1710.61 (carboxyl,C=O), (Supplementary Figure S20). X-ray crystallography: X-ray crystallography structure for thiscompound is shown in Supplementary Figure S19 with R = 0.2.
Reference:
[1] Journal of Organic Chemistry, 2013, vol. 78, # 15, p. 7436 - 7444
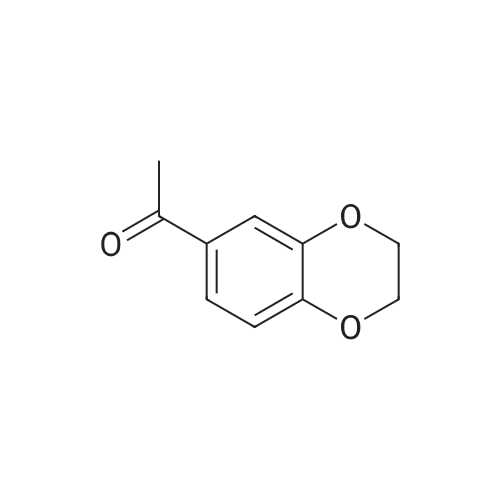
21
[ 619-44-3 ]
[ 158938-08-0 ]
Reference:
[1] Journal of Medicinal Chemistry, 1995, vol. 38, # 17, p. 3271 - 3281
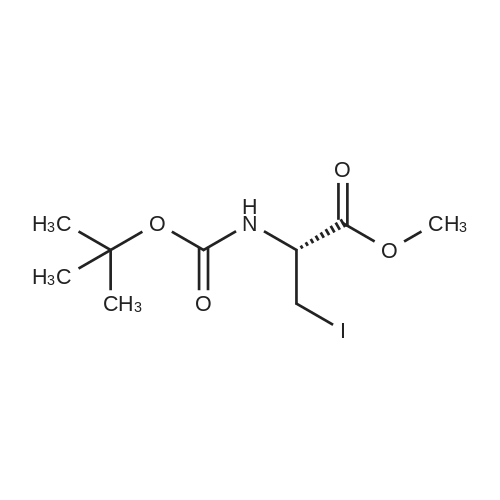
22
[ 619-44-3 ]
[ 113100-86-0 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2006, vol. 16, # 7, p. 1799 - 1802
[2] Journal of Medicinal Chemistry, 1997, vol. 40, # 16, p. 2502 - 2524
23
[ 5419-55-6 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Yield
Reaction Conditions
Operation in experiment
94.8%
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
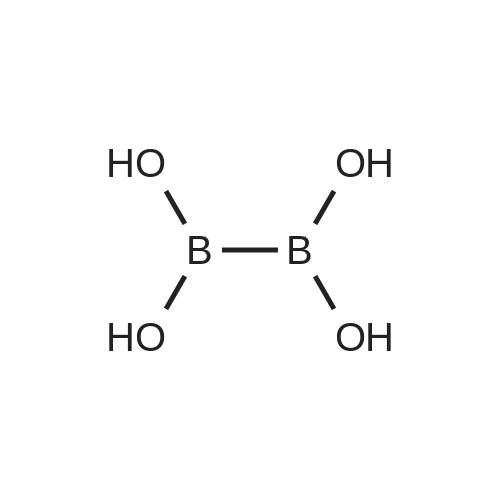
In 500mL three-necked flask,Methyl 4-iodobenzoate (20.1 g, 76.7 mmol) was dissolved in dry THF (200.0 mL)Triisopropyl borate (18.0 g, 95.9 mmol) was added,Cool to -78 ° C,N-Butyllithium (6.1 g, 95.9 mmol) was added dropwise,Maintain the reaction temperature 0.5h.The reaction was completed, quenched with saturated aqueous ammonium chloride solution,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL x 3)The combined organic phase,Saturated brine (60 mL × 1)Dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure,The residue was beaten with n-hexane, and filtered to obtain 13.1 g of 4-methoxycarbonylphenylboronic acid,Yield 94.8percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0023; 0024; 0079; 0080
24
[ 688-74-4 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Yield
Reaction Conditions
Operation in experiment
91%
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
In 500mL three-necked flask,Methyl 4-iodobenzoate (20.4 g, 77.8 mmol) was dissolved in dry THF (200.0 mL)Tri-n-butyl borate (21.5 g, 93.4 mmol) was added,Cool to -78 ° C,N-Butyllithium (5.5 g, 85.6 mmol) was added dropwise,Maintain the reaction temperature 0.5h.The reaction was completed, quenched with saturated aqueous ammonium chloride solution,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL x 3)The combined organic phases were washed with saturated brine (60 mL × 1)Dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure,The residue was beaten with n-hexane,Filtered to give 4-methoxycarbonyl phenylboronic acid 12.7g, the yield was 91.0percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0039; 0040
25
[ 121-43-7 ]
[ 619-44-3 ]
[ 99768-12-4 ]
Yield
Reaction Conditions
Operation in experiment
94.5%
With n-butyllithium In tetrahydrofuran at -78℃; for 0.5 h;
In 500mL three-necked flask,Methyl 4-iodobenzoate (20.3 g, 77.4 mmol) was dissolved in dry THF (200.0 mL)Trimethyl borate (16.1 g, 154.8 mmol) was added,The temperature was lowered to -78 ° C and n-butyllithium (7.4 g, 116.1 mmol) was added dropwise.Maintain the reaction temperature 0.5h.The reaction was completed, quenched with saturated aqueous ammonium chloride solution,1mol / L hydrochloric acid to adjust the pH to 1,Ethyl acetate (100.0 mL × 3), the organic phases were combined,Saturated brine (60 mL × 1), dried over anhydrous sodium sulfate,The solvent was distilled off under reduced pressure, the residue was beaten with n-hexane,The filtrate was filtered to give 4-methoxycarbonyl phenylboronic acid 13.2g, a yield of 94.5percent.
Reference:
[1] Patent: CN106565761, 2017, A, . Location in patent: Paragraph 0047; 0048
With palladium diacetate; potassium carbonate; nicotin; In N,N-dimethyl-formamide; at 100℃; for 24h;
Add 0.011 g of palladium acetate (0.05 mmol) to a 50 mL round bottom flask.10mL organic solvent N, N-dimethylformamide,0.008g spicy ingredients such as nicotine,0.138 g of potassium carbonate (1 mmol),0.131 g of methyl 4-iodobenzoate (0.5 mmol),Heat 100 oC for 24 hours,Until the reaction is complete (thin layer chromatography TLC detection).The reaction mixture was poured into 20 mL of distilled water.It was extracted with ethyl acetate (10 mL × 3).Combine the organic phase,It was dried over anhydrous magnesium sulfate.After evaporating the solvent,The residue was separated by silica gel column chromatography (eluent ethyl acetate: petroleum ether = 1:3)Obtained a white solid of 0.0513 g, a yield of 76percent.The nuclear magnetic resonance spectrum is shown in Figure 3.
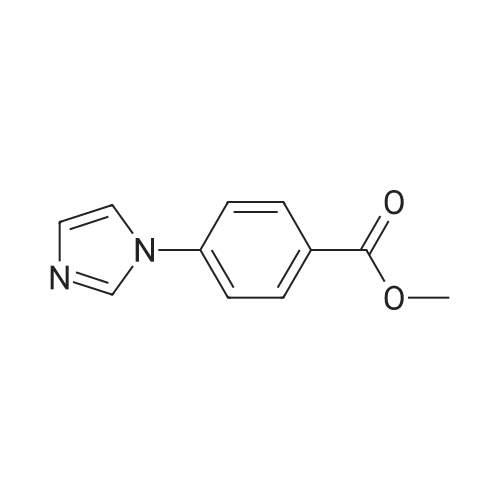
With caesium carbonate;copper(I) triflate; In DMF (N,N-dimethyl-formamide); at 100℃;
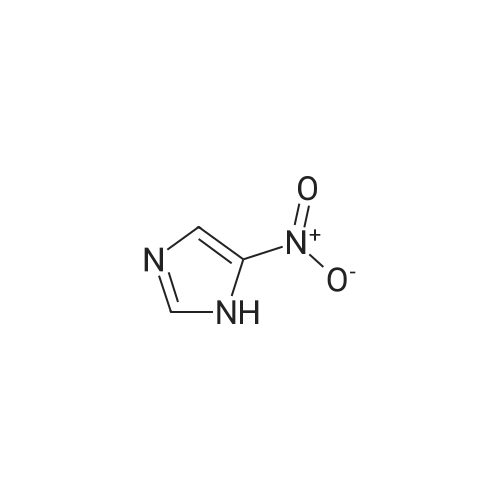
4-nitroimidazole (1 equiv) was combined with 4-iodo-methylbenzoate (1.2 equiv), cesium carbonate (1.0 equiv), copper (I) triflate (0.05 equiv), in DMF and heated overnight at 100C. The solid was filtered and washed with methylene chloride, the resultant solvent was concentrated, and residue purified by silica gel chromatography to provide the title compound: MS m/z 248.2 (M+1).
(1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; In 1,4-dioxane; at 85℃; for 16h;
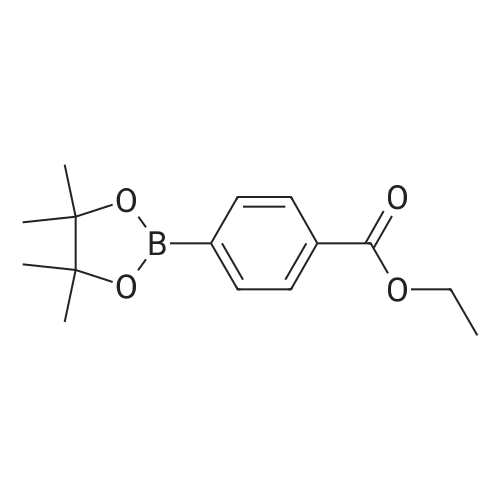
EXAMPLE 13 Preparation of Ethyl 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)Benzoate; [0105] EMI310.0[0106] A solution of methyl 4-iodobenzoate (2.00 g, 7.63 mmol) in 30 mL dioxane was degassed with Ar for 10 minutes. Then, Pd(dppf)Cl2 (171 mg, 3 mol %), triethylamine (3.27 mL), and pinacolborane (1.47 g, 11.45 mmol) were added. The resulting solution was stirred at 85[deg.] C. for 16 hours. The mixture was allowed to cool to ambient temperature, filtered through a pad of Celite(R), and concentrated in vacuo to obtain 3.97 g of product which was used without further purification. m/z=263 [M+H]<+>.
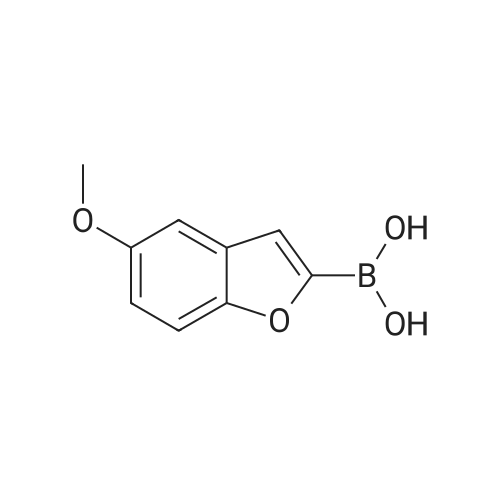
4-(5-Methoxy-benzofuran-2-yl)-benzoic acid methyl ester 127 A solution of <strong>[551001-79-7]5-Methoxy-benzofuran-2-boronic acid</strong> (1.9 g, 10 mmol), Methyl 4-iodobenzoate (2.61 g, 10 mmol), Pd(Ph3)4 (0.25 g), and 2M NaCO3 (10 ml) in toluene (40 ml) and EtOH (10 ml) was heated to reflux. After 2 hr, the reaction was cooled and the organic layer was seperated dried, and concentrated to give a solid. The solid was triturated with MeOH, filtered to give 127 (0.90 g, 32%) which was used without further purification or characterization for the next step.
With caesium carbonate;copper(l) iodide; 2-acetylcyclohexanone; In N,N-dimethyl-formamide; at 90℃; for 20h;
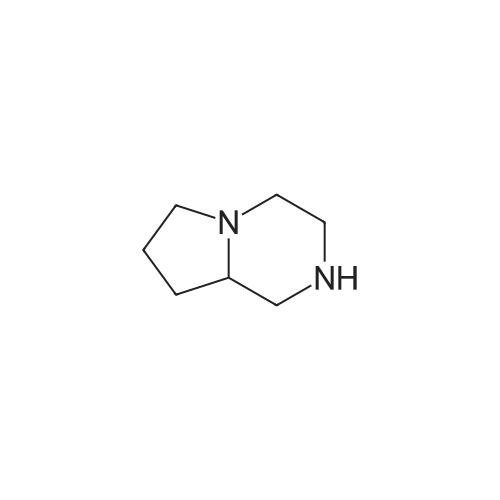
Methyl 4-iodobenzoate (2.076 g, 7.92 mmol), cesium carbonate (5.16 g, 15.85 mmol), 2-acetylcyclohexanone (0.209 mL, 1.58 mmol) and copper(I) iodide (0.075 g, 0.40 mmol) were added to a stirred solution 1,2,3,4,6,7,8,8a-<strong>[5654-83-1]octahydropyrrolo[1,2-a]pyrazine</strong> (1 g, 7.92 mmol) in DMF (20 mL) under nitrogen. The resulting suspension was stirred at 90 C. for 20 h. The reaction mixture was evaporated to dryness and redissolved in a mixture of methanol and water. The crude product was purified by ion exchange chromatography, using a SCX column. The desired product was eluted from the column using 3.5M NH3/MeOH and pure fractions were evaporated to dryness to afford the desired compound (1.243 g, 60.2%) as a brown gum. 1H NMR (399.9 MHz, DMSO-d6) delta 1.33-1.43 (1H, m), 1.57-1.79 (3H, m), 1.81-1.89 (1H, m), 1.99-2.03 (1H, m), 2.07 (1H, q), 2.16-2.23 (1H, m), 2.56 (1H, t), 2.83-2.95 (2H, m), 3.00-3.05 (2H, m), 3.06-3.09 (1H, m), 3.75-3.78 (3H, m), 3.86 (1H, t), 3.98-4.01 (1H, m), 6.98-7.02 (2H, m), 7.77-7.80 (2H, m). MS=m/z 261 (MH+).
With caesium carbonate;copper(l) iodide; 2-acetylcyclohexanone; In N,N-dimethyl-formamide; at 90℃; for 20h;Product distribution / selectivity;
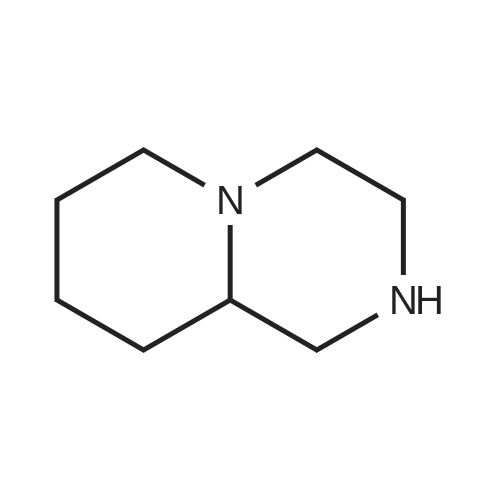
Methyl 4-iodobenzoate (2.84 g, 10.86 mmol), cesium carbonate (7.07 g, 21.71 mmol), 2-acetylcyclohexanone (0.286 mL, 2.17 mmol) and copper(I) iodide (0.103 g, 0.54 mmol) were added to a stirred solution of 2,3,4,6,7,8,9,9a-<strong>[4430-75-5]octahydro-1H-pyrido[1,2-a]pyrazine</strong> (1.5224 g, 10.86 mmol) in DMF (30 mL) under nitrogen. The resulting suspension was stirred at 90 C. for 20 h. The reaction mixture was evaporated to dryness and dissolved in methanol. The crude product was purified by ion exchange chromatography, using a SCX column. The desired product was eluted from the column using 3.5M NH3/MeOH and pure fractions were evaporated to dryness to afford a brown gum. This was purified again by silica column chromatography, eluting with 5% MeOH (containing 0.1% ammonium hydroxide) in DCM. Pure fractions were evaporated to dryness to afford the desired compound (0.503 g, 16.8%) as an orange solid 1H NMR (399.9 MHz, CDCl3) delta 1.29-1.33 (1H, m), 1.26-1.40 (1H, m), 1.62 (1H, d), 1.64 (1H, d), 1.65-1.68 (1H, m), 1.69 (1H, s), 1.80-1.82 (1H, m), 2.04-2.10 (2H, m), 2.32-2.39 (1H, m), 2.61 (1H, d), 2.89 (2H, d), 3.01-3.08 (1H, m), 3.58-3.63 (1H, m), 3.70-3.75 (1H, m), 3.86 (3H, s), 6.83-6.87 (2H, m), 7.89-7.93 (2H, m). MS m/z=275 (MH+).; Methyl -4-iodobenzoate (2.84 g, 10.86 mmol), cesium carbonate (7.07 g, 21.71 mmol), 2-acetylcyclohexanone (0.286 mL, 2.17 mmol) and copper(I) iodide (0.103 g, 0.54 mmol) were added to a stirred solution of 2,3,4,6,7,8,9,9a-<strong>[4430-75-5]octahydro-1H-pyrido[1,2-a]pyrazine</strong> (1.5224 g, 10.86 mmol) in DMF (30 mL) under nitrogen. The resulting suspension was stirred at 90 C. for 20 h. The reaction mixture was evaporated to dryness. The crude product was purified by ion exchange chromatography, using a SCX column. The desired product was eluted from the column using 3.5M NH3/MeOH and fractions were evaporated to dryness to afford the crude product as an impure brown gum. The crude product was purified by silica column chromatography, eluting with 5% MeOH, 0.1% aqueous ammonia in DCM. Pure fractions were evaporated to dryness to afford the desired compound (0.681 g, 22.87%) as an orange solid. 1H NMR (399.9 MHz, CDCl3) delta 1.29-1.33 (1H, m), 1.26-1.40 (1H, m), 1.62 (1H, d), 1.64 (1H, d), 1.65-1.68 (1H, m), 1.69 (1H, s), 1.80-1.82 (1H, m), 2.04-2.10 (2H, m), 2.32-2.39 (1H, m), 2.61 (1H, d), 2.89 (2H, d), 3.01-3.08 (1H, m), 3.58-3.63 (1H, m), 3.70-3.75 (1H, m), 3.86 (3H, s), 6.83-6.87 (2H, m), 7.89-7.93 (2H, m). MS: m/z 275 (MH+).
With caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene;palladium diacetate; In 1,4-dioxane;Sonographic reaction; Inert atmosphere;
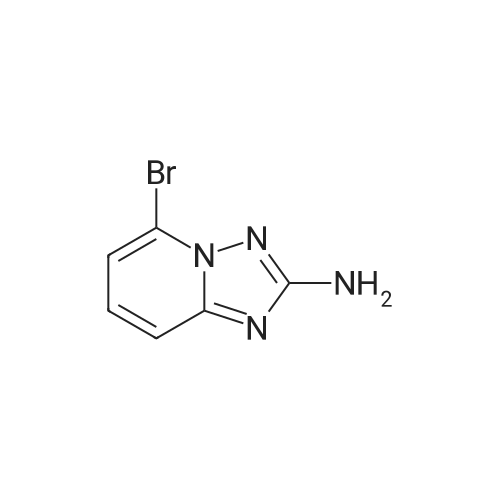
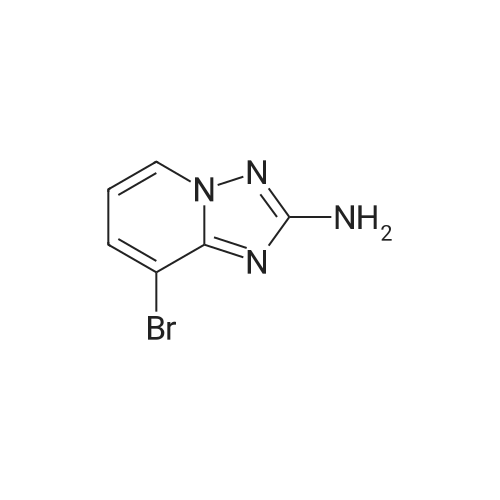
A mixture of 5-bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (1 eq), Cs2CO3 (5eq), Pd(OAc)2 (0.1 eq), Xantphos (0.1 eq.), 4-Iodo-benzoic acid methyl ester (1.5 eq) and 1,4-dioxane was sonicated for 5 minutes under nitrogen. Afterwards, the reaction was left in a sealed tube at 1000C or in a flasked equipped with a cooling system for 5 hrs. The crude mixture was extracted with ethyl acetate and the extracts were combined, washed with water and dried over anhydrous magnesium sulfate. The organic solvent was removed under high vacuum to yield the crude product. The crude product was purified by flash chromatography.
With caesium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene;palladium diacetate; In 1,4-dioxane;Sonographic reaction;
A mixture of 5-Bromo-[l,2,4]triazolo[l,5-a]pyridin-2-ylamine (1 eq), Cs2CO3 (5eq), Pd(OAc)2 (0.1 eq), Xantphos (0.1 eq.), 4-Iodo-benzoic acid methyl ester (1.5 eq) and 1,4-dioxane was sonicated for 5 minutes under nitrogen. Afterwards, the reaction was left in a sealed tube at 1000C or in a flasked equipped with a cooling system for 5 hrs. The crude mixture was extracted with ethyl acetate and the extracts were combined, washed with water and dried over anhydrous magnesium sulfate. The organic solvent was removed under high vacuum to yield the crude product. The crude product was purified by flash chromatography.
With caesium carbonate;palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 80℃; for 1h;
A suspension of 8-bromo-[l,2,4]triazolo[l,5-alpha]pyridin-2-amine (2.8 g,13.2 mmol, 1 equiv), methyl 4-iodobenzoate (3.4 g, 13 mmol, 1.0 equiv), cesium carbonate (8.4 g, 26 mmol, 2.0 equiv), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (763 mg, 1.32 mmol, 0.10 equiv), and palladium (II) acetate (300 mg, 1.32 mmol, 0.10 equiv) in dioxane (100 mL) was heated at <n="112"/>80 0C for 1 h. The reaction mixture was cooled to room temperature and diluted with dichloromethane (100 mL). The resulting solids were filtered and sequentially rinsed with water (3 x 50 mL) and methanol (2 X 20 mL). The solids were dried in vacuo to afford methyl 4-(8-bromo-[l,2,4]triazolo[l,5- a]pyridin-2-ylamino)benzoate (3.1 g). 1H NMR (400 MHz, DMSO-J6), delta:10.42 (s, 1 H), 8.87 (m, 1 H), 7.93 (m, 1 H), 7.92 (d, J= 8.8 Hz, 2 H), 7.77 (d, J= 8.8 Hz, 2 H), 7.00 (dd, J= 7.4, 6.9 Hz, 1 H), 3.81 (s, 3 H).
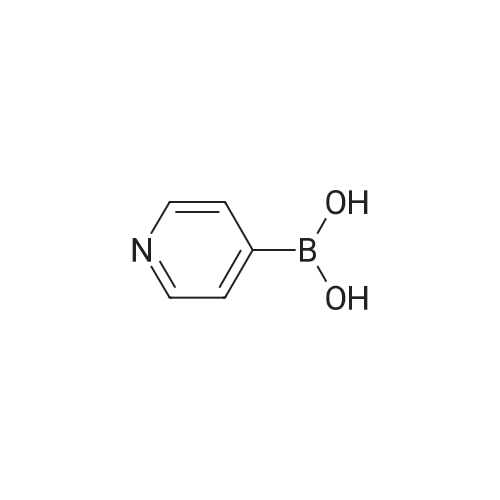
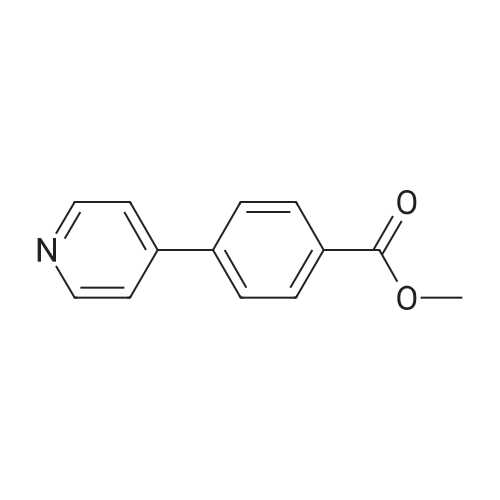
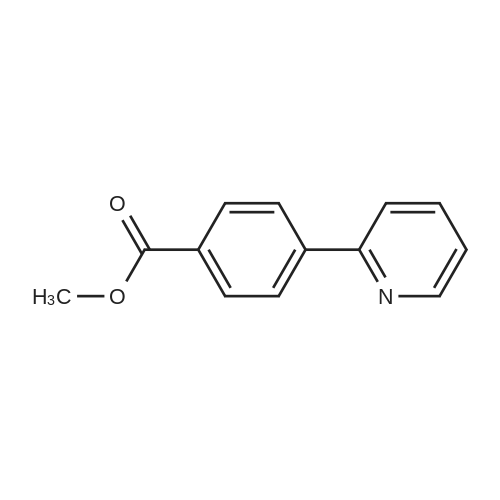
With potassium carbonate;Pd(PPh3)4; In ethanol; water; toluene;
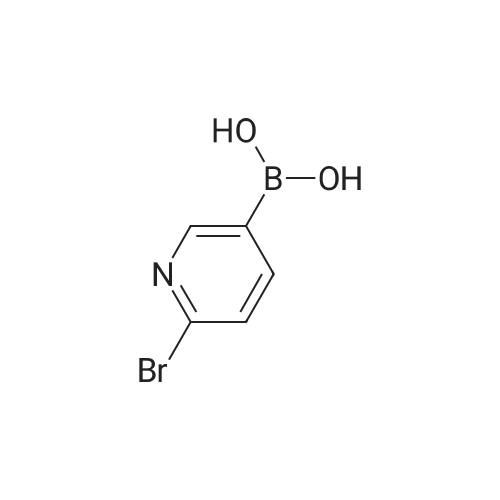
4-(6-(4-(hexyloxy)phenyl)pyridin-3-yl)benzoic acid A mixture of 2-Bromopyridyl-5-boronic acid (300 mg, 1.5 mmol), methyl-4-iodobenzoate (400 mg, 1.5 mmol) and K2CO3 (310 mg, 2.25 mmol) in toluene (10 mL) containing EtOH (1.5 mL) and water (0.4 mL) was degassed for 5 minutes using Argon. Pd(PPh3)4 (40 mg, 0.03 mmol) was added and the mixture refluxed overnight under Argon. After cooling to room temperature, the solvent was removed and the residue washed extracted with EtOAc. The organic layer was washed with water, dried and evaporated. The crude product was triturated with Et2O to afford pure product (300 mg, 70%). Mass: m/z 292 (M+1). 1H NMR (500 MHz, CDCl3): delta 3.98 (s, 3H), 7.61 (d, 1H, J=8.0 Hz), 7.64 (d, 2H, J=8.0 Hz), 7.79 (dd, 1H, J=2.5, 8.0 Hz), 8.19 (d, 2H, J=8.0 Hz), 8.65 (d, 1H, J=2.2 Hz).
With palladium diacetate; sodium carbonate; triphenylphosphine; In propan-1-ol; toluene; for 4h;Reflux; Inert atmosphere;
General procedure: A solution of compound 15c (3.23 g, 10.36 mmol) and methyl 4-iodobenzoate (2.78 g, 10.56 mmol) in toluene-propanol (8:1, 25 mL) was added 2 M Na2CO3 (6 mL), Pd(OAc)2 (0.24 g, 1.1 mmol) and Ph3P (0.84 g, 3.2 mmol). Then, the reaction mixture was refluxed for 4 h under nitrogen atmosphere. After filtration, the filter cake was washed by toluene, MTBE-EtOAc (2:1) and H2O. The residue was dried over P2O5 to give 16c (4.0 g, yield 96.1%) as white solid.
80.4%
With palladium diacetate; sodium carbonate; triphenylphosphine; In propan-1-ol; toluene; at 85℃;Inert atmosphere;
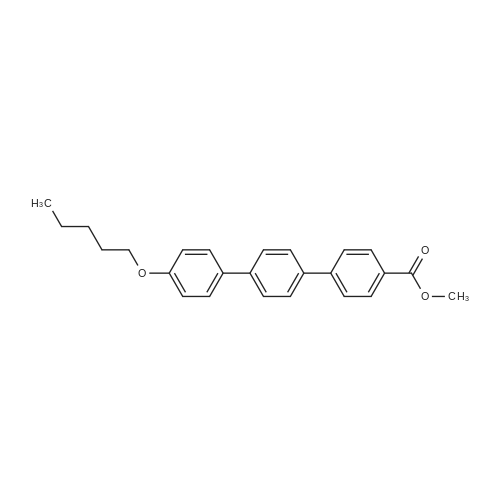
Example 6 4??-(pentyloxy)-[1,1',4',1??-terphenyl]-4-carboxylic acid Methyl ester Under the protection of synthetic nitrogen, 20 g of <strong>[158937-25-8]4-pentyloxy-4'-biphenylboronic acid</strong> was added to a 500 mL three-necked flask. 17.5 g 4-iodobenzoic acid Methyl ester 150 mL of toluene/n-propanol mixed solution (1:8) was stirred at room temperature, Add 40 mL of sodium carbonate solution (2 mol/L) in turn. 150 mg of palladium acetate and 526 mg of triphenylphosphine, Replaced with nitrogen three times, React at 85 C overnight, After the reaction of the raw materials is completed, it is cooled to room temperature and suction filtered. The filter cake was sequentially treated with 10 mL of toluene. Rinse with 20 mL of water and 15 mL of methyl tert-butyl ether. The filter cake was vacuum dried at 50 C for 5 hours to give a white solid 20.1 g. The yield was 80.4%.
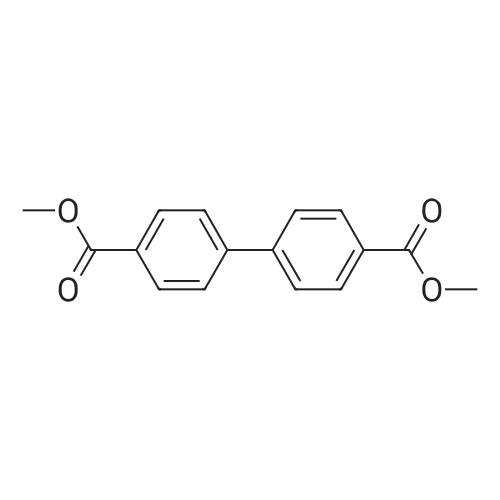
dimethyl-4,4′-(9,9-dimethyl-9H-fluorene-2,7-diyl)dibenzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
92%
With tetrakis(triphenylphosphine) palladium(0); potassium carbonate; triphenylphosphine; In tetrahydrofuran; methanol; at 80℃; for 72h;Inert atmosphere;
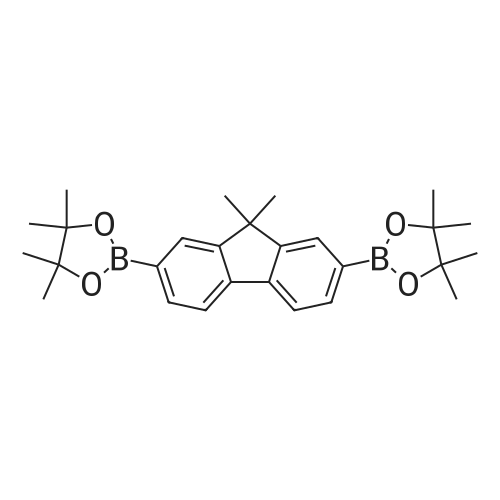
Dimethyl 4,4?-(9,9-dimethyl-9H-fluorene-2,7-diyl)dibenzoate: the mixture of 9,9-dimethyl-9H-2,7-dipinacolborylfluorene (8.9g,20 mmol),methyl 4-Iodolbenzoate (11.5g,44mmol), K2CO3 (13.8g,100mmol), 3% [Pd(PPh3)4] and 12%PPh3 were deaerated under nitrogen for 20min.Then fresh anhydrous THF (200mL) and methanol (100mL)were added to the mixture. The mixture was heated at 80 C for 3 days under N2. After cooling to room temperature, 50mL water was added,thes olid was separated,washed with water and extracted with dichloromethane. The CH2Cl2 solution was evaporated to dryness. White powders (8.5g)were obtained. Yield:92%. 1H NMR(CDCl3) delta 1.61(s,6H),3.96(s,6H),7.64(d,2H),7.70(s,2H),7.75(d,4H),7.84(d,2H),8.14(d,4H)(Fig.S3)
With bis-triphenylphosphine-palladium(II) chloride; sodium hydrogencarbonate; In tetrahydrofuran; water; for 4h;Inert atmosphere; Reflux;
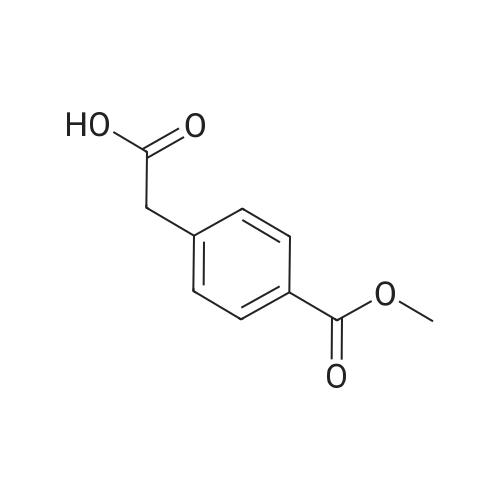
The synthesis has been carried out differing from the literature procedure [15] . A mixture of 4-(methoxycarbonyl)boronic acid (2.04 g, 11.4 mmol) and methyl 4-iodobenzoate (3.00 g, 11.4 mmol) in 75 ml THF and 90 ml H2O was degassed (10 min) under an argon atmosphere followed by the addition of Pd(PPh3)2Cl2 (92.4 mg, 0.132 mmol) and NaHCO3 (9.00 g, 107.1 mmol). After refluxing for 4 h and cooling down to room temperature, the resulting crystalline solid was collected by vacuum filtration (fraction 1). The residue obtained by the evaporation of the filtrate was extracted with 200 ml CH2Cl2. Drying of the organic phase (Na2SO4) and evaporation of the solvent gave a white solid (fraction 2). The two fractions were combined and crystallized from toluene to yield 7 as a white flaky solid (1.96 g, 64percent). Mp: 211-212 °C (lit. 212-213 °C [39] ). 1H NMR (DMSO-d6): deltaH = 3.89 (s, CH3), 7.91 (d, Ar-H, J = 8.55 Hz), 8.07 (d, Ar-H, J = 8.55 Hz). 13C NMR (DMSO-d6): deltaC = 52.2 (CH3), 127.3, 129.2, 129.9, 143.2 (Ar-C), 165.9 (COOMe).
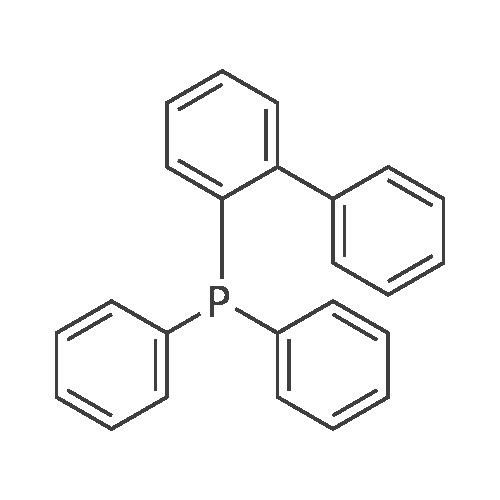
With tris-(dibenzylideneacetone)dipalladium(0); caesium carbonate; ruphos; In toluene; at 95℃;Inert atmosphere;
A mixture of methl 4-iodobenzoate (2.6 g, 10 mmol), compound 4 (6.4 g, 30 mmoi), Pd2(dba)3 (915 mg, I inmol), Ruphos (467 mg, 1 mmoi) and Cs2CO3 (975 g, 3Ommoi, 3eq) in to] (30 ml) was stirred al 95C under nitrogen atmosphere overnight. The mixture was cooled, and EA was added (lOOrni). The mixture was filtered and was concentrated to get a residue, which was purified by silica gel column to afford compound 5 (2.2 g, 60%) as light yellow solid.
With potassium phosphate tribasic hydrate; NiIICl(1-naphthyl)(tricyclohexylphosphine)2; tricyclohexylphosphine; In tetrahydrofuran; at 23℃; for 16h;Inert atmosphere; Glovebox; Sealed tube;
General procedure: Cross-Coupling of ortho-, meta-, and para-Substituted, Electron-Rich and Electron-Deficient Aryl Halides and Aryl Mesylates with Aryl Neopentylglycolboronates Catalyzed by NiIICl(1-naph-thyl)(PCy3)2/PCy3 in Anhydrous THF at 23 °C; General Procedure 2In an oven-dried test tube charged with a Teflon coated stirring barwere added aryl halide or aryl mesylate (0.3 mmol), aryl neopentyl-glycolboronates (0.315 mmol), K3PO4(H2O)3.2 (191.00 ± 1.00 mg, 0.9mmol), and NiIICl(1-naphthyl)(PCy3)2 (11.73 ± 0.0510 mg, 0.015mmol, 5percent catalyst loading). The test tube was brought into a N2 filledglove box (moisture level <2 ppm) through three degassing cycles andPCy3 (8.4 mg, 0.03 mmol, 10percent loading) ligand was added. Distilled sol-vent (1 mL) was added inside the glove box and the test tube wassealed by a rubber septum and left stirring at 23 °C. A sample was tak-en by syringe and transferred outside the glove box. The sample wasdiluted by distilled THF (0.2 mL) and filtered through a short columnof Al2O3. The filtrate was concentrated and the GC analysis was car-ried out. The reaction mixture was diluted with CH2Cl2 (2 mL), filteredthrough a layer of Al2O3, and washed with CH2Cl2 (3 1 mL). The fil-trate was collected and concentrated under vacuum. The crude prod-uct was purified by column chromatography on silica gel with EtO-Ac/hexane mixture as eluent. The reductive elimination side-productwas also isolated and characterized.
With (R,R)-N,N'-dimethyl-1,2-diaminocyclohexane; potassium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 100.0℃; for 48.0h;Inert atmosphere; Sealed tube;
To a round bottom flask was added CuBr (7.2 mg), (1R,2R)-N1,N2-dimethyl- cyclohexane-1,2-diamine (16 L), and (1H-pyrrol-3-yl) methanamine (58 mg). The mixture was sealed with septum,evacuated, and back-filled with nitrogen gas. Methyl 4-iodobenzoate (262 mg) and K2CO3 (357 mg)were then added. DMF (20 mL) was used as a solvent. Sodium carbonate was preferred because usingpotassium hydroxide (a hard base) resulted in hydrolysis of the ester moiety of the product. Theligand to copper ratio was 2:1. The mixture was heated for 2 days at 100 C. Reaction progress wasmonitored via TLC. Ethyl acetate was used to extract the organic phase from the aqueous layer. Flashchromatography was used to purify the product. The white-colored product was eluted with amixture of ethyl acetate to hexane with a yield of 20%. 1H-NMR (750 MHz, CDCl3) ae 8.37 (s, 1H, NH),7.85-7.86 (d, 2H, Ar-H), 7.11 (s, 1H, Ar-H), 6.76 (s, 1H, Ar-H), 6.58-6.59 (d, 2H, Ar-H), 6.22 (s, 1H, Ar-H), 4.23 (s, 2H, CH2, 3.84 (s, 3H, CH3), (Supplementary Figure S11). HRMS: calculated for C12H12N2O2 253.0953, found 253.0936 [M + Na]+, (Supplementary Figure S16).
With (R,R)-N,N'-dimethyl-1,2-diaminocyclohexane; potassium carbonate; copper(I) bromide; In N,N-dimethyl-formamide; at 100.0℃; for 48h;Inert atmosphere; Sealed tube;
For the synthesis of RK464 (Scheme 2), a round bottom flask with a magnetic stir bar was usedas a reaction vessel. To this vessel, CuBr (7.2 mg, 0.025 mmol), (1R,2R)-N1,N2-dimethyl-cyclohexane-1,2-diamine (16 L, 0.05 mmol), and imidazole (82 mg, 0.6 mmol), were added with DMF (20 mL) as asolvent. The vessel was sealed with a septum and nitrogen gas was passed through it.After reacting the initial mixture of compounds, methyl 4-iodobenzoate (262 mg, 0.5 mmol). K2CO3 (357 mg, 2.6 mmol) were added. Again, nitrogen gas was back-filled and the reaction mixturewas heated in a preheated oil bath for 48 h at constant temperature of 100 C. Reaction progress wasregularly monitored using TLC. As the reaction progressed, the color of the mixture changed from whiteto yellow. The product was extracted using ethyl acetate and purified using flash chromatography.A pure yellow viscous product (about 40 mg) was obtained and dried under vacuum. Yield: 19%.1H-NMR (750 MHz, CD3OD) delta: 6.75 (s, 1H, Ar-H), 6.63-6.64 (d, 2H, Ar-H), 6.19-6.20 (d, 2H, Ar-H),6.16 (s, 1H, Ar-H), 2.40 (s, 3H, CH3) (Supplementary Figure S9); HRMS (ESI) calcd C11H10N2O2 for203.0821, found 203.0808 [M+H]+, (Supplementary Figure S14). FT IR numax cm-1: 1710.61 (carboxyl, for 203.0821, found 203.0808 [M+H]+, (Supplementary Figure S14). FT IR max cm1: 1710.61 (carboxyl,C=O), (Supplementary Figure S20). X-ray crystallography: X-ray crystallography structure for thiscompound is shown in Supplementary Figure S19 with R = 0.2.
With acetic acid; palladium dichloride; for 24h;Reflux;
General procedure: Iodo methyl benzoate (0.5 g, 1.90 mmol), silver acetate (0.47 g, 2.85 mmol), sodium acetate (0.77g,9.50 mmol) and PdCl2 (34 mg, 0.19 mmol) were dissolved in AcOH (5 mL). Reaction mixture wasrefluxed for 24 h. The mixture was poured in the saturated NH4Cl and extracted with AcOEt. Theorganic phase was washed with water, dried over Na2SO4, filtered and concentrated under reducedpressure. The crude residue was purified with flash chromatography on silica gel, eluting with 50%AcOEt/Hex to give pure compound.
With pyridine; 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone; nickel(II) iodide hydrate; o-phenylenebis(diphenylphosphine); magnesium; 4,4'-di-tert-butyl-2,2'-bipyridine; at 120℃; for 1h;Microwave irradiation; Inert atmosphere;
NiI2.xH2O (87.7 mg, 0.198 mol), 1,2-bis(diphenylphosphino)benzene (42.6 mg, 0.093 mmol), 4,4-di-tert-butyl-2,2?-dipyridyl (25.4 mg, 0.093 mol), methyl 4-iodobenzoate (500 mg, 1.85 mmol), <strong>[461-17-6]1-iodo-4,4,4-trifluorobutane</strong> (454.1 mg, 1.85 mmol) were transferred to a 5 ml microwave vial equipped with a magnetic stir bar. The vial was then capped with a rubber septum and purged with argon gas. DMPU (2 ml), pyridine (1 drop) were injected into the vial. Manganese powder (204 mg, 3.7 mmol) was added and the vial was placed under microwave irradiation at 120 C. for 60 min. The mixture was diluted with water, extracted with ethyl acetate. The organic layer was concentrated and the residue was purified by flash column chromatography on silica gel (EtOAc/heptane: 0>>>10%>>>20%) to yield a white solid. 1H NMR (CHLOROFORM-d) delta: 7.98 (d, J=8.1 Hz, 2H), 7.24 (s, 2H), 3.91 (s, 3H), 2.75 (t, J=7.6 Hz, 2H), 2.00-2.19 (m, 2H), 1.85-1.98 (m, 2H).
With pyridine; 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone; manganese; nickel(II) iodide hydrate; o-phenylenebis(diphenylphosphine); 4,4'-di-tert-butyl-2,2'-bipyridine; at 120℃; for 1h;Inert atmosphere; Microwave irradiation;
Ni12.xH2O (87.7 mg, 0.198 mol), 1,2-bis(diphenyl- phosphino)benzene (42.6 mg, 0.093 mmol), 4,4-di-tert-bu- tyl-2,2?-dipyridyl (25.4 mg, 0.093 mol), methyl 4-iodoben- zoate (500 mg, 1.85 mmol), <strong>[461-17-6]1-iodo-4,4,4-trifluorobutane</strong> (454.1 mg, 1.85 mmol) were transferred to a 5 ml microwave vial equipped with a magnetic stir bar. The vial was then capped with a rubber septum and purged with argon gas. DMPU (2 ml), pyridine (1 drop) were injected into the vial. Manganese powder (204 mg, 3.7 mmol) was added and the vial was placed under microwave irradiation at 120 C. for 60 mm. The mixture was diluted with water, extracted with ethyl acetate. The organic layer was concentrated and the residue was purified by flash column chromatography on silica gel (EtOAc/heptane: 0>>>10%>>>20%) to yield a white solid. ?H NMR (CHLOROFORM-d) oe: 7.98 (d, J=8.1 Hz, 2H), 7.24 (s, 2H), 3.91 (s, 3H), 2.75 (t, J=7.6 Hz, 2H), 2.00-2.19 (m, 2H), 1.85-1.98 (m, 2H).
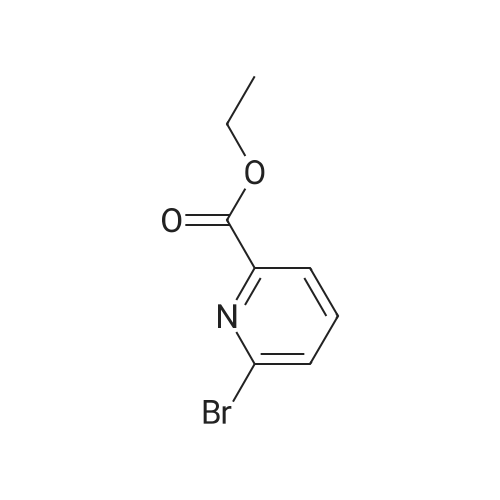
With potassium phosphate; In N,N-dimethyl acetamide; at 60℃; for 24h;Irradiation; Green chemistry;
General procedure: To a solution of 1 (0.3 mmol) and 2 (0.45mmol) in DMA (5 mL) was added 3wt% Pd/CeO2 (80 mg). The reaction mixture was stirred under incandescent light (0.79 Wcm-2) irradiation at 60C for 24h. After reaction (monitored by TLC), when the reaction is complete, in test tube extraction and centrifugal separation allows for isolation of the desired product, while the catalyst can be washed with ethanol and water, dried under vacuum, and reused for the next run. After completion of the reaction (monitored by TLC), water (5 mL) was added and the mixture was extracted with EtOAc (3 × 5 mL). The combined organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel (10:1 petroleum ether/EtOAc) to give the target product.
6-(4-(methoxycarbonyl)phenyl)pyridin-2-carboxylic acid ethyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
With potassium phosphate; In N,N-dimethyl acetamide; at 60℃; for 24h;Irradiation; Green chemistry;
General procedure: To a solution of 1 (0.3 mmol) and 2 (0.45mmol) in DMA (5 mL) was added 3wt% Pd/CeO2 (80 mg). The reaction mixture was stirred under incandescent light (0.79 Wcm-2) irradiation at 60C for 24h. After reaction (monitored by TLC), when the reaction is complete, in test tube extraction and centrifugal separation allows for isolation of the desired product, while the catalyst can be washed with ethanol and water, dried under vacuum, and reused for the next run. After completion of the reaction (monitored by TLC), water (5 mL) was added and the mixture was extracted with EtOAc (3 × 5 mL). The combined organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by chromatography on silica gel (10:1 petroleum ether/EtOAc) to give the target product.
methyl 4-(3-(2-methoxyphenyl)propioloyl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
86%
With nickel(II) acetate tetrahydrate; triethylamine; In 1,4-dioxane; at 100℃; under 1500.15 Torr; for 4h;
General procedure: A mixture of aryl iodide (2 mmol), aryl alkyne (2.4 mmol), Ni(OAc) 2 (10 mol %), Et3N (6 mmol) and 1,4-dioxane (15 mL) were charged into the reactor (steel bomb). The reactor was closed and pressurized to ~2bar CO and then stirred at 100 C for 4 h. The reaction Progress was monitored by TLC. The reaction mixture was cooled to room temperature, before being filtered through a pad of Celite. The filtrate was concentrated and purified by silica gel with a mixture of hexane and ethyl acetate to give the pure product. For all examples, reactions were performed on 100 mg scale of corresponding aryl iodides.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping