| 40% |
|
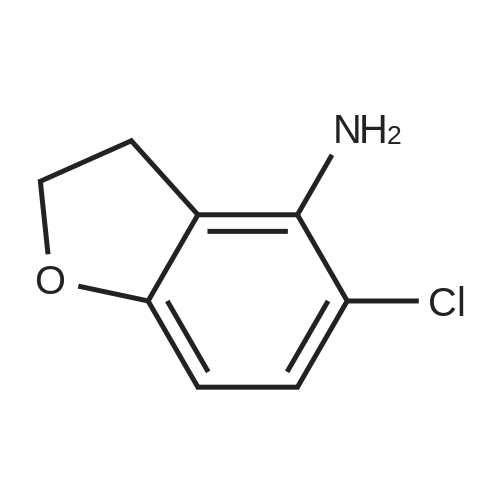
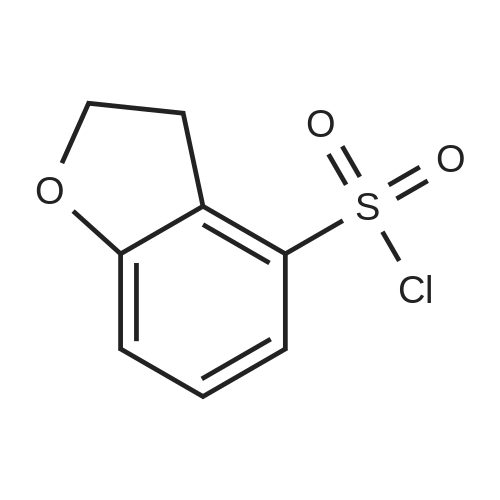
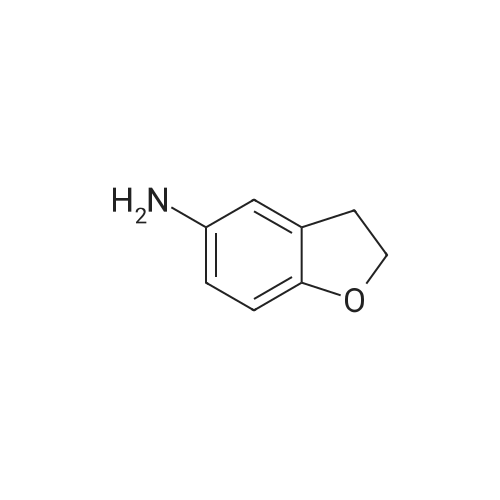
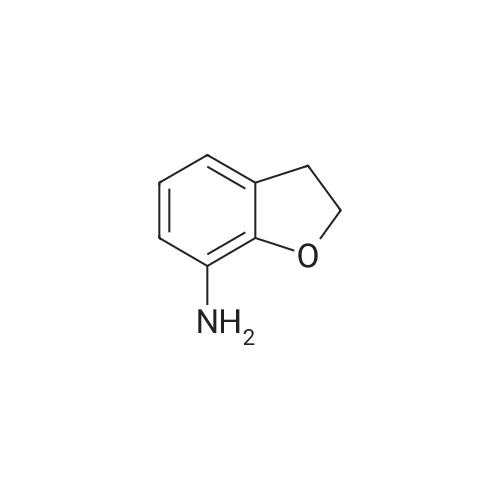
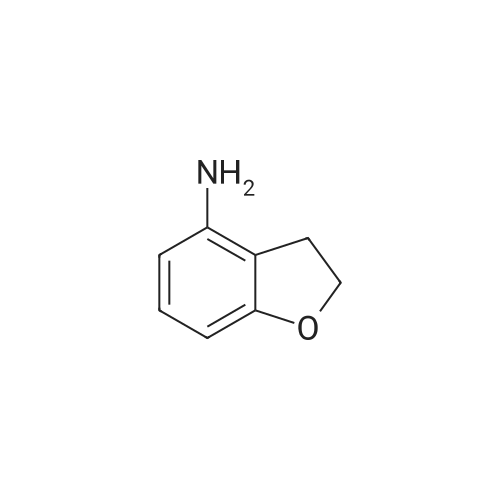
5. Synthesis of 2,3-dihydrobenzofuran-4-sulfonyl chloride; Hydrochloric acid (9.0 g) was added dropwise to a solution of <strong>[61090-37-7]2,3-dihydrobenzofuran-4-amine</strong> (16.3 mmol) and acetic acid (9.0 g) in acetonitrile (200 mL) at 0 C. A solution of sodium nitrite (22.0 mmol) in water (2 mL) was subsequently added and the mixture was maintained for 30 min at 0 C. Sulfur dioxide gas was passed through the reaction mixture for 2 h whereupon a solution of copper(II) chloride dihydrate (20.0 mmol) in water (3 mL) was added. Sulfur dioxide gas was passed through the reaction mixture for an additional 2 h. The reaction mixture was allowed to warm to rt and was maintained for 16 h. The reaction mixture was diluted with ice water (200 mL) and the resulting mixture was extracted with ethyl acetate (300 mL). The organic layer was washed with water (200 mL), dried (sodium sulfate), and concentrated. The residue was purified by Flash chromatography (1/70 ethyl acetate/petroleum ether) to provide 2,3-dihydrobenzofuran-4-sulfonyl chloride in 40% yield as a yellow solid. Data: 1H NMR (CDCl3) delta 7.40 (d, 1H), 7.30 (d, 1H), 7.10 (d, 1H), 4.70 (m, 2H), 3.60 (m, 2H). LC/MS (ES) m/z 283 [M+C5H11N2-Cl+H]+. |
| 40% |
|
5. Synthesis of 2,3-dihvdrobenzofuran-4-sulfonyl chloride.Hydrochloric acid (9.0 g) was added dropwise to a solution of 2,3-dihydrobenzofuran-4- amine (16.3 mmol) and acetic acid (9.0 g) in acetonitrile (200 mL) at 0 0C. A solution of sodium nitrite (22.0 mmol) in water (2 mL) was subsequently added and the mixture was maintained for 30 min at 0 0C. Sulfur dioxide gas was passed through the reaction mixture for 2 h whereupon a solution of copper(II) chloride dihydrate (20.0 mmol) in water (3 mL) was added. Sulfur dioxide gas was passed through the reaction mixture for an additional 2 h. The reaction mixture was allowed to warm to rt and was maintained for 16 h. The reaction mixture was diluted with ice water (200 mL) and the resulting mixture was extracted with ethyl acetate (300 mL). The organic layer was washed with water (200 mL), dried (sodium sulfate), and concentrated. The residue was purified by Flash chromatography (1/70 ethyl acetate/petroleum ether) to provide 2,3-dihydrobenzofuran-4-sulfonyl chloride in 40% yield as a yellow solid. Data: 1H NMR (CDCl3) delta 7.40 (d, IH), 7.30 (d, IH), 7.10 (d, IH), 4.70 (m, 2H), 3.60 (m, 2H). LC/MS (ES) m/z 283 [M+C5H11N2-C1+H]+. |
| 40% |
With hydrogenchloride; copper(II) choride dihydrate; sulfur dioxide; acetic acid; sodium nitrite; In water; acetonitrile; |
Hydrochloric acid (9.0 g) was added dropwise to a solution of <strong>[61090-37-7]2,3-dihydrobenzofuran-4-amine</strong> (16.3 mmol) and acetic acid (9.0 g) in acetonitrile (200 mL) at 0 C. A solution of sodium nitrite (22.0 mmol) in water (2 mL) was subsequently added and the mixture was maintained for 30 min at 0 C. Sulfur dioxide gas was passed through the reaction mixture for 2 h whereupon a solution of copper(II) chloride dihydrate (20.0 mmol) in water (3 mL) was added. Sulfur dioxide gas was passed through the reaction mixture for an additional 2 h. The reaction mixture was allowed to warm to rt and was maintained for 16 h. The reaction mixture was diluted with ice water (200 mL) and the resulting mixture was extracted with ethyl acetate (300 mL). The organic layer was washed with water (200 mL), dried (sodium sulfate), and concentrated. The residue was purified by Flash chromatography (1/70 ethyl acetate/petroleum ether) to provide 2,3-dihydrobenzofuran-4-sulfonyl chloride in 40% yield as a yellow solid. Data: 1H NMR (CDCl3) delta 7.40 (d, 1H), 7.30 (d, 1H), 7.10 (d, 1H), 4.70 (m, 2H), 3.60 (m, 2H). LC/MS (ES) ml/283 [M+C5H11N2-Cl+H]+. |
| 40% |
|
5. Synthesis of 2,3-dihydrobenzofuran-4-sulfonyl chloride.Hydrochloric acid (9.0 g) was added dropwise to a solution of 2,3-dihydrobenzofuran-4- amine (16.3 mmol) and acetic acid (9.0 g) in acetonitrile (200 mL) at 0 0C. A solution of sodium nitrite (22.0 mmol) in water (2 mL) was subsequently added and the mixture was maintained for 30 min at 0 0C. Sulfur dioxide gas was passed through the reaction mixture for 2 h whereupon a solution of copper(II) chloride dihydrate (20.0 mmol) in water (3 mL) was added. Sulfur dioxide gas was passed through the reaction mixture for an additional 2 h. The reaction mixture was allowed to warm to rt and was maintained for 16 h. The reaction mixture was diluted with ice water (200 mL) and the resulting mixture was extracted with ethyl acetate (300 mL). The organic layer was washed with water (200 mL), dried (sodium sulfate), and concentrated. The residue was purified by Flash chromatography (1/70 ethyl acetate/petroleum ether) to provide 2,3-dihydrobenzofuran-4-sulfonyl chloride in 40% yield as a yellow solid. Data: 1H NMR (CDCl3) delta 7.40 (d, IH), 7.30 (d, IH), 7.10 (d, IH), 4.70 (m, 2H), 3.60 (m, 2H). LC/MS (ES) m/z 283 [M+C5H11N2-C1+H]+. |
| 40% |
|
Hydrochloric acid (9.0 g) was added dropwise to a solution of <strong>[61090-37-7]2,3-dihydrobenzofuran-4-amine</strong> (16.3 mmol) and acetic acid (9.0 g) in acetonitrile (200 mL) at 0 C. A solution of sodium nitrite (22.0 mmol) in water (2 mL) was subsequently added and the mixture was maintained for 30 min at 0 C. Sulfur dioxide gas was passed through the reaction mixture for 2 h whereupon a solution of copper(II) chloride dihydrate (20.0 mmol) in water (3 mL) was added. Sulfur dioxide gas was passed through the reaction mixture for an additional 2 h. The reaction mixture was allowed to warm to rt and was maintained for 16 h. The reaction mixture was diluted with ice water (200 mL) and the resulting mixture was extracted with ethyl acetate (300 mL). The organic layer was washed with water (200 mL), dried (sodium sulfate), and concentrated. The residue was purified by Flash chromatography (1/70 ethyl acetate/petroleum ether) to provide 2,3-dihydrobenzofuran-4-sulfonyl chloride in 40% yield as a yellow solid. Data: 1H NMR (CDCl3) delta 7.40 (d, 1H), 7.30 (d, 1H), 7.10 (d, 1H), 4.70 (m, 2H), 3.60 (m, 2H). LC/MS (ES) m/z 283 [M+C5H11N2-Cl+H]+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping