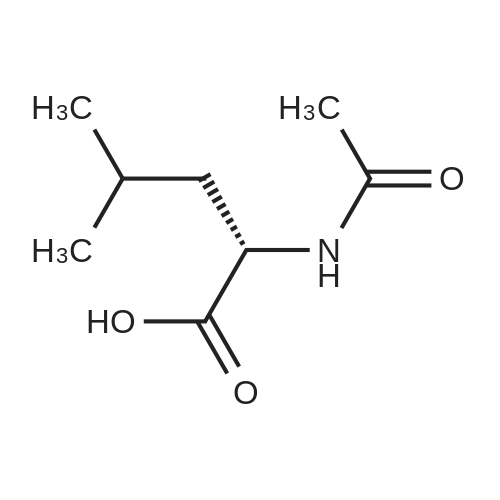
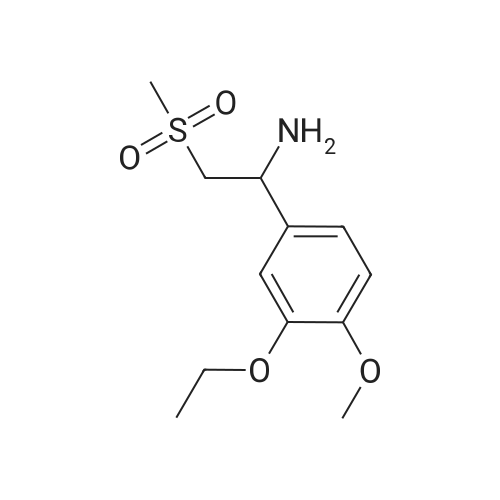
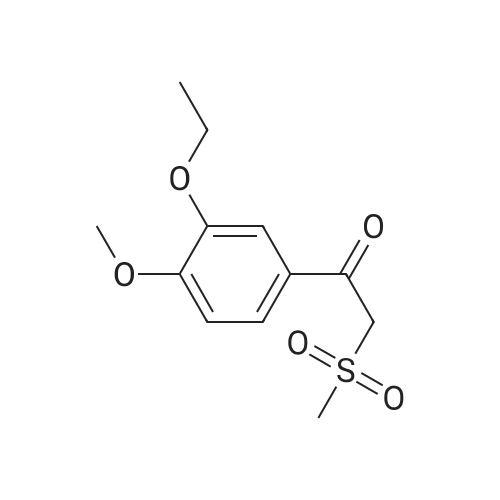
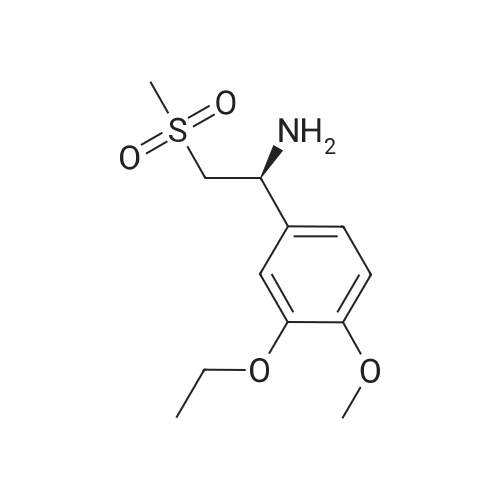
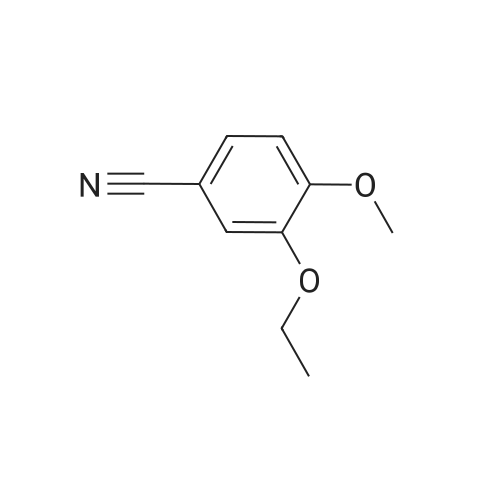
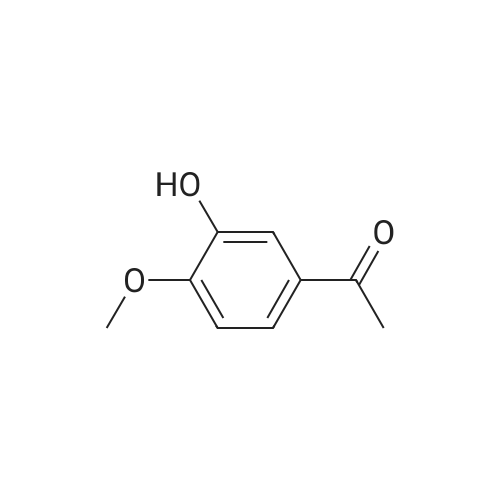
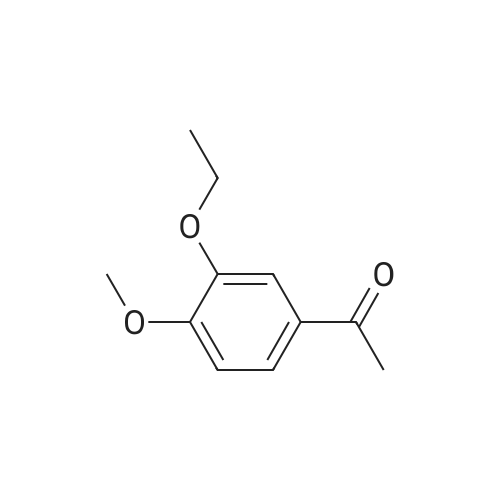
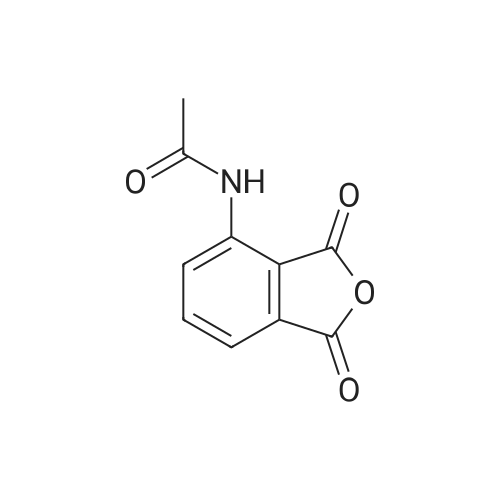
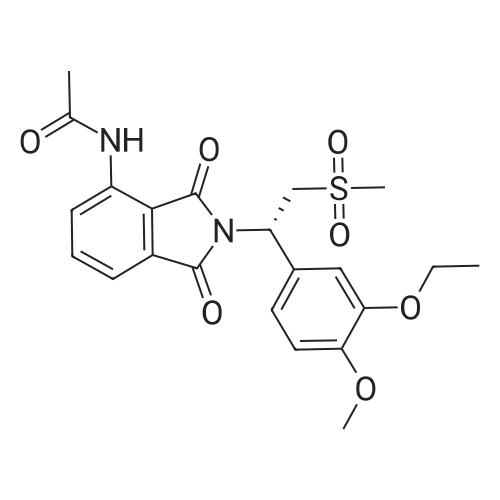
| 85.2% |
With magnesium sulfate; acetic acid; at 70℃; for 4h; |
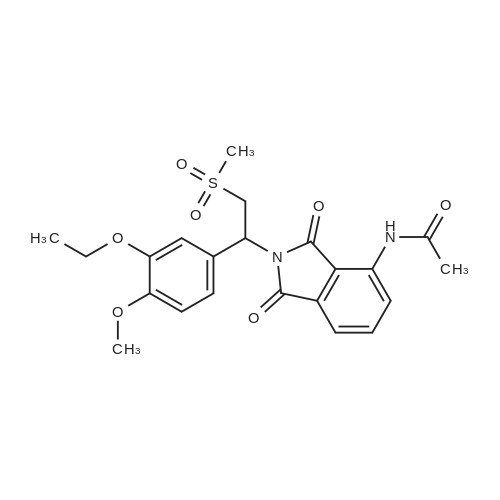
To a 2 L reaction flask was added Compound IV 100 g (0.224 mol, 98.4% ee), Anhydrous magnesium sulfate (13.5 g, 0.112 mol) and glacial acetic acid (600 mL) Mechanical stirring,Compound V 46 g (0.224 mol) was added.The reaction was stirred at 70 C for 4 h. Cooling to below 50 ,Filtration, solvent evaporation under reduced pressure, Washed with 1 L of ethyl acetate, washed with saturated sodium bicarbonate solution (2 x 800 mL), saturated sodium chloride solution (2 x 500 mL), and then dried over anhydrous sodium sulfate. Filtered and concentrated under reduced pressure. At room temperature, 300mL of acetone was added to the residue, and 600mL of ethanol was added after dissolving. The mixture was stirred for 30 minutes, 600mL of ethanol was added, and the mixture was stirred for 1 hour. Filtration and drying afforded 85.2 g (99.4% ee) of an off-white solid, 85.2% weight yield, 99.8% HPLC purity. |
| 75% |
|
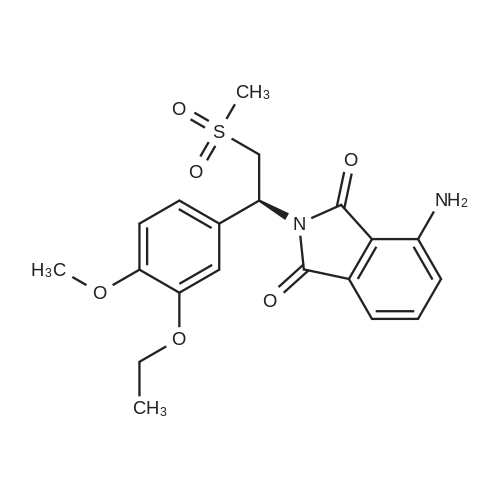
A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4-methoxyphenyl)-1-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50 C. After the solvent was removed in vacuum, the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL×2), saturated aqeous NaHCO3 (250 mL×2), and brine (250 mL×2), and then dried over anhydrous sodium sulfate. After the solvent was evaporated in vacuum, the residue was recrystallized from a binary solvent containing a mixture of ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL×2). The product was dried in vacuum at 60 C. to a constant weight, affording 19.4 g (75% yield) of (S)-{2-[1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-inoisoindoline-1,3-dione with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2PO4 (at)pH 0.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm×4.6 mm, 0.4 mL/min., (at)240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%). The product in CDCl3 was characterized by a 1H NMR spectrum showing the following chemical shifts (delta in ppm): 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). The product in DMSO-d6 was characterized by a 13C NMR spectrum showing the following chemical shifts (delta in ppm): 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48. |
| 75% |
In acetic acid;Reflux; |
A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)- 1 -(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98%ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <5 0C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x 2), saturated aqeous NaHCO3 (250 mL 2), brine (250 mL 2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solventcontaining ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL 2). The product was dried in vacuo at 60C. to a constant weight, affording 19.4 g (75% yield) of Compound 3 with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2PO4 pH 3.5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., 240 nm): 25.4 mm (S-isomer, 98.7%), 29.5 mm (R-isomer, 1.2%). ?H-NMR (CDC13) oe:1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61(dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). ?3C-NMR(DMSO-d6) oe: 14.66, 24.92,41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02,136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48. |
| 75% |
In acetic acid;Reflux; |
: A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (5)-2-(3-ethoxy-4-methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98%> ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to 3(250 mL x 2), brine (250 mL x 2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL x 2). The product was dried in vacuo at 60 C to a constant weight, affording 19.4 g (75% yield) of apremilast with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 pH 5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min, 240 nm): 25.4 min (^-isomer, 98.7%), 29.5 min (R- isomer, 1.2%). 1H-NMR (CDC13) delta: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H);13C-NMR (DMSO-d6) delta: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48. |
| 75% |
With acetic acid;Reflux; |
6.2.4. Preparation of (+)-2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-acetylaminoisoindoline-1,3-dione A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4-methoxyphenyl)-1-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50 C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL*2), saturated aqueous NaHCO3 (250 mL*2), brine (250 mL*2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL*2). The product was dried in vacuo at 60 C. to a constant weight, affording 19.4 g (75% yield) of (S)-{2-[1-(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-inoisoindoline-1,3-dione with 98% ee. |
| 75% |
With acetic acid;Reflux; |
[0189] A 500 mL 3 -necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4- methoxyphenyl)-l-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, (0620) 98% ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g 58.8 mmol), and glacial acetic acid (250 mL). (0621) The mixture was refluxed over night and then cooled to < 50 C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x 2), saturated aqeous NaHC03 (250 mL x 2), brine (250 mL x 2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL x 2). The product was dried in vacuo at 60 (0622) C to a constant weight, affording 19.4 g (75% yield) of (0623) (S)- {2- [ 1 -(3 -ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl] -4-inoisoindoline- 1 ,3 -dione with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2P04 pH .5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min., 240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (i?-isomer, 1.2%). 1H-NMR (CDC13) 5: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68- 3.75 (dd, IH), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, IH), 5.84-5.90 (dd, IH), 6.82-8.77 (m, 6H), 9.46 (s, IH). 13C-NMR (DMSO-d6) delta: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48. |
| 75% |
With acetic acid;Reflux; |
10217] A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3- ethoxy-4-methoxyphenyl)- 1 -(methylsulphonyl)-eth-2-ylamine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% cc), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed overnight and then cooledto <50C. The solvent was removed invacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mLx2), saturated aqueous NaHCO3 (250 mLx2), brine (250 mLx2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solidwas isolated by vacuum filtration and washed with ethanol (100 mLx2). The product was dried in vacuo at 60C. to a constant weight, affording 19.4 g (75% yield) of S-{2-[1-(3-ethoxy-4-meth- oxyphenyl)-2-methylsulfonylethyl]-4-acetamidoisoindo- line-1,3-dione} with 98% cc. Chiral HPLC (15/85 EtOH/20mM KH2PO4 pH 5, Ultron Chiral ES-OVS from Agilent Technology, 150 mmx4.6 mm, 0.4 mL/min, 240 nm): 25.4 mm (S-isomer, 98.7%), 29.5 mm (R-isomer, 1.2%). ?H-NMR (CDC13) oe: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H); ?3C- NMR (DMSO-d6) oe: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46,169.14, 169.48. |
| 75% |
With acetic acid;Reflux; |
A 500 mE 3-necked round bottom flask was equipped with amechanical stirrer, thermometer, and condenset The reaction vessel was charged with (S)-2-(3-ethoxy-4-methoxyphenyl)- 1 -(methylsulphonyl)-eth-2-ylamine N-acetyl-E-leucine salt(25 g, 56 mmol, 98% cc), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g 58.8 mmol), and glacial acetic acid (250 mE). Themixture was refluxed over night and then cooled to <50 C. Afier the solvent was removed in vacuum, the residue was dissolved in ethyl acetate. The resulting solution was washedwith water (250 mEx2), saturated aqueous NaHCO3 (250 mEx2), andbrine (250 mEx2), and then dried over anhydroussodium sulfate. After the solvent was evaporated in vacuum, the residue was recrystallized from a binary solvent containing a mixture of ethanol (150 mE) and acetone (75 mE). The solid was isolated by vacuum filtration and washed with ethanol (100 mEx2). The product was dried in vacuum at 60 C.to a constant weight, affording 19.4 g (75% yield) of(S)-{2- [1 -(3-ethoxy-4-methoxyphenyl)-2-methylsulfonylethyl]-4-inoisoindoline-i,3-dione with 98% cc. Chiral HPEC (15/85 EtOH/20 mM KH2PO4 pH 0.5, Ultron Chiral ES-OVSfrom Agilent Technology, 150 mmx4.6 mm, 0.4 mE/mm.,240 nm): 25.4 mm (S-isomer, 98.7%), 29.5 mm (R-isomer,1.2%). The product in CDC13 was characterized by a ?H NMR spectrum showing the following chemical shifts (6 in ppm):1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H),3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90(dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H). The product in DMSO-d5 was characterized by a ?3C NMR spectrum showing the following chemical shifis (oe in ppm): 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, ii 1.44, 112.40, 115.10,118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60,148.62 149.74 167.46 169.14 169.48. |
| 75% |
With acetic acid; at 110 - 115℃; for 1h; |
To a 500 mL RBF the product of example 4, <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> and acetic acidwere charged and stirred at 110 C to 115 C for 1 hour and the solution was cooled to 60-65C. Acetic acid was distilled off completely and the residue was dissolved indichloromethane. The dichloromethane layer was washed with water followed by washing with aq. sodium bicarbonate solution. The dichloromethane layer was separated and distilled off completely. The product was isolated by recrystallization in acetone:ethanol mixture to give apremilast as a light yellow solid with chiral purity of 99.9 % and HPLC purity of 99.8%. Yield: 75 % |
| 75% |
With acetic acid;Reflux; |
A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4-methoxyphenyl)-1-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x 2), saturated aqueous NaHCO3 (250 mL x 2), brine (250 mL x 2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL x 2). The product was dried in vacuo at 60 C to a constant weight, affording 19.4 g (75% yield) of apremilast with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2PO4 pH 5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min, 240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%). 1H-NMR (CDCl3) delta: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H); 13C-NMR (DMSO-d6) delta: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48. |
| 75% |
With acetic acid;Reflux; |
A 500 mL 3-necked round bottom flask was equipped with a mechanical stirrer, thermometer, and condenser. The reaction vessel was charged with (S)-2-(3-ethoxy-4-methoxyphenyl)-1-(methylsulphonyl)-eth-2-yl amine N-acetyl-L-leucine salt (25 g, 56 mmol, 98% ee), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (12.1 g, 58.8 mmol), and glacial acetic acid (250 mL). The mixture was refluxed over night and then cooled to <50 C. The solvent was removed in vacuo, and the residue was dissolved in ethyl acetate. The resulting solution was washed with water (250 mL x 2), saturated aqueous NaHCO3 (250 mL x 2), brine (250 mL x 2), and dried over sodium sulphate. The solvent was evaporated in vacuo, and the residue recrystallized from a binary solvent containing ethanol (150 mL) and acetone (75 mL). The solid was isolated by vacuum filtration and washed with ethanol (100 mL x 2). The product was dried in vacuo at 60 C to a constant weight, affording 19.4 g (75% yield) of apremilast with 98% ee. Chiral HPLC (15/85 EtOH/20 mM KH2PO4 pH 5, Ultron Chiral ES-OVS from Agilent Technology, 150 mm x 4.6 mm, 0.4 mL/min, 240 nm): 25.4 min (S-isomer, 98.7%), 29.5 min (R-isomer, 1.2%). 1H-NMR (CDCl3) delta: 1.47 (t, 3H), 2.26 (s, 3H), 2.87 (s, 3H), 3.68-3.75 (dd, 1H), 3.85 (s, 3H), 4.07-4.15 (q, 2H), 4.51-4.61 (dd, 1H), 5.84-5.90 (dd, 1H), 6.82-8.77 (m, 6H), 9.46 (s, 1H); 13C-NMR (DMSO-d6) delta: 14.66, 24.92, 41.61, 48.53, 54.46, 55.91, 64.51, 111.44, 112.40, 115.10, 118.20, 120.28, 124.94, 129.22, 131.02, 136.09, 137.60, 148.62, 149.74, 167.46, 169.14, 169.48. |
| 74% |
With N-Ac-Leu; acetic acid;Reflux; |
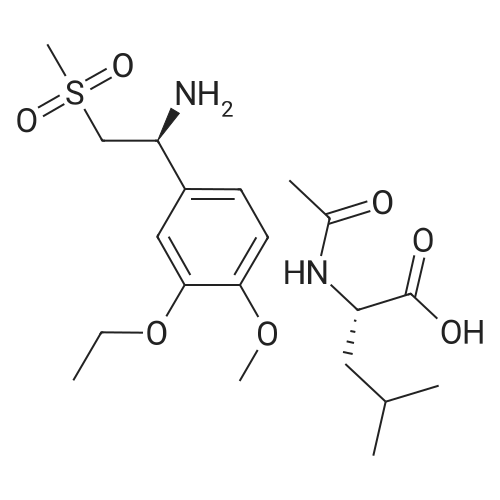
A 500ml three-neck flask was fitted with a mechanical stirrer, thermometer and cooler. 25 g (56 mmol) of the salt of (5)-2-(3-emoxy-4-methoxyphenyl)-l-(methylsulfonyl)-eth-2-ylamine with N-acetyl-Z-leucine, 12.1 g (58.8 mmol) of 3-acetamidophtalic anhydride and 250 ml of glacial acetic acid were charged into a reaction vessel. The mixture was refluxed overnight and then cooled down to <50C. The solvent (250 ml of acetic acid) was removed in vacuo and the residue, having rich yellow colour, was dissolved in 800 ml of ethyl acetate. The obtained solution was washed with water (2 x 250), saturated aqueous NaHC03 (2 x 250 ml), brine (2 x 250 ml) and dried with sodium sulphate. The solvent (800 ml of ethyl acetate) was evaporated in vacuo and the residue was recrystallized from a binary solvent containing 150 ml of ethanol and 75 ml of acetone. The solid was isolated by filtration in vacuo and washed with 2 x 100 ml of ethanol. The product was dried in vacuo at 60C until constant weight, which provided 19.1 g of the title compound; yield 74%, HPLC purity 99.29%, XRPD confirmed Form B. |
| 72.8% |
|
In a 250 ml round bottom flask, 50 ml acetic acid was charged followed by 10 gms of 2-( -3-ethoxy-4-methoxyphenyl)- 1 -(methanesulfonyl)-eth-2-ylamine N-acetyl-Lleucine salt and it was stirred at room temperature for a few minutes. Then 4.82 gms of 3- acetamidophthalic anhydride was added and reaction mass was heated for 11 to 12 hours at 80 to 90 C. The solvent was removed under vacuum and ethyl acetate was addedfollowed by sodium bicarbonate solution. The layers were separated. The organic layer was washed and solvent was evaporated under vacuum. In the distilled residue, 90 ml ethanol and 30 ml acetone added and was stirred it for 2 hours at room temperature. The solid was precipitated. The solid was filtered and washed with ethanol. The material was unloaded and dried under vacuum for longer hours at 60 C. Yield: 6.41 gm. Thismaterial will be crystalized using a solvent or mixture of solvents known in the art. |
| 70% |
|
Charged 50 mL of 1,4-Dioxane in a flask followed by the addition of 5 g of N-acetyl L- Leucine salt of l-(3-Ethoxy-4-methoxyphenyl)-2-methylsulfonylethylamine (Compound (AL)), 5 mL of glacial acetic acid and 2.3 g of <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (B). The reaction mixture was heated to a temperature of 85-90 C and stirred for about 8-10 h. The reaction mixture was cooled and solvent was distilled out under vacuum. To the residue was added 50 mL of dichloromethane followed by the addition of 0.014 g of Iodine (I2) and 0.88 g of Acetyl chloride. The reaction mixture further stirred for 2-4 hr at a temperature of about 25-30 C. The reaction mixture was washed with aqueous solution of sodium bicarbonate (7- 8% solution) and sodium thiosulfate solution (10%). The organic layer was separated and evaporated under vacuum and the residue was dissolved in 40 mL of methyl isobutyl ketone. The reaction mixture was heated at temperature 80-90 C, followed with subsequent cooling to 25-30 C temperature and stirred for about 14-16 h. The reaction mixture was further cooled to 0-5 C temperature and the precipitated solid was isolated. Yield: 3.6 g (70%). |
| 60.3% |
|
The compound of formula VI 40. 08mmol, a compound of formula IX 42. 06mmol acetic acid and a 500 mL, added to the reaction flask and the reaction was refluxed for 15H, the solvent was distilled off under reduced pressure, the residue was added water and EA each 100mL, extract separated, and extracted with EA ( 100mLX 2), the combined organic phases were washed with 100mL 2M aqueous sodium bicarbonate solution, dried over anhydrous sodium sulfate, filtered, and the filtrate was evaporated under reduced pressure to give a pale yellow solid, clear solution at reflux was added 35mL of acetone, and then adding 70mL absolute ethanol , refluxed for 15min, allowed to cool, 25 C crystallization was stirred 3h, filtered, washed with a small amount of anhydrous ethanol, the filter cake was dried in vacuo at 40 C for 5 hours to give a white solid 11. 1g, yield 60. 3%, HPLC: 99 · 7%, less than one-hybrid 0 ? 1%, : 98 · 6% |
|
With acetic acid; In toluene; at 120℃; |
Compound (S)-2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-4-acetylaminoisoindoline-1,3-dione specific preparation method: Select 480ml toluene as solvent, 48ml acetic acid as a catalyst. 24g of purified compound (S)-2-(3-ethoxy-4-methoxyphenyl)-1-(methylsulfonyl)-eth-2-ylamine N-acetyl-L-leucine and 12g compound <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> under reflux temperature is reacted for 2 ~ 3h to obtain compound 24g (S)-2-[1-(3-ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-4-acetylaminoisoindoline-1,3-dione. The reflux temperature is 120 deg.C. Apremilast high chiral purity preparation specifically: 24g of the compound (S) -2- [1- (3- ethoxy-4-methoxyphenyl) -2-methanesulfonyl-ethyl] -4-acetyl-amino-isoindoline-1,3-dione into the solvent at 50 ~ 60 conditions recrystallization of high purity Apremilast, the solvent is a mixture of 96ml of acetone and 192ml of ethanol. |
| 425 g |
In acetonitrile; for 3h;Reflux; |
A stirred solution of N-acetyl L-leucine salt of (S)-2-(3-efhoxy-4-methoxyphenyl)-l- (methylsulphonyl)eth-2-ylamine (500.0gm) and <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (229.85gm) in 4.01it acetonitrile was refluxed for 3.0 hr and then cooled to 50C. The solvent was removed under reduced pressure to obtain a semisolid residue. To the residue water was added and pH adjusted to 7-8 using saturated sodium bicarbonate solution. The product was extracted into ethyl acetate and the solvent was removed under reduced pressure to obtain apremilast. HPLC purity: 99% The apremilast thus obtained was heated at 60-65 C in a solvent mixture of acetone and ethanol to get clear solution. The reaction mixture was cooled to 25-30C and stirred for 12 hr. The precipitated product was filtered and washed with ethanol and dried at 60C under vacuum for 12.0hr to obtain 425.0gm of (5)-N-{2-(l-(3-ethoxy-4- methoxyphenyl)-2-methylsulphonyl)ethyl)-l,3-dioxo-2,3-dihydro-lH-isoindol-4- yljacetamide. HPLC purity 100.0%, R isomer <0.5%, S isomer> 99.5%. |
|
With sodium acetate; acetic acid; at 80℃; |
A reaction flask was charged with (S)-1 -(3-Ethoxy-4-methoxyphenyl)-2- Methylsulfonylethylamine-N-actyl-L-leucine salt (1 1 g), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong>(5.3g), sodium acetate anhydrous (2.0g) and glacial acetic acid (22ml). The reaction mixture was heated to 80 ± 3C till completion of the reaction. After completion of the reaction, reaction mixture was cooled to room temperature. Solvent was removed under vacuum and the residue was dissolved in ethyl acetate. The resulting organic solution was washed with water, saturated aqueous sodium bicarbonate solution and brine solution. The solvent ethyl acetate was evaporated under vacuum to give amorphous Apremilast which was stirred with cyclohexane and filtered to obtain amorphous Apremilast. |
|
In N,N-dimethyl acetamide; at 110 - 120℃;Inert atmosphere; |
The below preparation serves as a non-limiting example for preparing APM. Any other method for preparing APM is also suitable in the context of the present invention. (0518) Reference is made to Figure 1. APM-1 (9.65g) was dissolved in dimethylacetamide (100ml) at ambient temperature. Then APM-2 (20g) was suspended and dimethylacetamide (100 ml) was charged. After warming up of the suspension to the temperature 10-120C under flow of inert gas the reaction mixture was stirred between 2-4 hours. When the reaction was finished (after 2-4h at 110-120C) the solution was cooled down to a temperature of 20-25C. Ethyl acetate (180ml) and water (180ml) were added to the reaction mixture and the reaction mass was stirred for 10 min at 20-30C. After separation, the ethyl acetate phase was left and the water phase was extracted with ethyl acetate (2x50ml). The combined ethyl acetate phases were washed with 8% aHCOs solution (2x50ml), 1N HCI (2x50ml), and water (1x50ml). The organic solvent was evaporated under reduced pressure (200-30mbar) at 35C giving compound APM (as an oil residue left after ethyl acetate distillation). (0519) 1.2 Crystallization of N-[2-[(1S)-1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl]-1 ,3- dioxo-2,3-dihydro-1 H-isoindol-4-yl]acetamide) (APM) (0520) Crystallization - Option I (0521) Reference is made to Figure 2. For crystallization, APM in any form and prepared by any method (e.g. any known literature method), for example the method of above Example 1.1 , can be used. (0522) 10g of Apremilast were suspended in diethyl ether (200ml) and stirred for 12h at 25±5C to give a white to cream color suspension. Obtained solid was filtered under reduced pressure and dried at 85C and gave product APM Form N (as confirmed by XRPD, DSC and TGA) with purity above 99.90% (80-90% yield). (0523) Crystallization - Option II (0524) Reference is made to Figure 2. 10g of Apremilast were dissolved in dichloromethane (30ml) and stirred for 30 min at 25±5C until a clear solution was obtained. Then, methyl tert butyl ether (200m) was added under stirring and seeds of Form N were added. The mass was stirred for 12h at 25±5C to give a white to cream color suspension. Obtained solid was filtered under reduced pressure and dried at 85C gave product APM Form N (as confirmed by XRPD, DSC and TGA) with purity above 99.90% (90-95% yield). (0525) Crystallization - Option III (0526) Reference is made to Figure 2. The product 10g (APM as an oil residue left after ethyl acetate distillation) was suspended in methyl tert-butyl ether (MTBE) (1000ml) at 20±5C and 0.5g of Apremilast N seeds was charged. Crystallization mixture was vigorously stirred for 16h at 20±5C to give a white to cream color suspension. Obtained solid was filtered under reduced pressure and dried at 85C gave product APM form N (as confirmed by XRPD, DSC and TGA) with purity above 99.90% (80-90% yield) |
| 40 g |
With acetic acid; at 110 - 115℃; for 1h; |
To a 500 mL RBF the product of example 5 (70 g; 0.1280 moles), <strong>[6296-53-3]3-acetamidophthalic anhydride</strong> (27.6 g; 0.1345 moles) and acetic acid (350 mL) were charged and stirred at 110 C to 115 C for 1 hour and the solution was cooled to 60-65 C. Acetic acid was distilled off completely and the residue was dissolved in dichloromethane. The dichloromethane layer was washed with DM water followed by sodium bicarbonate solution and distilled off completely and the product isolated by recrystallization in acetone :ethanol mixture to give 40 g of apremilast as a light yellow solid with chiral purity of 99.9 % and HPLC purity of 99.8 %. |
|
|
(S)- N-Acetyl Leucine salt of 2- (3-ethoxy-4-methoxyphenyl)-l-(methylsulfonyl)-eth-2-ylamine (529g, 1.183 moles) , 3- acetamidopthalic anhydride(256 g, 1.246 moles) and toluene (9051g) were refluxed under N2 atmosphere in a 20 L 4-necked RBF fitted with a Dean stark apparatus , condenser for 8 hours until HPLC shows absence of starting material. The reaction mixture was cooled to room temperature, and filtered. The wet cake was washed with sodium bicarbonate solution, water and filtered to afford 470 gm of crude APR-3 -Toluene solvate of (S) (+) Apremilast. The crude solid was dissolved in 16920 ml acetonitrile(36 volume wrt crude wt of Apremilast) at RT, treated with 25 gm Activated Carbon, and filtered through Hyflo, washed with 1880 ml acetonitrile(4 volume wrt crude wt of Apremilast) , followed by atmospheric distillation to about 4 volume. The in process residual toluene content was checked for each stripping by atmospheric distillation .When the in process residual toluene content had reached to around 100 ppm, the stripping was stopped. The reaction mass was fine filtered through Whatman filter paper , cooled to around 50 C and then poured into 37600 ml water(20 volume wrt 1880 ml ACN) , the precipitated solid was filtered, washed with water and then dried under vacuum-NLT 730 mm Hg to afford S(+) Apremilast as an amorphous solid. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping