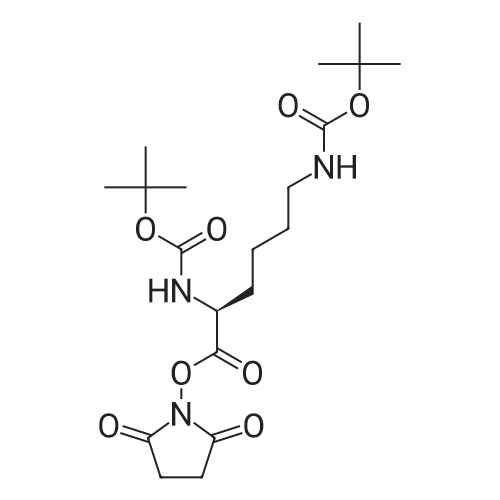
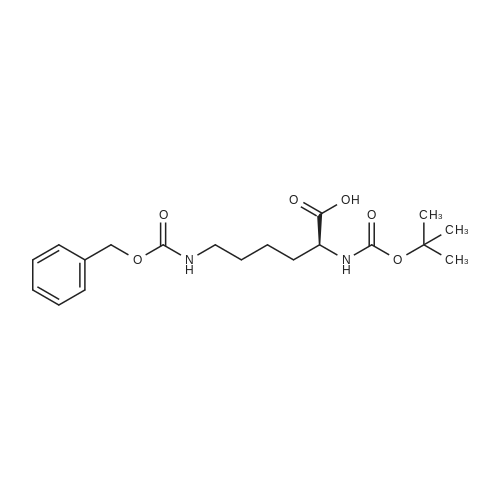
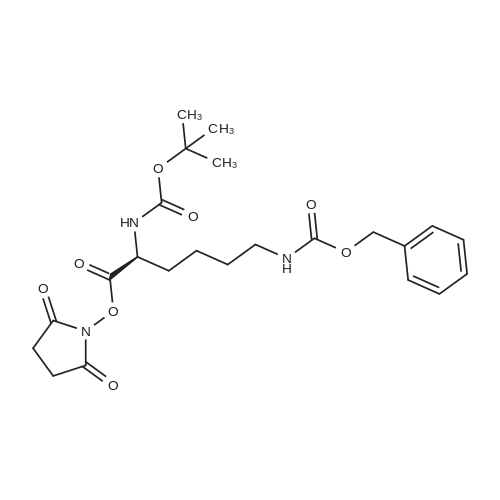
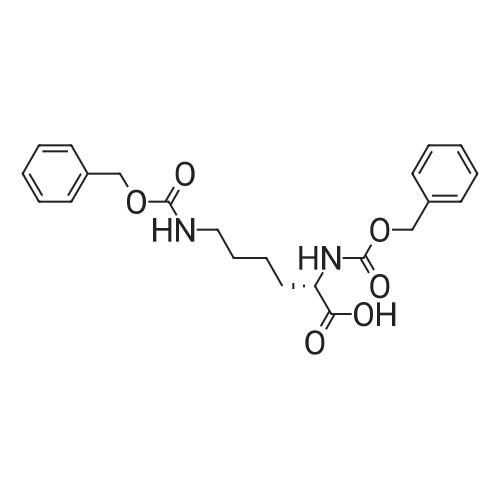
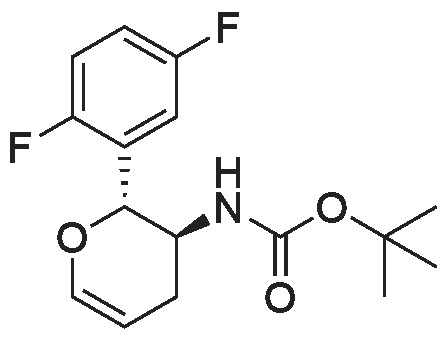
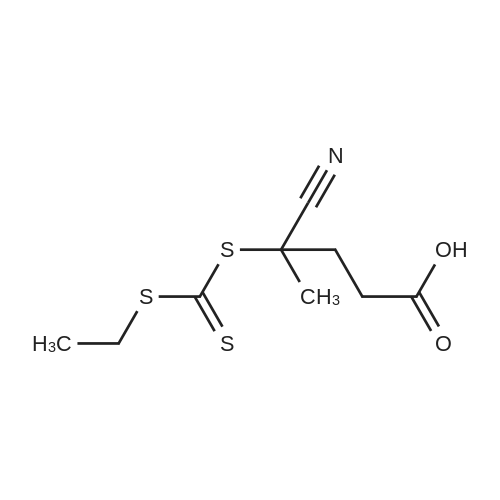
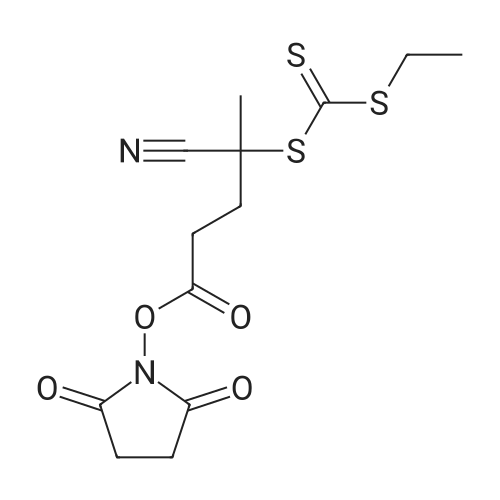
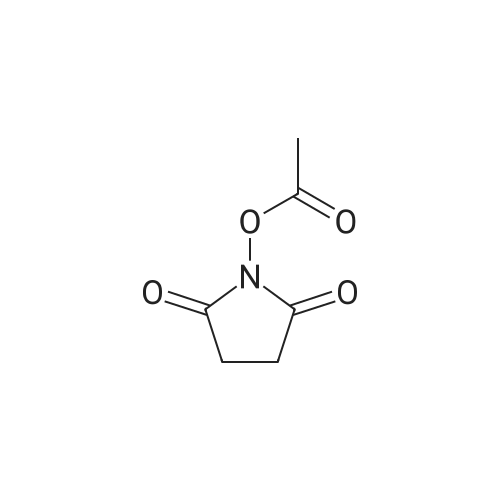
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
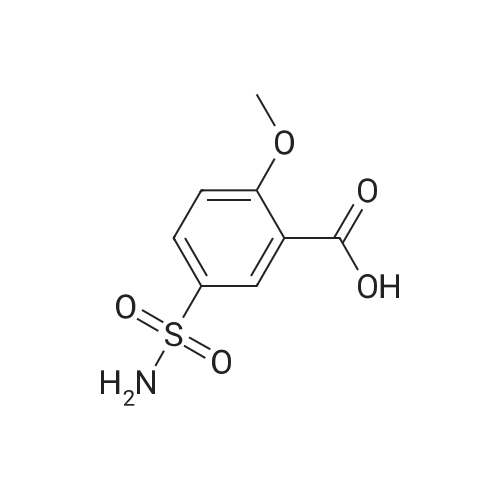
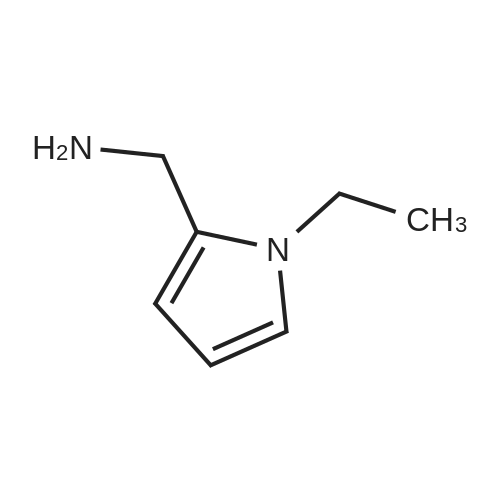
EXAMPLE 12 This example illustrates the synthesis of sulpiride with using as the condensation reagent N-succinimidyl diphenylphosphate (SDPP) which is prepared from N-hydroxysuccinimide (HOSu) and diphenylphospholy chloride. A solution of 462 mg (0.002 mol) of 2-methoxy-5-aminosulfonylbenzoic acid and 256 mg (0.002 mol) of N-ethyl-2-aminomethylpyrrole in 10 ml of acetonitrile was admixed with 694 mg (0.002 mol) of SDPP and 202 mg (0.002 mol) of triethylamine, and the admixture was stirred overnight at room temperature. The crystalline material deposited was collected by filtration and washed with acetonitrile and then with ethanol to give sulpiride, that is, 5-(aminosulfonyl)-N-[(1-ethyl-2-pyrrolidinyl) methyl]-2-methoxybenzamide. mp. 182°~185° C. Yield 24percent.
Reference:
[1] Patent: US4341707, 1982, A,
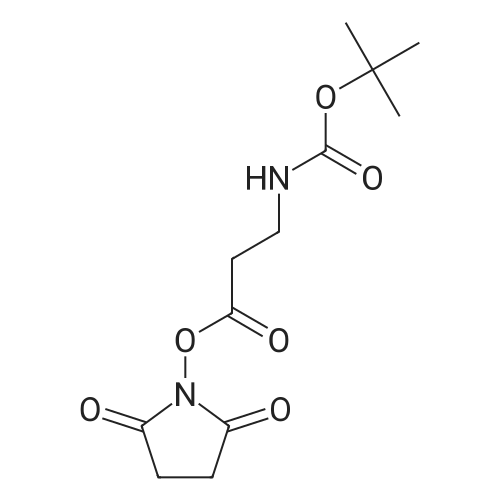
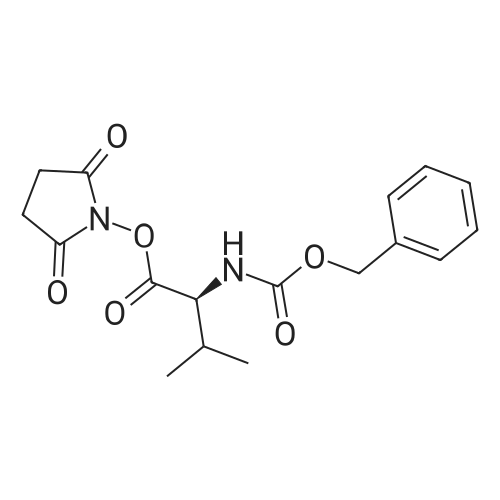
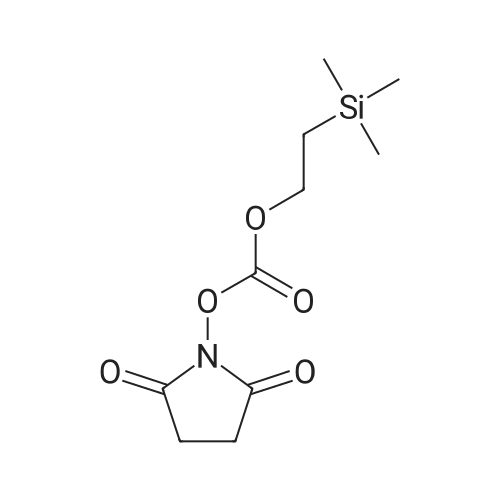
2
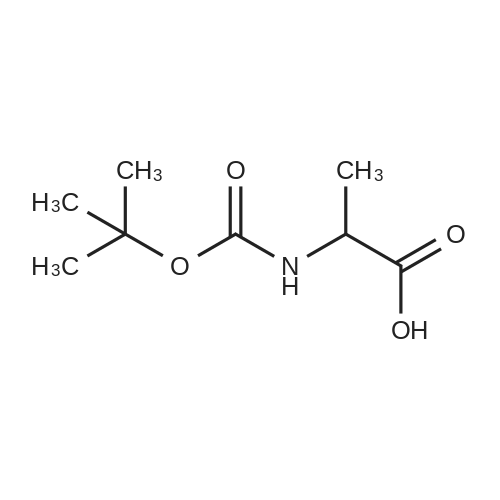
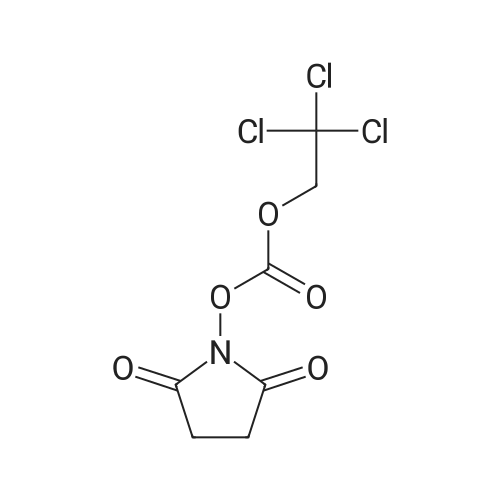
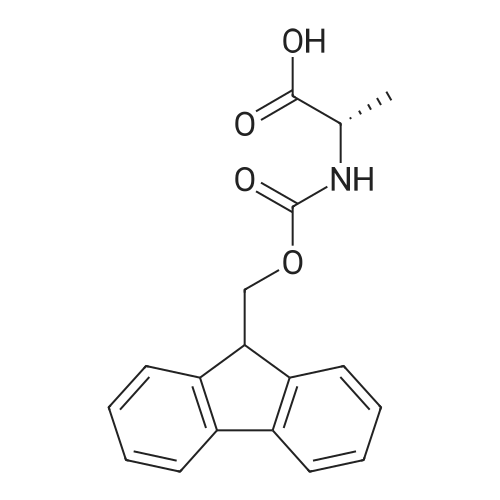
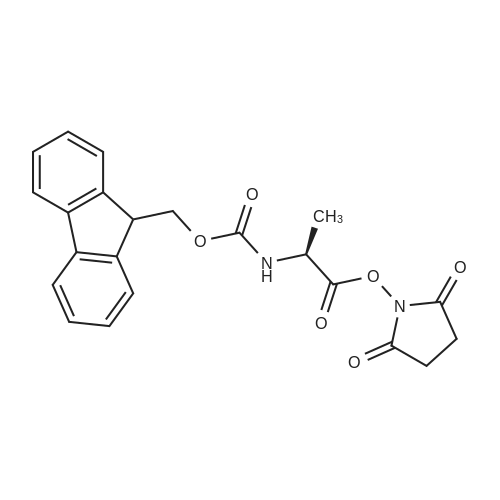
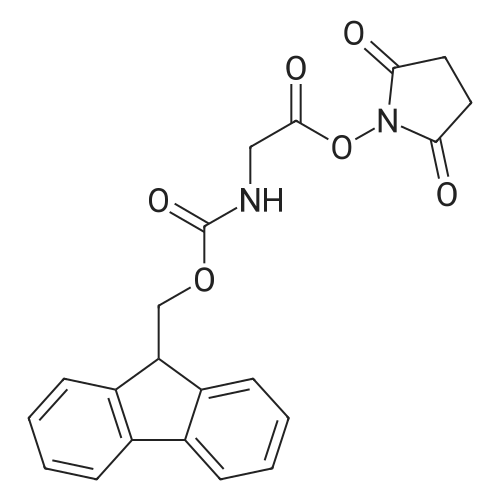
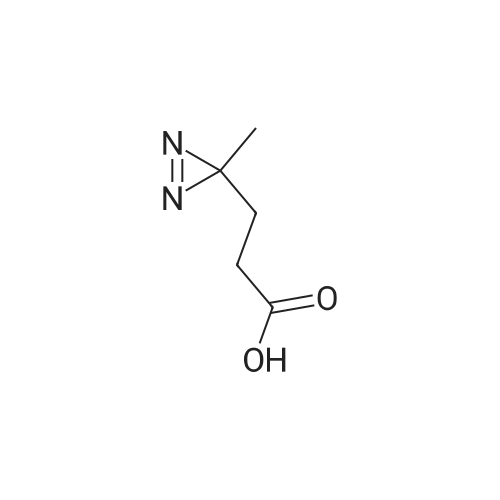
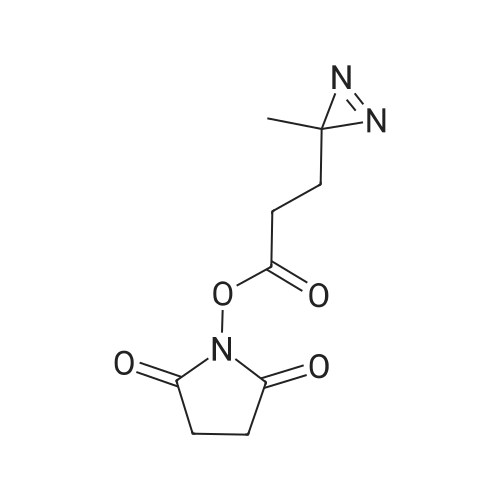
[ 6066-82-6 ]
[ 15761-38-3 ]
[ 3392-05-0 ]
Reference:
[1] Journal of Pharmaceutical Sciences, 1987, vol. 76, # 2, p. 134 - 140
[2] Chemical and pharmaceutical bulletin, 2002, vol. 50, # 2, p. 239 - 252
[3] Journal of the Chemical Society, Chemical Communications, 1985, # 8, p. 473
[4] Carbohydrate Research, 1984, vol. 132, p. 275 - 286
[5] Journal of the American Chemical Society, 1992, vol. 114, # 17, p. 6653 - 6661
[6] Journal of the American Chemical Society, 1996, vol. 118, # 29, p. 6975 - 6985
[7] Journal of Medicinal Chemistry, 1997, vol. 40, # 15, p. 2386 - 2397
[8] Chemistry of Natural Compounds, 1997, vol. 33, # 5, p. 568 - 570
[9] Chemistry of Natural Compounds, 1998, vol. 34, # 1, p. 78 - 79
[10] Journal of Medicinal Chemistry, 2003, vol. 46, # 21, p. 4543 - 4551
[11] Patent: EP2123283, 2009, A1, . Location in patent: Page/Page column 17-18
[12] Patent: US2009/325894, 2009, A1, . Location in patent: Page/Page column 11-12
[13] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 12, p. 3220 - 3224
[14] Biomacromolecules, 2010, vol. 11, # 2, p. 496 - 504
[15] Organic and Biomolecular Chemistry, 2011, vol. 9, # 11, p. 4182 - 4187
[16] Synlett, 2012, # 1, p. 142 - 144
[17] Chinese Chemical Letters, 2011, vol. 22, # 12, p. 1443 - 1446
[18] Biochimie, 2013, vol. 95, # 6, p. 1120 - 1126
[19] Chinese Chemical Letters, 2016, vol. 27, # 3, p. 357 - 360
[20] Patent: EP2612857, 2017, B1, . Location in patent: Paragraph 0137
[21] Journal of Molecular Biology, 2018, vol. 430, # 6, p. 842 - 852
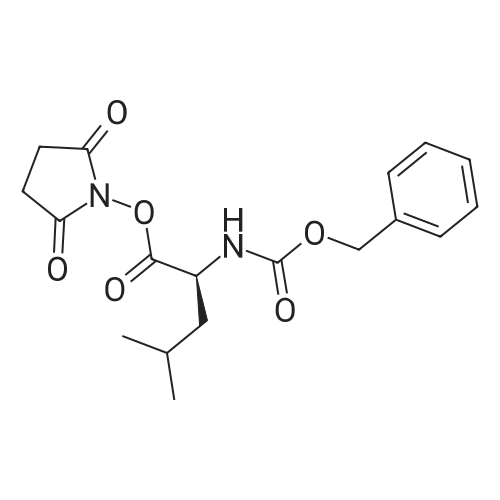
3
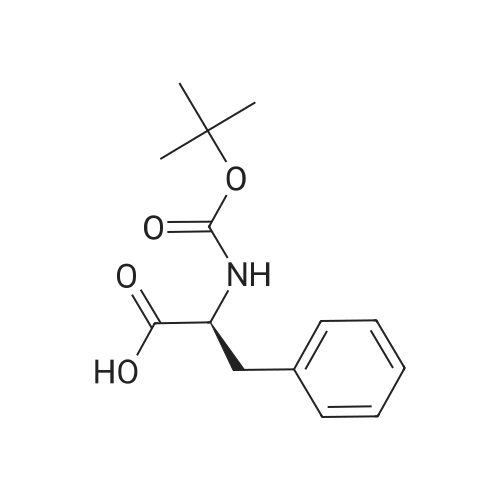
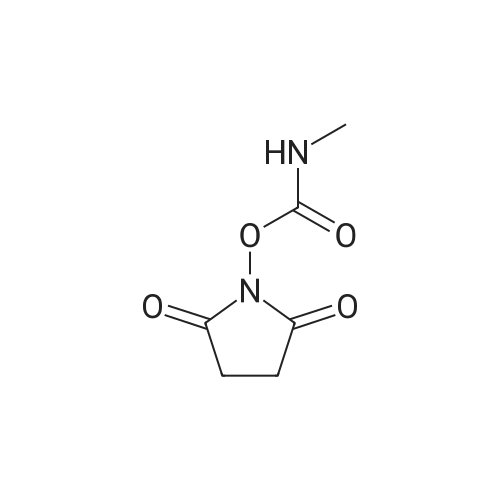
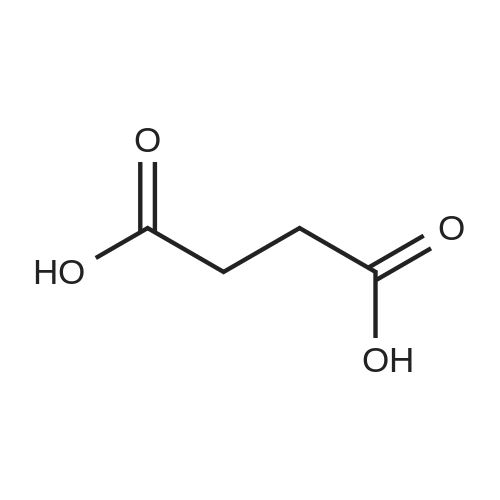
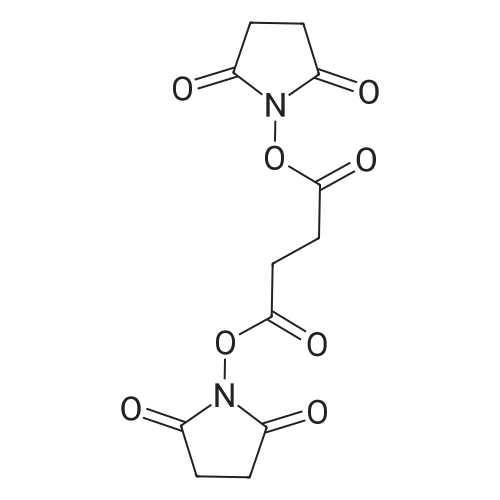
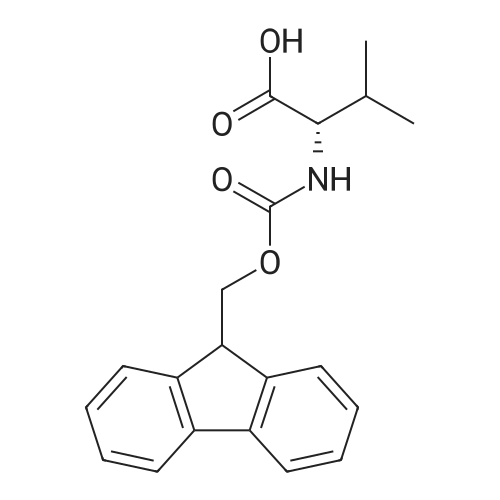
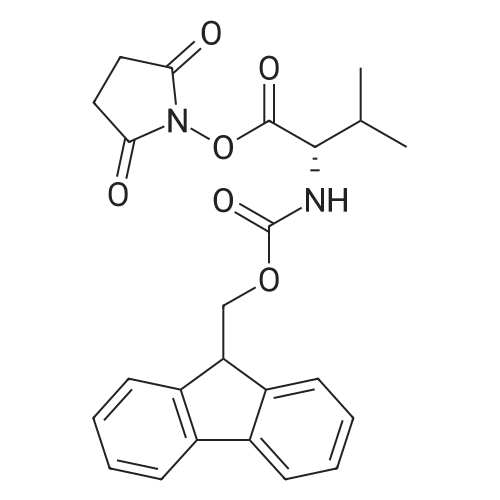
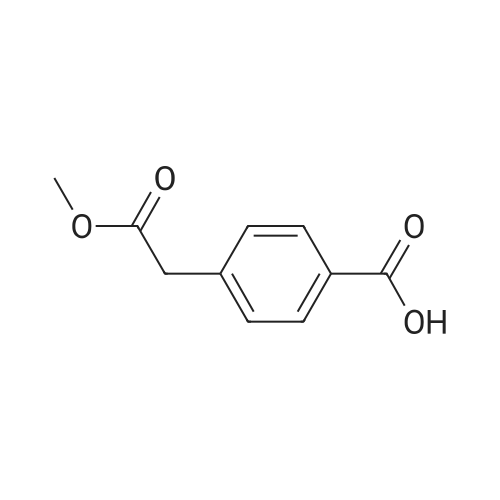
[ 6066-82-6 ]
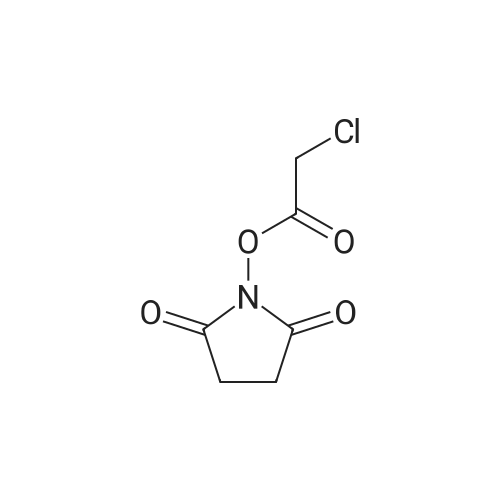
[ 3744-87-4 ]
[ 3392-05-0 ]
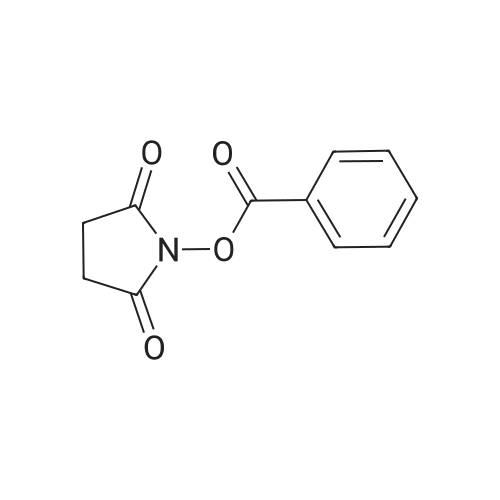
Reference:
[1] Collection of Czechoslovak Chemical Communications, 2005, vol. 70, # 10, p. 1615 - 1641
4
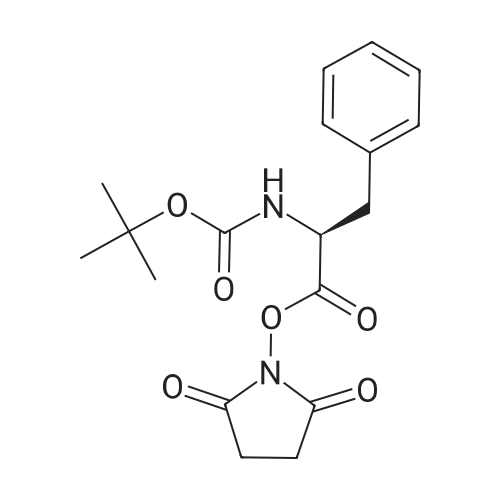
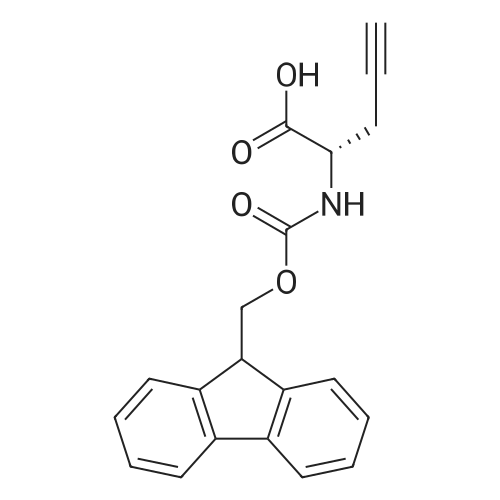
[ 6066-82-6 ]
[ 13734-34-4 ]
[ 3674-06-4 ]
Reference:
[1] European Journal of Organic Chemistry, 2014, vol. 2014, # 16, p. 3372 - 3378
[2] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1990, vol. 39, # 4.1, p. 720 - 726[3] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1990, # 4, p. 811 - 818
[4] Tetrahedron, 2007, vol. 63, # 43, p. 10637 - 10645
[5] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 15, p. 2443 - 2449
[6] Journal of Medicinal Chemistry, 2007, vol. 50, # 24, p. 6080 - 6094
[7] Angewandte Chemie - International Edition, 2018, vol. 57, # 22, p. 6475 - 6479[8] Angew. Chem., 2018, vol. 130, p. 6585 - 6589,5
[9] Organic and Biomolecular Chemistry, 2016, vol. 14, # 46, p. 10894 - 10905
[10] Chemistry Letters, 1981, p. 1331 - 1332
[11] Bioorganic and Medicinal Chemistry Letters, 1996, vol. 6, # 24, p. 2925 - 2930
[12] Journal of Medicinal Chemistry, 2003, vol. 46, # 21, p. 4543 - 4551
[13] Synlett, 2005, # 18, p. 2802 - 2804
[14] Chemistry - A European Journal, 2001, vol. 7, # 19, p. 4280 - 4295
[15] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 3, p. 1071 - 1078
[16] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 12, p. 3220 - 3224
[17] Chemistry - A European Journal, 2014, vol. 20, # 6, p. 1530 - 1538
[18] Chinese Chemical Letters, 2016, vol. 27, # 3, p. 357 - 360
[19] Research on Chemical Intermediates, 2017, vol. 43, # 10, p. 5305 - 5320
[20] Journal of Molecular Biology, 2018, vol. 430, # 6, p. 842 - 852
[21] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 8, p. 803 - 808
5
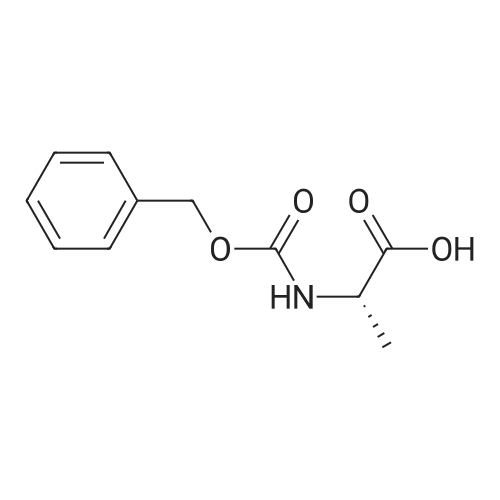
[ 6066-82-6 ]
[ 1142-20-7 ]
[ 3401-36-3 ]
Reference:
[1] Polish Journal of Chemistry, 2005, vol. 79, # 12, p. 1909 - 1917
[2] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 15, p. 2443 - 2449
[3] Liebigs Annales, 1996, # 3, p. 335 - 348
[4] Journal of Pharmaceutical Sciences, 1993, vol. 82, # 1, p. 1 - 10
[5] Chemische Berichte, 1980, vol. 113, # 11, p. 3511 - 3516
[6] Dalton Transactions, 2006, # 1, p. 137 - 148
[7] Chemistry Letters, 1985, p. 123 - 126
[8] Bulletin of the Chemical Society of Japan, 1987, vol. 60, # 7, p. 2409 - 2418
[9] Journal of Pharmaceutical Sciences, 1994, vol. 83, # 7, p. 989 - 998
[10] Journal of Medicinal Chemistry, 1994, vol. 37, # 18, p. 2841 - 2845
[11] Bioorganic & Medicinal Chemistry Letters, 1998, vol. 8, # 23, p. 3343 - 3346
[12] Chemistry - A European Journal, 2004, vol. 10, # 16, p. 3879 - 3890
[13] Tetrahedron Letters, 2008, vol. 49, # 48, p. 6885 - 6888
[14] Biochimie, 2013, vol. 95, # 6, p. 1120 - 1126
[15] Tetrahedron, 2013, vol. 69, # 2, p. 551 - 558
[16] Chemistry - A European Journal, 2013, vol. 19, # 48, p. 16256 - 16262
[17] Organic Preparations and Procedures International, 2014, vol. 46, # 4, p. 370 - 375
[18] Organic and Biomolecular Chemistry, 2016, vol. 14, # 47, p. 11125 - 11136
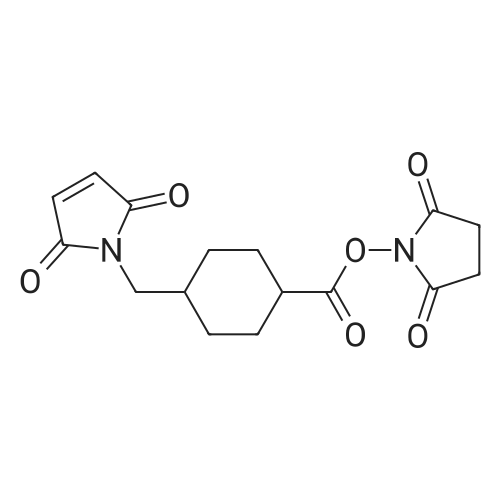
6
[ 30364-57-9 ]
[ 56-40-6 ]
[ 6066-82-6 ]
[ 13214-64-7 ]
Reference:
[1] Journal of Organic Chemistry, 1988, vol. 53, # 15, p. 3583 - 3586
7
[ 6066-82-6 ]
[ 64-69-7 ]
[ 39028-27-8 ]
Yield
Reaction Conditions
Operation in experiment
30%
at 0 - 20℃; for 4 h;
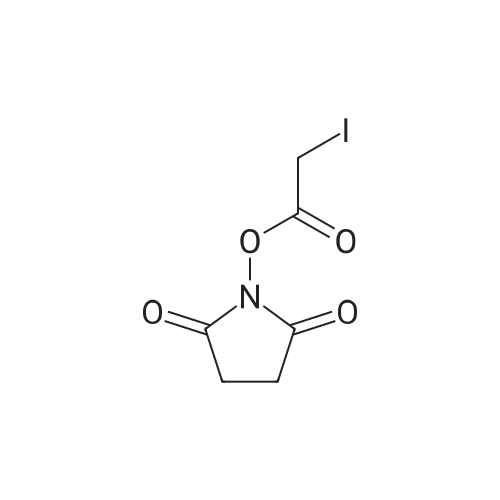
On a solution of N-hydroxysuccinimide (12.6 mmol) and dicyclohexylcarbodiimide (20.3 mmol) at 0° C., corresponding acid (6 mmol) was added and allowed to react for 4 hours at room temperature. In the case of compound VIa, a solution of maleic anhydride (10 mmol) and β-alanine was added in N,N-dimethylformamide, which has been previously made react for 1 hour. After 4 hours, mixture was evaporated at reduced pressure and the crude was dissolved in dichloromethane and washed with water. Organic extracts were dried with anhydrous magnesium sulfate, filtered and evaporated to dryness. Resulting residue was recrystallized to give desired compound. [0112] Using this methodology, and corresponding acid, the following compounds were prepared: Succinimidyl iodoacetate (VIb, 30percent yield). 1H NMR (CDCl3) δ ppm: 2.87 (2H, s), 3.96 (1H, s); 13C NMR (CDCl3) δ ppm: −12.47 (1C, s) 25.85 (2C, s) 164.78 (1C, s) 168.78 (2C, s).
Reference:
[1] Organic and Biomolecular Chemistry, 2016, vol. 14, # 32, p. 7777 - 7791
[2] Patent: US2013/273581, 2013, A1, . Location in patent: Page/Page column 0092; 0111-0112; 0114
[3] Farmaco, 1992, vol. 47, # 11, p. 1367 - 1383
8
[ 6066-82-6 ]
[ 501-53-1 ]
[ 13139-17-8 ]
Reference:
[1] Bulletin des Societes Chimiques Belges, 1987, vol. 96, # 10, p. 775 - 782
[2] Bulletin of the Chemical Society of Japan, 1987, vol. 60, # 7, p. 2409 - 2418
[3] Chemical and Pharmaceutical Bulletin, 1999, vol. 47, # 10, p. 1489 - 1490
[4] Organic Preparations and Procedures International, 2002, vol. 34, # 5, p. 531 - 537
[5] Patent: US3974137, 1976, A,
9
[ 108-31-6 ]
[ 6066-82-6 ]
[ 107-95-9 ]
[ 55750-62-4 ]
Yield
Reaction Conditions
Operation in experiment
80%
Stage #1: at 70℃; Inert atmosphere Stage #2: With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In acetonitrile at 0 - 70℃;
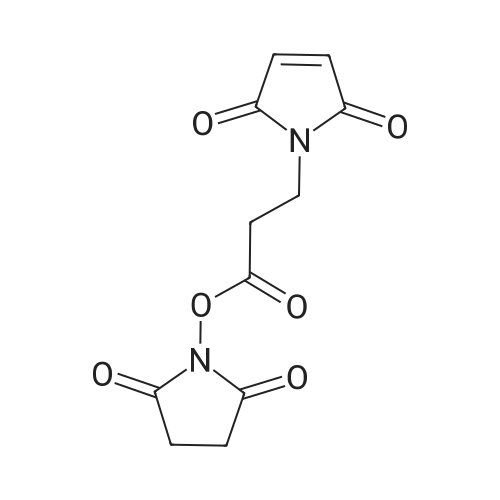
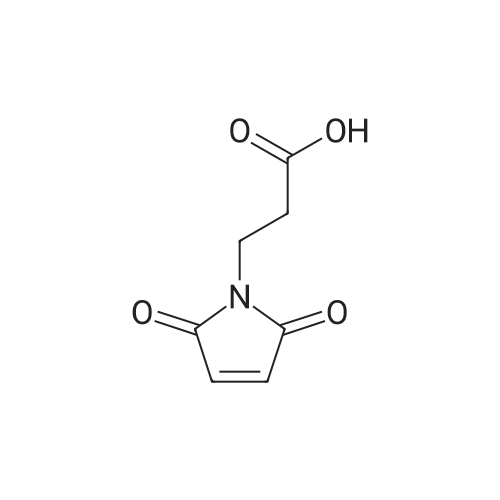
EXAMPLE 1N-Maleimidopropionic acid NHS ester[0083] Maleic anhydride, β-alanine (1 mol eq.), and acetonitrile (ACN, 25 volumes) were charged to a vessel. The slurry was stirred under nitrogen and heated to 70 0C. The temperature was maintained at 70 0C for a further 5 to 8 hours. After cooling to 5 0C, NHS (1 mol eq.) was charged, followed by Λ/-(3-dimethylaminopropyl)-Λf'-ethylcarbodiimide hydrochloride (EDCI, 1 mol eq.). The temperature was maintained at 0 - 5 0C for 1 hour before adding a further charge of EDCI (1 mol eq.). The mixture was heated to 70 0C and maintained at this temperature for 7 hours. The reaction mixture was cooled to 200C and concentrated under vacuum at a maximum temperature of 45°C until the rate of solvent distillation was negligible. Dichloromethane (DCM, 40 volumes) was added to the residue, which was stirred until a solution was formed. This solution was washed with aqueous ammonium chloride (12 percent w/w, 25 wt eq.) then with aqueous sodium chloride (24 percent w/w, 25 wt eq.). The organic solution was dried by stirring with magnesium sulphate (1 wt eq.) at ambient temperature for 1 to 2 hours. The inorganics were filtered off under vacuum and the filter cake was washed with DCM (4 volumes). The filtrates were concentrated under vacuum, whilst gradually replacing DCM with isopropylacetate (IPAC). The residual slurry was cooled and stirred at ambient temperature for 1 hour before filtering under vacuum. The product filter cake was washed with IPAC (4 volumes) and then dried to constant mass under vacuum with slow rotation at 40 0C to yield the desired compound as an off white solid. The typical yield is 60 to 80 percent of the maximum theoretical yield.[0084] The product was characerised by NMR as follows: 1H NMR (400MHz, d6-DMSO): δ 2.80 (4H, br s), δ 3.05 (2H, t, J ~ 7Hz), δ 3.75 (2H, t J ~ 7Hz), δ 7.05 (2H, s).
60%
Stage #1: at 20℃; for 2.5 h; Stage #2: With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 0 - 20℃;
To a solution of maleic anhydride (2.00 g, 20.4 mmol) in 24 mL of DMF was added β-alanine (1.82 g, 20.4 mmol)and the mixture was stirred for 2.5 h at room temperature in which the solid was gradually dissolved to afford clear solution. The reaction was cooled in an ice bath and N-hydroxysuccinimide (HOSu) (2.88 g, 25 mmol) andN,N'-dicyclohexylcarbodiimide (8.24 g. 40.0 mmol) were successively added to the solution. After 30 min, the reaction was warmed to room temperature and stirred for 14 h. The reaction mixture was filtered through a cottonplug and the residue was washed with DCM and water. The filtrates were gathered and the organic layer was washed with satd NaHCO3 (three times) and brine, dried over Na2SO4 and concentrated to afford 1 (3.20 g, 60percent) as a white solid. 1H NMR (400 MHz, d6-DMSO): 2.78 (s, 4H), 3.03 (t, J = 6.9 Hz, 2H), 3.73 (t, J = 6.9 Hz, 2H),7.02 (s, 2H); 13C NMR (100 MHz, d6-DMSO): 25.4, 29.0, 32.7, 134.6, 166.7, 169.9, 170.5.
25%
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 0 - 20℃; for 1 h;
On a solution of N-hydroxysuccinimide (12.6 mmol) and dicyclohexylcarbodiimide (20.3 mmol) at 0° C., corresponding acid (6 mmol) was added and allowed to react for 4 hours at room temperature. In the case of compound VIa, a solution of maleic anhydride (10 mmol) and β-alanine was added in N,N-dimethylformamide, which has been previously made react for 1 hour. After 4 hours, mixture was evaporated at reduced pressure and the crude was dissolved in dichloromethane and washed with water. Organic extracts were dried with anhydrous magnesium sulfate, filtered and evaporated to dryness. Resulting residue was recrystallized to give desired compound. [0112] Using this methodology, and corresponding acid, the following compounds were prepared: [0113] 3-succinimidyl maleimidopropionate (VIa, 25percent yield). 1H NMR (CDCl3) δ ppm: 2.82 (4H, s), 3.02 (2H, t, J=7.07 Hz), 3.94 (2H, t, J=7.07 Hz), 6.74 (2H, s); 13C NMR (CDCl3) δ ppm: 25.5 (2C, s), 29.7 (1C, s), 32.9 (1C, s), 134.3 (2C, s), 166.0 (1C, s), 168.7 (2C, s), 170.1 (2C, s).
Reference:
[1] Patent: WO2011/23680, 2011, A2, . Location in patent: Page/Page column 13-14
[2] Angewandte Chemie - International Edition, 2015, vol. 54, # 35, p. 10198 - 10201[3] Angew. Chem., 2015, vol. 127, p. 10336 - 10339
[4] Journal of the American Chemical Society, 2013, vol. 135, # 29, p. 10582 - 10585
[5] Journal of the American Chemical Society, 2004, vol. 126, # 3, p. 734 - 735
[6] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 5, p. 1721 - 1728
[7] Macromolecular Bioscience, 2012, vol. 12, # 9, p. 1209 - 1219
[8] Patent: US2013/273581, 2013, A1, . Location in patent: Paragraph 0111-0113
[9] Synthesis, 1991, # 10, p. 819 - 821
[10] Patent: WO2011/9047, 2011, A2, . Location in patent: Page/Page column 13
[11] Patent: US2013/157375, 2013, A1, . Location in patent: Paragraph 0103; 0104
[12] Chemical Communications, 2014, vol. 50, # 26, p. 3473 - 3475
[13] Patent: WO2014/100762, 2014, A1, . Location in patent: Paragraph 248
[14] Chemical Communications, 2015, vol. 51, # 53, p. 10758 - 10761
[15] Angewandte Chemie - International Edition, 2017, vol. 56, # 9, p. 2356 - 2360[16] Angew. Chem., 2017, vol. 129, # 9, p. 2396 - 2400,5
10
[ 6066-82-6 ]
[ 7423-55-4 ]
[ 55750-62-4 ]
Reference:
[1] Analytical Chemistry, 2016, vol. 88, # 22, p. 10837 - 10841
[2] Angewandte Chemie - International Edition, 2016, vol. 55, # 40, p. 12243 - 12247[3] Angew. Chem., 2016, vol. 128, # 40, p. 12431 - 12435,5
[4] International Journal of Molecular Sciences, 2017, vol. 18, # 9,
[5] Tetrahedron Letters, 1998, vol. 39, # 11, p. 1321 - 1324
[6] Organic Letters, 2004, vol. 6, # 20, p. 3561 - 3564
[7] Journal of Medicinal Chemistry, 2006, vol. 49, # 21, p. 6400 - 6407
[8] Chemical Communications, 2011, vol. 47, # 25, p. 7068 - 7070
[9] Chemistry - A European Journal, 2011, vol. 17, # 46, p. 13059 - 13067
[10] Patent: US2014/56810, 2014, A1, . Location in patent: Paragraph 0181
[11] Organic and Biomolecular Chemistry, 2014, vol. 12, # 34, p. 6624 - 6633
[12] Journal of Controlled Release, 2015, vol. 220, p. 660 - 670
[13] Patent: WO2015/196167, 2015, A1, . Location in patent: Paragraph 0269; 0277
11
[ 108-31-6 ]
[ 6066-82-6 ]
[ 56-41-7 ]
[ 55750-62-4 ]
Yield
Reaction Conditions
Operation in experiment
64%
Stage #1: at 60℃; for 3 h; Stage #2: With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 0 - 22℃; for 18.5 h;
To a solution of maleic anhydride (8; 86.0 g, 877 mmol) in DMF(1.03 L) was added β-alanine (9; 78.14 g, 877 mmol). The resultingsuspension was heated to 60 °C and, after 1 h, a solution was obtained.After 2 h, the mixture was cooled to 0–5 °C and N-hydroxysuccinimide(29; 126.17 g, 1.09 mol) was added followed by DCC(361.9 g, 1.75 mol) over 30 min in several portions while the internaltemperature was held below 22 °C. The thick, white slurry wasthen stirred at 20 °C for 18 h. The slurry was filtered and the solid(dicyclohexyl urea) was washed with H2O (1 L) and CH2Cl2 (1 L)and discarded. To the filtrates was added CH2Cl2 (1 L) and the phaseswere separated. The aqueous layer was extracted with CH2Cl2(2 × 300 mL) and the combined organic extracts were washed withH2O [500 mL; a small amount of brine (50 mL) was added to facilitatephase separation], sat. aq NaHCO3 (2 × 300 mL), and dried(MgSO4). The solvent was removed to give an oily, light tan solidthat was slurried in EtOH (520 mL) for 2 h at 20 °C. The solid wasfiltered, washed with EtOH (2 × 75 mL) and dried under vacuum at30 °C for 72 h to give 186.35 g (64percent) of NHS ester 30 as a whitesolid;32 HPLC purity: 96.6percent (areapercent); mp 169–171 °C.IR (ATR cell): 3088, 2954, 1825, 1783, 1705, 1583, 1446, 1433,1382, 1325, 1298, 1250, 1211, 1149, 1070, 1049, 998, 956, 901,836, 813, 786, 767, 756, 696, 651, 634, 596, 561, 544, 528 cm–1.1H NMR (400 MHz, DMSO-d6): δ = 2.80 (s, 4 H), 3.05 (t, J = 6.90Hz, 2 H), 3.75 (t, J = 6.78 Hz, 2 H), 7.05 (s, 2 H).13C NMR (100 MHz, DMSO-d6): δ = 25.38, 29.00, 32.67, 134.63,166.72, 169.93, 170.52.HRMS (ESI): m/z [M + H]+ calcd for C11H11N2O6: 267.06116;found: 267.06147.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 20, p. 5032 - 5038
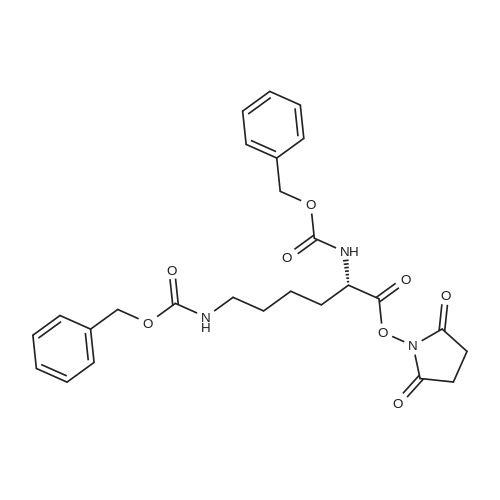
14
[ 6066-82-6 ]
[ 25021-08-3 ]
[ 55750-61-3 ]
Reference:
[1] Bioconjugate Chemistry, 2012, vol. 23, # 12, p. 2417 - 2433
[2] Journal of Medicinal Chemistry, 1984, vol. 27, # 10, p. 1325 - 1335
15
[ 6066-82-6 ]
[ 17686-36-1 ]
[ 55750-61-3 ]
Yield
Reaction Conditions
Operation in experiment
116 g
With triethylamine In dichloromethane at 20℃; for 2 h;
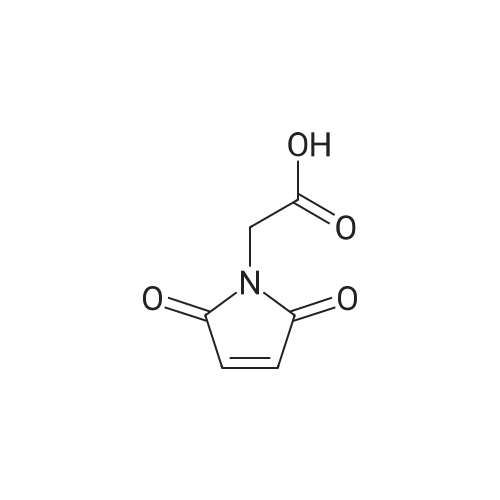
To a 500 ml three-necked flask, N-maleimidyl acetic acid (94 g, 0.6 mol)And dichloromethane 300ml,Oxalyl chloride (90 g, 0.72 mol) was then added,The reaction was warmed to reflux, a large amount of gas was released.After 2h the reaction became clear,Concentrate to remove methylene chloride and excess oxalyl chloride,Addition of 200 ml of methylene chloride to remove the residual acid in one pass afforded crude N-maleimidoacetyl chloride.The crude product was dissolved in 200 ml of dichloromethane,Was added dropwise to N-hydroxysuccinimide (69 g, 0.6 mol)Triethylamine (80.8 g, 0.8 mol)Dissolved in 300 ml of dichloromethane,Ice water cooling control temperature does not exceed 20 , drip finished room temperature 2h.The reaction solution was washed with 200 ml of water to remove triethylamine hydrochloride,1N hydrochloric acid 100ml washing to remove excess triethylamine,200ml saturated salt water, washed and dried,Concentrated to dry off like white solid 137g,After recrystallization from a mixed solvent of ethyl acetate and petroleum ether, 116 g of a white solid was obtained,HPLC purity 98.7percent, 76.7percent yield.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In 1,4-dioxane at 20℃; Inert atmosphere
In a dry flask containing a magnetic stir bar, 0.50 g of 1 (2.64 mmol) and 0.31 g of N-hydroxysuccinimide (SuOH) (2.72 mmol, 1.03 equivalent) were dissolved in 3mL of distilled dioxane. Subsequently, 1.25 equivalent of 3-(ethyliminomethyleneamino)-N,N-dimethylpropan-l-amine in hydrogen chloride form (EDC) (0.63 g, 3.3 mmol) was added into the flask. The solution was cloudy, and 2mL of freshly distilled dioxane was added into the reaction mixture. The reaction mixture was stirred under nitrogen for 30 minutes and then left for stirring overnight at room temperature. Dioxane was removed under vacuum. The residue was dissolved in chloroform and washed with water (3x). The organic layer was dried of over Na2S04 and concentrated under vacuum. The desire product was precipitated in ethanol and dried in a desiccator, yielding 0.47g of product (62percent). The 1H NMR spectral data matched the previously reported values for the compound.
Reference:
[1] ACS Medicinal Chemistry Letters, 2018, vol. 9, # 6, p. 557 - 561
[2] Patent: WO2017/53486, 2017, A1, . Location in patent: Page/Page column 11
[3] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1981, p. 529 - 537
[4] Bioorganic and Medicinal Chemistry Letters, 1998, vol. 8, # 24, p. 3603 - 3608
[5] Chemical Communications, 2010, vol. 46, # 20, p. 3553 - 3555
[6] Patent: WO2011/133875, 2011, A2, . Location in patent: Page/Page column 46-47
[7] Patent: WO2013/110002, 2013, A1, . Location in patent: Page/Page column 45-46
[8] Patent: WO2013/109998, 2013, A1, . Location in patent: Page/Page column 45
[9] Bioconjugate Chemistry, 2018, vol. 29, # 4, p. 1454 - 1465
18
[ 6066-82-6 ]
[ 28920-43-6 ]
[ 74124-79-1 ]
[ 102774-86-7 ]
Yield
Reaction Conditions
Operation in experiment
95 kg
With tributyl-amine In tetrahydrofuran at -2 - 5℃; Large scale
From the reactor solid feed port one - time investment HosuL43. 8Kg (l. 25KmOl),With stirring,So that the kettle liquid at -2 ° C ~ 0 ° C.The tributylamine300Kg (l. 62 kP)And tetrahydrofuran450Kg into the high slot,After mixing,In 10 ~ llhr slowly added to the reactor in the reaction,The amount of added per hour for the 50 ~ 57Kg;And keep the kettle temperature not to exceed 5 ° C,Because of the exothermic reaction,After adding tributylamine in tetrahydrofuran solution,The reaction vessel was allowed to warm to room temperature and the reaction was continued for 7.5 h.A large amount of white solid crystals were observed to be precipitated by DSC from the reaction vessel.(3) filtered DSC crude: the reactor material in lhr evenly introduced into a fully enclosed centrifuge in N2Under the protection of throwing dry, and then twice the introduction of about 0 ° C tetrahydrofuran solvent 100Kg in the centrifuge rinse, throw dry,The DSC wet goods 120Kg. The wet goods in a vacuum oven at 40 ~ 45 ° C drying 7 ~ 8hr to constant weight, get out of the white crystalline powder 95Kg; HPLC analysis of 99. 1percent. The resulting filtrate was centrifuged to proceed to the next step. (3) filtered to obtain DSC crude: the reactor material in 1hr evenly introduced into a fully enclosed centrifuge, in the N2Under the protection of throwing dry, and then twice the introduction of about 0 ° C tetrahydrofuran solvent 100Kg in the centrifuge rinse, throw dry, get DSC wet goods 120Kg.The wet product in a vacuum oven at 40 ~ 45 dry 7 ~ 8hr to constant weight, get out of the white crystalline powder 95Kg; HPLC analysis of 99.1percent.The resulting filtrate was centrifuged to proceed to the next step.(4) The centrifugal filtrate in step (3) is combined with two washing solvents and introduced into a 2000L distilling apparatus. The tetrahydrofuran solvent is removed at a vacuum control temperature of 0 to 5 DEG C, and the inside temperature of the kettle is kept at 40 to 42 DEG C .After the tetrahydrofuran was removed, 230 kg of dichloroethane was introduced, stirred at 40-50 ° C for 0.5 hr, and washed with 160 kg of water 3 times. The phases were separated and the dichloromethane was introduced into a 500 L autoclave.
With dicyclohexyl-carbodiimide In acetonitrile at 0 - 20℃; Inert atmosphere
Under nitrogen protection, 4.7 g (22 mmol) of MC and 25 g (22 mmol) of HOSu were added to 50 ml of acetonitrile.Another 4.5 g (22 mmol) of DCC was dissolved in 25 ml of acetonitrile,Keeping the internal temperature around 0°C,It was slowly dropped into the reaction solution.The reaction solution was reacted at 0°C for 2 hours and then reacted overnight at room temperature.filter,The filter cake was washed with acetonitrile 10ml×3.The filtrate was concentrated to dryness under reduced pressure.The resulting oil was dried under reduced pressure at room temperature for 6 h to give 6.4 g of light brown solid.Yield 95percent.
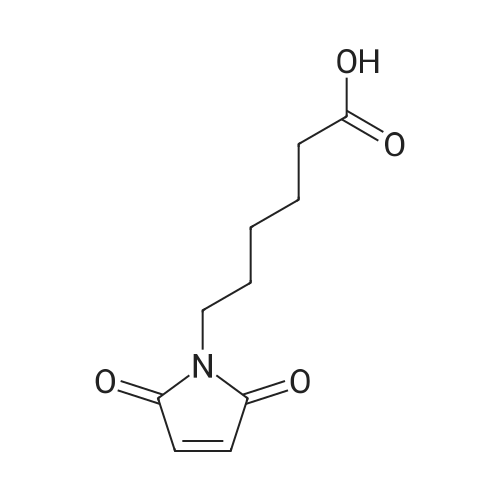
70%
With dicyclohexyl-carbodiimide In dichloromethane at 20℃; for 3 h;
Dicyclohexyl-carbodiimide (2.68 g, 13.02 mmol) and N-hydroxysuccinimide (1 .36 g, 1 1 .84 mmol) are added at room temperature to a stirrer solution of compound [4] (2.5 g, 1 1 .84 mmol) in anhydrous dichloromethane (15 mL) and the mixture stirred at room temperature for 3 hours. The white solid is filtered with dichloromethane to remove the dicyclohexylurea, the organic phase is washed with HCI 0.1 N and water, then dried over anhydrous sodium sulfate and the solvent removed by rotatory evaporation. The resulting residue is subjected to flash column chromatography with a medium pressure system Sepacore® Buchi (silica gel; gradient A: petroleum ether/B: ethyl acetate; Bpercent 0-80 in 15 minutes) to give the activated acid [5] as a white solid, 2.4 g (70percent). MS: m/z 309 [M+H]+.1H NMR (400 MHz, MeOD) δ 6.78 (s, 2H), 3.49 - 3.46 (m, 2H), 2.82 (s, 4H), 2.62 - 2.59 (m, 2H), 1 .78 - 1 .65 (m, 2H), 1 .64 - 1 .50 (m, 2H), 1 .46 - 1 .26 (m, 2H).13C NMR (100 MHz, MeOD) δ 169.8, 169.0, 167.3, 132.5, 35.4, 28.6, 26.1 , 23.8, 23.6, 22.3.
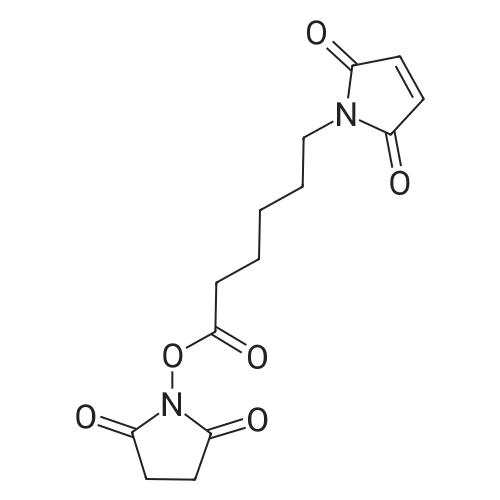
48%
With dicyclohexyl-carbodiimide In ethyl acetate at 0 - 20℃; for 20 h;
To a stirred solution of 6-(2,5-dioxo-2,5-dihydro-lH-pyrrol-l-yl)hexanoate (3.08 g, 14.6 mmol, 1.0 eq) and N-hydroxysuccinimide (1.76 g, 15.3 mmol, 1.05 eq) in EtOAc (30 mL) at 0 °C, was added dicyclohexylcarbodiimide (3.16 g, 15.3 mmol, 1.05 eq). The reaction was then allowed to warm to rt. After 20 h, the reaction was filtered and washed with EtOAc and the filtrate concentrated. The residue was purified by flash chromatography to yield the title compound (2.16 g, 48percent) as a clear oil that solidified slowly to a waxy white solid. NMR (400 MHz, Chloroform-i/) δ 6.71 (s, 2H), 3.56 (t, J = 7.2 Hz, 2H), 2.86 (s, 4H), 2.63 (t, J = 7.4 Hz, 2H), 1.80 (p, J= 7.4 Hz, 2H), 1.73 - 1.57 (m, 2H), 1.50 - 1.35 (m, 2H). m/z calcd. for C14H16N206 = 308.10. Found [M+H]+ = 309.13. Rf = 0.28 (50percent EtOAc/Hex).
48%
With dicyclohexyl-carbodiimide In ethyl acetate at 0 - 20℃; for 20 h;
To a stirred solution of 6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-l-yl)hexanoate (3.08 g, 14.6 mmol, 1.0 eq) and N-hydroxysuccinimide (1.76 g, 15.3 mmol, 1.05 eq) in EtOAc (30 mL) at 0 °C,was added dicyclohexylcarbodiimide (3.16 g, 15.3 mmol, 1.05 eq). The reaction was then allowed towarm to rt. After 20 h, the reaction was filtered and washed with EtOAc and the filtrate concentrated. The residue was purified by flash chromatography to yield the title compound (2.16 g, 48percent) as a clear oil that solidified slowly to a waxy white solid. ‘H NMR (400 MHz, Chloroform-d) ö 6.71 (s, 2H), 3.56 (t, J = 7.2 Hz, 2H), 2.86 (s, 4H), 2.63 (t, J = 7.4 Hz, 2H), 1.80 (p, J = 7.4 Hz, 2H), 1.73 — 1.57 (m,2H), 1.50 1.35 (m, 2H). m/z calcd. for C,4H,6N206 = 308.10. Found [M+H] = 309.13. Rf= 0.28 (50percent EtOAc/Hex)
Reference:
[1] Patent: CN107789630, 2018, A, . Location in patent: Paragraph 0064; 0065; 0066
[2] Patent: WO2018/178060, 2018, A1, . Location in patent: Page/Page column 58; 60; 61
[3] International Journal of Molecular Sciences, 2017, vol. 18, # 9,
[4] Patent: WO2015/95953, 2015, A1, . Location in patent: Page/Page column 118
[5] Patent: WO2016/41082, 2016, A1, . Location in patent: Page/Page column 97; 98
[6] Organic and Biomolecular Chemistry, 2014, vol. 12, # 34, p. 6624 - 6633
[7] Patent: CN108743968, 2018, A, . Location in patent: Paragraph 0007; 0008; 0009
22
[ 108-31-6 ]
[ 6066-82-6 ]
[ 60-32-2 ]
[ 55750-63-5 ]
Reference:
[1] Chemistry - A European Journal, 2012, vol. 18, # 41, p. 13091 - 13096
[2] Journal of Medicinal Chemistry, 2012, vol. 55, # 9, p. 4516 - 4520
[3] Synthesis (Germany), 2014, vol. 46, # 22, p. 3085 - 3096
23
[ 6066-82-6 ]
[ 57079-14-8 ]
[ 55750-63-5 ]
Reference:
[1] Journal fuer Praktische Chemie (Leipzig), 1985, vol. 327, # 5, p. 789 - 798
[2] Journal of Organic Chemistry, 2011, vol. 76, # 21, p. 9169 - 9174
24
[ 6066-82-6 ]
[ 55750-53-3 ]
[ 55750-63-5 ]
[ 57-13-6 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2009, vol. 7, # 17, p. 3400 - 3406
25
[ 6066-82-6 ]
[ 57079-14-8 ]
[ 55750-63-5 ]
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 21, p. 9169 - 9174
26
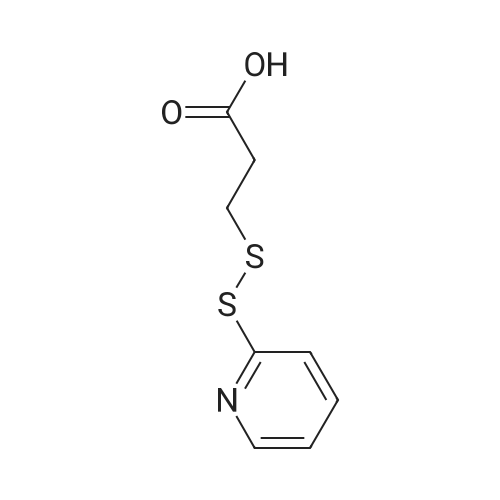
[ 6066-82-6 ]
[ 1119-62-6 ]
[ 57757-57-0 ]
Yield
Reaction Conditions
Operation in experiment
85%
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane at 20℃; for 2 h;
To the solution of 3,3'-dithioldipropionic acid (1 g, 4.75 mmol) in dichloromethane (20 mL), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (WSC) (2.16 g, 10.5 mmol) and N-hydroxysuccinimide (1.21 g, 10.5 mmol) were added. The mixture was stirred for 2 h at room temperature. The solution was concentrated in vacuo, and the obtained residue was subjected to silica column chromatography (ethyl acetate/hexane = 1:5) to give the NHS ester 2 (1.63 g, 85percent). 1H NMR (DMSO-d6, 400 MHz) δ 3.16 (m, 8H), 2.82 (s, 8H). FAB (DMSO-d6) [(M+H)+] calcd 405, found 405.
70%
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane at 20℃;
One kind of bis(succinimidyl) 3,3'-dithiodipropionate synthesis, the following steps: -; Weigh 3,3'-thiodipropionic acid (6.0g, 0.028mol), N-hydroxysuccinimide (8.0g, 0.06mol), EDC hydrochloride (13g, 0.06 mol), into a 100mL round bottom flask, 40mL of methylene chloride was added, stirred at room temperature, the solid gradually dissolved overnight reaction, there is generated a white solid. Filtered and dried to give the product (8.05g, white solid), yield 70percent.
49%
With dicyclohexyl-carbodiimide In dichloromethane; acetone at 20℃; for 24 h;
. 3,3'-Dithiobis(propanoic acid) (9.5 g, 45.2 mmol) was dissolved in a mixture of acetone (600 mL) and dichloromethane (600 mL) with stirring at room temperature. N-Hydroxysuccinimide (12.68 g, 110.2 mmol, 2.44 eq) was dissolved in the solution, then 1,3-dicyclohexylcarbodiimide (25.9 g, 125.5 mmol, 2.78 eq) was cautiously added. The mixture was allowed to stir at room temperature for 24 hours, after which time the solution was vacuum filtered and the residual solid discarded. The solvent was removed from the filtrate under vacuum, and the oily residue was redissolved in dichloromethane (ca 200 mL). The solution was reduced in volume (ca. 50 mL) and cooled, giving the product as a colourless, crystalline solid (8.90 g, 49percent yield). 1H-NMR (d6-DMSO): δ 2.80 (s, 8H), 3.05 (m, 8H). 13C-NMR (d6-DMSO): δ 25.8, 30.7, 32.3, 167.9, 170.4 ppm.
Reference:
[1] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 1, p. 96 - 100
[2] Organic and Biomolecular Chemistry, 2005, vol. 3, # 12, p. 2255 - 2261
[3] Patent: CN105315191, 2016, A, . Location in patent: Paragraph 0021; 0022; 0023
[4] Patent: US7074766, 2006, B1, . Location in patent: Page/Page column 44
[5] Angewandte Chemie - International Edition, 2010, vol. 49, # 26, p. 4405 - 4408
[6] Organic Letters, 2007, vol. 9, # 6, p. 1009 - 1012
[7] Rapid Communications in Mass Spectrometry, 2013, vol. 27, # 1, p. 238 - 248
[8] Patent: CN104667297, 2017, B, . Location in patent: Paragraph 0056-0058
[9] Nanomedicine: Nanotechnology, Biology, and Medicine, 2018, vol. 14, # 4, p. 1123 - 1136
27
[ 6066-82-6 ]
[ 107-96-0 ]
[ 57757-57-0 ]
Reference:
[1] Chemistry - A European Journal, 2014, vol. 21, # 10, p. 3956 - 3967
28
[ 6066-82-6 ]
[ 814-68-6 ]
[ 38862-24-7 ]
Yield
Reaction Conditions
Operation in experiment
70%
With triethylamine In chloroform at 0℃; for 2.5 h;
N-hydroxysuccinimide (10 g, 0.086 mol) and triethylamine (13.2 g, 0.129 mol) were dissolved in chloroform (130 mL) at 0 °C. Acryloyl chloride (8.6 g, 0.094 mol) was added dropwise over a period of 2 hours to the stirred reaction mixture. The reaction is described in Scheme 1, . After being stirred an additional 30 minutes at 0 °C, the solution was washed twice with 60 mL saturated NaCl aqueous solution, dried over MgSO4, filtered and concentrated so as to get a residual volume of 30 mL. An ethyl acetate/pentane mixture (14 mL, 1:3 v/v) was added and the temperature was maintained at 0 °C to induce NAS crystallization overnight (70percent yield). 1 H NMR (CDCl3) d (ppm): 2.95 (S, 4H, CH2CH2), 6.20 (m, 1 H, CH=CH2), 6.4 (m, 1H, CH=CH2) and 6.75 (m, 1 H, CH=CH2).
46%
With triethylamine In chloroform at 12℃; for 2.5 h; Inert atmosphere
2,5-Dioxopyrrolidin-1 -yl acrylate (28) To a cooled (ice-bath) solution of /V-hydroxysuccinimide (27) (6.41 g, 55.8 mmol) and NEt3 (8.5 ml_, 61.0 mmol) in CHCI3 (100 mL) was added acryloyl chloride (26) dropwise under argon. The temperature of the mixture was kept below 12°C during the addition. After stirring for 2.5 h, the reaction mixture was subsequently washed with ice-water (100 mL), water (100 mL), and brine (100 mL). The organic phase was dried over Na2S04, filtered, concentrated in vacuo to 15 mL, and filtered through a pad of celite. The celite was washed with CHCI3 (15 mL), the filtrate was diluted with EtOAc (2 mL) and petroleum ether (1 1 mL), and stored at -20 °C overnight. The formed precipitate was filtered off and dried in vacuo to yield 28 (4.30 g, 25.4 mmol, 46percent) as white needles.
Reference:
[1] Journal of Medicinal Chemistry, 2018, vol. 61, # 10, p. 4528 - 4560
[2] Chemical Communications, 2014, vol. 50, # 95, p. 15045 - 15048
[3] Organic Letters, 2002, vol. 4, # 5, p. 737 - 740
[4] Chemical Communications, 2009, # 46, p. 7107 - 7109
[5] Journal of Medicinal Chemistry, 1995, vol. 38, # 21, p. 4179 - 4190
[6] Analytical Chemistry, 2003, vol. 75, # 7, p. 1658 - 1663
[7] Journal of Materials Chemistry, 2011, vol. 21, # 34, p. 12917 - 12926
[8] Patent: EP2960263, 2015, A1, . Location in patent: Paragraph 0098
[9] Angewandte Chemie - International Edition, 2018, vol. 57, # 6, p. 1557 - 1562[10] Angew. Chem., 2018, vol. 130, p. 1573 - 1578,6
[11] Organic Letters, 2014, vol. 16, # 15, p. 3974 - 3977
[12] Macromolecules, 2004, vol. 37, # 7, p. 2371 - 2382
[13] Patent: WO2015/136027, 2015, A1, . Location in patent: Page/Page column 48
[14] Journal of the American Chemical Society, 2003, vol. 125, # 2, p. 320 - 321
[15] Angewandte Chemie - International Edition, 2008, vol. 47, # 10, p. 1875 - 1878
[16] International Journal of Pharmaceutics, 2011, vol. 420, # 2, p. 333 - 340
[17] Angewandte Chemie - International Edition, 2017, vol. 56, # 14, p. 3880 - 3885[18] Angew. Chem., 2017, vol. 129, # 14, p. 3938 - 3943,6
29
[ 6066-82-6 ]
[ 79-10-7 ]
[ 38862-24-7 ]
Reference:
[1] Tetrahedron, 1998, vol. 54, # 14, p. 3489 - 3494
[2] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 19, p. 6841 - 6850
[3] Journal of the American Chemical Society, 2010, vol. 132, # 2, p. 472 - 483
30
[ 6066-82-6 ]
[ 79-10-7 ]
[ 2524-64-3 ]
[ 38862-24-7 ]
Reference:
[1] Patent: US5734064, 1998, A,
31
[ 6066-82-6 ]
[ 624-83-9 ]
[ 18342-66-0 ]
Yield
Reaction Conditions
Operation in experiment
86%
With triethylamine In ethyl acetate at 0 - 20℃; for 24 h;
A dried round-bottom flask was charged with N-hydroxysuccinimide (5.83 g, 50.7 mmol), ethyl acetate (20 mL) as a solvent and cooled at 0 °C. To the mixture were slowly added triethylamine (4.76 g) and methylisocyanate (6.39 g, 0.112 mmol) at the same temperature. The reaction temperature was slowly raised to rt, and the mixture was stirred for 24 h at rt. After the volatile materials were removed under reduced pressure the crude residue was re-crystallized from ethyl acetate/diethyl ether to give 51a (86 percent). Colorless needles, mp 148.0–149.0 °C (lit [19]. 148.0–152.0°C); 1H NMR (300 MHz, CDCl3): δ 8.15 (1H, br, NH), 2.76 (4H, s, CH2CH2), 2.67 (3H, s-like, CH3); 13C NMR (22.5 MHz, CDCl3): δ 170.66, 151.98, 27.98, 25.47; MS (FAB+) m/z 173 (M+H).
Reference:
[1] European Journal of Medicinal Chemistry, 2014, vol. 82, p. 16 - 35
[2] Journal of Medicinal Chemistry, 1982, vol. 25, # 2, p. 178 - 182
[3] Journal of the American Chemical Society, 1995, vol. 117, # 4, p. 1240 - 1245
32
[ 6066-82-6 ]
[ 79-04-9 ]
[ 27243-15-8 ]
Yield
Reaction Conditions
Operation in experiment
53%
With triethylamine In chloroform at 0℃; for 0.416667 h;
To a solution of N-hydroxysuccinimide (640.3 mg, 5.56 mmol) in chloroform (8.5 mL) was added triethylamine (861.6 μ, 6.18 mmol) 0 C. Then, a-chloroacetyl chloride was added dropwise over a 5 minute period and stirred for an additional 20 minutes at 0°C. The reaction mixture was washed with ice-cold water (15 mL) and brine (15 mL), concentrated to a volume of 1.7 mL in vacuo, then dried with sodium sulfate and filtered. To the resulting solution were added ethyl acetate (170 μ) and hexanes (1.2 mL), and the mixture was cooled down to 0 °C stirred for 2 h, and a white solid was precipitated. It was filtered and washed first with ice-cold 10 mL portion of hexanes/ethyl acetate (4: 1), then with 10 mL hexanes/ethyl acetate (9: 1), and finally with hexanes (10 mL, twice). The resulting white solid was dried under house vacuum to yield 2,5-dioxopyrrolidin-l-yl 2-chloroacetate (563.9 mg, 53percent). NMR 400 MHz (CDC13) δ 4.37 (s, 2H), 2.87 (s, 4H).
Reference:
[1] Angewandte Chemie - International Edition, 2014, vol. 53, # 1, p. 199 - 204[2] Angew. Chem., 2013, vol. 126, # 1, p. 203 - 208,6
[3] Patent: WO2014/160200, 2014, A1, . Location in patent: Paragraph 000273
[4] Polish Journal of Chemistry, 1992, vol. 66, # 1, p. 111 - 118
[5] Journal of Medicinal Chemistry, 2014, vol. 57, # 6, p. 2380 - 2392
33
[ 6066-82-6 ]
[ 79-11-8 ]
[ 27243-15-8 ]
Reference:
[1] Polish Journal of Chemistry, 2009, vol. 83, # 11, p. 1959 - 1966
[2] Helvetica Chimica Acta, 1996, vol. 79, p. 133 - 136
[3] Bioorganic and Medicinal Chemistry Letters, 1998, vol. 8, # 24, p. 3671 - 3676
[4] Patent: US6335437, 2002, B1, . Location in patent: Page column 22
[5] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 11, p. 6156 - 6166
[6] Organic and Biomolecular Chemistry, 2011, vol. 9, # 21, p. 7296 - 7299
[7] Journal of Medicinal Chemistry, 2015, vol. 58, # 16, p. 6516 - 6532
34
[ 6066-82-6 ]
[ 65-85-0 ]
[ 23405-15-4 ]
Yield
Reaction Conditions
Operation in experiment
93%
With dicyclohexyl-carbodiimide In tetrahydrofuran at 20℃; for 48 h;
General procedure: benzoic acid (0.353 g, 2.89 mmol) was dissolved in 50 mL of dry THF, and N-hydroxysuccinimide (0.40 g, 3.47 mmol) and DCC (0.776 g, 3.76 mmol) were added. The mixture was stirred at room temperature for 2 days. The soluble portion was separated from the viscous residue and the solvent was removed under reduced pressure. The residue was dissolved in CHCl3separated and dried over anhydrous Na2SO4, the solvent was removed by evaporation to dryness. The crude product was purified by column chromatography with 40percent ethyl acetate in hexane to afford a white solid (0.586 g, 93percent). The white solid wasrecrystallized from 2-propanol.
78%
With triethylamine; dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20℃; for 3 h;
General procedure: Benzoic acid (ring d-5, heavy isotope, d-Bz) (127 mg, 1.00 mmol) or benzoic acid (ring d-0, light isotope, Bz) (122 mg, 1.00 mmol), N-hydroxysuccinimide (230 mg, 2.00 mmol) and triethylamine (167 μL, 1.20 mmol) were dissolved in anhydrous dimethylformamide (DMF) (1 mL). After addition of dicyclohexylcarbodiimide (DCC) (206 mg, 1.20 mmol), the solution was stirred at room temperature for 3 h. The reaction mixture was filtered to remove the precipitate, and concentrated in vacuo. The resulting syrup was crystallized from isopropanol (5 mL) to yield benzoic acid N-succinimidyl ester (d-Bz labeling reagent, 184 mg, 82percent) (Bz labeling reagent, 170 mg, 78percent). Rf = 0.42 (toluene-EtOAc = 10:1); 1H-NMR (600 MHz, CDCl3): δ 2.91 (s, 4H) for the heavy isotope, δ 2.91 (s, 4H), 7.52 (dd, 2H), 7.69 (dd, 1H), 8.14 (dd, 2H) for the light isotope; 13C-NMR (125 MHz, CDCl3): δ 25.63, 124.88, 128.31, 130.13, 134.38, 161.82, 169.24 for the heavy isotope, δ 25.65, 125.08, 128.83, 130.55, 134.91, 161.84, 169.23 for the light isotope.
61%
With dicyclohexyl-carbodiimide In dichloromethane at 0℃; for 6 h;
Benzoic acid (3 g, 24.6 mmol) and N-hydroxysuccinimide (4.24 g, 36.8 mmol, 1.5 eq) were dissolved in 50 mL CH2Cl2, and the mixture was cooled to 0 °C. To this solution was added 7.6 g of DCC (36.8 mmol, 1.5 eq) in 25 mL CH2Cl2. The reaction mixture was stirred vigorously for 6 h. After filtration, the solvent was evaporated, and the crude product was purified by column chromatography to afford 7 as a white solid with a yield 61percent. m.p.:134-136 °C.1H NMR (400 MHz, CDCl3): δ 8.14 (400 MHz, d, J = 7.8 Hz, 2H, Ar-H), 7.69 (t, J = 7.4 Hz, 1H, Ar-H), 7.52 (t, J = 7.7 Hz, 2H, Ar-H), 2.91 (s, 4H, CH2).
Reference:
[1] Chemical and Pharmaceutical Bulletin, 2007, vol. 55, # 1, p. 124 - 127
[2] Chemistry Letters, 1980, p. 1161 - 1164
[3] Bulletin of the Chemical Society of Japan, 1984, vol. 57, # 3, p. 781 - 786
[4] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 15, p. 4629 - 4632
[5] Journal of the Chemical Society, Chemical Communications, 1985, # 8, p. 473
[6] ACS Catalysis, 2013, vol. 3, # 8, p. 1685 - 1692
[7] Bioorganic and Medicinal Chemistry, 2000, vol. 8, # 6, p. 1317 - 1329
[8] Molecules, 2014, vol. 19, # 7, p. 9944 - 9961
[9] Journal of Medicinal Chemistry, 2006, vol. 49, # 1, p. 31 - 34
[10] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 4, p. 949 - 954
[11] Angewandte Chemie - International Edition, 2014, vol. 53, # 40, p. 10718 - 10722[12] Angew. Chem., 2014, vol. 126, # 40, p. 10894 - 10898,5
[13] Angewandte Chemie - International Edition, 2008, vol. 47, # 14, p. 2700 - 2703
[14] Tetrahedron, 1991, vol. 47, # 39, p. 8407 - 8416
[15] Journal of Medicinal Chemistry, 1992, vol. 35, # 7, p. 1246 - 1259
[16] Tetrahedron Letters, 1998, vol. 39, # 11, p. 1321 - 1324
[17] Helvetica Chimica Acta, 2007, vol. 90, # 6, p. 1043 - 1068
[18] Patent: WO2007/42775, 2007, A2, . Location in patent: Page/Page column 45-46
[19] Patent: WO2007/2135, 2007, A2, . Location in patent: Page/Page column 12-13
[20] Patent: WO2012/17400, 2012, A1, . Location in patent: Page/Page column 18
[21] Carbohydrate Research, 2013, vol. 378, p. 45 - 55
[22] Organic Preparations and Procedures International, 2014, vol. 46, # 4, p. 370 - 375
[23] Journal of the American Chemical Society, 2015, vol. 137, # 9, p. 3271 - 3275
[24] Patent: US2015/335759, 2015, A1, . Location in patent: Sheet 13A
[25] European Journal of Medicinal Chemistry, 2017, vol. 138, p. 13 - 25
[26] Patent: CN107963978, 2018, A, . Location in patent: Paragraph 0014
[27] Patent: WO2018/71879, 2018, A1, . Location in patent: Sheet 5
35
[ 6066-82-6 ]
[ 100-52-7 ]
[ 23405-15-4 ]
Yield
Reaction Conditions
Operation in experiment
80%
With oxygen In acetonitrile at 80℃; for 10 h; Green chemistry
General procedure: p-Fluorobenzaldehyde 1a (124.0 mg, 1.0 mmol, 1.0 equiv.) and NHSI (2b) (62.5 mg, 0.5 mmol, 0.5 equiv.) were dissolved in DEC/CH3CN (1:1 3 mL) and stirred under 80oC for 10 h. After the reaction was completed (monitored by TLC), the reaction mixture was concentrated under vacuum. The residue was purified by ethanol recrystallization to give the 4a as a white solid, m. p. 100-102 °C.
Reference:
[1] Angewandte Chemie - International Edition, 2012, vol. 51, # 50, p. 12538 - 12541[2] Angew. Chem., 2012, vol. 124, # 50, p. 12706 - 12709,4
[3] New Journal of Chemistry, 2018, vol. 42, # 2, p. 807 - 811
[4] Tetrahedron Letters, 2017, vol. 58, # 18, p. 1742 - 1746
[5] RSC Advances, 2015, vol. 5, # 56, p. 44928 - 44932
[6] Tetrahedron Letters, 2012, vol. 53, # 38, p. 5094 - 5098
[7] Organic and Biomolecular Chemistry, 2013, vol. 11, # 47, p. 8241 - 8246
[8] Advanced Synthesis and Catalysis, 2014, vol. 356, # 11-12, p. 2709 - 2713
36
[ 6066-82-6 ]
[ 94-36-0 ]
[ 23405-15-4 ]
Yield
Reaction Conditions
Operation in experiment
84%
With 1,4-diaza-bicyclo[2.2.2]octane In acetonitrile at 20℃; for 1 h;
General procedure: To a test tube charged with a stir bar, 0.20 mmol of1a and 0.20 mmol of BPO weresequentially added MeCN 1.0 mL and 0.20 mmol of DABCO. The resulting reactionmixture was stirred at room temperature for 3 h and diluted by addition of EtOAc,the organic layer was briefly washed by saturated aqueous sodium bicarbonatesolution, brine and dried over anhydrous sodium sulfate. The bulk solvent wasremoved in vacuo, and the residue waspurified by silica gel flash chromatography (hexane/EtOAc = 5:1) to affordproduct 2a 48.8 mg, 90percent yield.
Reference:
[1] Journal of Organic Chemistry, 2015, vol. 80, # 3, p. 1920 - 1928
46
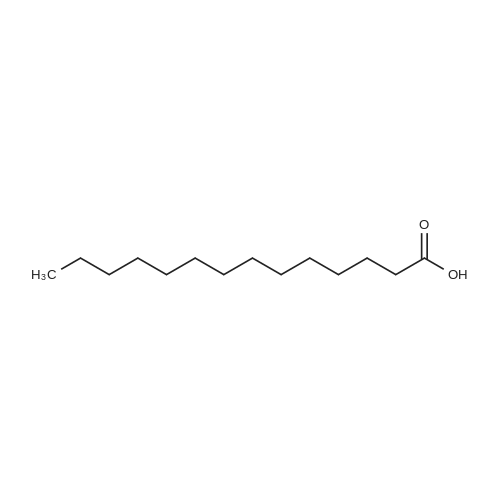
[ 143-07-7 ]
[ 6066-82-6 ]
[ 14565-47-0 ]
Yield
Reaction Conditions
Operation in experiment
95%
With dicyclohexyl-carbodiimide In ethyl acetate at 20℃; for 12 h;
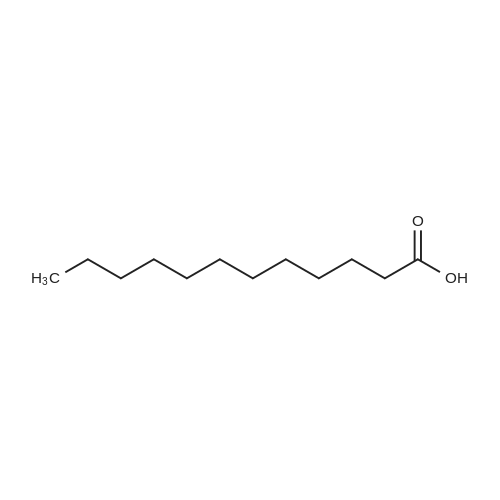
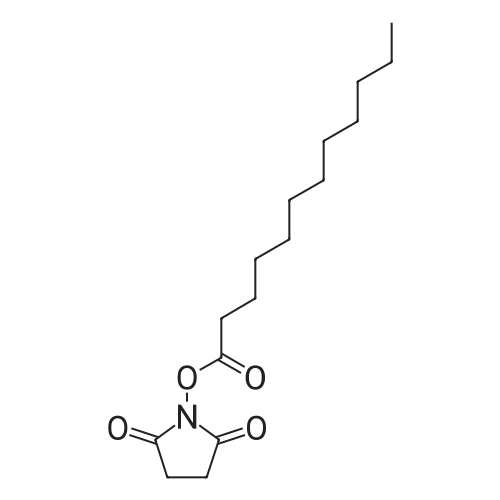
Lauric acid (12.0 g, 60 mmol) dissolved in dry ethyl acetate (100 mL) was added to a solution of N-hydroxysuccinimide (NHS) (6.9 g, 60 mmol) and N,N’-dicyclohexylcarbodiimide (DCC) (12.4 g, 60 mmol) in dry ethyl acetate (300 mL). The reaction mixture was left overnight at room temperature. Dicyclohexylurea (DCU) was removed by filtration and the filtrate was evaporated under reduced pressure to yield white crystals (17.4 g, 95 percent yield). Recrystallization from ethanol yielded 16.6g of pure dodecanoic acid 2,5-dioxo-pyrrolidine-1-yl ester.
Reference:
[1] Journal of the American Chemical Society, 2015, vol. 137, # 24, p. 7692 - 7705
[2] Journal of the American Chemical Society, 2003, vol. 125, # 40, p. 12110 - 12111
[3] RSC Advances, 2015, vol. 5, # 81, p. 66339 - 66354
[4] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 6, p. 1763 - 1767
[5] Synthetic Communications, 2009, vol. 39, # 24, p. 4467 - 4472
[6] Bioorganic and Medicinal Chemistry, 2003, vol. 11, # 24, p. 5381 - 5390
[7] Liebigs Annalen der Chemie, 1990, # 2, p. 145 - 150
[8] Journal of Mass Spectrometry, 1995, vol. 30, # 6, p. 900 - 910
[9] Tetrahedron Letters, 2008, vol. 49, # 48, p. 6838 - 6840
[10] Bioorganic and Medicinal Chemistry Letters, 2003, vol. 13, # 14, p. 2351 - 2353
[11] New Journal of Chemistry, 2013, vol. 37, # 4, p. 961 - 964
[12] Agricultural and Biological Chemistry, 1982, vol. 46, # 2, p. 597 - 600
[13] Journal of the American Chemical Society, 2008, vol. 130, # 41, p. 13555 - 13557
[14] Chemistry - A European Journal, 2014, vol. 20, # 4, p. 941 - 945
[15] Journal of the American Chemical Society, 1990, vol. 112, # 7, p. 2498 - 2506
[16] Journal of Molecular Structure, 1992, vol. 273, # 1, p. 215 - 226
[17] Agricultural and Biological Chemistry, 1986, vol. 50, # 10, p. 2561 - 2572
[18] Russian Journal of Bioorganic Chemistry, 2006, vol. 32, # 5, p. 413 - 419
[19] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 10, p. 3117 - 3121
[20] Molecules, 2009, vol. 14, # 10, p. 4051 - 4064
[21] Australian Journal of Chemistry, 2009, vol. 62, # 7, p. 653 - 656
[22] Chemical Communications, 2012, vol. 48, # 58, p. 7265 - 7267
[23] Organic and Biomolecular Chemistry, 2012, vol. 10, # 30, p. 6121 - 6129
[24] International Journal of Pharmaceutics, 2013, vol. 457, # 1, p. 124 - 135
[25] Organic Preparations and Procedures International, 2014, vol. 46, # 4, p. 370 - 375
[26] Journal of Drug Targeting, 2016, vol. 24, # 1, p. 68 - 79
Reference:
[1] Synthesis, 1983, # 8, p. 671 - 673
[2] Bulletin of the Chemical Society of Japan, 1989, vol. 62, # 2, p. 524 - 531
49
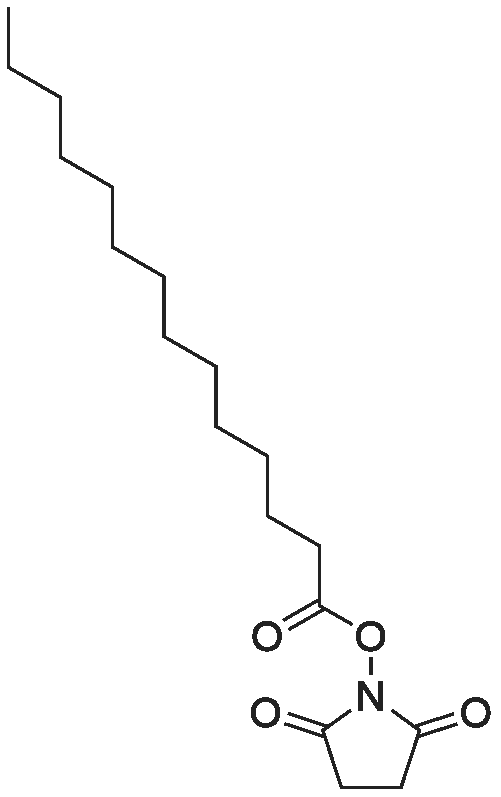
[ 6066-82-6 ]
[ 544-63-8 ]
[ 69888-86-4 ]
Yield
Reaction Conditions
Operation in experiment
72%
Stage #1: With dicyclohexyl-carbodiimide In tetrahydrofuran at 20℃; for 0.166667 h; Stage #2: at 20℃; for 22.5 h;
Tetradecanoic acid [3.00 g (13.1 mmol)] and 3.24 g of N,N′-dicyclohexylcarbodiimide (15.7 mmol) were dissolved in 26.2 ml of dry tetrahydrofuran and stirred at room temperature for 10 min. Then, 1.81 (15.7 mmol) of N-hydroxysuccinimide in 14.1 ml of the same solvent were added and the reaction mixture was stirred at room temperature for 22.5 h. TLC analysis [CHCl3/MeOH (50:1, v/v)] revealed a complete conversion of the educt. After this, the reaction mixture was filtrated by suction. The solvent was removed under reduced pressure and the residue was recrystallized from ethanol containing a trace of H2O (34). The product was obtained as a colorless solid (3.08 g, 72percent), 1H-NMR (300 MHz,CDCl3): [ppm] = 2.83 (s, 4H, H-3, 4), 2.60 (t, 2H,3 J H2 ′ -H3 ′ = 7.5 Hz, H-2 ′ ), 1.74 (tt, 2H, 3 J H3′-H4′ = 7.5 Hz, 3 JH3′-H2′ = 7.5Hz, H-3 ′ ), 1.46-1.18 (m, 20H), 0.88 (t, 3H, 3 J H14 ′ -H13 ′ = 6.7 Hz,H-14 ′ ), 13 C-NMR (75 MHz, CDCl3): [ppm] = 169.15 (R C ON,C-2, 5), 168.68 (R C O 2 R ′ , C-1 ′ ), 31.90 (CH 2 , C-2 ′ ), 30.93 (CH 2 ,C-12 ′ ), 29.63-28.77 (8 CH 2 ), 25.57 (CH 2 , C-3, 4), 24.56 (CH 2 ,C-3 ′ ), 22.67 (CH 2 , C-13 ′ ), 14.09 (CH 3 , C-14 ′ ), EI-MS: m/z 211.3[M+-C4H4NO3], 129.1 [C8H17O+ ], 98.2 [C7H14+· ], 84.1 [C6H12+· ], CHN analysis: calc.: C, 66.43; H, 9.60; N, 4.30, found: C, 66.03; H,9.63; N, 4.29.
67%
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane
Myristic acid (1.5 g, 6.6 mmol) was dissolved in dichloromethane (30 ml) and the mixture was stirred. To this solution were added N-hydroxysuccinimide (0.91 g, 1.2 eq.) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) (1.5 g, 1.2 eq.), and the mixture was stirred overnight. After washing twice with water, the mixture was washed once with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate/hexane=1/3) to give the object product (1.4 g, yield 67percent). The NMR measurement results of the obtained myristic acid-N-hydroxysuccinimide ester (C14-NHS) are shown below. Myristic Acid-N-Hydroxysuccinimide Ester (C14-NHS) (0195) 1H-NMR (CDCl3) δ: 2.83 (4H, s), 2.60 (2H, t, J=7.6 Hz), 1.74 (2H, q, J=7.6 Hz), 1.44 (2H, q, J=6.9 Hz), 1.48-1.22 (18H, m), 0.88 (3H, t, J=6.8 Hz).
Reference:
[1] RSC Advances, 2015, vol. 5, # 81, p. 66339 - 66354
[2] Journal of Pharmaceutical Sciences, 1994, vol. 83, # 2, p. 233 - 238
[3] Journal of Pharmacology and Experimental Therapeutics, 2004, vol. 309, # 1, p. 340 - 347
[4] Angewandte Chemie, 1996, vol. 108, # 15, p. 1791 - 1794
[5] Journal of Mass Spectrometry, 1995, vol. 30, # 6, p. 900 - 910
[6] Journal of Lipid Research, 2014, vol. 55, # 12, p. 2692 - 2704
[7] Patent: US9663784, 2017, B2, . Location in patent: Page/Page column 41
[8] Agricultural and Biological Chemistry, 1982, vol. 46, # 2, p. 597 - 600
[9] Tetrahedron, 1991, vol. 47, # 39, p. 8407 - 8416
[10] Agricultural and Biological Chemistry, 1986, vol. 50, # 10, p. 2561 - 2572
[11] Journal of Medicinal Chemistry, 2001, vol. 44, # 13, p. 2188 - 2203
[12] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 10, p. 3117 - 3121
[13] Organic and Biomolecular Chemistry, 2011, vol. 9, # 3, p. 820 - 833
[14] Organic and Biomolecular Chemistry, 2012, vol. 10, # 30, p. 6121 - 6129
[15] Chemistry Letters, 2013, vol. 42, # 2, p. 127 - 129
[16] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 6, p. 1763 - 1767
[17] ACS Medicinal Chemistry Letters, 2015, vol. 6, # 4, p. 476 - 480
[18] RSC Advances, 2016, vol. 6, # 30, p. 24808 - 24819
[19] Patent: CN105461681, 2016, A, . Location in patent: Paragraph 0024; 0025
[20] Patent: US2018/222960, 2018, A1, . Location in patent: Paragraph 0395-0396
50
[ 6066-82-6 ]
[ 112-64-1 ]
[ 69888-86-4 ]
Reference:
[1] Liebigs Annalen der Chemie, 1990, # 11, p. 1111 - 1114
Reference:
[1] Angewandte Chemie - International Edition, 2016, vol. 55, # 42, p. 13023 - 13027[2] Angew. Chem., 2016, vol. 128, p. 13217 - 13221,5
[3] J. Gen. Chem. USSR (Engl. Transl.), 1985, vol. 55, # 9, p. 1909 - 1910[4] Zhurnal Obshchei Khimii, 1985, vol. 55, # 9, p. 2152
[5] Journal of the American Chemical Society, 2007, vol. 129, # 31, p. 9640 - 9649
[6] Journal of the American Chemical Society, 2017, vol. 139, # 11, p. 4009 - 4018
[7] Chemical Communications, 2016, vol. 52, # 90, p. 13229 - 13232
[8] Chemical Communications, 2015, vol. 51, # 26, p. 5622 - 5625
53
[ 6066-82-6 ]
[ 119023-08-4 ]
[ 68181-17-9 ]
Reference:
[1] Bulletin des Societes Chimiques Belges, 1988, vol. 97, # 7, p. 535 - 540
54
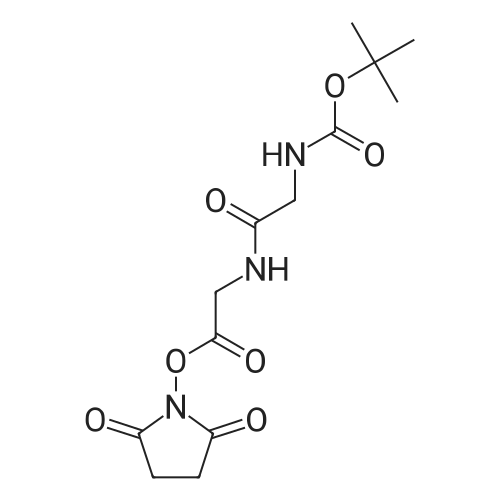
[ 4530-20-5 ]
[ 6066-82-6 ]
[ 3392-07-2 ]
Yield
Reaction Conditions
Operation in experiment
61.8%
With dicyclohexyl-carbodiimide In dichloromethane at 15 - 20℃;
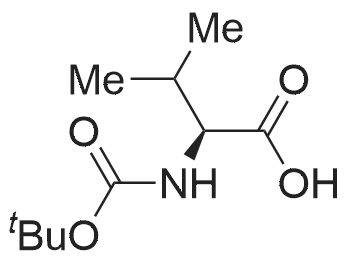
Step2: 2,5-Dioxopyrrolidine-1-yl 2-(fert-butoxycarbonylamino)acetate. /V-Boc-glycine (7.31 g, 41 .7 mmol) was dissolved in 100 mL of DCM and to the cooled (15°C) solution /V-hydroxysuccinimide (5.28 g, 45.9 mmol) was added. Λ/,Λ/'-dicyclohexylcarbodiimide (9.47 g, 45.9 mmol) was added to the formed suspension under vigorous stirring. After a few seconds, a cloudy white suspension formed, the mixture was allowed to reach room temperature and stirred for 1 h. It was subsequently filtrated over celite, washed with 50m L saturated sodium - - bicarbonate, dried over sodium sulfate and concentrated in vacuo to yield a crystalline powder. Yield: 7.02 g, 61 .8percent 1 H NMR (400 MHz, DMSO -cfe) : δ 1 .38 (s, 9H), 2.80 (s, 4H), 4.07 (d, J = 6 Hz, 2H), 7.43 (br s, 1 H). MS (ESI) m/z 567.2 [2M + Na]+.
61.8%
With dicyclohexyl-carbodiimide In dichloromethane at 15℃;
N-Boc-glycine (7.31 g, 41.7 mmol) was dissolved in 100 mL of DCM and to the cooled (15° C.) solution N-hydroxysuccinimide (5.28 g, 45.9 mmol) was added. N,N'-dicyclohexylcarbodiimide (9.47 g, 45.9 mmol) was added to the formed suspension under vigorous stirring. After a few seconds, a cloudy white suspension formed, the mixture was allowed to reach room temperature and stirred for 1 h. It was subsequently filtrated over celite, washed with 50 mL saturated sodium bicarbonate, dried over sodium sulfate and concentrated in vacuo to yield a crystalline powder. Yield: 7.02 g, 61.8percent 1H NMR (400 MHz, DMSO-d6): δ 1.38 (s, 9H), 2.80 (s, 4H), 4.07 (d, J=6 Hz, 2H), 7.43 (br s, 1H). MS (ESI) m/z 567.2 [2M+Na]+.
Reference:
[1] Chemistry - A European Journal, 2016, vol. 22, # 52, p. 18865 - 18872
[2] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 15, p. 2443 - 2449
[3] Chemical communications (Cambridge, England), 2003, # 23, p. 2870 - 2871
[4] ACS Medicinal Chemistry Letters, 2013, vol. 4, # 4, p. 402 - 407
[5] Helvetica Chimica Acta, 2004, vol. 87, # 5, p. 1077 - 1089
[6] Bioconjugate Chemistry, 2010, vol. 21, # 9, p. 1642 - 1655
[7] Patent: WO2013/107820, 2013, A1, . Location in patent: Page/Page column 42; 43
[8] Patent: US2014/357650, 2014, A1, . Location in patent: Paragraph 0265; 0268; 0269
[9] Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry (1972-1999), 1984, p. 2305 - 2308
[10] Journal of the American Chemical Society, 1993, vol. 115, # 10, p. 4228 - 4245
[11] Pharmaceutical Chemistry Journal, 1992, vol. 26, # 9-10, p. 753 - 756[12] Khimiko-Farmatsevticheskii Zhurnal, 1992, vol. 26, # 9-10, p. 72 - 74
[13] Journal of the American Chemical Society, 1996, vol. 118, # 29, p. 6975 - 6985
[14] Nucleosides and Nucleotides, 1998, vol. 17, # 9-11, p. 2135 - 2141
[15] Chemistry - A European Journal, 2001, vol. 7, # 19, p. 4280 - 4295
[16] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 12, p. 3220 - 3224
[17] Organic and Biomolecular Chemistry, 2015, vol. 13, # 13, p. 3950 - 3962
[18] Organic Process Research and Development, 2015, vol. 19, # 9, p. 1257 - 1262
[19] Journal of Molecular Biology, 2018, vol. 430, # 6, p. 842 - 852
Reference:
[1] Liebigs Annalen der Chemie, 1981, vol. No. 7, p. 1294 - 1302
57
[ 6066-82-6 ]
[ 3744-87-4 ]
[ 34404-33-6 ]
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1987, # 15, p. 1155 - 1156
[2] Journal of the American Chemical Society, 1996, vol. 118, # 29, p. 6975 - 6985
[3] Journal of Molecular Biology, 2018, vol. 430, # 6, p. 842 - 852
58
[ 6066-82-6 ]
[ 13734-41-3 ]
[ 3392-12-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
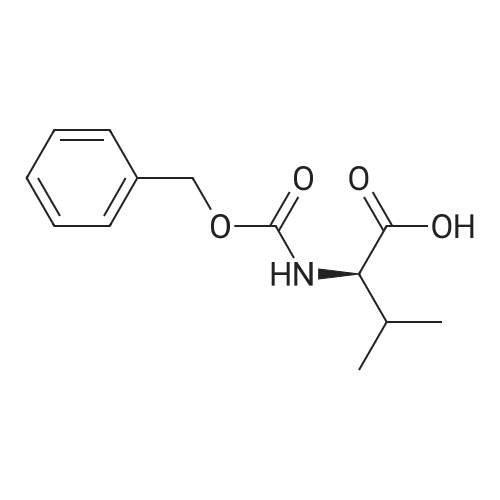
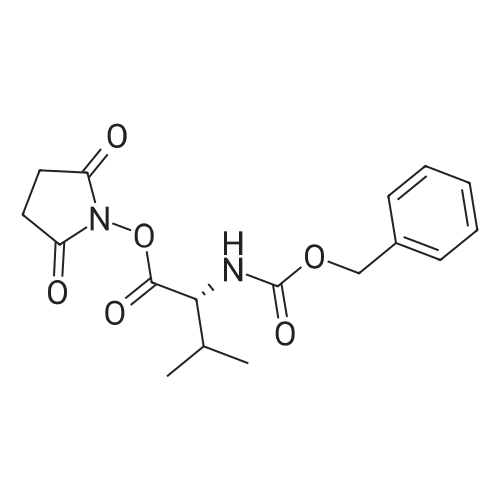
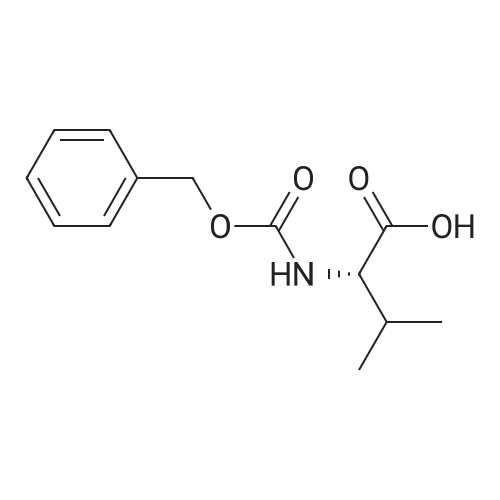
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In dichloromethane at 20℃; for 72 h; Inert atmosphere; Sealed tube
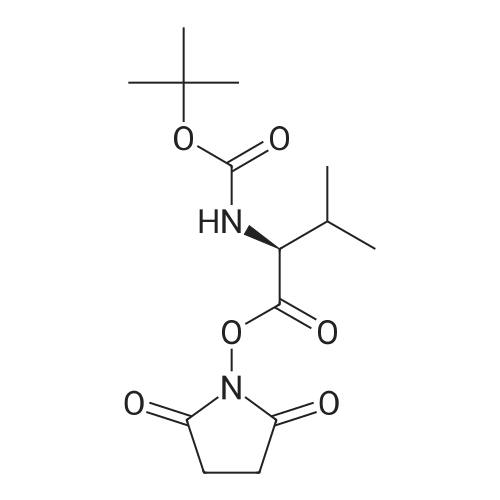
Step A: To a round-bottom flask was weighed Boc-L-valine (1.03 g, 4.74 mmol), N-hydroxysuccinimide (1.22 g, 10.6 mmol), and EDC (1.60 g, 8.35 mmol). The reagents were dissolved in dry DCM (30 mL), the flask sealed via rubber septum, purged with argon, and the reaction stirred at ambient temperature. After 3 days no Boc-valine remained by TLC (after staining with ninhydrin), so the reaction was washed with water and sat. aq. NaHCO3, the aqueous layer extracted with DCM, combined organic layers washed with brine, dried over Na2SO4, and filtered. The filtrate was then evaporated and dried in vacuo giving Boc-L-valine-succinate as a white solid (1.52 g, 100percent). 1H-NMR (300 MHz, CDCl3): ^ 4.98 (br d, 1H), 4.58 (dd, 1H), 2.82 (m, 4H), 2.27 (m, 1H), 1.44 (s, 9H), 1.03 (dd, 6H). Boc-L-valine-L-citrulline (3):
13.28 g
Stage #1: at 20℃; for 0.05 h; Stage #2: With dicyclohexyl-carbodiimide In tetrahydrofuran; dichloromethane at 0 - 20℃; for 18 h;
[000701 A solution of N-Hydroxysuccinimide (5.0 g, 43.44 mmol) and Boc-Val-OH (la, 9.462 g, 43.44 mmol ) in THF (83 mL) was stirred in room temperature for 3.0 minutes. Then DCC (9.856 g, 45.57 mmol) in CH2C12 (83 mL) was slowly added to the solution at 0°C and then warmed to room temperature. The reaction mixture was stirred for further 18 hours. The solution was cooled to 0°C and the precipitate was filtered and washed with EA (100 mL), dried over reduced pressure to give Boc-Val-OSu (ib, 13.28 g). ‘H NMR (500 MHz, DMSO-d6): ö: 5.02 (d, 1H), 4.60 (d, 1H), 2.85 (s, 4H), 2.31 (m, 1H), 1.48 (m, 9H), 1.05 (m, 6H). MS (M+1 ): 314.8.
Reference:
[1] Patent: WO2016/160615, 2016, A1, . Location in patent: Paragraph 0444; 0445
[2] Journal of Medicinal Chemistry, 1998, vol. 41, # 14, p. 2461 - 2480
[3] Journal of Medicinal Chemistry, 1991, vol. 34, # 9, p. 2852 - 2857
[4] Journal of the Chemical Society, Dalton Transactions: Inorganic Chemistry (1972-1999), 1984, p. 2305 - 2308
[5] Journal of Organic Chemistry, 1986, vol. 51, # 24, p. 4580 - 4585
[6] Journal of the American Chemical Society, 1992, vol. 114, # 17, p. 6653 - 6661
[7] Nucleosides and Nucleotides, 1998, vol. 17, # 9-11, p. 2135 - 2141
[8] Synlett, 2005, # 18, p. 2802 - 2804
[9] Chemistry - A European Journal, 2001, vol. 7, # 19, p. 4280 - 4295
[10] Patent: US2011/65796, 2011, A1, . Location in patent: Page/Page column 23-24
[11] Organic and Biomolecular Chemistry, 2011, vol. 9, # 11, p. 4182 - 4187
[12] Patent: WO2014/100762, 2014, A1, . Location in patent: Paragraph 240; 244
[13] Biochimica et Biophysica Acta - General Subjects, 2015, vol. 1850, # 9, p. 1849 - 1854
[14] Patent: WO2015/196167, 2015, A1, . Location in patent: Paragraph 0269; 0273
[15] Patent: WO2016/109802, 2016, A1, . Location in patent: Paragraph 00068; 00069; 00070
[16] Journal of Molecular Biology, 2018, vol. 430, # 6, p. 842 - 852
59
[ 6066-82-6 ]
[ 1138-80-3 ]
[ 2899-60-7 ]
Reference:
[1] Journal of the Chemical Society - Perkin Transactions 1, 1998, # 15, p. 2443 - 2449
[2] Journal of Mass Spectrometry, 2010, vol. 45, # 2, p. 178 - 189
[3] Tetrahedron Letters, 1995, vol. 36, # 12, p. 2105 - 2108
[4] Journal of Medicinal Chemistry, 1986, vol. 29, # 10, p. 1922 - 1929
[5] Journal of Medicinal Chemistry, 1991, vol. 34, # 6, p. 1845 - 1849
[6] Journal of Medicinal Chemistry, 1986, vol. 29, # 6, p. 912 - 917
[7] Bioorganic and Medicinal Chemistry Letters, 1996, vol. 6, # 13, p. 1543 - 1546
[8] Tetrahedron Letters, 1997, vol. 38, # 17, p. 3039 - 3042
[9] Chemistry - A European Journal, 2004, vol. 10, # 16, p. 3879 - 3890
[10] Patent: US6423869, 2002, B1,
[11] Synlett, 2009, # 8, p. 1233 - 1236
[12] Tetrahedron Letters, 2012, vol. 53, # 6, p. 623 - 626
[13] Patent: WO2006/127702, 2006, A2, . Location in patent: Page/Page column 76
60
[ 6066-82-6 ]
[ 598-45-8 ]
[ 1138-80-3 ]
[ 2899-60-7 ]
Reference:
[1] Patent: US3933783, 1976, A,
61
[ 6066-82-6 ]
[ 19506-72-0 ]
[ 56-40-6 ]
[ 2899-60-7 ]
Reference:
[1] Liebigs Annalen der Chemie, 1981, vol. No. 7, p. 1294 - 1302
62
[ 6066-82-6 ]
[ 64987-85-5 ]
Reference:
[1] Journal of Pharmaceutical Sciences, 1986, vol. 75, # 7, p. 711 - 713
63
[ 6066-82-6 ]
[ 1685-33-2 ]
[ 4467-55-4 ]
Reference:
[1] Journal of Pharmaceutical Sciences, 1993, vol. 82, # 1, p. 1 - 10
[2] Tetrahedron, 2008, vol. 64, # 41, p. 9717 - 9724
[3] Chemistry - A European Journal, 2013, vol. 19, # 48, p. 16256 - 16262
64
[ 6066-82-6 ]
[ 1149-26-4 ]
[ 3496-11-5 ]
Yield
Reaction Conditions
Operation in experiment
90%
With dicyclohexyl-carbodiimide In tetrahydrofuran at 0 - 20℃; for 16 h;
Step 1. To a stirred solution of compound 39-1 (1 g, 4 mmol), compound 39-2 (459 mg, 4 mmol) in THF (20 mL) was added DCC (908 mg, 4.4 mmol) at 0°C. The reaction mixture was stirred at r.t. for 16 h. The mixture was filtered and the filtrate was concentrated to give compound 39-3 (Yield: 90 percent).
85.5 g
With dmap; dicyclohexyl-carbodiimide In tetrahydrofuran at 5℃; for 20.75 h;
2-(((benzyloxy)carbonyl)amino)-3-methylbutanoic acid (65g, 259mMol), tetrahydrofuran (450ml) were charged to a flask. N-hydroxy succinimide (33.3g 285mMol) was added to the reaction mass. Reaction mass was stirred for lOmins. Dimethyl amino pyridine (DMAP, lg) was added to the reaction mass. Reaction mass was stirred and temperature of reaction mass was decreased to 5°C. Solution of DCC in THF (42.5percent, 200ml) was added dropwise to the reaction mass over a period of 45 minutes at 5°C. Reaction mass was stirred for 20 hours. Obtained solid was filtered; solvent was stripped off under reduced pressure. Solid was washed with heptane (3x250ml ), to get crude material (2,5- dioxopyrrolidin-l-yl 2-(((benzyloxy)carbonyl)amino)-3-methylbutanoate) with sufficient purity. Yield 85.5 g. (0091) JH NMR (DMSO-D6, 400 MHz): δ 1.02(d,6H), 2.24(m,lH), 2.82(s,4H), 4.37(q,lH), 5.09 (s,2H), 7.34-7.44 (m,5H), 8.09 (d,lH). (0092) 13C NMR (DMSO D6, 100MHz): δ 18.3, 19.3, 25.4, 30.1, 58.1, 65.9, 127.7, 128.4,136.7, 156.3, 168.0, 170.0, 172.8 (0093) FTIR (KBr): 3327, 2933, 2117, 1816, 1784, 1741, 1527, 1204, 893 cm"1. (0094) MS (EI): Q7H20N2O6 Exact mass: 348.35, observed mass: 366.2 (+ H20).
Reference:
[1] Chemical Communications, 2015, vol. 51, # 67, p. 13213 - 13216
[2] Organic and Biomolecular Chemistry, 2015, vol. 13, # 28, p. 7736 - 7749
[3] Patent: WO2015/95227, 2015, A2, . Location in patent: Page/Page column 133
[4] Journal of Medicinal Chemistry, 2018, vol. 61, # 3, p. 989 - 1000
[5] Journal of the American Chemical Society, 2003, vol. 125, # 22, p. 6677 - 6686
[6] RSC Advances, 2015, vol. 5, # 89, p. 72579 - 72589
[7] Tetrahedron, 1998, vol. 54, # 14, p. 3489 - 3494
[8] Journal of Pharmaceutical Sciences, 1993, vol. 82, # 1, p. 1 - 10
[9] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1981, vol. 30, # 5, p. 806 - 812[10] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1981, # 5, p. 1045 - 1051
[11] Journal of Medicinal Chemistry, 1994, vol. 37, # 18, p. 2841 - 2845
[12] Bioorganic & Medicinal Chemistry Letters, 1998, vol. 8, # 23, p. 3343 - 3346
[13] Organic and Biomolecular Chemistry, 2006, vol. 4, # 5, p. 853 - 859
[14] Chemistry - A European Journal, 2004, vol. 10, # 16, p. 3879 - 3890
[15] Synlett, 2005, # 18, p. 2802 - 2804
[16] Chemical Communications, 2002, # 7, p. 738 - 739
[17] Tetrahedron, 2007, vol. 63, # 38, p. 9493 - 9501
[18] Tetrahedron, 2008, vol. 64, # 41, p. 9717 - 9724
[19] Tetrahedron Letters, 2008, vol. 49, # 48, p. 6885 - 6888
[20] Inorganic Chemistry, 2010, vol. 49, # 17, p. 7841 - 7852
[21] European Journal of Organic Chemistry, 2010, # 31, p. 5967 - 5979
[22] Patent: US2011/224455, 2011, A1, . Location in patent: Page/Page column 11
[23] Tetrahedron, 2013, vol. 69, # 2, p. 551 - 558
[24] Chemistry - A European Journal, 2013, vol. 19, # 48, p. 16256 - 16262
[25] Organic and Biomolecular Chemistry, 2014, vol. 12, # 5, p. 823 - 831
[26] Chemical and Pharmaceutical Bulletin, 2016, vol. 64, # 8, p. 1181 - 1189
[27] Organic and Biomolecular Chemistry, 2016, vol. 14, # 47, p. 11125 - 11136
[28] Organic and Biomolecular Chemistry, 2017, vol. 15, # 14, p. 3013 - 3024
[29] Patent: WO2017/134548, 2017, A1, . Location in patent: Page/Page column 15-16
65
[ 6066-82-6 ]
[ 2018-66-8 ]
[ 3397-35-1 ]
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1985, # 8, p. 473
[2] Journal fuer Praktische Chemie (Leipzig), 1987, vol. 329, # 5, p. 915 - 922
[3] Tetrahedron Letters, 1986, vol. 27, # 29, p. 3403 - 3406
[4] Bioorganic & Medicinal Chemistry Letters, 1998, vol. 8, # 23, p. 3343 - 3346
[5] Chemistry - A European Journal, 2004, vol. 10, # 16, p. 3879 - 3890
[6] Journal of Medicinal Chemistry, 2005, vol. 48, # 24, p. 7520 - 7534
[7] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 8, p. 2518 - 2526
[8] Synlett, 2005, # 18, p. 2802 - 2804
[9] Journal of Medicinal Chemistry, 1991, vol. 34, # 6, p. 1845 - 1849
[10] Tetrahedron Asymmetry, 2010, vol. 21, # 8, p. 982 - 989
[11] Patent: WO2010/146164, 2010, A1, . Location in patent: Page/Page column 15-16
[12] Tetrahedron Letters, 2012, vol. 53, # 6, p. 623 - 626
[13] Chemistry - A European Journal, 2013, vol. 19, # 48, p. 16256 - 16262
[14] Organic and Biomolecular Chemistry, 2016, vol. 14, # 47, p. 11125 - 11136
66
[ 6066-82-6 ]
[ 2483-46-7 ]
[ 30189-36-7 ]
Reference:
[1] Journal of the American Chemical Society, 2011, vol. 133, # 31, p. 12220 - 12228
[2] Journal of Materials Chemistry, 2012, vol. 22, # 19, p. 10035 - 10041
[3] Russian Journal of Bioorganic Chemistry, 2006, vol. 32, # 5, p. 407 - 412
[4] Journal of the American Chemical Society, 2015, vol. 137, # 32, p. 10056 - 10059
[5] Chemical Communications, 2015, vol. 51, # 31, p. 6832 - 6835
[6] Patent: WO2006/121552, 2006, A2, . Location in patent: Page/Page column 28-29; 2/70
[7] Bioconjugate Chemistry, 2010, vol. 21, # 8, p. 1473 - 1478
[8] Macromolecules, 2015, vol. 48, # 6, p. 1688 - 1702
[9] Journal of the American Chemical Society, 2015, vol. 137, # 18, p. 6000 - 6010
[10] Patent: US2015/259535, 2015, A1, . Location in patent: Paragraph 0106
[11] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 2, p. 672 - 676
67
[ 6066-82-6 ]
[ 2389-45-9 ]
[ 34404-36-9 ]
Reference:
[1] Journal of Organic Chemistry, 1989, vol. 54, # 23, p. 5551 - 5558
[2] Chemical and pharmaceutical bulletin, 1987, vol. 35, # 6, p. 2327 - 2333
[3] Chinese Chemical Letters, 2011, vol. 22, # 12, p. 1443 - 1446
[4] Organic and Biomolecular Chemistry, 2018, vol. 16, # 20, p. 3732 - 3740
68
[ 6066-82-6 ]
[ 405-39-0 ]
[ 21160-83-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 1987, vol. 30, # 9, p. 1658 - 1663
[2] Journal of Organic Chemistry, 1995, vol. 60, # 7, p. 1932 - 1935
[3] Journal of Organic Chemistry, 1986, vol. 51, # 17, p. 3320 - 3324
[4] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 8, p. 2518 - 2526
[5] Synlett, 2005, # 18, p. 2802 - 2804
[6] Patent: WO2014/194030, 2014, A2, . Location in patent: Paragraph 245
69
[ 6066-82-6 ]
[ 4286-55-9 ]
[ 4224-70-8 ]
[ 42014-54-0 ]
Reference:
[1] Angewandte Chemie - International Edition, 2002, vol. 41, # 21, p. 4059 - 4061
Reference:
[1] Tetrahedron Letters, 1983, vol. 24, # 41, p. 4451 - 4454
[2] Patent: US3978079, 1976, A,
80
[ 6066-82-6 ]
[ 58-85-5 ]
[ 35013-72-0 ]
Yield
Reaction Conditions
Operation in experiment
91%
at 20℃;
To a solution of (+)-biotin (0.82 mmol, 200 mg) and N-hydroxysuccinimide (0.89 mmol, 102 mg) was added 3-(ethyliminomethylidenamino)-N,N-dimethylpropan-1-aminehydrochloride (0.96 mmol, 184 mg). The reaction mixture was stirred overnight at room temperature and concentrated to give a white solid. The crude solid was mixed with isopropanol, heated up to 70 °C and cooled down. The pure product was filtered off as white powder in 91percent yield (0.75 mmol, 255 mg).
90%
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20℃; for 12 h; Heating
D-Biotin (5.00 g, 2.05 mmol) and N-hydroxysuccinimide (2.36 g, 2.05 mmol) were dissolved in hot DMF (150 mL). N,N′-Dicyclohexylcarbodiimide (5.50 g, 2.67 mmol) was added to the above reaction mixture, and was stirred overnight at room temperature. The dicyclohexyl urea was filtered, and the filtrate was evaporated and triturated with ether. The white precipitate was obtained and washed with ether to give a white powder in 90percent. (6.29 g) yield (1H NMR(400 MHz, CDCl3): 1.5–1.6 (m, 2H)), 1.6–1.9 (m, 4H) 2.58–2.70 (m, 2H),2.75 (d, 1H,) 2.86 (s, 4H), 2.87–2.97 (m, 1H), 3.16 (m, 1H), 4.33 (m, 1H), 4.52 (m, 1H), 4.96 (s, 1H), 5.23 (s, 1H). m/z (ESI) found 342 (M + H)+.C14H19N3O5S calculated 341.
79%
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20 - 70℃;
D-Biotin (0.819 mmol) and N-hydroxysuccinimide (0.819 mmol) were dissolved into anhydrous DMF (6 ml) at 70 00 while stirring. DCC (1.064 mmol) was added and the solution was stirred overnight at room temperature. The formed DCU was filtered off and the solvent was removed under reduced pressure. The residue was taken up into boiling isopropanol and the solution was allowed to cooldown to RT. The target compound precipitated and the product was filtered off as a white solid.Yield: 79percent, MS (ESI) m/z 364.1 [M ÷ Na]NMR (DMSO-d6, 400 MHz): 6 1.42-1.67 (m, 6H), 2.57-2.60 (d, J=12.4 Hz, 1H), 2.68 (t, J=7.3 Hz, 2H), 2.81-2.90(m, 5H), 3.11 (m, 1H), 4.14-4.17(m, 1H), 4.29-4.35 (m, 1H), 6.37(s, 1H), 6.42 (s, 1H)
70%
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20 - 50℃;
1. Synthesis of Biotinyl-N-hydroxysuccinimide (Scheme I) . D-Biotin (0.49 g, 2.0 mmol, Sigma B4501) was added to N-hydroxysuccinimide (0.3 g, 2.7 mmol, Pierce HC102040) into 20 mL DMF (Sigma 227056), heated to 50°C until most of the material dissolved. A solution of Ν,Ν'- Dicyclohexylcarbodiimide (0.45g, 2.1 mmol, Sigma D80002) in 5 mL DMF was added dropwise to the aforementioned solution. The resulting solution was stirred overnight at room temperature during which time a white precipitate was formed. The reaction mixture was filtered through celite, and the filtrate was triturated with diethyl ether (Sigma 309966) . The white precipitate was vacuum filtered and then washed with diethyl ether to give a white powder. The yield was 0.52 g (~ 70 percent final efficiency) .
68%
Stage #1: With 1,1'-carbonyldiimidazole In N,N-dimethyl-formamide at 78℃; Inert atmosphere Stage #2: at 20℃;
Example 2. Biotin N-Hydroxysuccinimide Ester (5); [0157] To a solution containing 101 mg (0.41 mmol) of biotin in 2 rnL of DMF at 78 0C was added 67 mg (0.41 mmol) of l,l '-carbonyldiimidazole with continued heating until CO2 evolution ceased. The solution was equilibrated to room temperature and stirred for an additional 3 h. To the reaction mixture was added a solution of 47 mg (0.41 mmol) of N- hydroxysuccinimide in 2 mL of DMF. The solution was stirred overnight at room temperature. The solvent was removed under diminished pressure and the product was crystallized from 2-propanol and then DMF-ether to afford 5 as a fine white powder: yield 95 mg (68percent); 1H NMR (CDCl3) δ 1.43-1.66 (m, 7H), 2.58 (m, IH), 2.66 (t, 2H), 2.75 (s, 4H), 3.05 (m, IH), 4.11 (m, IH), 4.27 (m, IH), and 6.36 (d, 2H, J = 18.4 Hz); 13C NMR (CDCl3) δ 55.19, 60.68, 63.46, 65.43, 69.11, 70.08, 73.25, 75.85, 79.05, 101.35, 101.83, 126.32, 127.20, 127.81, 128.11, 128.16, 128.48, 128.72, 129.18, 137.81, and 138.24; mass spectrum (MALDI-TOF), m/z 342.1 (M + H)+.
65%
With diisopropyl-carbodiimide In N,N-dimethyl-formamide at 45℃;
[0074] To 2 grams of biotin was added 2 eq of N,N’-diisopropylcarbodiimide and 3 eq of Nhydroxysuccinimide in 0.2 M DMF at 45 °C to yield compound 6. One eq of t-Boc protected PEG amine (3) was then added in methylene chloride at room temperature to give compound 7 and deprotected using TFA to afford compound 8, followed be activation using 2 eq of N,N’-diisopropylcarbodiimide and 3 eq of N-hydroxysuccinimide to form its activated ester, compound 9. G3 dendrimer DNT-296 was added to compound 9 in methanol at room temperature to yield compound 10.
61%
With CDI In N,N-dimethyl-formamide; isopropyl alcohol
1. Preparation of Biotin-N-hydroxysuccinimide Biotin (1.0 g, 4.1 mmole) was dissolved in 10 ml DMF (dry) with heating at 80° C. in oil bath. CDI (665 mg, 4.1 mmoles) was added and the mixture was heated at 80° C. The reaction mixture was stirred at 80° C. for 30 minutes, then at room temperature for 2 hours; a white precipitate formed. N-hydroxysuccinimide (475 mg, 4.1 mmoles) was added and the reaction mixture was stirred at room temperature overnight. DMF was removed under vacuum on rotary evaporator. The solid residue was dissolved in 250 ml of refluxing isopropanol, filtered, and stored in the cold room overnight. The precipitate was filtered, washed one time with cold isopropanol and dried in vacuo at 45° C. overnight to give 870 mg (61percent yield) of the desired product.
57%
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20℃; Inert atmosphere
Biotin (498mg, 2mmol) was dissolved in anhydrous DMF (10mL) by heating to approximately 70°C for 10min or until fully dissolved. The reaction mixture was allowed to cool to room temperature before adding N-hydroxysuccinimide (240mg, 2.1mmol) with stirring at room temperature. A solution of N,N′-dicyclohexylcarbodiimide (438mg, 2.13mmol) in anhydrous DMF (2mL) was added dropwise to the stirred solution. The reaction was then stirred overnight at room temperature over which time a white precipitate formed (N,N′-dicyclohexylurea). The precipitate was removed by filtration and washed with DMF. The filtrate was diluted with EtO2 until a white precipitate formed. The precipitate was collected by filtration and rinsed with EtO2 then dried to give the crude product 26 as a white solid (395mg, 57percent): m.p. (decomp.) 178–190°C; IR (νmax, ATR): 3227, 2941, 2876, 1818, 1788, 1729, 1698, 1465, 1369, 1210, 1071, 861, 739, 656cm−1; 1H NMR (600MHz, DMSO-d6): δ=6.40 (1H, s, NH), 6.35 (1H, s, NH), 4.30 (1H, m, HNCHCHNH), 4.14 (1H, m, HNCHCHNH), 3.10 (1H, m, SCH), 2.84–2.78 (5H, contains NCOCH2CH2 and SCHAHB), 2.67 (2H, t, J=7.7Hz, CH2CH2CO2N), 2.57 (1H, d, J=12.4Hz, SCHAHB), 1.64 (3H, contains CHAHBCH2CH2CH2CO2N), 1.52–1.36 (3H, contains CHAHBCH2CH2CH2COON); 13C NMR (150MHz, DMSO-d6): δ=170.3, 169.0, 162.7, 61.0, 59.2, 55.3, 40.1 (overlaps with NMR solvent peak), 30.0, 27.9, 27.6, 25.5, 24.3; HRMS (ESI): M+H+, found 342.1128. C14H20N3O5S+ requires 342.1118.
30%
Stage #1: With pyridine; dicyclohexyl-carbodiimide In N,N-dimethyl-formamide for 0.5 h; Stage #2: for 24 h;
Biotin (24.4mg, 0.1mmol) was dissolved to N, N- dimethylformamide (DMF) (10mL),Pyridine (Py) (2mL) and N, N'- dicyclohexylcarbodiimide imide (DCC) (41.2mg, 0.2mmol), after half an hour the reaction was added N- hydroxysuccinimide (N-Hydroxysuccinimide ) (13.8mg, 0.12mmol), the reaction is stopped after 24h the reaction, after removal of DMF by filtration and the filtrate evaporated under reduced pressure, then dissolved in hot isopropanol solvent and then filtered and the filtrate precipitated 2 days in a refrigerator at 4 finally filtered to give a white solid precipitated 10.2mg, yield 30percent.
1 g
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 50℃; for 24 h; Inert atmosphere
Into a 50 mL reaction flask, 1.0 g of biotin [manufactured by Wako Pure Chemical Industries, Ltd.], 0.59 g of NHS, and 19 g of DMF were charged and the mixture was dissolved with stirring at 50° C. To this solution, 1.0 g of DCC was added and the resultant reaction liquid was stirred at 50° C. for 24 hours in a nitrogen atmosphere. Subsequently, insoluble substances were removed from the reaction liquid by filtration and then DMF was removed by distillation. The obtained residue was washed with 3 g of diethyl ether and then recrystallized with 15 g of IPA. The obtained white crystal was filtered under reduced pressure and dried under vacuum to obtain 1.0 g of biotin N-hydroxysuccinimide ester.
Reference:
[1] Journal of the American Chemical Society, 2007, vol. 129, # 45, p. 13987 - 13996
[2] New Journal of Chemistry, 2013, vol. 37, # 11, p. 3762 - 3769
[3] Organic Letters, 2013, vol. 15, # 16, p. 4130 - 4133
[4] Journal of Materials Chemistry, 2004, vol. 14, # 17, p. 2638 - 2642
[5] Beilstein Journal of Organic Chemistry, 2015, vol. 11, p. 784 - 791
[6] MedChemComm, 2015, vol. 6, # 2, p. 363 - 371
[7] Spectrochimica Acta - Part A: Molecular and Biomolecular Spectroscopy, 2016, vol. 153, p. 566 - 571
[8] Organometallics, 2014, vol. 33, # 21, p. 6154 - 6164
[9] Chemistry - A European Journal, 2017, vol. 23, # 45, p. 10906 - 10914
[10] Chemical Communications, 2006, # 7, p. 717 - 719
[11] Journal of Medicinal Chemistry, 2013, vol. 56, # 11, p. 4300 - 4319
[12] Angewandte Chemie - International Edition, 2015, vol. 54, # 22, p. 6641 - 6644[13] Angew. Chem., 2015,
[14] Journal of the American Chemical Society, 2005, vol. 127, # 48, p. 16912 - 16920
[15] Bioconjugate Chemistry, 2012, vol. 23, # 12, p. 2417 - 2433
[16] Chemical Communications, 2009, # 33, p. 5030 - 5032
[17] Angewandte Chemie - International Edition, 2014, vol. 53, # 2, p. 575 - 581[18] Angew. Chem., 2014, vol. 126, # 2, p. 586 - 592,7
[19] Nucleosides, Nucleotides and Nucleic Acids, 2011, vol. 30, # 7-8, p. 490 - 502
[20] Patent: WO2015/144933, 2015, A1, . Location in patent: Page/Page column 29; 40
[21] Nucleosides and Nucleotides, 1999, vol. 18, # 6-7, p. 1231 - 1234
[22] Biochimie, 2011, vol. 93, # 8, p. 1357 - 1367
[23] Journal of Medicinal Chemistry, 2008, vol. 51, # 9, p. 2816 - 2832
[24] Chemical Communications, 2012, vol. 48, # 14, p. 2003 - 2005
[25] Patent: WO2017/13547, 2017, A1, . Location in patent: Page/Page column 45-46
[26] Angewandte Chemie - International Edition, 2017, vol. 56, # 32, p. 9449 - 9453[27] Angew. Chem., 2017, vol. 129, # 32, p. 9577 - 9581,5
[28] Macromolecules, 2011, vol. 44, # 7, p. 2016 - 2024
[29] Patent: WO2011/19419, 2011, A1, . Location in patent: Page/Page column 47
[30] RSC Advances, 2017, vol. 7, # 11, p. 6259 - 6265
[31] Patent: WO2015/38493, 2015, A1, . Location in patent: Page/Page column 0074
[32] Journal of Natural Products, 2016, vol. 79, # 1, p. 180 - 188
[33] Chemical Communications, 2017, vol. 53, # 72, p. 9971 - 9974
[34] Patent: US4828979, 1989, A,
[35] Chemical Communications, 2012, vol. 48, # 65, p. 8129 - 8131
[36] Journal of Drug Targeting, 2011, vol. 19, # 6, p. 418 - 426
[37] Patent: US2016/361436, 2016, A1, . Location in patent: Paragraph 0206
[38] Tetrahedron, 2018, vol. 74, # 12, p. 1220 - 1228
[39] Chemical Communications, 2018, vol. 54, # 29, p. 3613 - 3616
[40] European Journal of Organic Chemistry, 2007, # 28, p. 4711 - 4720
[41] Patent: CN104031055, 2016, B, . Location in patent: Paragraph 0094; 0115; 0116; 0117; 0118
[42] Angewandte Chemie - International Edition, 2018, vol. 57, # 38, p. 12478 - 12482[43] Angew. Chem., 2018, vol. 130, p. 12658 - 12662,5
[44] Molecular Crystals and Liquid Crystals Science and Technology, Section A: Molecular Crystals and Liquid Crystals, 1993, vol. 235, p. 121 - 126
[45] Journal of the American Chemical Society, 2004, vol. 126, # 44, p. 14435 - 14446
[46] Organic and Biomolecular Chemistry, 2005, vol. 3, # 13, p. 2463 - 2468
[47] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2006, vol. 45, # 11, p. 2512 - 2522
[48] Organic Letters, 2007, vol. 9, # 11, p. 2131 - 2134
[49] Chemistry of Natural Compounds, 2006, vol. 42, # 3, p. 325 - 327
[50] Patent: US2005/118252, 2005, A1, . Location in patent: Page/Page column 7; sheet 7
[51] Journal of Medicinal Chemistry, 2009, vol. 52, # 22, p. 7003 - 7013
[52] Organic and Biomolecular Chemistry, 2011, vol. 9, # 21, p. 7296 - 7299
[53] Analytical Biochemistry, 2011, vol. 413, # 1, p. 30 - 35
[54] Bioorganic and Medicinal Chemistry Letters, 2012, vol. 22, # 1, p. 586 - 590
[55] European Journal of Organic Chemistry, 2013, # 3, p. 465 - 473
[56] Chemistry and Physics of Lipids, 2015, vol. 188, p. 27 - 36
[57] Asian Journal of Chemistry, 2013, vol. 25, # 2, p. 811 - 816
[58] Bioconjugate Chemistry, 2016, vol. 27, # 11, p. 2780 - 2790
[59] European Journal of Medicinal Chemistry, 2014, vol. 71, p. 219 - 228
[60] Organic and Biomolecular Chemistry, 2014, vol. 12, # 34, p. 6624 - 6633
[61] Patent: WO2014/203156, 2014, A2, . Location in patent: Paragraph 0001-0002
[62] Journal of Nanoscience and Nanotechnology, 2015, vol. 15, # 1, p. 176 - 180
[63] Journal of Polymer Science, Part A: Polymer Chemistry, 2015, vol. 53, # 17, p. 2036 - 2049
[64] Journal of Materials Chemistry B, 2015, vol. 3, # 41, p. 8170 - 8179
[65] Patent: US9284393, 2016, B2, . Location in patent: Page/Page column 15
[66] Organic and Biomolecular Chemistry, 2014, vol. 12, # 47, p. 9638 - 9643
[67] Molecules, 2016, vol. 21, # 5,
[68] Patent: US2016/272669, 2016, A1, . Location in patent: Paragraph 0056-0058
[69] Journal of the American Chemical Society, 2017, vol. 139, # 1, p. 156 - 170
[70] Chemical Communications, 2017, vol. 53, # 36, p. 5020 - 5023
[71] Patent: CN106866743, 2017, A, . Location in patent: Paragraph 0016; 0119
[72] Chemistry - A European Journal, 2017, vol. 23, # 49, p. 11945 - 11954
[73] Angewandte Chemie - International Edition, 2018, vol. 57, # 13, p. 3406 - 3410[74] Angew. Chem., 2018, vol. 130, p. 3464 - 3468,5
[75] Chemical Communications, 2018, vol. 54, # 68, p. 9506 - 9509
[76] Organic Letters, 2018, vol. 20, # 17, p. 5344 - 5347
[77] RSC Advances, 2018, vol. 8, # 57, p. 32775 - 32793
81
[ 6066-82-6 ]
[ 35013-72-0 ]
Yield
Reaction Conditions
Operation in experiment
2.7 g
With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20℃; Heating
Step 1: To a round bottom flask with a stirring bar, biotin (2.0 g, 8.2 mmol) and DMF (60 mL) were added. After the solid was dissolved with heat, N-hydroxysuccinimide (0.944 g, 8.2 mmol) and DCC (2.2 g, 10.7 mmol) were added. The reaction mixture was stirred at room temperature overnight. The white solid was filtered, and the DMF was evaporated under reduced pressure. The resulting residue was further purified by re-crystallization from isopropanol to give the desired product 2 (2.7 g, M+H+ = 342.5) as white crystals.
Reference:
[1] Journal of Medicinal Chemistry, 1984, vol. 27, # 11, p. 1406 - 1410
[2] Journal of the American Chemical Society, 2009, vol. 131, # 7, p. 2438 - 2439
83
[ 6066-82-6 ]
[ 20160-60-5 ]
[ 78269-85-9 ]
Yield
Reaction Conditions
Operation in experiment
85%
With triethylamine In acetonitrile at 0 - 20℃; for 16 h; Inert atmosphere
TeocCl reagent S13 was dissolved in CH3CN (105 mL) at 0 °C. Then, NHS (5.22 g,45.4 mmol, 1.30 equiv) and Et3N (4.59 g, 45.4 mmol, 1.30 equiv; in 11.0 mL CH3CN) wereadded. The reaction mixture was stirred at 0 °C rt. After 16 h, the reaction mixture waspoured into H2O. The aqueous layer was extracted with Et2O (6×). The combined organiclayers were washed with H2O (2×), HCl (1.0 M), followed by H2O and subsequent dried overMgSO4, filtered a well as concentrated under reduced pressure. The desired TeocOSu S14(7.72 g, 29.8 mmol, 85percent over two steps) was collected as colourless solid.
Stage #1: With 2,4,6-tripropyl-1,3,5,2,4,6-trioxatriphosphinane-2,4,6-trioxide; triethylamine In dichloromethane at 20℃; for 0.333333 h; Stage #2: for 48 h;
To a solution of Gly-Gly (1.00 g, 7.57 mmol) in dioxane:water (30:5 mL) at room temperature was added triethylamine (1.15 g,11.4 mmol) and di-tert-butyl-dicarbonate (1.84 g, 8.33 mmol) consecutively. The mixture was stirred at room temperature overnight, then diluted with water, acidified to approximately pH 2via the addition of solid KHSO4, extracted with EtOAc, dried(Na2SO4), and concentrated in vacuo to afford (tert-butoxycarbonyl)glycylglycine [8] as a white solid. To a solution of crude (tertbutoxycarbonyl)glycylglycine (0.106 g, 0.451 mmol) in anhydrous dichloromethane (5.00 mL) was added triethylamine (45.6 mg,0.451 mmol) and propylphosphonic anhydride solution (T3P®) [9] (0.344 g, 0.541 mmol) at room temperature. The mixture was stirred for 20 min, followed by the addition of N-hydroxy succinimide (51.9 mg, 0.451 mmol). The reaction was stirred for 48 h and upon completion, the organic layer was washed with brine (3),dried (Na2SO4), and concentrated in vacuo to afford the product as a white solid. The crude solid was triturated from diethyl ether toafford 65.4 mg (44percent) of the product as a white solid: 1H NMR(400 MHz, CDCl3) d 6.76 (brs, 1 H), 5.15 (brs, 1 H), 4.43 (d, 2 H,J 5.7 Hz), 3.87 (d, 2 H, J 5.3 Hz), 2.85 (s, 4 H), 1.45 (s, 9 H); ESIMSm/z 330 [MH]. Direct infusion ESI-MS on a high resolution accurate mass measurements (Exactive Plus, Thermo Fisher Scientific) in the negative ion mode yielded an elemental composition of the expected product within 5 ppm (see Fig. 1).
Reference:
[1] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 22, p. 5973 - 5982
[2] Journal of Pharmaceutical Sciences, 1984, vol. 73, # 2, p. 275 - 277
[3] Analytical Biochemistry, 2016, vol. 512, p. 114 - 119
85
[ 6066-82-6 ]
[ 372-09-8 ]
[ 56657-76-2 ]
Reference:
[1] Polish Journal of Chemistry, 1992, vol. 66, # 1, p. 111 - 118
[2] Farmaco, 1992, vol. 47, # 11, p. 1367 - 1383
[3] Journal of Medicinal Chemistry, 2005, vol. 48, # 24, p. 7781 - 7788
[4] Patent: WO2010/14930, 2010, A2, . Location in patent: Page/Page column 29-30
86
[ 6066-82-6 ]
[ 35661-39-3 ]
[ 73724-40-0 ]
Reference:
[1] Journal of Pharmaceutical Sciences, 1994, vol. 83, # 7, p. 999 - 1005
[2] Helvetica Chimica Acta, 2018, vol. 101, # 1,
[3] Journal of the American Chemical Society, 2003, vol. 125, # 45, p. 13680 - 13681
[4] Chemical Communications, 2009, # 4, p. 407 - 409
[5] Tetrahedron, 2009, vol. 65, # 19, p. 3871 - 3877
[6] Organic and Biomolecular Chemistry, 2011, vol. 9, # 11, p. 4182 - 4187
[7] Patent: WO2015/21092, 2015, A1, . Location in patent: Page/Page column 36
[8] Bioconjugate Chemistry, 2015, vol. 26, # 11, p. 2261 - 2278
[9] Patent: WO2015/187540, 2015, A1, . Location in patent: Page/Page column 22; 23
87
[ 6066-82-6 ]
[ 110-15-6 ]
[ 30364-60-4 ]
Yield
Reaction Conditions
Operation in experiment
27%
With dicyclohexyl-carbodiimide In tetrahydrofuran at 0 - 20℃;
To succinic acid (3 0 g, 25 42 mmol, 1 OO equiv) m THF (50 mL) was added a solution of 1- hydroxypyrrohdine-2,5-dione (64 g, 55 65 mmol, 2 20 eqmv) This was followed by the addition of a solution of DCC (11 5 g, 55 83 mmol, 2 20 equiv) in THF (50 mL) dropwise with stirring at O0C The resulting solution was stirred overnight at room temperature The reaction progress was monitored by LCMS The solids were collected by filtration and the filtrate was concentrated to give the crude product The resulting solids were washed with THF and ethanol This resulted in 2 4 g (27percent) of bis(2,5- dioxopyrrohdin-1-yl) succinate as a white solid.
Reference:
[1] Patent: WO2010/78449, 2010, A2, . Location in patent: Page/Page column 291
[2] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1980, vol. 29, # 5, p. 785 - 789[3] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1980, # 5, p. 1078 - 1081
[4] Chemical Papers, 2016, vol. 70, # 4, p. 505 - 514
[5] Patent: WO2016/205488, 2016, A1, . Location in patent: Paragraph 0264
88
[ 6066-82-6 ]
[ 543-20-4 ]
[ 30364-60-4 ]
Reference:
[1] Journal of Inclusion Phenomena and Macrocyclic Chemistry, 2011, vol. 69, # 1-2, p. 75 - 84
[2] Patent: WO2015/155231, 2015, A1, . Location in patent: Page/Page column 67; 68
89
[ 6066-82-6 ]
[ 57078-98-5 ]
[ 80307-12-6 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 1999, vol. 7, # 5, p. 901 - 919
[2] Patent: WO2013/162757, 2013, A1, . Location in patent: Sheet 7
[3] Organic and Biomolecular Chemistry, 2014, vol. 12, # 34, p. 6624 - 6633
[4] Patent: US2016/228572, 2016, A9, . Location in patent: Paragraph 0185
90
[ 6066-82-6 ]
[ 116965-29-8 ]
[ 80307-12-6 ]
Yield
Reaction Conditions
Operation in experiment
104.2g
With tributyl-amine In acetonitrile at 20℃; for 2 h;
N-maleimidobutyric acid (99 g, 0.54 mol) and acetonitrile (300 ml) were placed in a500 ml three-necked flask andthionyl chloride (93.6 g, 0.82 mol) was added. The reaction was warmed to reflux and a large amount of gas was released .After the reaction was refluxed for 4h, the reaction became clear,acetonitrile and excess thionyl chloride were removed by concentration, and 200 ml of acetonitrile was added to remove the remaining acid in one pass to obtaincrudeN-maleimidobutyrylchloride.The crude product was dissolved in 200ml of acetonitrile was added dropwise to N- hydroxysuccinimide (69g, 0.6mol) and tri-n-butylamine (148g, 0.8mol) was dissolved in 300ml of acetonitrile, with ice water cooling to control the temperature does not exceed 20 , drip finished at room temperature for2h.The reaction solution was concentrated to remove acetonitrile, dissolved in 400 ml of methylene chloride, and dissolved in 200 ml of dichloromethane to remove tri-n-butylaminehydrochloride and 100 ml of 1N hydrochloric acid to remove excess tri-n-butylamine and 200 ml of saturated saline solution, Drywhite solid 139g, a mixed solvent of ethyl acetate and petroleum ether to give a white solid after recrystallization 104.2g, HPLC purity 98.9percent, 69.1percent yield.
Reference:
[1] Organic and Biomolecular Chemistry, 2009, vol. 7, # 17, p. 3400 - 3406
92
[ 6066-82-6 ]
[ 29022-11-5 ]
[ 113484-74-5 ]
Reference:
[1] Journal of Pharmaceutical Sciences, 1994, vol. 83, # 7, p. 999 - 1005
[2] Journal of the American Chemical Society, 2003, vol. 125, # 45, p. 13680 - 13681
[3] Tetrahedron, 2009, vol. 65, # 19, p. 3871 - 3877
[4] Patent: WO2015/21092, 2015, A1, . Location in patent: Page/Page column 36
93
[ 6066-82-6 ]
[ 68858-20-8 ]
[ 130878-68-1 ]
Yield
Reaction Conditions
Operation in experiment
89%
With dicyclohexyl-carbodiimide In dichloromethane at 20℃; for 3 h;
Dicyclocarbodiimide (1 .55 g, 7.52 mmol) and N-hydroxysuccinimide (762 mg, 6.63 mmol) are added at room temperature to a stirrer solution of Fmoc-Val-OH [9] (1 .5 g, 4.42 mmol) in anhydrous dichloromethane (25 ml_). The mixture is kept at room temperature for 3 hours. The white solid formed in this reaction is filtrated with dichloromethane to remove the dicyclohexylurea, the organic phase is washed with HCI 0.1 N and water, then dried over anhydrous sodium sulfate and the solvent removed by rotatory evaporation. The residue is subjected to a flash column chromatography in 1 percent methanol in dichloromethane to afford product [10] as a white solid, 1 .7 g (89percent yield). MS: m/z 459 [M+Na]+.1H NMR (400 MHz, CDCIs) δ 7.80 (d, J = 7.5 Hz, 2H), 7.68 - 7.55 (m, 2H), 7.43 (t, J = 7.4 Hz, 2H), 7.34 (dd, J = 15.9, 8.5 Hz, 2H), 4.70 (d, J = 4.6 Hz, 1 H), 4.56 - 4.40 (m, 3H), 4.28 (t, J = 6.6 Hz, 1 H), 2.85 (s, 4H), 2.37 (dd, J = 12.3, 6.5 Hz, 1 H), 1 .08 (dd, J = 1 1 .0, 6.9 Hz, 6H).
65%
With dicyclohexyl-carbodiimide In 1,2-dimethoxyethane at 20℃; for 23 h; Cooling with ice; Inert atmosphere
Fmoc-Valine (1.02 g, 3 mmol) and N-hydroxysuccinimide (0.345 g, 3 mmol) were dissolved in dimethoxy ethane (35 ml) and cooled in an ice bath, then DCC (0.681 g, 3.3 mmol) was added. The resulting mixture was stirred in the ice bath for 3 hours, then at room temperature for 20 hours. The precipitate formed was filtered off and the filtrate concentrated under vacuo. The crude product was further purified by flash chromatography (ethyl acetate/hexane, v:v, 4:6) to afford SI20B as a white solid. Isolated yield: 65percent. TLC (EtOAc:Hexane 3:2). Rf=0.57, irradiated by a UV lamp. HPLC: 0.1percent TFA (v/v) in water (solvent A):acetonitrile (solvent B); gradient 45-85percent in 30 min, flow rate=0.5 mL/min. Retention time (Rt)=15.77 min. 1H NMR (FIG. 26A): (400 MHz, CDCl3) δ 7.76-7.78 (d, J=7.20 Hz, 2H), 7.59-7.60 (d, J=7.20 Hz, 2H), 7.38-7.42 (t, J=7.20 Hz, 2H), 7.30-7.34 (m, 2H), 5.26-5.28 (d, J=9.20 Hz, 1H), 4.67-4.71 (dd, J=4.8, 5.2 Hz, 1H), 4.42-4.46 (dd, J=6.8, 6.4 Hz, 2H), 4.23-4.26 (t, J=6.8 Hz, 1H), 2.84 (s, 4H), 2.04-2.36 (m, 1H), 0.83-088 (m, 6H). 13C NMR (FIG. 26B): (101 MHz, CDCl3) δ 168.7, 141.5, 127.9, 127.3, 127.3, 125.3, 120.2, 120.2, 67.5, 57.7, 47.4, 31.9, 25.8, 18.9, 17.5.
26.51 g
With dicyclohexyl-carbodiimide In tetrahydrofuran at 0 - 20℃; for 6 h; Inert atmosphere
[00125] This compound is prepared according to R. A. Firestone et al, US 6,214,345. Fmoc-Val-OH (20.24 g; 59.64 mmol) and N-hydroxysuccinimide (6.86 g =1 .0 eq.) in tetrahydrofuran (200 ml) at 000 were treated with N,N’dicyclohexylcarbodiimide (12.30 g; 1.0 eq.). The mixture was stirred at RT under argon atmosphere for 6 h and then the solid dicyclohexyl urea (DCU) by-product was filtered off and washed with THF and the solvent was removed by rotavap. The residue was dissolved in 300 ml dichloromethane, cooled in an ice bath for 1 h and filtered again to remove additional DCU. The dichloromethane was evaporated and the solid foam (26.51 g) was used in the next step without further purification.
Reference:
[1] Journal of Pharmaceutical Sciences, 1994, vol. 83, # 7, p. 999 - 1005
[2] Langmuir, 2010, vol. 26, # 7, p. 4990 - 4998
[3] Patent: WO2018/178060, 2018, A1, . Location in patent: Page/Page column 64-66
[4] Journal of the American Chemical Society, 2015, vol. 137, # 21, p. 6932 - 6940
[5] Patent: US2017/247324, 2017, A1, . Location in patent: Paragraph 0284; 0285
[6] Tetrahedron, 2009, vol. 65, # 19, p. 3871 - 3877
[7] Organic and Biomolecular Chemistry, 2011, vol. 9, # 11, p. 4182 - 4187
[8] Journal of Controlled Release, 2012, vol. 160, # 3, p. 618 - 629
[9] Patent: WO2013/67597, 2013, A1, . Location in patent: Page/Page column 80
[10] Patent: WO2014/80251, 2014, A1, . Location in patent: Sheet 16/23
[11] Patent: WO2015/21092, 2015, A1, . Location in patent: Page/Page column 36
[12] Bioconjugate Chemistry, 2015, vol. 26, # 11, p. 2261 - 2278
[13] Patent: WO2017/149077, 2017, A1, . Location in patent: Paragraph 00128-00129
[14] Patent: CN107789630, 2018, A, . Location in patent: Paragraph 0067; 0068; 0069
[15] Patent: WO2018/115466, 2018, A1, . Location in patent: Paragraph 00103; 00124; 00125
94
[ 6066-82-6 ]
[ 133081-25-1 ]
[ 133081-26-2 ]
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 16, p. 4764 - 4767
[2] Journal of Labelled Compounds and Radiopharmaceuticals, 1997, vol. 40, p. 455 - 457
[3] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 12, p. 3440 - 3444
[4] RSC Advances, 2015, vol. 5, # 113, p. 93374 - 93385
[5] Journal of Labelled Compounds and Radiopharmaceuticals, 2017, vol. 60, # 9, p. 431 - 438
95
[ 6066-82-6 ]
[ 253265-97-3 ]
Yield
Reaction Conditions
Operation in experiment
99.6%
With triethylamine In tetrahydrofuran for 2 h; Reflux
The compounds of formula 1 obtained in Example 3 (3.8g, 0.02mol) was dissolved in 30ml of tetrahydrofuran, was added dropwise to N- hydroxyalkylSuccinimide (2.3g, 0.02mol) in 45ml of tetrahydrofuran was added triethylamine (2.02g, 0.02mol), the addition was complete, reflux, 2 hours reaction, TLC detection material disappeared, water was added to brine and extracted , take the upper concentrated to give 5.4g compound of formula 6-1 in a yield of 99.6percent.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In 1,4-dioxane; at 20℃;Inert atmosphere;
In a dry flask containing a magnetic stir bar, 0.50 g of 1 (2.64 mmol) and 0.31 g of N-hydroxysuccinimide (SuOH) (2.72 mmol, 1.03 equivalent) were dissolved in 3mL of distilled dioxane. Subsequently, 1.25 equivalent of 3-(ethyliminomethyleneamino)-N,N-dimethylpropan-l-amine in hydrogen chloride form (EDC) (0.63 g, 3.3 mmol) was added into the flask. The solution was cloudy, and 2mL of freshly distilled dioxane was added into the reaction mixture. The reaction mixture was stirred under nitrogen for 30 minutes and then left for stirring overnight at room temperature. Dioxane was removed under vacuum. The residue was dissolved in chloroform and washed with water (3x). The organic layer was dried of over Na2S04 and concentrated under vacuum. The desire product was precipitated in ethanol and dried in a desiccator, yielding 0.47g of product (62%). The 1H NMR spectral data matched the previously reported values for the compound.
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide;
A mixture of N-Boc- -Ala-OH (1 g, 5.29 mmol), iV-hydroxysuccinimide (0.9 g, 7.82 mmol) in DMF (10 mL) were added HOBt (0.7 g, 5.25 mmol) and EDCI.HC1 (1 g, 5.23 mmol) at 5 C. The reaction mixture was warmed to RT and stirred for 16 h. The progress of the reaction was monitored by TLC. The reaction was quenched with water and extracted with ethyl acetate (2 x 150 mL). The combined organic layers were washed with water (3 x 100 mL), brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was triturated with ether (2 x 25 mL) to afford N-Boc- -Ala-OSu (1.1 g, crude) as white solid. 1H NMR (500 MHz, CDC13): delta 5.10 (bs, 1H), 3.52 (q, / = 6.0 Hz, 2H), 2.85-2.82 (m, 6H), 1.31 (s, 9H).
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 5 - 20℃; for 16h;
To mixture of Boc- -alanine (N-Boc- -Ala-OH; 1 g, 5.29 mmol) and N-hydroxysuccinimide (0.9 g, 7.82 mmol) in DMF (10 mL) were added 1-hydroxybenzotriazole hydrate (HOBt FontWeight="Bold" FontSize="10" xH20; 0.7 g, 5.25 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI'HCl; 1 g, 5.23 mmol) at 5 C. The reaction mixture was warmed to RT and stirred for 16 h. The progress of the reaction was monitored by TLC. The reaction was quenched with water and the mixture was extracted with EtOAc (2 x 150 mL). The combined organic layers were washed with water (3 x 100 mL) and brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was triturated with ether (2 x 25 mL) to afford N-Boc- -Ala-OSu (1.1 g, crude) as a white solid. 1H NMR (500 MHz, CDC13): delta 5.10 (br s, 1H), 3.52 (q, J= 6.0 Hz, 2H), 2.85-2.82 (m, 6H), 1.31 (s, 9H).
With 1-hydroxybenzotriazol-hydrate; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 5 - 20℃; for 16h;
To a mixture of Boc- -alanine (N-Boc- -Ala-OH; 1 g, 5.29 mmol) and N- hydroxysuccinimide (0.9 g, 7.82 mmol) in DMF (10 mL) were added 1 -hydroxybenzotriazole hydrate (EtaOmicronBeta·chiEta20; 0.7 g, 5.25 mmol) and l-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI'HCl; 1 g, 5.23 mmol) at 5 C. The reaction mixture was warmed to RT and stirred for 16 h. The progress of the reaction was monitored by TLC. The reaction was quenched with water and the mixture was extracted with EtOAc (2 x 150 mL). The combined organic layers were washed with water (3 x 100 mL) and brine (150 mL), dried over anhydrous Na2S04 and concentrated under reduced pressure. The crude compound was triturated with ether (2 x 25 mL) to afford N-Boc- -Ala-OSu (1.1 g, crude) as a white solid. 1H NMR (500 MHz, CDC13): delta 5.10 (br s, 1H), 3.52 (q, J= 6.0 Hz, 2H), 2.85-2.82 (m, 6H), 1.31 (s, 9H).
General procedure: The N-Cbz-L-amino acid (1 mmol) and N-hydroxysuccinimide(1 mmol) were dissolved in dry THF at 0 C. Once a clear solutionhad been obtained, DCC (1 mmol) in anhydrous THF was added inseveral aliquots and the resulting solution was stirred at 0e5 C for3 h. The dicyclohexylurea formed was filtered off and the filtratewas concentrated to dryness. The crude product was recrystallizedfrom 2-propanol to furnish the pure product
With bis(trichloromethyl) carbonate; triethylamine; In dichloromethane; at 0℃; for 0.5h;
General procedure: To a stirred solution of the carboxylic acid (2.0 mmol, 1.0 eq) in CH2Cl2 (10.0 ml) at 0C were added triphosgene (296.8 mg, 1.0 mmol, 1.5 eq), triethylamine (1.40 ml, 10.0 mmol,5.0 eq) and N-hydroxysuccinimide (230.2 mg, 2.0 mmol, 1.0 eq). After 30 min, amine(2.0 mmol, 1.0 eq) was added at 0C to the reaction mixture. The reaction mixture was stirred for 1 h at room temperature. The progress of the reaction was monitored by TLC. Removal of the solvent by rotary evaporation followed by short path silica gel column chromatography purification using 50% ethyl acetate in hexane as themobile phase afforded the pure product in high yields.
With dicyclohexyl-carbodiimide; In 1,4-dioxane; at 5 - 20℃; for 20h;
2.2 ml (25 mmol) DCCD was added at 5C to a solution of 1.43 ml (25 mmol) acetic acid and 2.2 ml (0.25 mmol)N-hydroxysuccinimide in 50 ml dioxane. The reaction mixture was stirred at 20C for 20 h. The resulting precipitate wasfiltered off and washed with dry dioxane. The filtrates were combined and evaporated to yield 3.65 g (93%) of the productas white crystals. Rf 0.41 (C), Rf 0.75 (I); m.p. 103-105C. 1H NMR (DMSO-d6): 2.34 (3H, s, -CH3CO-), 2.80 (4H, m, -CH2CH2- OSu).
With 1,2-dichloro-ethane; In dichloromethane; for 3h;
General procedure: To a stirred solution of appropriate acid 44a-f (1.96mmol) in CH2Cl2 (3mL) were added NHS (0.361g, 3.14mmol) and EDC (0.601g, 3.14mmol). After 3h, the mixture was diluted with CH2Cl2 (40mL). The organic layer was washed with brine (2×40mL), saturated aqueous NaHCO3 (40mL) and brine (2×40mL), dried over Na2SO4, filtered and concentrated to give NHS activated esters (80-100% yield) that were used immediately without further purification.
With dicyclohexyl-carbodiimide; In ethyl acetate; at 20℃;Inert atmosphere;
The syntheses of GlcNAc and GlcNGc from glucosamine were based on the method described by Lapidot [31]. The procedure for the synthesis of GlcNAc was performed as follows: acetic acid (0.286 mL, 5 mmol) was added to a solution of N-hydroxysuccinimide (0.575 g, 5 mmol) in dry ethyl acetate (23 mL). A solution of dicyclohexylcarbodiimide (1.03 g, 5 mmol) in ethyl acetate (2 mL)was then added. The reaction mixture was stirred at room temperature overnight under a nitrogen atmosphere. Then, the precipitate of the reaction mixture was removed by centrifugation at 13,400 g for 10 min. The supernatant contained an NHS-ester of acetic acid was dried by rotary evaporation under reduced pressure and re-dissolved in dry 17 mL of methanol. D-Glucosamine hydrochloride(0.862 g, 4 mmol) and triethylamine (0.56 mL) were then added and dissolved by placing the sample ina heated sonication bath (50 C for 1 h). The reaction mixture was then stirred at room temperature for 16 h. The sample was again dried using rotary evaporation and washed three times with ethyl acetate(1 mL) to remove the unreacted starting material. After vacuum drying of the sample using centrifugal evaporation, the product was dissolved in 8 mL of 10% methanol in water (v/v). The supernatant was collected and concentrated using a rotary evaporator under reduced pressure. For the synthesisof GlcNGc, glycolic acid (0.3 mL, 5 mmol) was used instead of acetic acid. As judged by TLC analysis, the conversion of D-glucosamine to the reaction products 4a or 4b reached approximately 70%. These amidated reaction products were then used directly as starting material for the enzymatic sialylation reactions.
With dicyclohexyl-carbodiimide; In tetrahydrofuran; at 0 - 20℃;
To succinic acid (3 0 g, 25 42 mmol, 1 OO equiv) m THF (50 mL) was added a solution of 1- hydroxypyrrohdine-2,5-dione (64 g, 55 65 mmol, 2 20 eqmv) This was followed by the addition of a solution of DCC (11 5 g, 55 83 mmol, 2 20 equiv) in THF (50 mL) dropwise with stirring at O0C The resulting solution was stirred overnight at room temperature The reaction progress was monitored by LCMS The solids were collected by filtration and the filtrate was concentrated to give the crude product The resulting solids were washed with THF and ethanol This resulted in 2 4 g (27%) of bis(2,5- dioxopyrrohdin-1-yl) succinate as a white solid.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In acetone; at 25℃; for 24.0h;
General procedure: NHS-Cn was prepared as described by Chen et al. (2011) with a small modification; end-bit binary acid (15 mmol each; C2, C3, C4, C5, C6, C8, C10, C14) and NHS (40 mmol) were dissolved in acetone (25 mL), and then EDC (30 mmol) was added to the solution. The clear mixture was gently stirred at 25C for 24 h. After the acetone was removed by rotary evaporation under reduced pressure, the residue was washed several times with deionised water and then dried under vacuum at 50C. The crosslinker thus prepared was characterised by the 1H NMR and FT-IR spectra.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride;
[0264] To a solution of diacid la (2 mmol) and hydroxysuccinimide (0.55 g, 4.8 mmol) in 30 mL of DMF was added EDC (0.75 g, 4.8 mmol) over 4 min. The reaction mixture was stirred for 22 h and the solvent was removed under reduced pressure. The residue was taken up in EtOAc (150 mL) and transferred to a separatory funnel. The organic phase was washed (I N HCl, brine, sat. NaHC03, brine), dried (Na2S04), filtered, and concentrated to afford the product as a white solid, which was used without additional purification or characterization.
With dicyclohexyl-carbodiimide; In tetrahydrofuran; at 25 - 28℃; for 2h;
Step (1): 1.3 mmoles of <strong>[69-78-3]5,5'-dithiobis(2-nitrobenzoic acid)</strong> (DNBA), 2.5 mmole of dicyclohexylcarbodiimide (DCCD), 2.6 mmoles of N-hydroxysuccinimide Amine (NHS) and 20 mL of tetrahydrofuran (THF) to give a mixture.Step (2): The mixture was stirred at 25 to 28 C for 2 hours to allow the mixture to react to obtain a reaction solution.Step (3): The reaction solution was filtered to remove the residual solid to give a filtered solution.Step (4): The filtered solution was added to 3 g (24.91 mmole) of magnesium sulfate desiccant, and the filtered solution was dehydrated and dried to obtain a dehydrated solution.Step (5): THF in the dewatered solution was removed by means of reduced pressure concentrating to obtain a crude product as a solid.Step (6): The DSNB compound (white solid, yield 97%) was prepared by dissolving the crude solid with 10 mL of ethyl acetate and then adding 400 mL of n-hexane dropwise under supersonic shaking.
With diisopropyl-carbodiimide; In dichloromethane; at 20℃;
Compound 124 (400 g, 2.18 mol, 1.0 eq. ) was dissolved in CH2Cl2(1.5 L) , to which N-hydroxysuccinimide (276 g, 2.40 mmol, 1.1 eq. ) and DIC (303 g, 2.40 mol, 1.1 eq. ) were added at r.t. and stirred overnight. The reaction was concentrated and purified by column chromatography (1: 2 petroleum ether/EtOAc) to give NHS ester 125 as a white solid (382 g, 63%yield) .1H NMR (500 MHz, CDCl3) delta 6.74 (s, 2H) , 3.67 (t, J = 6.8 Hz, 2H) , 2.85 (s, 4H) , 2.68 (t, J = 7.5 Hz, 2H) , 2.13 -2.03 (m, 2H) .
With dicyclohexyl-carbodiimide; In dichloromethane; at 0℃;
4-maleimidobutyric acid (0.50 g, 2.7 mmol) and N-hydroxysuccinimide (NHS) (0.32 g, 2.7 mmol) was dissolved in 3 mL of dichloromethane and stirred at 0 C. A solution of N,N?-dicyclohexylcarbodiimide (0.56 g, 2.7 mmol) in 3 mL of dichloromethane was added slowly to the reaction mixture at 0 C. The reaction mixture was stirred over night and the byproducts were filtered of and the NHS activated 4-maleimidobutyric acid was concentrated and dried under vacuum.
With dmap; dicyclohexyl-carbodiimide; In dichloromethane; at 0 - 20℃; for 16h;Inert atmosphere;
To a round bottom flask were added acid 33 (640 mg, 2.43 mmol), N- hydroxysuccinimide (419 mg, 3.64 mmol), DMAP (30 mg, 0.25 mmol), and 10 mL CH2CI2 under N2. The mixture was cooled down to 0 C and then a solution of DCC (752 mg, 3.64 mmol) in 10 mL CH2CI2 was added dropwise. The mixture was allowed to warm up to room temperature slowly overnight (16 h). Lots of white precipitates formed during the reaction. The precipitates were filtered off and the filtrate was washed with 20 mL 0.05 N HCl(aq) twice. The organic layer was dried with Na2S04 and concentrated under vacuum. The residue was dispersed in 20 mL EtOAc, and the insoluble precipitates were filtered off. The filtrate was concentrated to give T2 as a colorless oil (840 mg, 96%). NMR (400 MHz, CDCL): d 4.99 (s, 1H), 4.51 (s, 2H), 3.81-3.64 (m, 4H), 3.54 (t, J= 5.2 Hz, 2H), 3.32 (br q, J= 5.2 Hz, 2H), 2.86 (s, 4H), 1.44 (m, 9H).
91%
With diisopropyl-carbodiimide; In tetrahydrofuran; at 20℃; for 2h;
Boc-AEEA-OH (26.4 g, 100 mmol) and HOSu (12.6 g, 110 mmol) were dissolved in 200 ml tetrahydrofuran. DIC (13.9 g, 110 mmol) was added dropwise under a condition of ice bath, and the reaction was continued at room temperature for 2 h after the dropwise addition. TLC showed that the reaction of the raw materials was completed. A vacuum concentration was performed, and the residue was recrystallized with EA to obtain 33.0 g of Boc-AEEA-OSu with yield: 91%, purity: 96.7%, MS: 361.4 (M+1).
With N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; In dichloromethane; at 20℃; for 8h;
To 2-chloro-4-methylsulfonylbenzoic acid (20 g, 86 mmmol) and N-hydroxysuccinimide (11.5 g, 100 mmol) in 300 mL of dichloromethane was added EDC (19.8 g, 100 mmol) and the resulting mixture was stirred at room temperature for 8 h. The mixture was then quenched with 100 mL of saturated aqueous sodium bicarbonate solution then diluted with 200 mL of water. The mixture was then extracted with ethyl acetate (3 x 300 mL), and the organic layers were washed with saturated aqueous sodium bicarbonate solution and saturated aqueous brine (100 mL each). The organic phase was dried over magnesium sulfate, filtered, and evaporated in vacuo to yield the crude ester. The ester was then dissolved into 200 mL of dioxane then treated with 100 mL of ammonium hydroxide. After stirring for 2 h, the reaction mixture was evaporated in vacuo, diluted with 700 mL of ethyl acetate and then washed with saturated aqueous brine (2 x 200 mL). The organic phase was then dried over magnesium sulfate, filtered, and evaporated in vacuo to yield the primary amide, which was used without further purification.
With acetic acid; triethylamine; In ethanol; ethyl acetate; isopropyl alcohol;
EXAMPLE 12 1.27 g (0.011 mol) of N-hydroxysuccinimide were initially introduced into 10 ml of ethyl acetate, a solution of 3.22 g (0.012 mol) of diphenyl chlorophosphate in 10 ml of ethyl acetate was added and then a solution of 2.53 g (0.025 mol) of triethylamine in 10 ml of ethyl acetate was added dropwise in the course of 10 minutes, the temperature rising to 41° C. A thick, white suspension resulted, which was treated with a solution of 0.6 g (0.01 mol) of acetic acid in 10 ml of ethyl acetate and heated at 45°-55° C. for 16 hours. After ending of the reaction, the solid was filtered off and washed with 2*3 ml of ethyl acetate. The combined filtrates were evaporated in vacuo and the semicrystalline residue which remained was washed by stirring with 20 ml of ethanol for 30 minutes. The white solid thus obtained was filtered off, washed with 2*3 ml of ethanol, sucked dry and then washed by stirring with 10 ml of isopropanol. After filtering off, washing with 2*2 ml of isopropanol and drying in vacuo, 1.1 g (70percent) of white acetic acid succinimidyl ester were obtained. M.p.: 130°-134° C.
4-pyridylformimidoylglycyl-d-phenylglycine[ No CAS ]
[ 541-41-3 ]
[ 551-16-6 ]
[ 110-86-1 ]
Yield
Reaction Conditions
Operation in experiment
In N-methyl-acetamide; diethyl ether;
EXAMPLE 22 A solution of 3.12 g. of 4-pyridylformimidoylglycyl-D-phenylglycine in 30 ml. of dimethylformamide is cooled to -15° C. and treated with 1.18 g. of ethyl chloroformate. After stirring for 15 min. 1.15 g. of N-hydroxysuccinimide is added and the solution allowed to warm to room temperature. The reaction mixture is diluted with 60 ml. of diethyl ether, and the N-hydroxysuccinimide ester hydrochloride allowed to crystallize. The intermediate product is filtered and washed with cold ether. A solution of 3.5 g. of ester hydrochloride in 40 ml. of dimethylformamide is treated with 2.16 g. of <strong>[551-16-6]6-aminopenicillanic acid</strong>, and the resulting slurry treated with 850 mg. of pyridine. The reaction mixture is stirred at -10° C. for 30 min. and then allowed to stir at room temperature for 2 hrs. The reaction is diluted with 200 ml. of methanol and the pH adjusted to 5.5 with dilute hydrochloric acid.
1-(2-aminoethan-1-yl)-2,6-dioxo-4-(3-phenylpropan-1-yl)piperazine dihydrochloride[ No CAS ]
[ 1892-57-5 ]
[ 198895-49-7 ]
N-[2-(2,6-dioxo-4-(3-phenylpropan-1-yl)piperazin-1-yl)ethan-1-yl]-5-thia-1,8b-diazaacenaphthylene-4-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In chloroform; water; acetonitrile;
1) Synthesis of N-[2-(2,6-dioxo-4-(3-phenylpropan-1-yl)piperazin-1-yl)ethan-1-yl]-5-thia-1,8b-diazaacenaphthylene-4-carboxamide To a suspension of 0.8 g (3.67 mmol.) of ethyl 5-thia-1,8b-diazaacenaphthylene-4-carboxylate and 0.84 g (7.30 mmol.) of N-hydroxysuccinimide in acetonitrile (10 ml) was added, at room temperature, 1.41 g (7.36 mmol.) of N-ethyl-N'-3-(N,N-dimethylamino)propylcarbodiimide. The mixture was stirred for 2 hours. To the reaction system was added a solution of 1.66 g (4.77 mmol.) of 1-(2-aminoethan-1-yl)-2,6-dioxo-4-(3-phenylpropan-1-yl)piperazine.dihydrochloride and 2.7 ml (19.4 mmol.) of triethylamine in chloroform (5 ml). The mixture was stirred for further one hour. The solvent was distilled off under reduced pressure. To the residue was added water, and the mixture was subjected to extraction with chloroform. The organic layer was washed with a saturated aqueous saline solution, which was dried over magnesium sulfate. The concentrate was purified by means of a column chromatography (methanol/ethyl acetate 20%) to afford the object compound as a reddish amorphous product. The yield was 1.56 g (89%). 1H-NMR (CDCl3, 200 MHz) delta: 1.72-1.92 (2H, m), 2.40-2.54 (2H, m), 2.60-2.73 (2H, m), 3.24-3.59 (6H, m), 3.97-4.06 (2H, m), 5.73 (1H, dd, J=6.2 & 1.4 Hz), 6.23-6.35 (1H, m), 6.54-6.66 (3H, m), 7.01 (1H, s), 7.11-7.35 (5H, m). IR (KBr): 3350, 2941, 1736, 1682, 1620, 1545, 1346, 1279, 1234, 1182, 1151, 748, 700, 644 cm-1.
1-(trans-4-aminomethyl-1-cyclohexylmethyl)-4-phenylpiperidine dihydrochloride[ No CAS ]
[ 1892-57-5 ]
[ 198895-68-0 ]
[ 198894-70-1 ]
Yield
Reaction Conditions
Operation in experiment
With sodium chloride; triethylamine; In chloroform; acetonitrile;
1) Synthesis of N-[trans-4-(4-phenylpiperidin-1-ylmethyl)-1-cyclohexylmethyl]-5-thia-1,8b-diazaacenaphthylene-4-carboxamide To a solution of 0.41 g (1.88 mM) of 5-thia-1,8b-diazaacenaphthylene-4-carboxylic acid and 0.37 g (3.21 mM) of N-hydroxysuccinimide in acetonitrile (5 ml) was added 0.62 g (3.23 mM) of N-ethyl-N'-3-(N,N-dimethylamino)propylcarbodiimide at room temperature, and the mixture was stirred for 1 hour. To this reaction mixture was added a suspension of 0.70 g (1.95 mM) of 1-(trans-4-aminomethyl-1-cyclohexylmethyl)-4-phenylpiperidine dihydrochloride and 1.0 ml (7.71 mM) of triethylamine in chloroform (10 ml) and the mixture was further stirred for 2 hours. The solvent was then distilled off under reduced pressure and the residue was diluted with water and a small amount of ethanol and extracted with ethyl acetate. The organic layer was washed with saturated aqueous solution of sodium chloride and dried over MgSO4. After concentration, the crude product obtained was purified by column chromatography (methanol/ethyl acetate 30-50%) and recrystallization from ethyl acetate to provide the title compound as red-purple crystals. Yield 576.9 mg (63%) 1H-NMR (200 MHz, CDCl3) delta: 0.78-1.03 (m, 4H), 1.62-2.03 (m, 12H), 2.20 (d, J=7.0 Hz, 2H), 2.39-2.59 (m, 1H), 2.94-3.08 (m, 2H), 3.11-3.23 (m, 2H), 5.72-5.84 (m, 1H), 5.79 (dd, J=6.2, 1.6 Hz, 1H), 6.58-6.74 (m, 3H), 7.06 (s, 1H), 7.12-7.36 (m, 5H). IR (KBr): 3336, 2922, 1618, 1533, 1277, 1161 cm-1.
3-(4-methoxyphenoxy)propanoic acid N-hydroxysuccinimide ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;Inert atmosphere;
Compound 163 was prepared by following Scheme 1A above. <strong>[20811-60-3]3-(4-methoxyphenoxy)propanoic acid</strong> (see Example 1D1, 34.47 g, 169 mmol, 96% purity by HPLC), DCC (34.78 g, 169 mmol) and N-hydroxysuccinimide (19.33, 169 mmol) were combined as dry powders and methylene chloride (500 mL) was added. The suspension was mechanically stirred overnight, ambient temperature, under a nitrogen atmosphere. HPLC analysis showed complete conversion of the acid to the NHS ester (N-hydroxy succinyl ester). To the mixture was added (1R,2R)-2-amino-1-(2,3-dihydro-benzo[1,4]dioxin-6-yl)-3-pyrrolidin-1-yl-propan-1-ol (50 g, 169 mmol) and stirring continued for 2.5 hours. HPLC showed conversion to the product and loss of both the NHS ester and step 5 amine. The reaction mixture was vacuum filtered on a Buechner funnel to remove DCC urea. The solid urea was washed with 500 mL of methylene chloride. The organic layers were combined, placed in a separatory funnel, and treated with 500 mL of 1.0M NaOH. The layers were separated, and the cloudy organic layer was recharged into a separatory funnel and treated with a 6% HCl solution (adjusted to pH=0.03-0.34, 100 mL of solution). Two clear layers formed. The resultant biphasic solution was poured into an Erlenmeyer flask and cautiously neutralized to a pH of 7.2-7.4 with a saturated solution of sodium bicarbonate (approx 200 mL of solution). The organic layer was separated from the aqueous layer, dried over sodium sulfate and evaporated to yield 83.6 g of yellow oil (theoretical yield: 77.03 g). The oil was dissolved in isopropyl alcohol (500 mL) with heating and transferred to a 1 L round bottom flask equipped with a mechanical stirrer and heating mantel. The solution was heated to 50 C. and the mechanical stirrer was set to a rate of 53-64 rpm. Tartaric acid (25.33 g, 168 mmol) was dissolved in deionized water (50 mL) and added to the stirred solution at 50 C. Once the solution turned from milky white to clear, seed crystals were added to the mixture and crystallization immediately began (temperature jumped to 56 C.). After 20 minutes, the mixture was set to cool to a temperature of 35 C. (cooling took 1.15 hours). Heating was removed and the solution was allowed to stir for 12 hours. The resulting thick slurry was filtered on a Buchner funnel. Any remaining solid in the flask was washed onto the funnel using ice-cold isopropyl alcohol (100 mL). The material was transferred to a drying tray and heated to 48 C. under vacuum for 3 days (after two days the material weighed 76 g and after three days it weighed 69.3 g). The solid was analyzed by LC and shown to be 98.1% pure (AUC), the residual solvent analysis showed the material to possess 3472 ppm of isopropyl alcohol, and the DSC (differential scanning calorimetery) showed a melting point of 134.89 C. A total of 69.3 g of white solid was collected (65.7% overall yield). 1H NMR (400 MHz, CDCl3) delta=1.8 (M, 4H), 2.4-2.6 (m, 4H), 2.6 (m, 1H), 2.85 (m, 2H), 3.0 (m, 1H), 3.65 (s, 3H), 3.8 (m, 2H), 3.86 (2, 2H), 4.18 (br s, 5H), 4.6 (s, 1H), 6.6-6.8 (m, 7H), 7.8 (d, 1H); MS for C29H40N2O13 m/z 457.3 [M+H] for main peak (free-base).
With dicyclohexyl-carbodiimide; In dichloromethane; at 20℃;
Example 2El . Preparation of Hemi-Hydrate of Compound 163 N-[2-Hydroxy-2-(2,3-dihvdrobenzo[beta1[1,41dioxin-6-yl)-1-pyrrolidin-1-ylmethyl-ethyll-3-(4- methoxy-phenoxy)-propionamide(Scheme IA); Compound 163 was prepared by following Scheme IA above. 3-(4- methoxyphenoxy)propanoic acid (see Example IDl, 34.47g, 169mmol, 96% purity by HPLC), DCC (34.78g, 169 mmol) and N-hydroxysuccinimide (19.33, 169mmol) were combined as dry powders and methylene chloride (50OmL) was added. The suspension was mechanically stirred overnight, ambient temperature, under a nitrogen atmosphere. HPLC analysis showed complete conversion of the acid to the NHS ester (N-hydroxy succinyl ester). To the mixture was added (IR, 2R)-2- amino- 1 -(2,3 -dihydro-benzo [ 1 ,4] dioxin-6-yl)-3 -pyrrolidin- 1 -yl-propan- 1 -ol (5Og, 169mmol) and stirring continued for 2.5 hours. HPLC showed conversion to the product and loss of both the NHS ester and step 5 amine. The reaction mixture was vacuum filtered on a Bchner funnel to remove DCC urea. The solid urea was washed with 50OmL of methylene chloride. The organic layers were combined, <n="96"/>placed in a separatory funnel, and treated with 50OmL of 1.0M NaOH. The layers were separated, and the cloudy organic layer was recharged into a separatory funnel and treated with a 6% HCl solution (adjusted to pH=0.03-0.34, 10OmL of solution). Two clear layers formed. The resultant biphasic solution was poured into an Erlenmeyer flask and cautiously neutralized to a pH of 1.2-1 A with a saturated solution of sodium bicarbonate (approx 20OmL of solution). The organic layer was separated from the aqueous layer, dried over sodium sulfate and evaporated to yield 83.6g of yellow oil (theoretical yield: 77.03g). The oil was dissolved in isopropyl alcohol (50OmL) with heating and transferred to a IL round bottom flask equipped with a mechanical stirrer and heating mantel. The solution was heated to 50C and the mechanical stirrer was set to a rate of 53-64 rpm. Tartaric acid (25.33g, 168mmol) was dissolved in deionized water (5OmL) and added to the stirred solution at 50C. Once the solution turned from milky white to clear, seed crystals were added to the mixture and crystallization immediately began (temperature jumped to 56C). After 20 minutes, the mixture was set to cool to a temperature of 35C (cooling took 1.15 hours). Heating was removed and the solution was allowed to stir for 12 hours. The resulting thick slurry was filtered on a Bchner funnel. Any remaining solid in the flask was washed onto the funnel using ice-cold isopropyl alcohol (10OmL). The material was transferred to a drying tray and heated to 48C under vacuum for 3 days (after two days the material weighed 76g and after three days it weighed 69.3g). The solid was analyzed by LC and shown to be 98.1% pure (AUC), the residual solvent analysis showed the material to possess 3472 ppm of isopropyl alcohol, and the DSC (differnetial scaaning calroimetery) showed a melting point of 134.89C. A total of 69.3g of white solid was collected (65.7% overall yield). 1H NMR (400 MHz, CDCl3) delta= 1.8 (M, 4H), 2.4-2.6 (m, 4H), 2.6 (m, 1H), 2.85 (m, 2H), 3.0 (m, 1H), 3.65 (s, 3H), 3.8 (m, 2H), 3.86 (2, 2H), 4.18 (br s, 5H), 4.6 (s, 1H), 6.6-6.8(m, 7 H), 7.8 (d, 1H); MS for C29H40N2O13 m/z 457.3 [M+H] for main peak (free-base).
10.0 mg (20 mumol) 25-hydroxyvitamin D3-3-hemisuccinate was dissolved in 7 ml anhydrous dichloromethane and admixed with 2.76 mg (24 mumol) N-hydroxysuccinimide and 3.72 mg (24 mumol) N(3-dimethylaminopropyl)-N'-ethyl-carbodiimide (EDC). It was stirred overnight under argon, the organic phase was then washed twice with 10 ml water, dried over about 1 g anhydrous sodium sulfate and filtered. The solvent was removed in a vacuum and the residual reaction product was dried for 3 h in a high vacuum. 11.3 mg (yield: 94%) N-hydroxysuccinimide ester was obtained which was used for the conjugation without further purification.
With dicyclohexyl-carbodiimide; In tetrahydrofuran; at 0 - 20℃;Inert atmosphere;
Into a 100- mL round-bottom flask purged and maintained with an inert atmosphere of nitrogen, was placed a solution of <strong>[23351-91-9]5-bromoisophthalic acid</strong> (3 g, 11 76 mmol, 1 00 equiv, 96percent) in THF (20 mL) followed by NHS (3 g, 2609 mmol, 220 equiv) at 0-50C To this was added a solution of DCC (5 6 g, 27 18 mmol, 2 20 equiv) m THF (20 mL) dropwise with stirring at 0-50C The resulting solution was stirred overnight at room temperature The solids were filtered out and the filtrate was concentrated under vacuum The crude product was re-crystalhzed from DCM/ethanol in the ratio of 1 10 This resulted m 4 g (75percent) of the title compound as a white solid.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In aq. phosphate buffer; at 20℃; for 1h;pH 7.4;Inert atmosphere; Darkness;
In order to develop MTX conjugated Alg-Ccm AuNPs, at first MTX was conjugated to Bis (aminopropyl) PEG to generate MTX-PEG conjugate (MP conjugate). For MP conjugate synthesis, MTX was dissolved in PBS (pH 7.4) and was activated using EDC and NHS (MTX:EDC:NHS = 1:1.3:1.1 molar ratio) at room temperature for one hour under the blanket of N2 (g) in dark. Then solution of bis(aminopropyl) PEG (MTX:PEG = 1:1 molar ratio) in PBS (pH 7.4) was gradually added to the activated MTX solution and the mixture was allowed to stir at room temperature for around four hours in dark. Next, the MP conjugate was dialyzed (using dialysis mem-brane of MWCO 1000) against PBS (pH 7.4) for one day and against distilled H2O for one day and was dried by lyophilization. In the second step, MP conjugate was conjugated to the Alg-Ccm AuNPs utilizing the EDC chemistry. The free carboxyl functionality on Alg-Ccm AuNPs (15 mg) was activated with EDC/NHS (in aqueous carbonate/bicarbonate buffer medium with pH 8.2) for one hour at 25 C followed by the addition of aqueous solution of MP conjugate (25 mg) and the reaction mixture was moderately stirred overnight at 25 C. The reaction mixture was then dialyzed (MWCO 3500) and lyophilized to get MPAlg-Ccm AuNPs.
With triethylamine; In dichloromethane; at 20℃; for 3.0h;
To a mixture of N-hydroxysuccinimide (3.00 g, 26.1 mmol) and Et3N (3.20 g, 31 .3 mmol) in CH2CI2 (60 ml_) was added dropwise succinyl chloride (2.00 g, 13.0 mmol). The mixture was stirred at room temperature for 3 hours before diluted with water (60 ml_). The resulting suspension was filtered, washed with water and CH2CI2. The cake was collected and dried to give bis(2,5-dioxopyrrolidin-1 -yl) succinate as a grey solid.
2,5-dioxopyrrolidin-1-yl tetrahydro-2H-pyran-3-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
Synthesis of <strong>[873397-34-3]tetrahydro-2H-pyran-3-carboxylic acid</strong>:DCC (7.85 g, 38.1 mmol) was added to <strong>[873397-34-3]tetrahydro-2H-pyran-3-carboxylic acid</strong> (4.5 g, 34.6 mmol) in THF (100 mL) at 0 °C. The reaction mixture was stirred at room temperature for 1 h and cooled down to 0 °C. N-Hydroxysuccinimide (4.78 g, 41.5 mmol) was added and stirring was continued overnight at room temperature. The reaction mixture was filtered, the solvent was evaporated, and the residue was purified by column chromatography (silica gel, EtOAc/Hept, 1 :1-1 :0) to prepare 2,5-dioxopyrrolidin-l-yl tetrahydro-2H-pyran-3-carboxylate (6.5 g, 83percent yield). 1H NMR (300 MHz, CDCI3/TMS): delta 4.14-4.06 (m, 1H), 3.88-3.79 (m, 1H), 3.71-3.64 (m, 1H), 3.54-3.44 (m, 1H), 2.99-2.89 (m, 1H), 2.81 (s, 4H), 22.2-2.14 (m, 1H), 1.95-1.84 (m, 1H), 1.78-1.64 (m, 3H).
With dicyclohexyl-carbodiimide; In tetrahydrofuran; at 0 - 20℃;
[0565] Synthesis of <strong>[873397-34-3]tetrahydro-2H-pyran-3-carboxylic acid</strong>: [0566] DCC (7.85 g, 38.1 mmol) was added to <strong>[873397-34-3]tetrahydro-2H-pyran-3-carboxylic acid</strong> (4.5 g, 34.6 mmol) in THF (100 mL) at 0 °C. The reaction mixture was stirred at room temperature for 1 h and cooled down to 0 °C. N-Hydroxysuccinimide (4.78 g, 41.5 mmol) was added and stirring was continued overnight at room temperature. The reaction mixture was filtered, the solvent was evaporated, and the residue was purified by column chromatography (silica gel, EtOAc/Hept, 1 : 1-1 :0) to prepare 2,5- dioxopyrrolidin-l-yl tetrahydro-2H-pyran-3-carboxylate (6.5 g, 83percent yield). 1H NMR (300 MHz, CDCI3/TMS): delta 4.14-4.06 (m, 1H), 3.88-3.79 (m, 1H), 3.71-3.64 (m, 1H), 3.54-3.44 (m, 1H), 2.99-2.89 (m, 1H), 2.81 (s, 4H), 22.2-2.14 (m, 1H), 1.95-1.84 (m, 1H), 1.78-1.64 (m, 3H).
With dicyclohexyl-carbodiimide; In tetrahydrofuran; at 0 - 20℃; for 2.5h;
General procedure: To a solution of carboxylic acid (1.0 equiv) in THF (10 mL) at 0 °C were added N-hydroxysuccinimide (1.0 equiv) and DCC (1.0 equiv). The resulting mixture was stirred for 30 min at 0 °C and 2 h at room temperature. The reaction mixture was filtered to remove the precipitates, and the filtrate was concentrated under reduced pressure. Purification by flash chromatography (hexane/EtOAc) afforded the desired N-hydroxysuccinimidyl esters.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 0 - 20℃; for 3.5h;Inert atmosphere;
EXAMPLE 4 SYNTHESIS OF PROBE COMPOUND, DYn-Naph [0147] The DYn-Naph compound (FIG. 5) may be synthesized by the following method (FIG. 6A, Scheme 2). To a dry round bottom flask flushed with nitrogen was added 6-amino-2-napthoic acid (200 mg, 1.07 mmol), NHS (135.18 mg, 1.17 mmol), and dry DMF (2 mL). The reaction mixture was cooled in an ice-bath. EDC-HC1 (224.71 mg, 1.17 mmol) was added and the reaction mixture was allowed to react at 0 °C for 30 min, then the reaction was brought up to room temperature and allowed to mix for an additional 3 hrs. The reaction was quenched with the addition of saturated sodium bicarbonate and extracted with EtOAc. The organic layer was dried over MgS04, filtered, and concentrated for the next step. To a dry round bottom flask flushed with nitrogen was added the NHS-ester of Intermediate Compound (1) (323 mg, 1.13 mmol), which was suspended in dry DMF (4 mL). 4-pentyneamine (292.5 mg, 3.52 mmol) and DIPEA (613 uL, 3.52 mmol) were subsequently added and the reaction mixture was allowed to mix at 75 °C overnight under nitrogen. The reaction was quenched with the addition of saturated sodium bicarbonate and extracted with EtOAc. The organic layer was dried over MgS04, filtered, and concentrated. Flash column chromatography was used for purification (1 : 1 EtOAc:Hexanes, 10percent MeOH in EtOAc spiked with 1percent TEA) to yield 188.2 mg of Intermediate Compound (2) (70percent yield). To a dry round bottom flask flushed with nitrogen was added 3-methoxy-5-oxocyclohex-3-9-enecarboxylic acid (115.39 mg, 0.678 mmol), EDC-HC1 (194.48 mg, 1.02 mmol), and DMAP (99.4 mg, .813 mmol). The flask was sealed under nitrogen. Intermediate Compound (2) (188.2 mg, 0.746 mmol) was resuspended in dry DMF (2 mL) and added to the reaction mixture followed by the addition of TEA (95 mu, 0.678 mmol). The reaction mixture was brought up to 85 °C and allowed to mix overnight under nitrogen. The reaction was quenched with the addition of water and extracted with EtOAc. The organic phases were combined and dried over MgS04, filtered, and concentrated. Flash column chromatography was used for purification (1 : 1 Hexanes:EtOAc, 7:3 EtOAc:Hexanes, 5percent MeOH in EtOAc) to yield 138.1.6 mg of product (46percent yield). In a round bottom flask, Intermediate Compound (3) (138.1 mg, 0.34 mmol) was suspended in a 1 : 1 Acetonitrile:Water solution (5 mL). 10percent> CAN (18.8 mg, 0.034 mmol) was added and the reaction mixture was refluxed at 95 °C for 3 hrs., resulting in the synthesis of DYn-Naph (FIG. 5). The reaction was then cooled and concentrated and directly purified by HPLC.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 0 - 20℃; for 3.5h;Inert atmosphere;
To a dry round bottom flask flushed with nitrogen was added 6-amino-2-napthoic acid (200 mg, 1.07 mmol), NHS (135.18 mg, 1.17 mmol), and dry DMF (2 mL). The reaction mixture was cooled in an ice-bath. EDC-HCl (224.71 mg, 1.17 mmol) was added and the reaction mixture was allowed to react at 0 °C for 30 min, then the reaction was brought up to rt and allowed to mix for an additional 3 hrs. The reaction was quenched with the addition of saturated sodium bicarbonate and extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and concentrated for the next step without further purification. To a dry round bottom flask flushed with nitrogen was added the NHS-ester (323 mg, 1.13 mmol) which was suspended in dry DMF (4 mL). 4-pentyneamine (292.5 mg, 3.52 mmol) and DIPEA (613 uL, 3.52 mmol) were subsequently added and the reaction mixture was allowed to mix at 75 °C overnight under nitrogen. The reaction was quenched with the addition of saturated sodium bicarbonate and extracted with EtOAc. The organic layer was dried over MgSO4, filtered, and concentrated. Flash column chromatography was used for purification (1:1 EtOAc:Hexanes, 10percent MeOH in EtOAc spiked with 1percent TEA) to yield 188.2 mg of (5) (70percent yield over 2 steps)
In a 50 mL round-bottomed flask <strong>[203866-15-3](S)-1-(tert-butoxycarbonyl)-4,4-difluoropyrrolidine-2-carboxylic acid</strong> (1.6 g, 6.37 mmol), obtained from step 4, was dissolved in 10 mL of dichloromethane at 15 C. Then 1-hydroxypyrrolidine-2,5-dione (0.806 g, 7.01 mmol) was added. To the formed suspension N,N-dicyclohexylcarbodiimide (1.445 g, 7.01 mmol) was added at vigorous stirring. In a few seconds a cloudy white suspension forms. The mixture was allowed to reach RT and stirred for 30 min, followed by the addition of 7N ammonia in methanol (2.002 ml, 14.01 mmol) and stirring for another 20 min. Before evaporation of volatile components 1 spoon of Celite was added to the flask. Cold ethyl acetate was added to the residue and filtered over Celite. The filtrate was washed with saturated sodium bicarbonate. The formed slightly yellowish crystals were used without further purification. Yield: 1.15 g, 72% 1H NMR (400 MHz, CDCl3): delta 1.45 (s, 9H), 2.77-2.44 (m, 1H), 2.82-3.02 (m, 1H), 3.56-3.78 (m, 1H), 3.80-4.00 (m, 1H), 4.52 (br_s, 1H), 5.54 (br s, 1H), 6.77 (br_s, 1H). LC-MS (I) Rt 1.34 min, m/z 251.5 [M+H]+ (89%). MS (ESI) m/z 251.2 [M+H]+
Hexacosanoic N-hydroxysuccinimidyl ester (11) To a solution of hexacosanoic acid (121 mg, 0.304 mmol) in CH2Cl2 (4 mL) were added <strong>[1892-57-5]1-ethyl-3-(3-dimethylaminopropyl)carbodiimide</strong> (0.058 mL, 0.33 mmol) and N-hydroxysuccinimide (42 mg, 0.37 mmol). The reaction mixture was heated to 40 C, stirred for 3 h and then quenched with water (4 mL). The solution was diluted with Et2O (8 mL) and the two layers were separated. The aqueous phase was extracted with Et2O (8 mL) and the combined organic layers were washed with sat aq. NaCl solution (5 mL), dried over MgSO4 and filtered. After removal of the solvent in vacuo, N-hydroxysuccinimidyl ester 11 was obtained as a white solid (85 mg, 57%).
1-(2,5-dioxopyrrolidin-1-yl) 1-ethyl cyclobutane-1,1-dicarboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate;
Commercially available 1 ,1-cyclobutanedicarboxylic acid, 1,1 -diethyl ester (CAS Reg. No. 3779-29-1) was converted by limited saponification with aqueous base to the half acid/ester 1 ,1-cyclobutanedicarboxylic acid, 1-ethyl ester (CAS Reg No. 54450-84-9) and activation with a coupling reagent such as TBTU (0-(Benzotriazol-l-yl)-NN,N',N'- tetramethyluronium tetrafluoroborate, also called: NN,N',N'-Tetramethyl-0-(benzotriazol-l- yl)uronium tetrafluoroborate, CAS No. 125700-67-6, Sigma-Aldrich B-2903), and N- hydroxysuccinimide to the NHS ester, l-(2,5-dioxopyrrolidin-l-yl) 1-ethyl cyclobutane-1 , 1- dicarboxylate.
With dicyclohexyl-carbodiimide; In dichloromethane; at -20 - 30℃; for 2h;
Di-tert-butyl octadecanedioate (9.1 g, 24.56 mmol) was dissolved in dichloromethane (180 ml).-N-hydroxysuccinimide (2.97 g, 25.85 mmol) and dicyclohexylcarbodiimide (5.59 g, 27.04 mmol) were added separately at -20C, the reaction was continued at -20C for 2 hours, and the stirring was resumed at 30C overnight. .The mixture was suction filtered, the solvent was evaporated under reduced pressure, recrystallized by adding isopropyl alcohol (80 ml), and the filter cake was collected by filtration.The filter cake was washed with n-heptane (30 ml) and the cake was collected, dried under reduced pressure until constant weight, weight 9.3 g, yield: 81%, HPLC purity: 99.3%.
With dicyclohexyl-carbodiimide; In N,N-dimethyl-formamide; at 20℃;
DCC (5.11 ml, 5.11 mmol) was added to a solution of <strong>[843666-40-0]18-(tert-butoxy)-18-oxooctadecanoic acid</strong> (1.72 g, 4.64 mmol) and 1-hydroxypyrrolidine-2,5-dione (0.588 g, 5.11 mmol) in DMF (48 mL). The mixture was stirred at rt overnight. The mixture was filtered and concentrated to get 1-tert-butyl 18-(2,5-dioxopyrrolidin-1- yl) octadecanedioate which was used as is in the next step.
2.6 g
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 2h;
To a 100mL three-necked flask were added 0.95 g N-hydroxy succinimide (HOSU), 2.0g compound 19 and 15 ml dichloromethane, into which 1.58g EDC?HCl was added and reacted for 2h at room temperature. After the completion of the reaction under the monitor of TLC, they were diluted with dichloromethane, and then washed with 50mmol/L aqueous solution of potassium dihydrogen phosphate at pH=6.0 for 2 times, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated to give 2.6g compound BP103n02 as a white solid.
2.13 g
With N-(3-dimethylaminopropyl)-N-ethylcarbodiimide; In dichloromethane; at 20℃; for 16h;
EDC (1.406 g, 7.33 mmol) was added to a mixture of <strong>[843666-40-0]18-(tert-butoxy)-18-oxooctadecanoic acid</strong> (28) (prepared by method similar to that used to prepare Compound 1 in Example 4) (2.09 g, 5.64 mmol), and 1-hydroxypyrrolidine-2,5-dione (0.844 g, 7.33 mmol) in CH2Cl2 (40 mL). The mixture was stirred at RT overnight, diluted with CH2Cl2 (100 mL) and washed with NaHCO3. After separation, the aqueous layer was extracted with CH2Cl2 (80 mL*2). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated under vacuum. The residue was subjected to flash chromatography eluting with CH2Cl2 to afford 1-tert-butyl 18-(2,5-dioxopyrrolidin-1-yl) octadecanedioate (29) (2.13 g, 4.55 mmol, 81% yield) as a white solid. 1H-NMR (400 MHz, CDCl3) delta 2.85 (br s, 4H), 2.62 (t, J=7.5 Hz, 2H), 2.22 (t, J=7.6 Hz, 2H), 1.77 (quin, J=7.5 Hz, 2H), 1.67-1.54 (m, 3H), 1.50-1.37 (m, 10H), 1.37-1.21 (m, 22H). 13C-NMR (101 MHz, CDCl3) delta ppm 173.33, 169.12, 168.67, 79.85, 35.67, 30.98, 29.65 (br s), 29.62 (br s), 25.61, 29.55, 29.50, 29.36, 29.31, 29.12, 29.09, 28.81, 28.15, 25.15, 24.60.
ω-carboxypentanedecanoic acid monosuccinimidyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With dicyclohexyl-carbodiimide; In 1,4-dioxane; ethyl acetate; at 20℃; for 12h;
Three-necked flask (300 mL) in hexadecane diacid 1.15g (4mmol), N-hydroxysuccinimide 550mg (4.4mmol), dioxane 200 mL, ethyl acetate 200 mL, placed in 50 mL of DMF, was stirred at room temperature. was added dropwise an ethyl acetate solution 60mL of N'N-dicyclohexylcarbodiimide (DCC) 908mg (4.4mmol), and stirred at room temperature for 12 hours. Acetic acid 0.5mL was added to the reaction solution, stirred for 30 minutes, to decompose excess DCC. The resulting dicyclohexyl amide was filtered off and distilled off dioxane, ethyl acetate. The resulting solid ethyl acetate 150mL and chloroform 15mL in addition to, the insoluble material was filtered off. Ethyl acetate, chloroform was distilled off, silica gel column (chloroform: methanol = 100: 0 ? 1) was purified the desired product at. Colorless solid yield 45%
41%
With dmap; dicyclohexyl-carbodiimide; In tetrahydrofuran; for 0.5h;
<strong>[505-54-4]Hexadecanedioic acid</strong> (1.43 g, 5 mmol), HOSu (0.58 g, 5 mmol) and DMAP (3.1 mg, 0.025 mmol) were dissolved in 50 ml of THF, a solution of DCC (1.03 g, 5 mmol) dissolved in THF (20 ml) was slowly added dropwise after stirring for 30 min, and after the addition was completed, stirring was performed overnight at room temperature. Then filtration was performed, the filtrate was subjected to vacuum concentration, and the residue was recrystallized with methanol to obtain 0.79 g of hexadecanedioic acid mono-N-hydroxysuccinimide ester with yield: 41%, purity: 96.2%, MS: 384.5 (M+1).
ω-carboxynonadecanoic acid monosuccinimidyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
45%
With dicyclohexyl-carbodiimide; In 1,4-dioxane; ethyl acetate; at 20℃; for 12h;
Three-necked flask (300 mL) to <strong>[2424-92-2]eicosanedioic acid</strong> 1.37g (4mmol), N-hydroxysuccinimide 550mg (4.4mmol), dioxane 200 mL, ethyl acetate 200 mL, placed in 50 mL of DMF, was stirred at room temperature was added dropwise an ethyl acetate solution 60mL of N'N-dicyclohexylcarbodiimide (DCC) 908mg (4.4mmol), and stirred at room temperature for 12 hours. Acetic acid 0.5mL was added to the reaction solution, stirred for 30 minutes, to decompose excess DCC. The resulting dicyclohexyl amide was filtered off and distilled off dioxane, ethyl acetate. The resulting solid ethyl acetate 200mL and chloroform 20mL was added to the insoluble material was filtered off. Ethyl acetate, chloroform was distilled off, silica gel column (chloroform: methanol = 100: 0 ? 1) was purified the desired product at. Colorless solid yield 45%
41%
With dmap; dicyclohexyl-carbodiimide; In tetrahydrofuran; for 0.5h;
Eicosanedioic acid (1.71 g, 5 mmol), HOSu (0.58 g, 5 mmol) and DMAP (3.1 mg, 0.025 mmol) were dissolved in 50 ml of THF, a solution of DCC (1.03 g, 5 mmol) dissolved in THF (20 ml) was slowly added dropwise after stirring for 30 min, and after the dropwise addition was completed, stirring was performed overnight at room temperature. Then filtration was performed, the filtrate was subjected to vacuum concentration, and the residue was recrystallized with methanol to obtain 0.90 g of <strong>[2424-92-2]eicosanedioic acid</strong> mono-N-hydroxysuccinimide ester with yield: 41%, purity: 95.8%, MS: 440.6 (M+1).
(S)-2,5-dioxopyrrolidin-1-yl 2-((S)-4-methyl-2-((S)-2-(2-morpholinoacetamido)-4-phenylbutanamido)pentanamido)-3-phenylpropanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
26.5 g
With diisopropyl-carbodiimide; In dichloromethane; at 0 - 30℃; for 6h;Inert atmosphere;
A solution of diisopropyl carbodiimide (6.12 gm) in dichloromethane (75 ml) was slowly added to a mixture of (S)-2-((S)-4-methyl-2-((S)-2-(2-mo holinoacetamido)-4- phenylbutanamido)pentanamido)-3-phenylpropanoic acid compound of formula-2 (25 gm), N-hydroxysuccinimide (5.6 gm) and dichloromethane (250 ml) at 25-30C under nitrogen atmosphere and stirred the reaction mixture for 4 hrs at the same temperature. Cooled the reaction mixture to 0-5C and stirred for 2 hrs at the same temperature. Filtered the reaction mixture and distilled off the solvent completely from the filtrate under reduced pressure to get the title compound as a solid. Yield: 26.5 gm
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; for 16.5h;Inert atmosphere;
<strong>[25055-86-1]3-(3-methyl-3H-diazirin-3-yl)propanoic acid</strong> (60.0 mg, 0.468 mmol) and N-hydroxysuccinimide (56.6 mg, 0.491 mmcl) dissolved in 600 pL anhydrous dimethylformamide with stirring under nitrogen. 1-ethyl-3-(3- dimethylaminopropyl)carbodiimide 107 mg, 0.562 mmcl) was subsequently added and the reaction stirred 16.5 hrs and then was diluted with chloroform. The reaction waswashed with water, aqueous iN HCI (3x) and aqueous saturated sodium chloride. The organic layer was dried with sodium sulfate and evaporated to yield a yellow, semi- viscous oil. Anhydrous diethyl ether was added to the oil to yield a white precipitate. The ether layer was decanted and the precipitate was dried. White powder (78.4 mg, 74%). 1H NMR (600 MHz, Chloroform - d) 2.83 (s, 4H, NHS methylene (x2)), 2.53 -2.50 (m, 2H, methylene), 1.82 - 1.75 (m, 2H, methylene), 1.07 (s, 3H, methyl). 13CNMR (151 MHz, Chloroform-d) oe 169.04 (NHS carbonyl (x2)), 167.71 (carbonyl), 29.64(methylene), 25.85 (methylene), 25.70 (NHS methylene (x2), 24.85 (diazirine), 19.61(methyl).
71%
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 0 - 20℃; for 2h;
(2)The intermediate (400 mg, 3.13 mmol) obtained in step (1) was added successively to the reaction flask at 0 C,Dichloromethane (10 mL), N-hydroxysuccinimide (NHS) (396 mg, 3.44 mmol) and1- (3-dimethylaminopropyl) -3-ethylcarbodiimide hydrochloride (EDC-HCl) (659 mg, 3.44 mmol) was added and stirred at room temperature for 2 h;After completion of the reaction, the mixture was diluted with methylene chloride and washed with ultrapure water (25 mL x 3)The organic phase was dried over anhydrous magnesium sulfate and concentrated to give an ultraviolet-sensitive cross-linker N-[3-(3-methyl-3H-diazirine-3-yl)propionyloxy]succinimide(Whose structure is shown in compound 2 in Figure 1)The crude product was purified by silica gel column chromatography (ethyl acetate: petroleum ether = 1: 3) to give pure UV-sensitive crosslinking agent,The yield was 71%.
2,5-dioxopyrrolidin-1-yl 4-(2-methoxy-2-oxoethyl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
97%
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃; for 3.5h;Inert atmosphere;
2,5-Dioxopyrrolidin-1-yl 4-(2-methoxy-2- oxoethyl)benzoate (13) To a solution of monoacid 12 (0.917 g, 4.72 mmol, 1 equiv.) in DMF (107 ml_), A/-hydroxysuccinimide (1.47 g, 12.8 mmol, 2.7 equiv.) and N-(3- dimethylamino-propyl)-/V-ethylcarbodiimide hydrochloride (2.44 g, 12.8 mmol, 2.7 equiv.) were added. The reaction mixture was stirred at room temperature for 3.5 h under an argon atmosphere. The colorless mixture was diluted with EtOAc and water. The solution was extracted three times with EtOAc. The combined organic layers were washed three times with water, dried over Na2S04 and concentrated under reduced pressure in order to get NHS-ester 13 (1.329 g, 97percent) as a pale yellow oil. NMR (CDCI3, 600 MHz) delta 8.10 (d, 2H, J = 8.1 Hz), 7.44 (d, 2H, J= 8.1 Hz), 3.72 (s, 2H), 3.71 (s, 3H), 2.90 (s, 4H); 13C NMR (CDCI3, 151 MHz) delta 169.9, 168.3, 160.8, 140.5, 130.0, 129.0, 123.2, 51.5, 40.4, 24.8; Ci4Hi4N06 [M+H]+ 292.0816, found 292.0824; C14H17N2O6 [M+NH4]+ 309.1081 , found 309.1084; Purity: 100percent, tR = 2.4 min.
With tributyl-amine; In acetonitrile; at 20℃; for 2h;
N-maleimidobutyric acid (99 g, 0.54 mol) and acetonitrile (300 ml) were placed in a500 ml three-necked flask andthionyl chloride (93.6 g, 0.82 mol) was added. The reaction was warmed to reflux and a large amount of gas was released .After the reaction was refluxed for 4h, the reaction became clear,acetonitrile and excess thionyl chloride were removed by concentration, and 200 ml of acetonitrile was added to remove the remaining acid in one pass to obtaincrudeN-maleimidobutyrylchloride.The crude product was dissolved in 200ml of acetonitrile was added dropwise to N- hydroxysuccinimide (69g, 0.6mol) and tri-n-butylamine (148g, 0.8mol) was dissolved in 300ml of acetonitrile, with ice water cooling to control the temperature does not exceed 20 , drip finished at room temperature for2h.The reaction solution was concentrated to remove acetonitrile, dissolved in 400 ml of methylene chloride, and dissolved in 200 ml of dichloromethane to remove tri-n-butylaminehydrochloride and 100 ml of 1N hydrochloric acid to remove excess tri-n-butylamine and 200 ml of saturated saline solution, Drywhite solid 139g, a mixed solvent of ethyl acetate and petroleum ether to give a white solid after recrystallization 104.2g, HPLC purity 98.9%, 69.1% yield.
N-tert-butoxycarbonyl-N-methyl-beta-alanine succinate ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane;
N-tert-Butoxycarbonyl-N-methyl-beta-alanine succinate ester (Compound 4, FIG. 19) The title compound was prepared from commercial Boc-N-Me-beta-Ala-OH by a method well known in the art (cf.-Widdison et al., J. Med. Chem., 2006, 49 (14), 4401). 1H NMR (300 MHz, CDCl3): delta 3.62 (bm, 2H), 2.88 (m, 9H), 1.47 (s, 9H).
With dmap; dicyclohexyl-carbodiimide; In dichloromethane; at 0 - 20℃; for 16h;Inert atmosphere;
To a round bottom flask were added acid 9 (307 mg, 1.0 mmol), N- hydroxysuccinimide (173 mg, 1.5 mmol), DMAP (12 mg, 0.1 mmol), and 10 mL CH2CI2 under N2. The mixture was cooled down to 0 C and then a solution of DCC (310 mg, 1.5 mmol) in 10 mL CH2CI2 was added dropwise. The mixture was allowed to warm up to room temperature slowly overnight (16 h). Lots of white precipitates formed during the reaction. The precipitates were filtered off and the filtrate was washed with 20 mL 0.05 N HCl(aq) twice. The organic layer was dried with Na2S04 and concentrated under vacuum. The residue was dispersed in 20 mL EtOAc, and the insoluble precipitates were filtered off. The filtrate was concentrated to give T3 as a colorless oil (0.4 g, 99%). NMR (400 MHz, CDCh): d 5.02 (s, 1H), 4.53 (s, 2H), 3.83-3.79 (m, 2H), 3.73-3.69 (m, 2H), 3.67-3.60 (m, 4H), 3.54 (t, J = 5.2 Hz, 2H), 3.31 (br q, J= 5.2 Hz, 2H), 2.86 (s, 4H), 1.44 (m, 9H).
0.72 g
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 2h;
To a 100mL three-necked flask were added 286mg N-hydroxy succinimide (HOSU), 0.50g BP103a05 and 5 ml dichloromethane, into which 477mg EDC?HCl was added and reacted at room temperature for 2h. After the completion of the reaction under the monitor of TLC, they were diluted with dichloromethane, and then washed with 50mmol/L aqueous solution of potassium dihydrogen phosphate at pH=6.0 for 2 times, washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated to give 0.72g compound BP103m50 as an oil.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃; for 48h;
The commercial product 6-Biotinylamino-hexanoic acid (Biotin-Ahx-OH, 101 mg, 0.28 mmol) was dissolved in 4 ml of DMF (light suspension). N-hydroxy succinimide (NHS, 36 mg, 0.31 mmol) and N-(3-Dimethylaminopropyl)-N' ethyl-carbodiimide (EDC x HCI, 59 mg, 0.31 mmol.) were added to reaction mixture and stirred at room temperature for 2 days following disappearance of the acid on TLC analysis. Then, the organic solvent was evaporated under high vacuum and the obtained residue was dissolved in CH2CI2 (20 ml) and rapidly washed with 0.1 N HCI solution to remove excess reagents. The combined organic phase were washed with brine (20 ml), dried over anhydrous Na2S04, filtered and concentrated to give a white solid Biotin-OSu activated (91 mg, yield 71 percent), used immediately without further purification.
2,5-dioxopyrrolidin-1-yl 2-morpholinoacetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
75%
With N-ethyl-N,N-diisopropylamine; dicyclohexyl-carbodiimide; In dichloromethane; at 15℃; for 12h;
A mixture of the product of the previous step (150 g, 826 mmol), 67 1-hydroxypyrrolidine-2,5-dione (95 g, 826 mmol), 68 DCC (256 g, 1.24 mol) and 69 DIPEA (160 g, 1.24 mol) in 70 DCM (2 L) was stirred at 15° C. for 12 h and filtered. The filtrate was concentrated and washed with EtOAc (800 mL). The solid was collected by filtration and concentrated to give the 71 title compound (150 g, 75percent yield) as a white solid. 1H NMR (400 MHz, DMSO-d6) delta 3.68 (s, 2H), 3.58 (t, J=4.8 Hz, 4H), 2.82 (s, 4H), 2.57 (t, J=5.2 Hz, 4H).
(S)-N-boc-(3-hydroxyadamantan-1-yl)glycine succinimide ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With triethylamine; In dichloromethane; at 25℃; for 1h;
In 150 mL of dichloromethane, 8.14 g (25 mmol) of (S)-N-Boc-(3-hydroxyadamantan-1-yl)glycine and 3.45 g (30 mmol) of N-hydroxysuccinimide, 5.04 g ( 50 mmol) triethylamine, stirring at 25 ° C for 1 hour, adjusting the pH to 7.5 with hydrochloric acid, adding a water layer, concentrating the organic phase under reduced pressure, column chromatography,(S)-N-Boc-(3-hydroxyadamantan-1-yl)glycine succinimide ester 9.55 g (22.6 mmol) was obtained in a yield of 90percent.
With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃;Cooling with ice;
FPBA-NHS was prepared by mixing 4-Carboxy-3-fluorophenylboronic acid (2 g, 12 mmol) and N-hydroxysuccinimide (NHS, 2 g, 17 mmol) in DMF (100 ml_), which was cooled in an ice bath while stirring. N-{ 3- dimethylaminopropyl)-/V'-ethylcarbodiimide hydrochloride (3 g, 18 mmol) was added, and the reaction was stirred overnight at room temperature. The mixture was poured into CH2CI2 (200 ml_) and washed with HCI (0.1 N, 3x50 ml_) and NaHC03 (0.1 N, 3x50 ml_) successively and dried over anhydrous MgS04. The solvent was evaporated under reduced pressure, and pure white FPBA-NHS was obtained (2.7 g, yield 80%). 1H-NMR (400 MHz, in DMSO-de, d): 8.59 (s, 2H), 8.0 (t, 1 H), 7.79 (q, 2H), 2.9 (s, 4H).
2,5-dioxopyrrolidin-1-yl 2-fluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62%
Mixture of <strong>[120153-08-4]4-borono-2-fluorobenzoic acid</strong> (1, 1.14 g, 6.20 mmol), pinacol (0.74 6.20 mmol) and molecular sieves (2.60 g) in dry toluene (40 mL) was stirred overnight at room temperature. The suspension was filtered; solid residue was suspended in dichloromethane (50 mL). Subsequently hydroxysuccinimide (HOSu, 1.43 g, 12.4 mmol) and A/-(3-dimethylaminopropyl)-A/'-ethylcarbodiimide hydrochloride (EDC.HCI, 2.38 g, 12.4 mmol) were added. Resulting mixture was stirred overnight at room temperature. The suspension was filtered and the filtrate was partitioned between ethyl acetate (80 mL) and 0.1 M aqueous solution of hydrochloric acid (50 mL). Organic layer was washed with 0.1 M aqueous solution of hydrochloric acid (2 x 75 mL) and brine (1 x 75 mL), dried over anhydrous sodium sulfate, filtered and evaporated to yield 2,5-dioxopyrrolidin-l-yl 2-fluoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)benzoate (2) as white powder. Yield : 1.36 g (62%). JH NMR spectrum (400 MHz, CDCI3, deltaEta) : 8.07-8.03 (m, 1 H); 7.69-7.65 (m, 1 H); 7.65-7.60 (m, 1 H); 2.92 (bs, 4 H); 1.37 (s, 12 H).
4-aminobutyric acid (compound 38) (200 mg, 2.04 mmol, 1.0 eq) was added to a solution of maleic anhydride (compound 39) (10 mg, 2.04 mmol, 1.0 eq) in DMF (2 mL) and the mixture was added to 2 Stir for hours. The resulting solution was cooled in an ice bath, and N-hydroxysuccinimide (288.7 mg, 2.51 mmol, 1.23 eq) and DCC (883.7 mg, 4.28 mmol, 2.1 eq) were sequentially added. After 5 minutes, the ice bath was removed and the solution was stirred vigorously overnight. The white precipitate that formed was filtered off, washed with DMF, and the filtrate was poured onto ice. The aqueous layer was extracted with CH2Cl2. The CH2Cl2 layer was dried over Na2 SO4 and concentrated under reduced pressure. Silica gel column chromatography gave compound 41 as a colorless solid (157.1 mg, 0.56 mmol, 28%) as product.
17-{(S)-1-tert-butoxycarbonyl-3-[2-(2-[2-(2-carboxymethoxyethoxyl)ethylcarbamoyl]methoxy}ethoxy)ethylcarbamoyl]propylcarbamoyl}heptadecanoic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
The activated fatty acid side chain, Moiety A-OSu was prepared by solid phase synthesis using 2-chlorotrityl chloride resin as schematically represented in FIG. 1A. 2-[2-(2-Fmoc-aminoethoxy)ethoxy]acetic acid (Intermediate-1) was attached to 2-chlorotrityl chloride resin in presence of N,N?-di-isopropylethylamine (DIPEA) which yielded 2-[2-(2-Fmoc-aminoethoxy)ethoxy] acetic acid-2-Cl-Trt-Resin. The intermediate-1 can be prepared by coupling of 2-[2-(2-amino ethoxy)-ethoxy] acetic acid with Fmoc N-hydroxysuccinimide ester. Alternatively, intermediate-1 is available commercially and can be procured as such. The Fmoc protecting group was removed by selective de-blocking of amino group of 2-[2-(2-Fmoc-aminoethoxy)ethoxy] acetic acid-2-Cl-Trt-Resin using piperidine and the free amino group was then coupled to 2-[2-(2-Fmoc-aminoethoxy)ethoxy]acetic acid using 1-hydroxybenztriazole(HOBt) and N,N?-di-isopropylcarbodiimide (DIPC) which yielded 2-[2-[2-[[2-[2-(2-Fmoc-aminoethoxy)ethoxy]acetyl] amino]ethoxy]ethoxy]acetic acid-2-Cl-Trt-Resin. The Fmoc group was then removed by selective de-blocking of amino group of 2-[2-[2-[[2-[2-(2-Fmoc-aminoethoxy)ethoxy]acetyl] amino]ethoxy]ethoxy]acetic acid-2-Cl-Trt-Resin using piperidine and the free amino group was then coupled to Fmoc-Glu-OtBu using HOBt and DIPC to obtain 2-[2-[2-[[2-[2-[2-[[(4S)-4-Fmoc-amino-5-tert-butoxy-5-oxo-pentanoyl]amino]ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy] acetic acid-2-Cl-Trt-Resin. The resultant 2-[2-[2-[[2-[2-[2-[[(4S)-4-Fmoc-amino-5-tert-butoxy-5-oxo-pentanoyl]amino]ethoxy] ethoxy]acetyl]amino]ethoxy]ethoxy]acetic acid-2-Cl-Trt-Resin was selectively deblocked using piperidine and then coupled with octadecanedioic acid mono tert-butyl ester to give intermediate-2 namely [2-[2-[2-[[2-[2-[2-[[(4S)-5-tert-butoxy-4-[(18-tert-butoxy-18-oxo-octadecanoyl)amino]-5-oxo-pentanoyl]amino]ethoxy]ethoxy]acetyl]amino]ethoxy]ethoxy] acetic acid]-2-Cl-Trt-Resin. The intermediate 2 was then cleaved from 2-Cl-Trt-Resin using trifluoroethanol: DCM (1:1). The resultant compound was then reacted with N-hydroxysuccinimide (HOSu) in presence of isobutyl chloroformate (IBCF) and N-methylmorpholine (NMM) followed by de-protection with trifluoroacetic acid to yield the title compound (Moiety A-OSu, intermediate-3). The whole process can also be depicted as schematically represented in FIG. 1B.
N-hydroxysuccinimide 2-(4-methylpiperazin-1-yl)acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With thionyl chloride; N,N-dimethyl-formamide; In tetrahydrofuran; at 20℃;
General procedure: Briefly, 0.7 g (0.0096 mol) of dimethylformamide and 1.14 g (0.0096 mol) of thionyl chloride were dissolved in 12 ml and 8ml of tetrahydrofuran, respectively. Thionyl chloride solution was cooled in an ice bath and dimethylformamide solution was added dropwise. The reaction mixture was left in an ice bath for 30min. After that, the ice bath was removed and 0.8 g (0.0068mol) of pre-powdered NHS was added to a stirred solution, followed by the addition of (4-methyl-1-piperazinyl) acetic acid (1.004 g, 0.0064mol) or 4-morpholinylacetic acid (0.93 g, 0.0064 g). The reaction mixture was stirred overnight at room temperature. The solids were filtered and washed with tetrahydrofuran. The presence of active NHS esters was confirmed by the reactions of synthesized products with tryptamine.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping