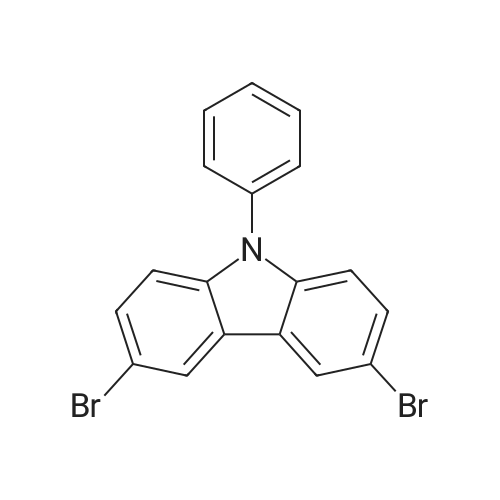
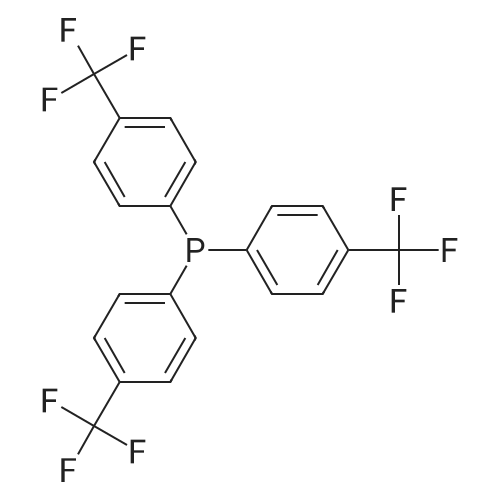
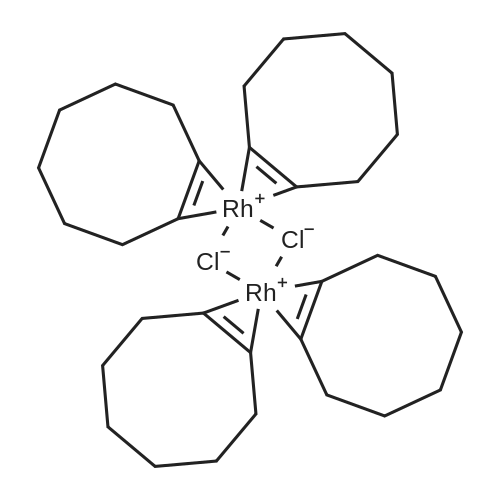
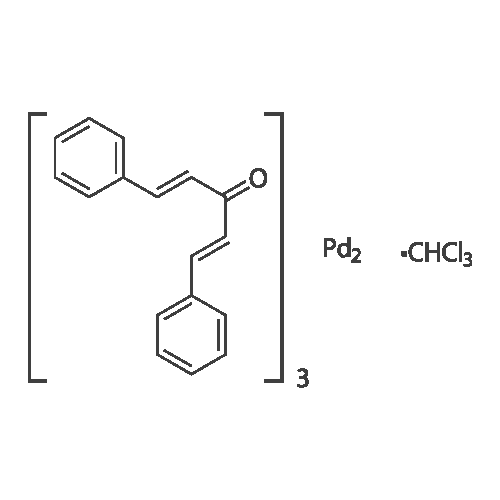
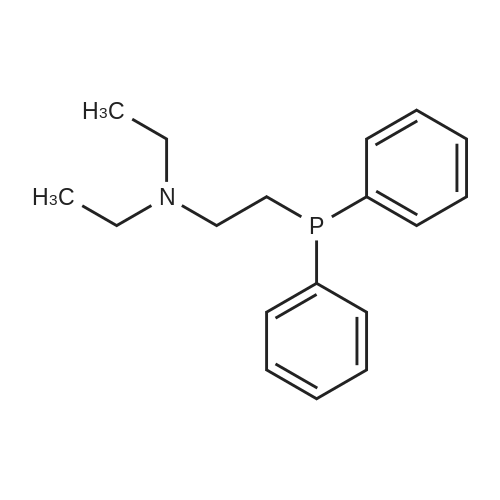
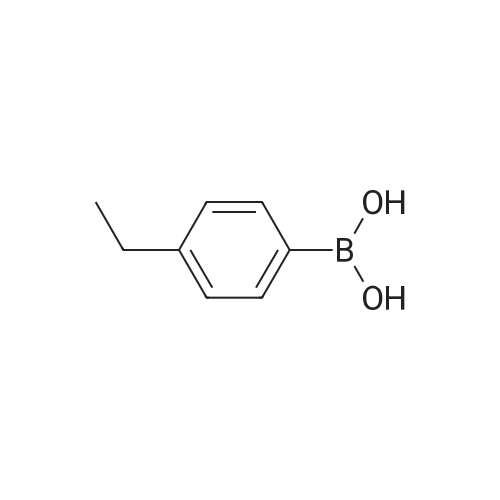
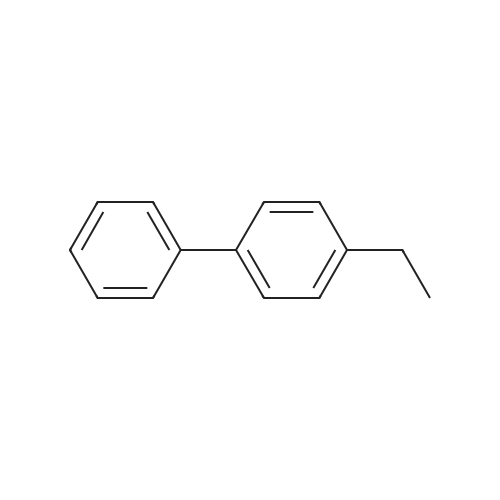
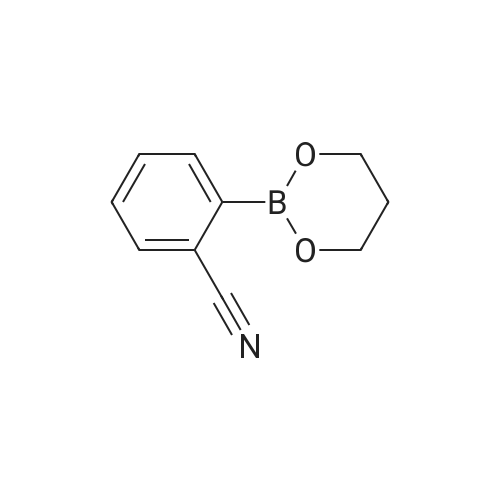
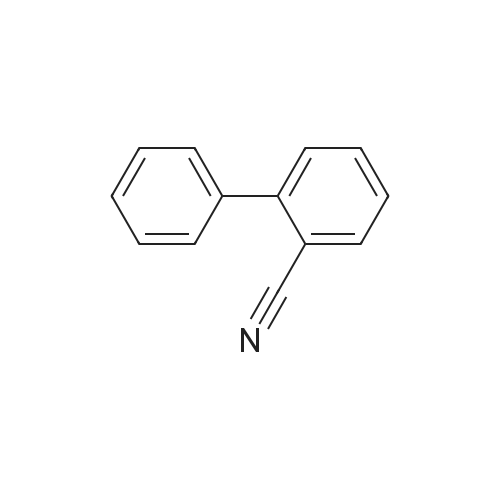
| 96% |
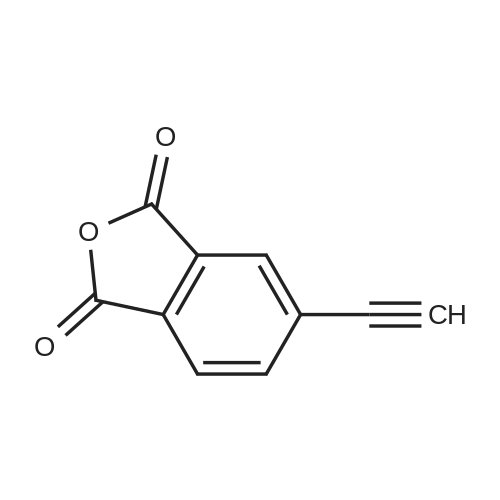
With potassium phosphate; copper(l) iodide; N,N`-dimethylethylenediamine; In N,N-dimethyl-formamide; at 110℃; for 72h;Inert atmosphere; |
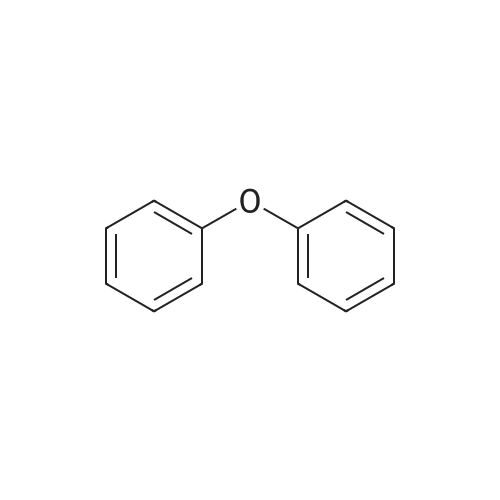
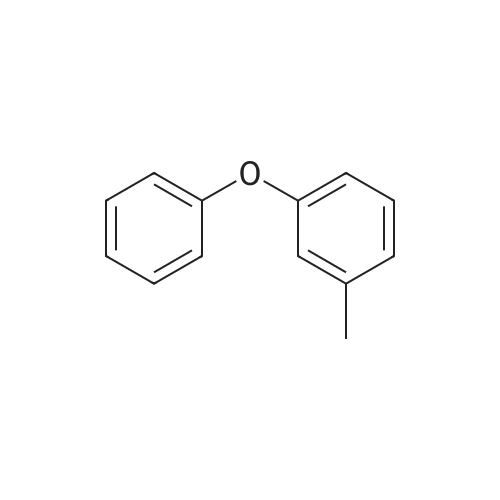
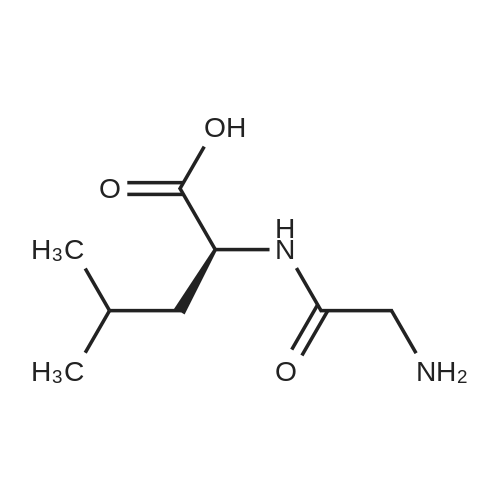
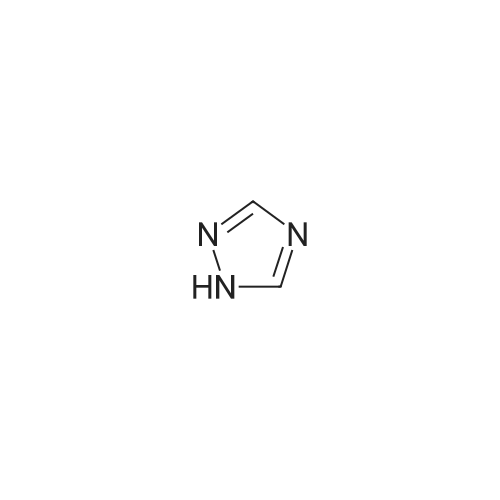
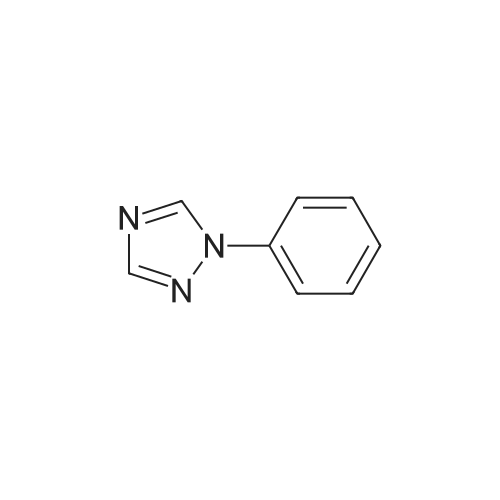
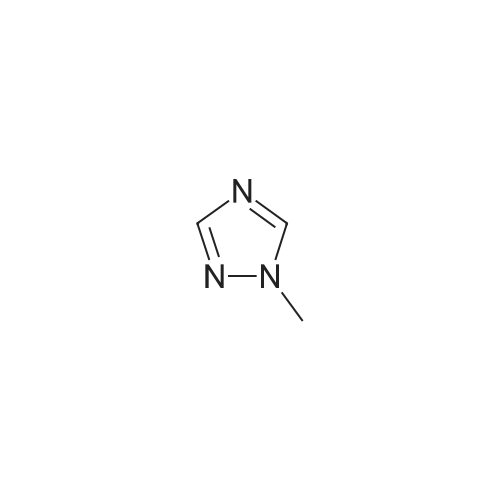
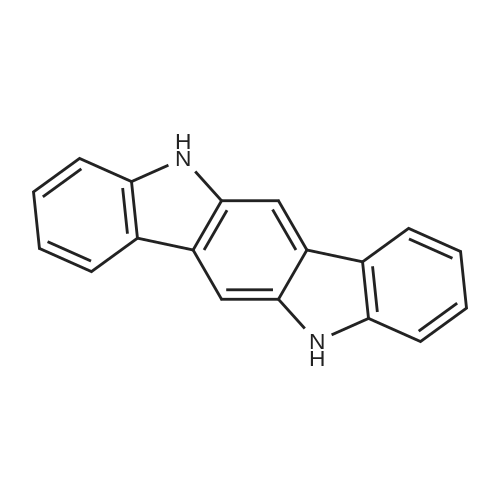
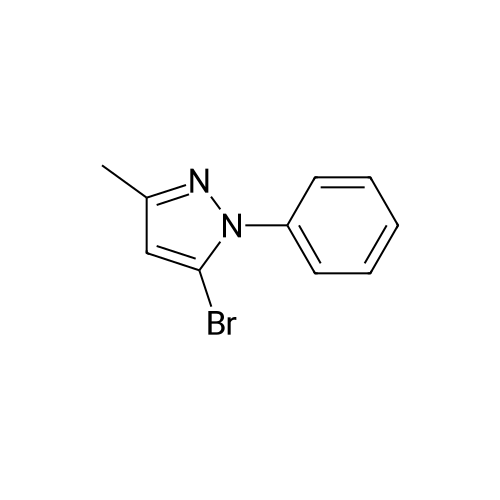
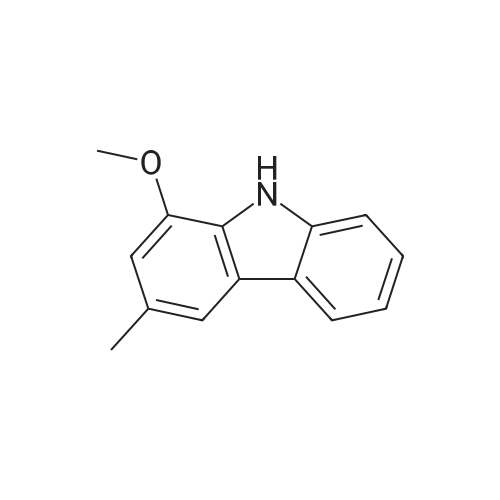
General procedure: A mixture of CuI (0.10 g, 0.50 mmol), the required azole(10 mmol), K3PO4 (4.4 g, 20 mmol), the required halide (12 mmol)and N,N0-dimethylethylenediamine (0.11 mL, 1.0 mmol) in DMF(5 mL) was degased and heated under argon at 110 C for 72 h.After filtration over celite (washing using AcOEt) and removal ofthe solvents, the crude product is purified by chromatography oversilica gel (the eluent is given in the product description). 4.3.1 1-Phenyl-1H-1,2,4-triazole (2a) Compound 2a was prepared from 1,2,4-triazole (0.69 g) and iodobenzene (1.4 mL) using the general procedure 1, and was isolated (eluent: heptane/AcOEt 7:3) in 96% yield as a yellow powder: mp 48 C (lit. 28 46 C); 1H NMR (CDCl3, 300 MHz) 7.42 (m, 1H), 7.53 (m, 2H), 7.71 (m, 2H), 8.15 (s, 1H), 8.74 (s, 1H); 13C NMR (CDCl3, 75 MHz) 120.1 (2CH), 128.3 (CH), 129.9 (2CH), 137.1 (C), 140.9 (CH), 152.7 (CH). |
| 91% |
With caesium carbonate;copper(I) oxide; trans-N,N'-bis(pyridin-2-ylmethylene)cyclohexane-1,2-diamine; In DMF (N,N-dimethyl-formamide); at 50 - 82℃; for 24 - 72h;Conversion of starting material; |
Example 1.17 [0636] Preparation of 1-phenyl-1H-1,2,41 triazole [0637] Operating protocol A (82 C., 48 hours) was followed using 117 mg of Chxn-Py-Al (0.4 mmoles), 336 ?l of iodobenzene (3 mmoles), 138 mg of 1,2,4-triazole (2 mmoles), 1.042 g of caesium carbonate (3.2 mmoles) and 1.2 ml of DMF. [0638] The degree of transformation and selectivity were 100% and 98% respectively. [0639] The residue obtained following treatment was purified by silica gel chromatography (eluent: hexane/dichloromethane, 100/0 to 50/50). [0640] 264 mg of a dark yellow solid was obtained in a yield of 91%. [0641] Pale yellow needles were obtained after re-crystallisation from chloroform. [0642] The compound obtained had the following formula: [CHEMMOL-00056] [0643] The characteristics were as follows: [0644] MPt: 46 C. (CHCl3) (Lit: 46-47 C. given by Micetich, R G; Spevak, P; Hall, T W; Bains, B K; Heterocycles 1985, 23, 1645-1649); [0645] H NMR/CDCl3:? 8.52 (wide s, 1H, HI), 8.04 (wide s, 1H, H2), 7.53-7.65 (m, 2H, H4,8), 7.26-7.51 (m, 3H, H5,6,7); [0646] 13C NMR/CDCl3: ? 152.55 (C1), 140.88 (C2), 139.96 (C3), 129.73 (C5 and C7), 128.15 (C6), 119.99 (C4 and C8); [0647] GC/MS: Rt=14.02 min, M/Z=145, purity=100%; [0648] Rf=0.21 (eluent: dichloromethane/ethyl acetate, 90/10). Example 1.18 [0649] Preparation of 1-phenyl-1H-[1,2,4]triazole [0650] Operating protocol A (82 C., 24 hours) was followed using 117 mg of Chxn-Py-Al (0.4 mmoles), 336 ?l of iodobenzene (3 mmoles), 138 mg of 1,2,4-triazole (2 mmoles), 1.042 g of caesium carbonate (3.2 mmoles) and 1.2 ml of DMF. [0651] The degree of transformation and selectivity were 79% and 99% respectively. [CHEMMOL-00057] Example 1.19 [0652] Preparation of 1-phenyl-1H-[1,2,4]triazole Example 1.18 was repeated, operating at 50 C. (72 hours). The degree of transformation and selectivity for 1-phenyl-1H-[1,2,4-triazole] were 75% and 99% respectively. [0653] [CHEMMOL-00058] |
| 91% |
|
Operating protocol A (82 C., 48 hours) was followed using 117 mg of Chxn-Py-Al (0.4 mmoles), 336 mul of iodobenzene (3 mmoles), 138 mg of 1,2,4-triazole (2 mmoles), 1.042 g of caesium carbonate (3.2 mmoles) and 1.2 ml of DMF. The degree of transformation and selectivity were 100% and 98% respectively. The residue obtained following treatment was purified by silica gel chromatography (eluent: hexane/dichloromethane, 100/0 to 50/50). 264 mg of a dark yellow solid was obtained in a yield of 91%. Pale yellow needles were obtained after re-crystallisation from chloroform. The compound obtained had the following formula: The characteristics were as follows: MPt: 46 C. (CHCl3) (Lit: 46-47 C. given by Micetich, R G; Spevak, P; Hall, T W; Bains, B K; Heterocycles 1985, 23, 1645-1649); 1H NMR/CDCl3: delta8.52 (wide s, 1H, H1), 8.04 (wide s, 1H, H2), 7.53-7.65 (m, 2H, H4,8), 7.26-7.51 (m, 3H, H5,6,7); 13C NMR/CDCl3: delta 152.55 (C1), 140.88 (C2), 139.96 (C3), 129.73 (C5 and C7), 128.15 (C6), 119.99 (C4 and C8); GC/MS: Rt=14.02 min, M/Z=145, purity=100%; Rf=0.21 (eluent: dichloromethane/ethyl acetate, 90/10). Example 1.18; Preparation of 1-phenyl-1H-[1,2,4]triazole; Operating protocol A (82 C., 24 hours) was followed using 117 mg of Chxn-Py-Al (0.4 mmoles), 336 mul of iodobenzene (3 mmoles), 138 mg of 1,2,4-triazole (2 mmoles), 1.042 g of caesium carbonate (3.2 mmoles) and 1.2 ml of DMF. The degree of transformation and selectivity were 79% and 99% respectively. ; Example 1.19; Preparation of 1-phenyl-1H-[1,2,4]triazole; Example 1.18 was repeated, operating at 50 C. (72 hours). The degree of transformation and selectivity for 1-phenyl-1H-[1,2,4-triazole] were 75% and 99% respectively. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science








 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping