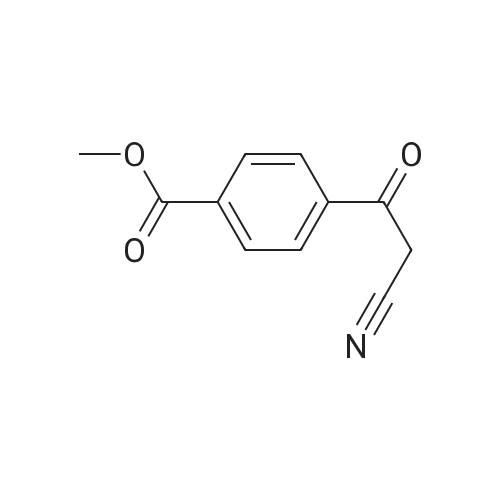
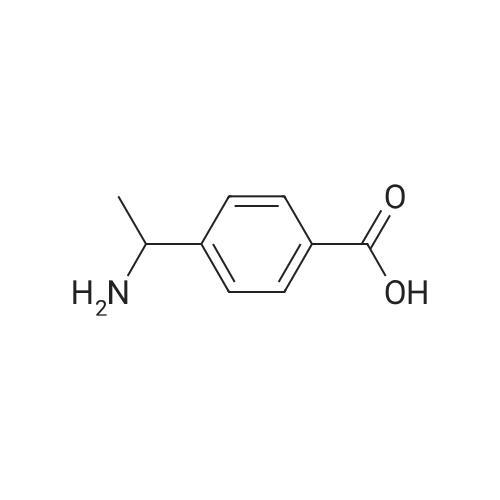
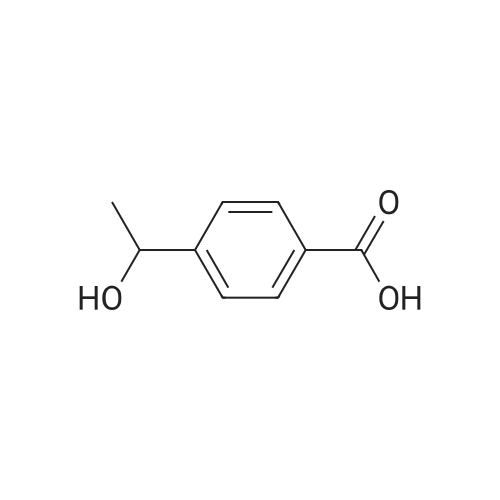
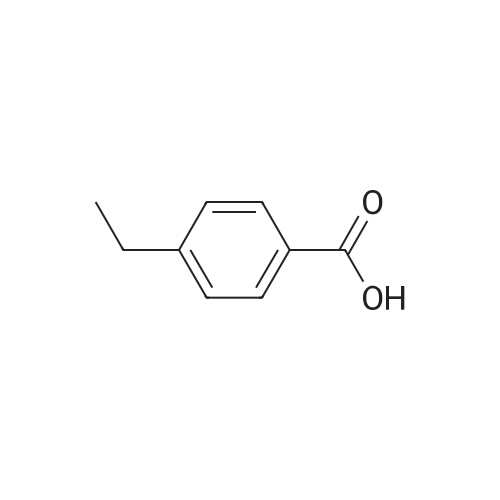
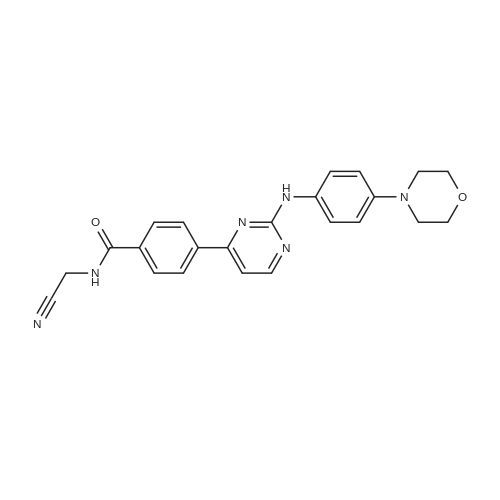
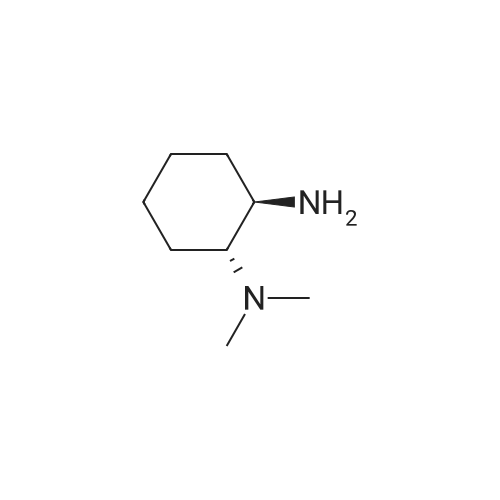
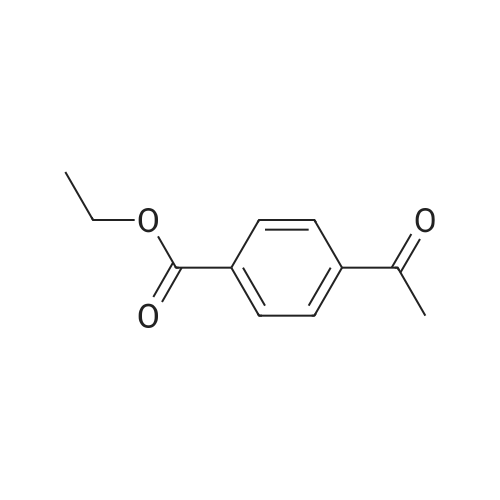
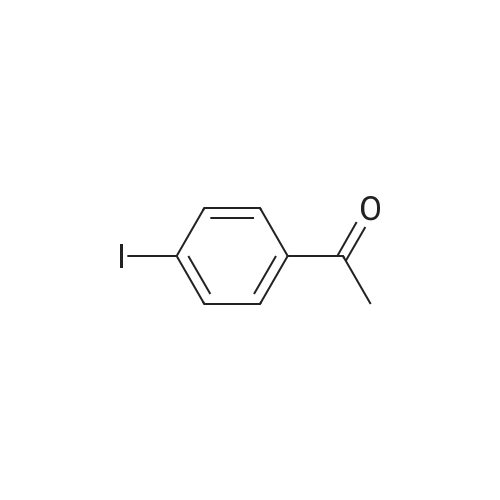
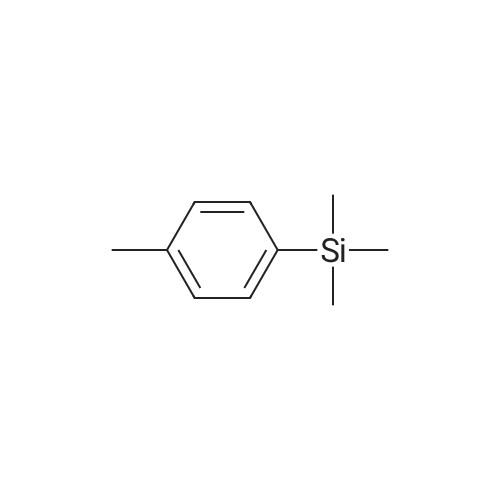
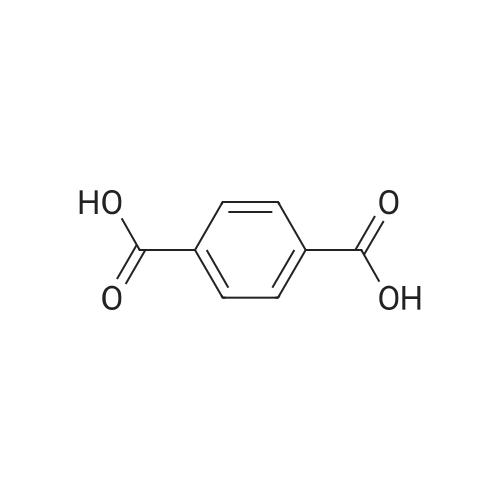
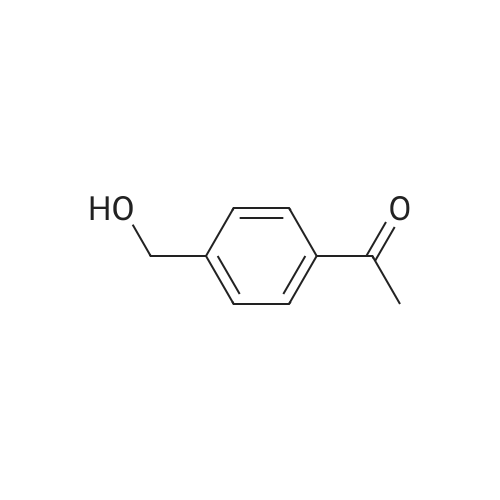
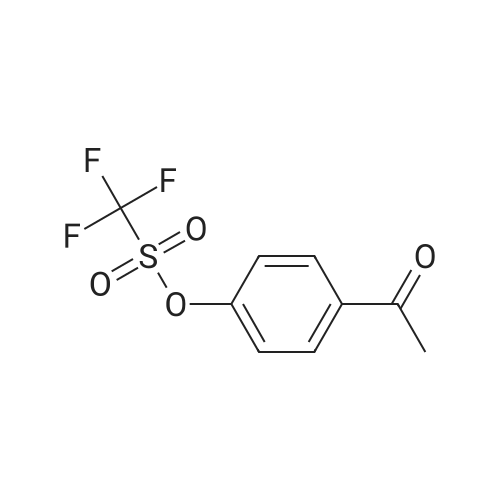
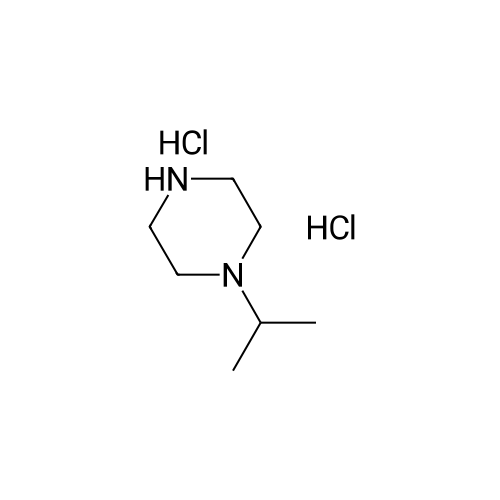
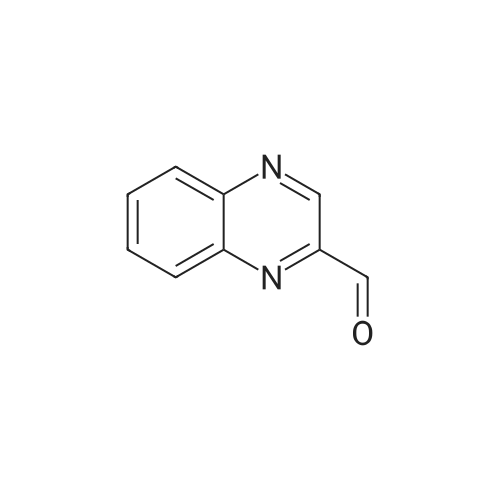
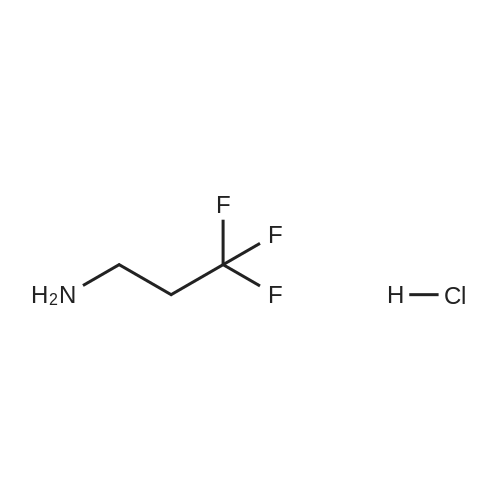
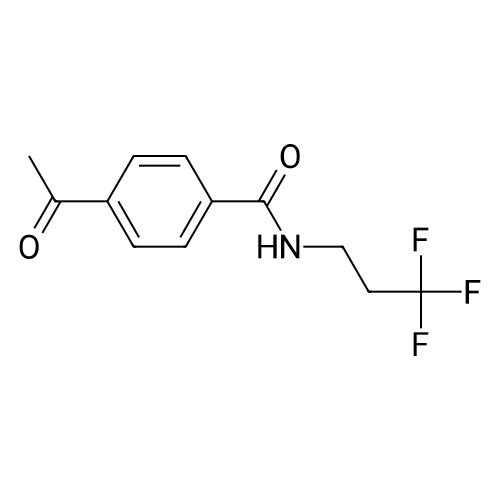
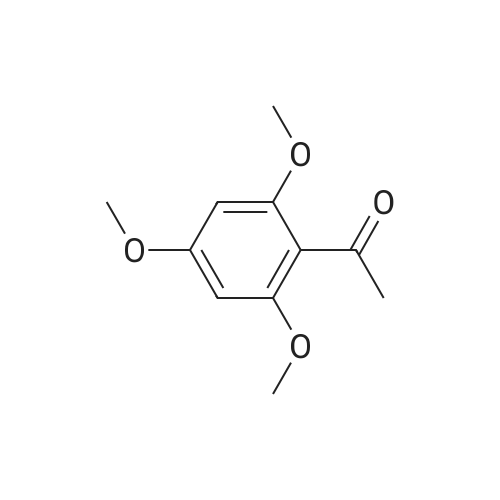
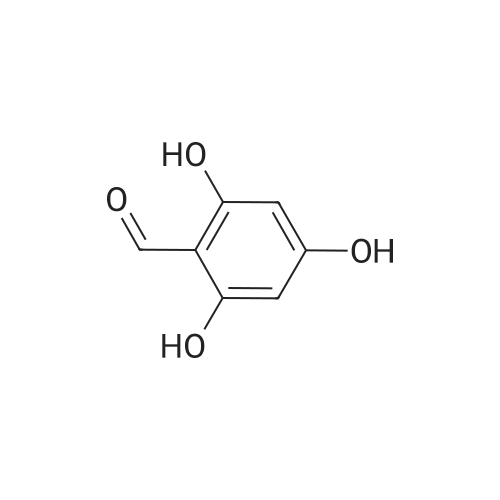
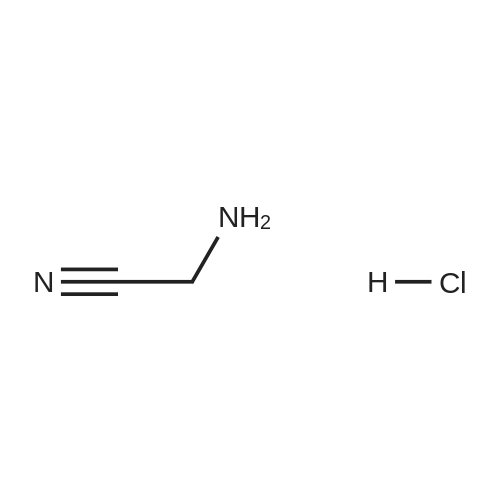
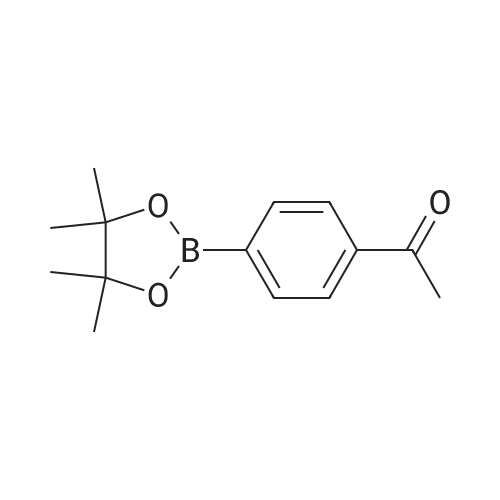
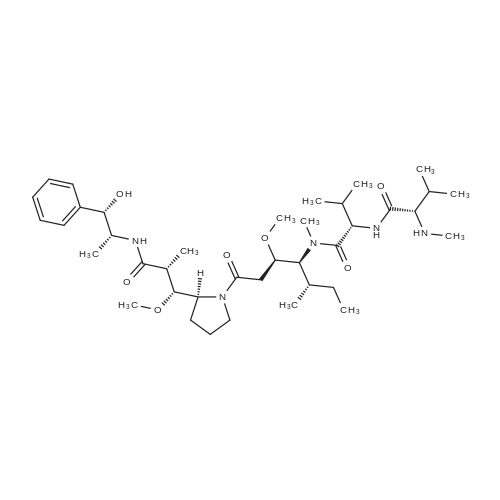
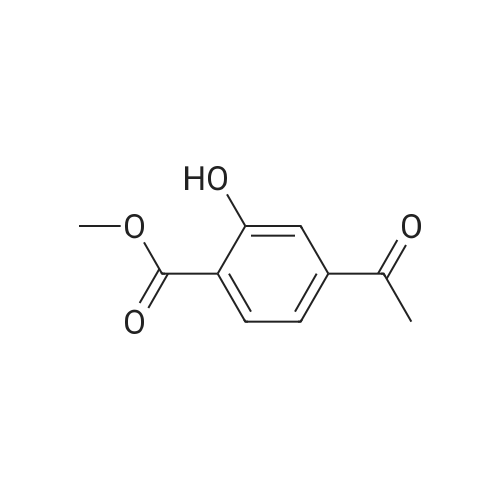
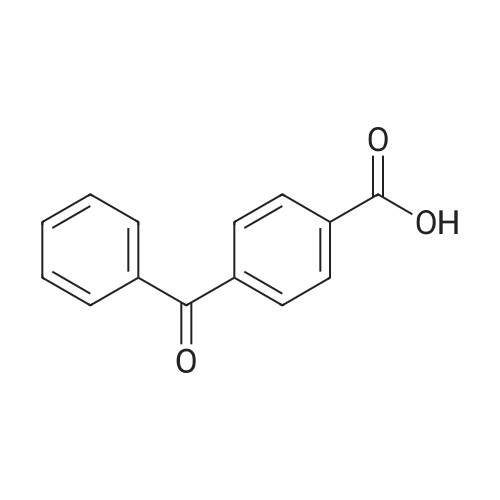
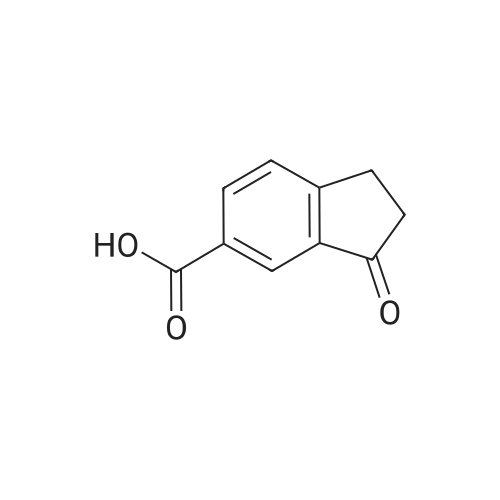
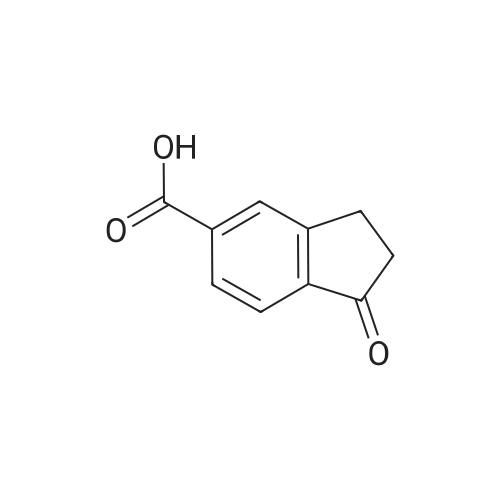
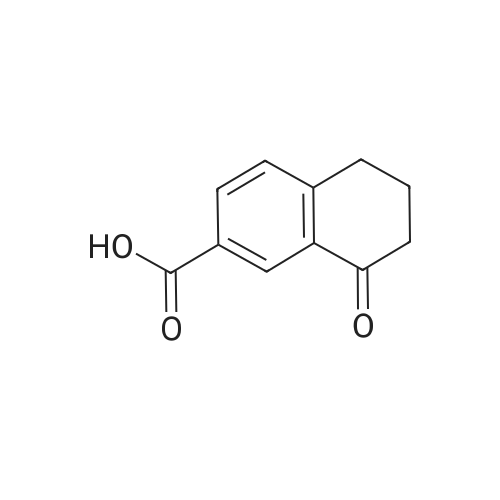
|
With phosphorus(V) chloride |
|
|
With thionyl chloride |
|
|
With thionyl chloride In toluene for 1h; Heating; |
|
|
With thionyl chloride In dichloromethane Heating; |
|
|
With thionyl chloride In ethyl acetate at 77℃; for 4h; |
|
|
With thionyl chloride In toluene |
P PREPARATION P:
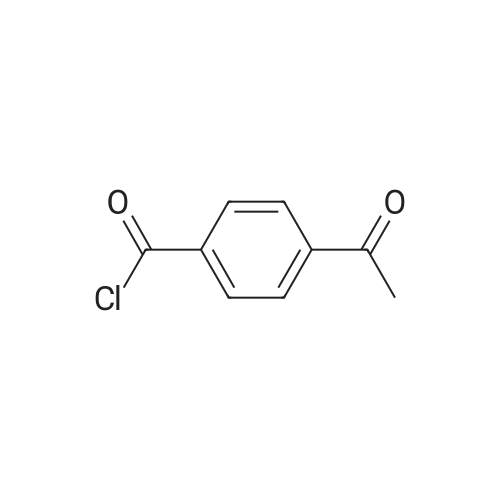
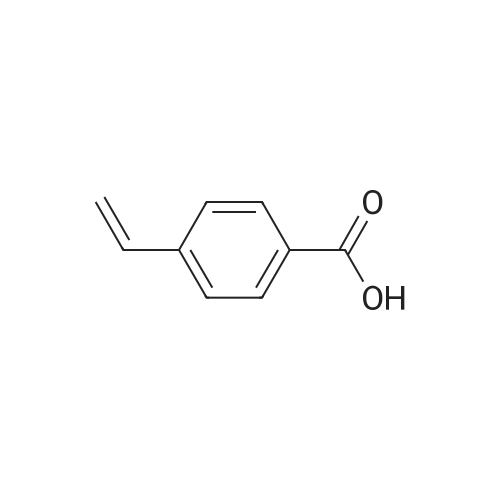
PREPARATION P: 4-Acetylbenzoyl chloride 120 mmol of thionyl chloride are added to 600 mmol of 4-acetylbenzoic acid in 150 ml of toluene. The reaction mixture is heated at reflux for 3 hours and then cooled. The solvent is evaporated off. The product is used without any other treatment. |
|
With oxalyl dichloride In dichloromethane at 20℃; for 1h; Heating / reflux; |
1.1
Example 1; Preparation of N- (4-tert-butylphenyl) -4- (2-methyl-l, 3- dioxane-2-yl) benzamide (Ia-2); (the 1st step); To a mixture of 4-acetylbenzoic acid (compound 1: 1.64g, lOmmole) , oxalyl chloride (1.07mL, 12mmole) and dichloromethane (3OmL) was added a drop of N, N- dimethylformamide, and the mixture was stirred at rt for 1 hour and then heated to reflux for 1 hour. The reaction mixture was concentrated in vacuo to give a crude product of 4-acetylbenzoyl chloride (compound 2c). |
|
With oxalyl dichloride In dichloromethane at 20℃; for 1h; |
A.1
4-Acetyl-benzoic acid tert-butyl ester. To a slurry of 4-acetyl-benzoic acid (21.3 g, 130 mmol) inCH2Cl2 (200 mL) at RT was added oxalyl chloride (11.93 mL, 136 mmol), and then catalytic DMF (0.7 mL) was added drop wise. After Ih, the solvent was removed from the resultant solution. The solid was dissolved in CH2Cl2 and washed with 0.5 N HCl and H2O. The combined organic extracts were washed with brine, dried over MgSOψ filtered, and concentrated in vacuo. The solid was dissolved in pyridine (100 mL) and to the solution was added DMAP (0.793 g, 6.49 mmol) and t-butyl alcohol (31.0 mL, 324 mmol). After 18h, the solvent was removed. The residue was dissolved in EtOAc, washed with IN HCl, 0.5 N NaOH, dried over MgSOφ filtered, and concentrated in vacuo. The solid was used without further purification.1HNMR (CDCl3) δ 8.06 (d, J= 8.1 Hz, 2H), 7.97 (d, J= 8.1 Hz, 2H)5 2.63 (s, 3H), 1.60 (s, 9H). cal'd 221 (MH+), exp 221 (MH+). |
|
With pivaloyl chloride |
|
|
With thionyl chloride In toluene for 16h; Reflux; |
|
|
With oxalyl dichloride In chloroform; N,N-dimethyl-formamide at 0 - 20℃; Inert atmosphere; |
|
|
With thionyl chloride Heating / reflux; |
22 EXAMPLE 22; 1-[4-(5H,11H-Pyrrolo[2,1-c][1,4]benzodiazepine-10-carbonyl)-phenyl]-ethanone
EXAMPLE 22 1-[4-(5H,11H-Pyrrolo[2,1-c][1,4]benzodiazepine-10-carbonyl)-phenyl]-ethanone 4-Acetylbenzoic acid (5.0 g) and thionyl chloride (10 ml) were heated on a steam bath under argon for 0.75 hour, and the volatile material was removed under reduced pressure.. toluene was added and the volatiles were removed again to give the crude acid chloride as a red-orange oil.. This compound tended to solidify and was used as such for further transformations. The acid chloride (4.56 g) in dichloromethane (25 ml) was added portionwise to an ice-cooled solution of 10,11-dihydro-5H-pyrrolo[2,1-c][1,4]-benzodiazepine (3.68 g) and diisopropylethylamine (3.25 g) in dichloromethane (100 ml).. After stirring at room temperature for 18 hours, the reaction mixture was washed with water and saturated aqueous sodium bicarbonate.. The dichloromethane solution was dried over anhydrous sodium sulfate and filtered through a short column of hydrous sodium magnesium silicate eluding with several volumes of dichloromethane.. The combined organic phase was concentrated on a hot plate with the gradual addition of hexane until crystallization occurred.. After cooling, the crystals were collected by filtration to yield the title compound (1.75 g), m.p. 135-137 °C. |
|
With thionyl chloride at 100℃; for 0.75h; |
22 EXAMPLE 22; 1-[4-(5H,11H-Pyrrolo[2,1-c][1,4]benzodiazepine-10-carbonyl)-phenyl]-ethanone
4-Acetylbenzoic acid (5.0 g) and thionyl chloride (10 ml) were heated on a steam bath under argon for 0.75 hour, and the volatile material was removed under reduced pressure. Toluene was added and the volatiles were removed again to give the crude acid chloride as a red-orange oil. This compound tended to solidify and was used as such for further transformations. |
|
With oxalyl dichloride In dichloromethane at 20℃; for 1h; Inert atmosphere; |
3.A
Example 3; Preparation of intermediates 4-acetyl-N-(5-bromothiazolo[5,4-b]pyridin-2-yl)benzamide (3C) and N-(5-bromothiazolo[5,4-b]pyridin-2-yl)-4-(1,1,1-trifluoro-2-hydroxypropan-2-yl)benzamide (3D); Step A: 4-acetylbenzoic acid 3A (500 mg, 1.0 equivalent) was dissolved in DCM (0.3 M), and then followed by addition of oxalyl chloride (1.2 equivalent) and DMF (0.3 equivalent). The mixture was stirred at room temperature for 1 h. Volatiles were removed in vacuo. The residue 3B was obtained as a white solid and was used without further purification. 3B: ESI-MS: m/z 183.0 (M+H)-. |
|
With oxalyl dichloride In dichloromethane; N,N-dimethyl-formamide at 20℃; |
3
Oxalyl Chloride was added to 4-acetyl-benzoic acid in dichloromethane and DMF below 20° C., and the reaction mass concentrated to remove excess reagent. The residue was dissolved in fresh dichloromethane and the secondary amine was then added. Still below 20° C., triethyl amine was added and stirred for an hour. The reaction was quenched with water, washed with 5% HCl to remove excess triethyl amine, and then washed with 5% sodium bicarbonate to remove unreacted starting material. The organic layer was washed with water, dried and concentrated to give 4-acetyl-dialkyl benzamide 116 in yields of 85-90%. Treatment of the benzamide with pre-forming sodium ethyl methyl-ortho-trifluoroacetate from NaOMe and ethyl trifluoroacetate in benzene and reacting the preformed ortho-alkoxide with 4-acetyl-N,N-dialkyl benzamide and subsequent acidification followed by extraction gave compounds 117 in yields of 50-55%. The final pyrazole compounds 118a/b were synthesize by the addition of the appropriate substituted hydrazine to compound 117 in ethanol, acidified ethanol, or acetic acid, depending on the specific hydrazine used. |
|
With oxalyl dichloride |
|
|
at 0 - 20℃; Inert atmosphere; |
|
|
With thionyl chloride at 90℃; |
|
|
With oxalyl dichloride In chloroform; N,N-dimethyl-formamide at 0 - 20℃; Inert atmosphere; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; for 2h; |
|
|
With oxalyl dichloride In dichloromethane; N,N-dimethyl-formamide |
|
|
With thionyl chloride; N,N-dimethyl-formamide In dichloromethane at 0℃; Inert atmosphere; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 20℃; for 12h; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane |
|
|
With thionyl chloride for 2h; Reflux; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 20℃; for 3h; Inert atmosphere; |
|
|
With oxalyl dichloride |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; for 1h; Inert atmosphere; |
|
|
With oxalyl dichloride In dichloromethane; N,N-dimethyl-formamide at 0 - 20℃; Inert atmosphere; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; Inert atmosphere; |
1
Oxalyl chloride (2.0 M in CH2Cl2; 3.0 mL, 6.0 mmol) was added dropwise over 2-3 min to a stirred suspension of 4-acetylbenzoic acid (493 mg, 3.0 mmol) and DMF (1-2 drops) in CH2Cl2 (6 mL) at 0° C. under nitrogen. After complete addition, the mixture was allowed to warm to room temperature and stirred overnight. The mixture was concentrated under vacuum to leave the acid chloride 5l, which was used directly in the tetrazolone-forming step below (yield assumed quantitative=548 mg). |
|
With oxalyl dichloride In dichloromethane; N,N-dimethyl-formamide at 0 - 20℃; for 12h; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; for 2h; |
|
|
Multi-step reaction with 2 steps
1: magnesium(II) sulfate; sulfuric acid / dichloromethane / 20 °C / Inert atmosphere
2: thionyl chloride; water monomer / toluene / 16 h / 100 °C / Sealed tube |
|
|
With thionyl chloride for 2h; Reflux; Inert atmosphere; |
|
|
With thionyl chloride; N,N-dimethyl-formamide In dichloromethane at 55℃; for 5h; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In 1,4-dioxane for 1h; |
|
|
With thionyl chloride; N,N-dimethyl-formamide In toluene at 75℃; for 4h; Sealed tube; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; for 2h; |
6 Synthesis of 4-acetyl-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)benzamide (B)
To a stirred solution of 4-acetylbenzoic acid (8.25 g, 50.30 mmol) in CH2Cl2 (165 mL) were added oxalyl chloride (8.31 mL, 95.57 mmol) and DMF (catalytic amount) at 0° C. The resulting mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated to dryness under high vacuum to give the 4-acetylbenzoyl chloride as a pale yellow solid. The crude material was used as such in the next step without any further purification. Yield: 9.15 g, 99.67%. To a stirred solution of Gemcitabine hydrochloride (15 g, 50 mmol) in pyridine (150 mL), was added chlorotrimethylsilane (31.6 mL, 250 mmol) dropwise [˜1.0 mL/min] over 30 min at 0° C. The resulting mixture was stirred at room temperature for 2 h. 4-acetylbenzoyl chloride (9.12 g, 50 mmol) was added to the reaction mixture portionwise (3 portions, ˜3.04 g/5.0 min) over 15 min. The resulting mixture was stirred at 45° C. for 16 h. Then ethanol (150 mL) was added to the above reaction mixture and stirred for 30 min. Subsequently desalted water (75 mL) was added and stirred for additional 5 h. Then the reaction mixture was concentrated to dryness and the residue was quenched with ice water (300 mL). The resulting solid was filtered through Whatman filter paper (11 μm) and dried under high vacuum for 5 h. The crude product was purified by silica gel flash chromatography. The crude mass was dissolved in CH2Cl2 and adsorbed on silica gel (60-120 mesh, 60 g) and purified over 60*12.5 cm flash column using 240 g of 60-120 mesh silica gel and ˜20 L of 60% ethyl acetate in petrol ether as an eluent. The resulting solid was washed with methanol (100 mL), filtered and dried under high vacuum for 5 h to give the 4-acetyl-N-(1-((2R,4R,5R)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)-tetrahydrofuran-2-yl)-2-oxo-1,2-dihydropyrimidin-4-yl)benzamide (B) as a white solid. Yield: 5.2 g, 12.71 mmol, 25.42%. |
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 35℃; for 3h; |
I.1.1 1.1 ) Preparation of 4-acetyl-N-phenyl-benzamide
1 1.4 g (90.0 mmol) oxalyl chloride were added dropwise to a solution of 4.9 g (30.0 mmol) 4- acetyl benzoic acid and two drops of A ,Ndimethylformamide in dichloromethane. After stirring for 3 hours at 35 °C all volatile components were removed under reduced pressure and the resulting acid chloride was used without further purification. (0620) 2.4 g (13.1 mmol) of the crude acid chloride were added dropwise to a solution of 1 .24 g (13.1 mmol) aniline in 60 ml. of tetrahydrofurane. Upon completion, the reaction mixture was diluted with methyl fe/f-butylether and washed twice with an aqueous solution of sodium hydroxide and twice with a saturated sodium chloride solution. Drying over sodium sulfate, evaporation of the solvent and recrystallization in methyl fe/f-butylether resulted in 2.5 g (10 mmol, 80 %) of the title amide. H-NMR [400 MHz, DMSO-a] δ = 10.4 (s, 1 H), 8.1 (m, 4H), 7.8 (d, 2H), 7.4 (t, 2H), 7.1 (t, 1 H), 2.7 (s, 3H) ppm. |
|
With oxalyl dichloride |
|
|
With oxalyl dichloride In dichloromethane; N,N-dimethyl-formamide |
|
|
Stage #1: acetophenone-4-carboxylic acid With N,N-dimethyl-formamide In dichloromethane at 0℃; for 0.0833333h; Inert atmosphere;
Stage #2: With oxalyl dichloride In dichloromethane Inert atmosphere; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; for 2h; Inert atmosphere; |
|
|
With thionyl chloride Reflux; Inert atmosphere; |
|
|
With oxalyl dichloride; N,N-dimethyl-formamide In dichloromethane at 0 - 20℃; for 2.58333h; |
A dried round-bottom flask was charged with the acid (4.5 mmol), DCM (15 mL), and 2 drops of DMF. Then, oxalyl chloride (0.60 mL, 6.9 mmol) was added dropwise within 5 min at 0 °C. The resulting mixture was stirred at rt. for 3.5 h and concentrated underreduced pressure. The residue was dissolved in EA (40 mL) and K2CO3 (1.24 g, 9.0mmol), MeONH2 .HCl (458 mg, 5.4 mmol) and water (20 mL) were added sequentially.The resulting mixture was stirred for 20 h at rt. and extracted with EA (50 mL × 2). Theorganic layer was washed with saturated aqueous NaHCO3 (15 mL × 2), brine (15 mL× 2), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure.The crude product was recrystallized (EA) to afford the product. In the case of oil, thecrude product was purified by silica gel column chromatography, using EA/PE as theeluent |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping