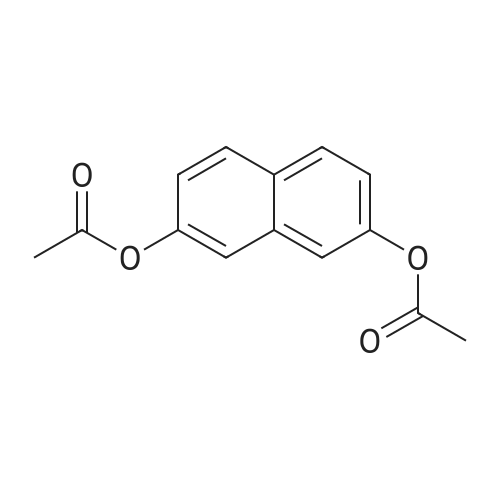
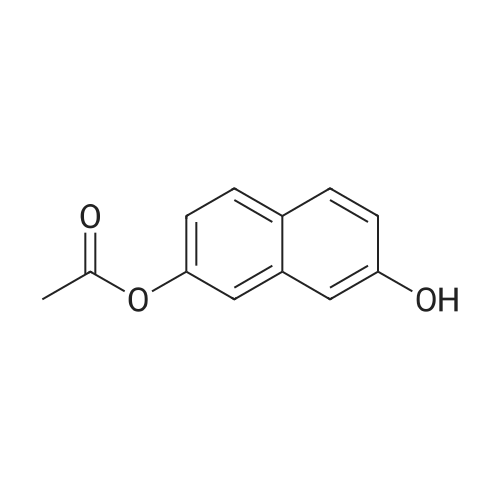
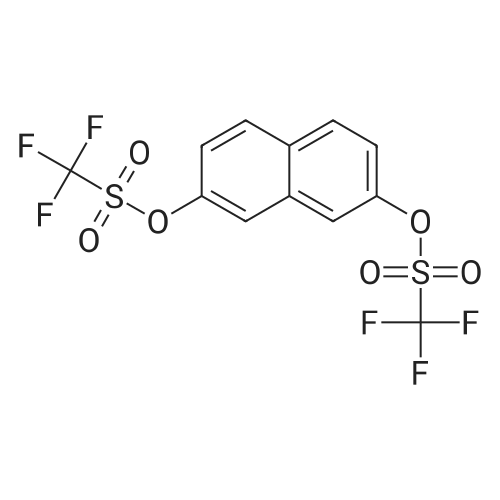
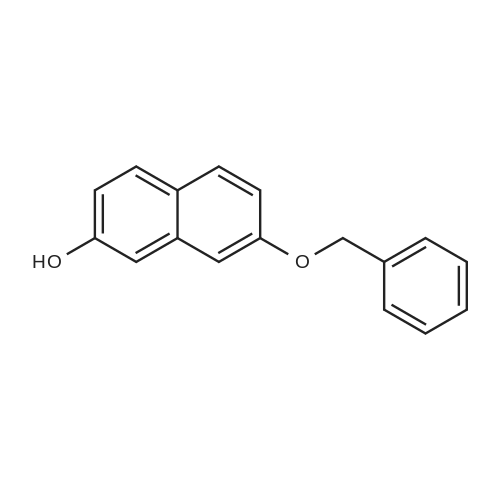
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Chemical Communications, 2012, vol. 48, # 47, p. 5847 - 5849
[2] Synthetic Communications, 2005, vol. 35, # 1, p. 71 - 78
[3] Journal of Organic Chemistry, 2004, vol. 69, # 21, p. 7123 - 7133
[4] Canadian Journal of Chemistry, 1987, vol. 65, p. 2397 - 2404
[5] European Journal of Organic Chemistry, 2016, vol. 2016, # 6, p. 1119 - 1131
[6] Journal fuer Praktische Chemie (Leipzig), 1916, vol. <2> 94, p. 44
[7] Roczniki Chemii, 1961, vol. 35, p. 953 - 966
[8] Canadian Journal of Chemistry, 1981, vol. 59, p. 3034 - 3038
[9] Bulletin of the Chemical Society of Japan, 1989, vol. 62, # 2, p. 545 - 550
[10] Australian Journal of Chemistry, 1983, vol. 36, # 12, p. 2575 - 2580
[11] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2891 - 2898
[12] Journal of Medicinal Chemistry, 2007, vol. 50, # 22, p. 5293 - 5300
[13] Journal of Medicinal Chemistry, 2012, vol. 55, # 12, p. 5720 - 5733
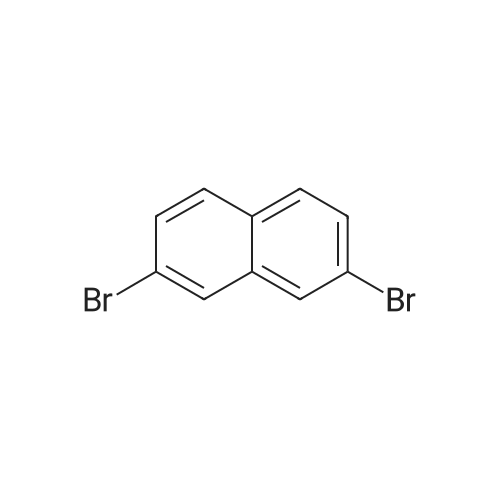
7
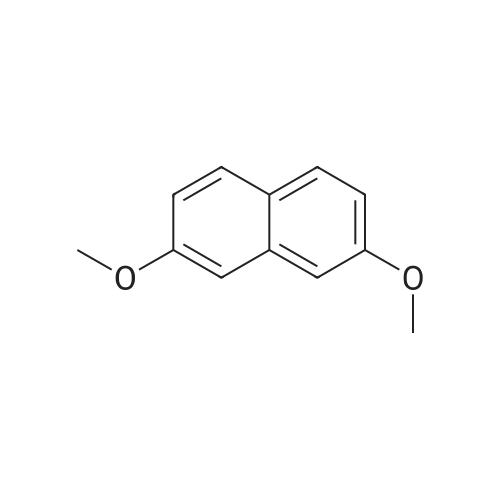
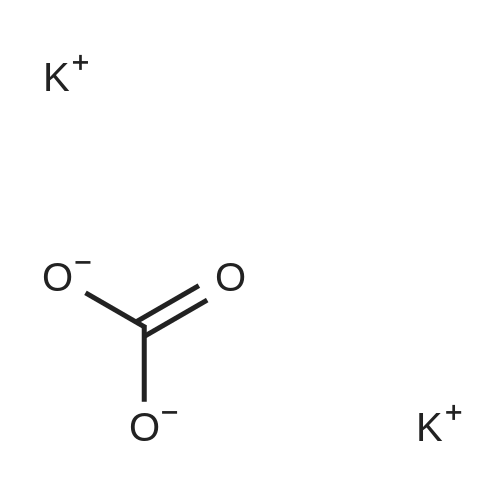
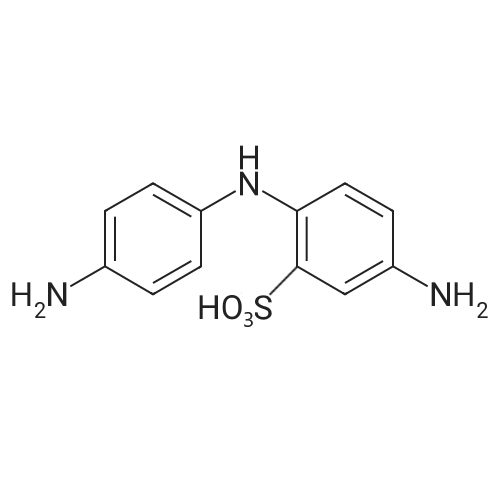
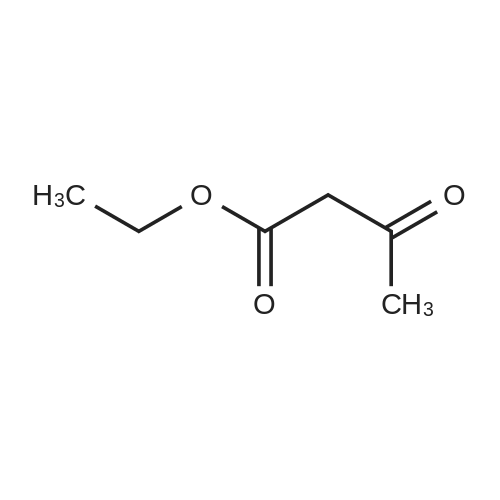
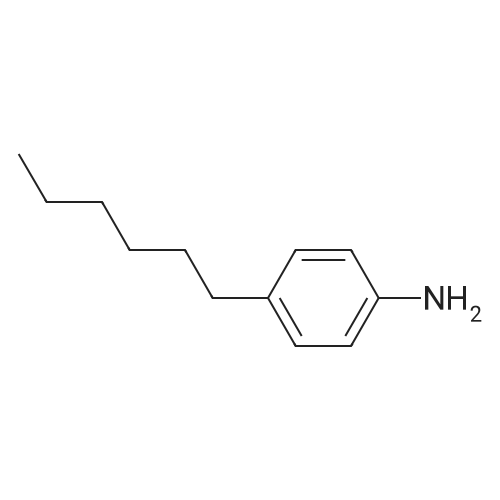
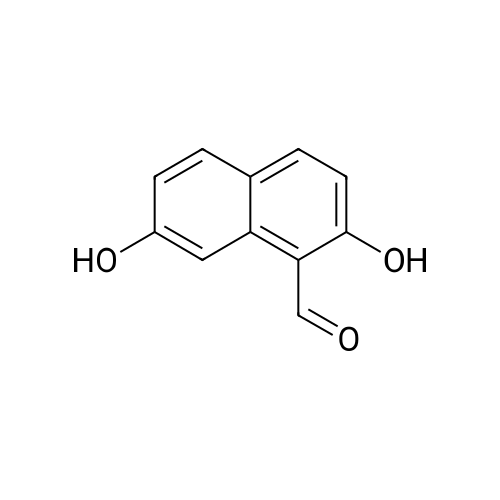
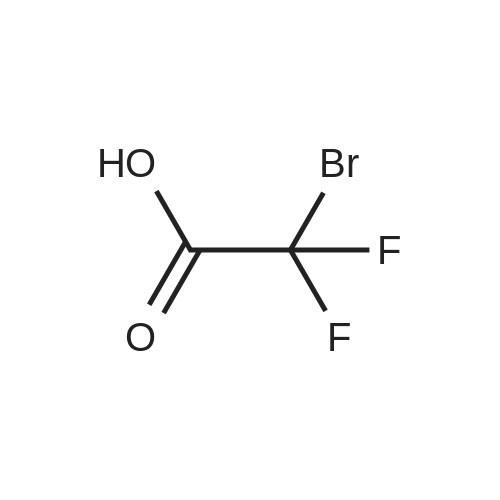
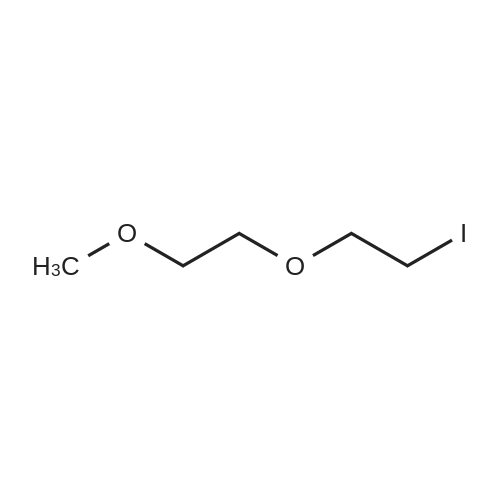
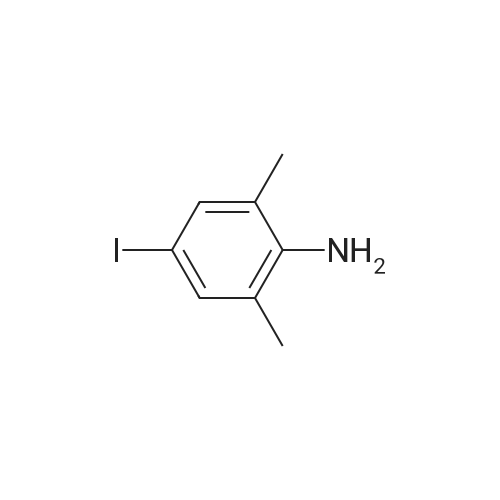
[ 582-17-2 ]
[ 74-88-4 ]
[ 3469-26-9 ]
Reference:
[1] Organic and Biomolecular Chemistry, 2005, vol. 3, # 10, p. 1911 - 1921
[2] Chemische Berichte, 1881, vol. 14, p. 2206
[3] Journal of Organic Chemistry, 1996, vol. 61, # 2, p. 539 - 548
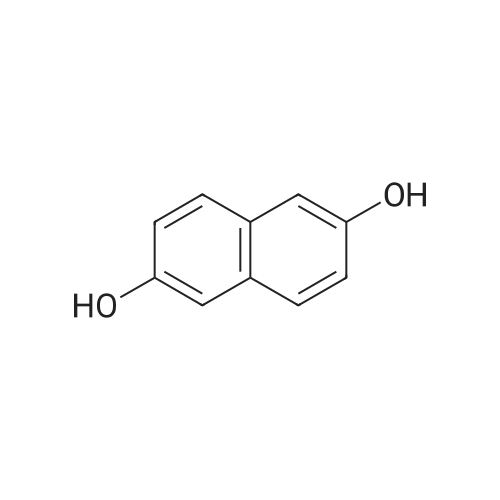
8
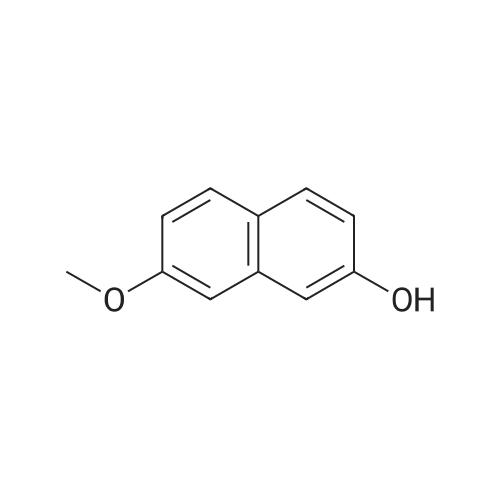
[ 582-17-2 ]
[ 3469-26-9 ]
Reference:
[1] Patent: US6313160, 2001, B1,
9
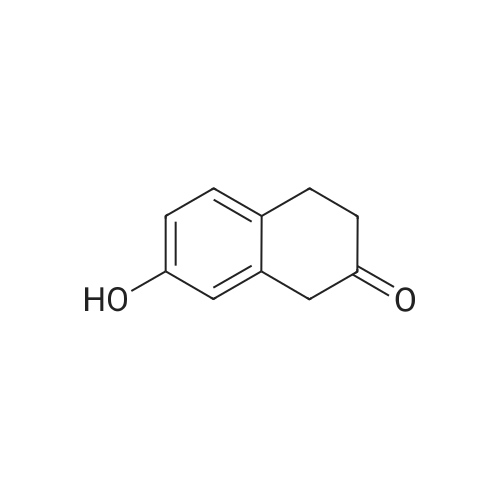
[ 582-17-2 ]
[ 584-08-7 ]
[ 3469-26-9 ]
Reference:
[1] Patent: US5712312, 1998, A,
10
[ 582-17-2 ]
[ 77-78-1 ]
[ 3469-26-9 ]
[ 5060-82-2 ]
Reference:
[1] Chemische Berichte, 1905, vol. 38, p. 3272
[2] Tetrahedron, 1982, vol. 38, # 15, p. 2347 - 2354
11
[ 67-56-1 ]
[ 582-17-2 ]
[ 3469-26-9 ]
[ 5060-82-2 ]
Reference:
[1] Australian Journal of Chemistry, 1993, vol. 46, # 5, p. 731 - 737
12
[ 74-83-9 ]
[ 582-17-2 ]
[ 3469-26-9 ]
Reference:
[1] Journal of Fluorine Chemistry, 2014, vol. 168, p. 193 - 197
13
[ 67-56-1 ]
[ 582-17-2 ]
[ 3469-26-9 ]
Reference:
[1] Chemische Berichte, 1902, vol. 35, p. 1323
14
[ 582-17-2 ]
[ 77-78-1 ]
[ 5060-82-2 ]
Yield
Reaction Conditions
Operation in experiment
63.4%
With potassium carbonate In acetonitrile for 1 h; Reflux
General procedure: A mixture of 2-hydroxy-1-nitronaphthalene (500 mg,2.64 mmol), dimethyl sulfate (0.5 ml, 5.3 mmol), and K2CO3 (73.1 mg, 5.3 mmol) inacetonitrile (10 ml) was refluxed for 1hrs. The solvent was evaporated and the residue waspoured into water and extracted with ethyl acetate. The organic layer was washed with waterand brine and dried over anhydrous MgSO4 and concentrated in vacuo. The residue waspurified by column chromatography on silica gel with nhexane/ethyl acetate (15:1) as theeluent to obtain the product
30%
With sodium hydroxide In dichloromethane; water at 20℃; for 2 h;
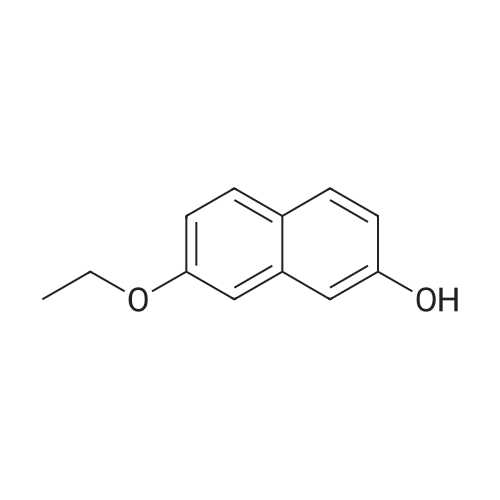
Step A: Sodium hydroxide (20 ml, 1M) and dimethyl sulfate (15.0 ml, 158 mmol) were added dropwise to a solution of 2,7-dihydroxynaphthalene (6.6 g, 40 mmol) in dichloromethane (100 ml) and water (60 ml). Additional sodium hydroxide (20 ml, 1M) and dimethyl sulfate (15.0 ml, 158 mmol) were added and the resulting mixture was stirred at room temperature for 2 hours. The two phases were separated and the aqueous layer was extracted with dichloromethane twice. The combined organic extracts were washed with 1 M HCl, dried over magnesium sulfate and concentrated under vacuum. The crude material was purified by column chromatography (5:1 heptane/ethyl acetate) to give 2-hydroxy-7-methoxynaphthalene (2.1 g, 30percent, 100percent AUC HPLC): 1H NMR (300 MHz, CDCl3) δ 7.66-7.70 (m, 2H), 6.94-7.08 (m, 4H), 4.90 (s, 1H), 3.92 (s, 3H).
[0709] To a mixture of naphthalene-2,7-diol (25 g, 156.08 mmol) and K2CO3 (32.3 g, 232.02 mmol) in acetone (300 ml) was added iodomethane (22.2 g, 156.41 mmol) dropwise with stirring at 0° C. The resulting solution was stirred overnight at room temperature. The solids were filtered off and the filtrate was concentrated under vacuum to give a residue, which was purified by silica gel column chromatography using 1percent-10percent ethyl acetate in petroleum ether to afford 7-methoxynaphthalen-2-ol as a light yellow solid (10 g, 37percent). (ES, m/z): [M+H]+ 175.1; 1H NMR (400 MHz, DMSO-d6): δ 9.65 (s, 1H), 7.65 (d, J=8.8 Hz, 2H), 6.79 (dd, J=13.6, 1.6 Hz, 2H), 6.92-6.89 (m, 2H), 3.84 (s, 3H).
Reference:
[1] Patent: US2014/142114, 2014, A1, . Location in patent: Paragraph 0708; 0709
[2] Journal of Medicinal Chemistry, 1992, vol. 35, # 25, p. 4665 - 4675
[3] Journal of Organic Chemistry, 1981, vol. 46, p. 4988 - 4991
16
[ 582-17-2 ]
[ 77-78-1 ]
[ 3469-26-9 ]
[ 5060-82-2 ]
Reference:
[1] Chemische Berichte, 1905, vol. 38, p. 3272
[2] Tetrahedron, 1982, vol. 38, # 15, p. 2347 - 2354
17
[ 67-56-1 ]
[ 582-17-2 ]
[ 3469-26-9 ]
[ 5060-82-2 ]
Reference:
[1] Australian Journal of Chemistry, 1993, vol. 46, # 5, p. 731 - 737
18
[ 582-17-2 ]
[ 37827-68-2 ]
Reference:
[1] Russian Journal of Organic Chemistry, 2000, vol. 36, # 10, p. 1474 - 1477
19
[ 64-67-5 ]
[ 582-17-2 ]
[ 57944-44-2 ]
Reference:
[1] Farmaco, Edizione Scientifica, 1975, vol. 30, # 11, p. 870 - 883
20
[ 582-17-2 ]
[ 4003-89-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2891 - 2898
21
[ 582-17-2 ]
[ 116230-30-9 ]
Yield
Reaction Conditions
Operation in experiment
84%
With bromine; triphenylphosphine In acetonitrile at 0 - 250℃; for 1.5 h;
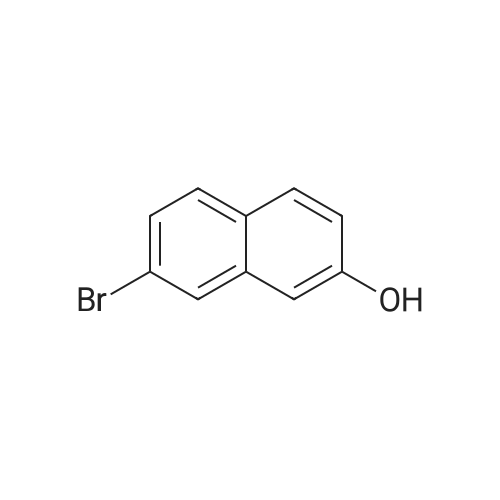
A synthetic procedure was based on the literature method [12]. To a vigorously stirred mixture of triphenylphosphine (31.5g, 120mmol) in acetonitrile (50.0mL), bromine (19.2g, 120mmol) was added dropwise at 0°C. The reaction mixture was allowed to reach room temperature, and 2,7-dihydroxynaphthalene 1 (16.0g, 100mmol) was added in one portion. The mixture was heated to 70°C for 30min, after which the solvent was removed by rotary evaporation. The flask was equipped with a gas trap, and the black residue was heated to 250°C for 1h. After cooling to room temperature, the mixture was dissolved in 200mL of dichloromethane and the viscous liquid was obtained after column chromatography (silica gel, petroleum ether/dichloromethane 1:1). The crude product was purified by column chromatography (silica gel, petroleum ether/dichloromethane 3:2) to give the compound 2 (18.8g, 84.3mmol, 84percent) as a beige powder. 1H NMR (400MHz, CDCl3) δ (ppm): 5.09 (s, 1H, –OH), 7.06 (d, 1H, J 2.4Hz, naphthalene-H), 7.10 (dd, 1H, J 8.8, 2.8Hz, naphthalene-H), 7.24 (dd, 1H, J 8.8, 2.0Hz, naphthalene-H), 7.63 (d, 1H, J 8.8Hz, naphthalene-H), 7.72 (d, 1H, J 8.8Hz, naphthalene-H), 7.84 (d, 1H, J 1.2Hz, naphthalene-H). 13C NMR (100MHz, CDCl3) δ (ppm): 108.7, 118.1, 120.8, 127.0, 127.3, 128.3, 129.4, 129.9, 135.7, 154.0. HR-ESI-MS m/z: [M−H]− calcd. for C10H6BrO, 220.9602; found, 220.9601.
66%
With bromine; triphenylphosphine In acetonitrile at 70℃; for 1 h; Schlenk technique
To a stirring suspension of Triphenyl phosphine (31.5 g) in Acetonitrile (50 mL) in a 250 mL10 Schlenk-flaskwas added carefully Bromine (6.2 mL) at 0 00 with a syringe over 30 mm. The yellow solution was warmed to room temperature and 2,7-Dihydroxynaphthalene (16 g) was added in one portion. The reaction was refluxed at 70 00 for one hour. After cooling to room temperature, the solvent was removed under reduced pressure. The reaction flask was connected to a gas-washing bottle filled with a concentrated sodium hydroxide solution. The flask was heatedto 250 00 for two hours and the black residue dissolved in 100 mL Dichloromethane and purified via column chromatography (DCM:Pentan 1:1 to pure DCM). Product 101 was received as a beige powder (14.7 g, 66percent)DC: Dichloromethane:Pentane, 1:1, R=0,21H-NMR: (300 MHz, ODd3) = 7.76 (d, 1 H), 7.63 (d, 1 H), 7.54 (d, 1 H), 7.32 (dd, 1 H), 7.03 (dd, 1H), 6.98 (d, 1H), 4.98 (b, 1H)
43%
Stage #1: With bromine; triphenylphosphine In acetonitrile at 10℃; for 0.166667 h; Stage #2: for 3 h; Heating / reflux Stage #3: at 280 - 310℃; for 1.08333 h; Neat (no solvent)
Preparation 2 [0100] Preparation of 2-Bromo-7-methoxy-naphthalene. [CHEMMOL-00021] [0101] Preparation of 7-Bromo-2-naphthalenol. [0102] Triphenyl phosphine (89.7 g, 0.342 mol) and acetonitrile (350 mL) were combined in a 1-L flask under N2 atmosphere. The mixture was cooled to 10° C. Bromine (17.6 mL, 0.342 mol) was added dropwise over 10 minutes. The cooling bath was removed and 2,7-dihydroxynaphthalene (50.0 g, 0.312 mol) was added along with 350 mL of CH3CN rinse. The resulting yellow tan mixture was heated at reflux for 3 hours. Acetonitrile was distilled off under a water aspirator pressure over 2 hours, resulting in a grayish white solid. The solid was heated to 280° C. over 30 minutes giving a black liquid. The liquid was heated to 310° C. over 20 minutes and the temperature was maintained at 310° C. for an additional 15 minutes until gas evolution ceased. The black mixture was cooled to room temperature. Chromatography yielded 34.5 g of the intermediate title compound as an off-white solid which was 87percent pure by HPLC (43percent yield).
43%
Stage #1: With bromine; triphenylphosphine In acetonitrile for 3 h; Heating / reflux Stage #2: at 280 - 310℃; for 1.08333 h;
Triphenyl phosphine (89.7 g, 0.342 mol) and acetonitrile (350 mL) were combined in a 1-L flask under N2 atmosphere. The mixture was cooled to 10° C. Bromine (17.6 mL, 0.342 mol) was added dropwise over 10 minutes. The cooling bath was removed and 2,7-dihydroxynaphthalene (50.0 g, 0.312 mol) was added along with 350 mL of CH3CN rinse. The resulting yellow tan mixture was heated at reflux for 3 hours. Acetonitrile was distilled off using a water aspirator over 2 hours resulting in a grayish white solid. The solid was heated to 280° C. over 30 minutes giving a black liquid. The liquid was heated to 310° C. over 20 minutes and the temperature was maintained at 310° C. for an additional 15 minutes until gas evolution ceased. The black mixture was cooled to room temperature. Chromatography yielded 34.5 g of the intermediate title compound as an off-white solid which was 87percent pure by HPLC (43percent yield).
43%
Stage #1: With bromine; triphenylphosphine In acetonitrile at 10 - 20℃; Stage #2: for 3 h; Heating / reflux Stage #3: at 20 - 310℃; for 1.08333 h; Neat (no solvent)
Triphenyl phosphine (89.7 g, 0.342 mol) and acetonitrile (350 mL) were combined in a 1-L flask under N2 atmosphere. The mixture was cooled to 10 C. Bromine (17.6 mL, 0.342 mol) was added dropwise over 10 minutes. The cooling bath was removed and 2,7-dihydroxynaphthalene (50.0 g, 0.312 mol) was added along with 350 mL of CH3CN rinse. The resulting yellow tan mixture was heated at reflux for 3 hours. Acetonitrile was distilled off using a water aspirator over 2 hours resulting in a grayish white solid. The solid was heated to 280 C. over 30 minutes giving a black liquid. The liquid was heated to 310 C. over 20 minutes and the temperature was maintained at 310 C. for an additional 15 minutes until gas evolution ceased. The black mixture was cooled to room temperature. Chromatography yielded 34.5 g of the intermediate title compound as an off-white solid which was 87percent pure by HPLC (43percent yield).
Reference:
[1] Journal of the American Chemical Society, 2008, vol. 130, # 13, p. 4541 - 4552
[2] Dyes and Pigments, 2014, vol. 107, p. 174 - 181
[3] Patent: WO2015/121785, 2015, A1, . Location in patent: Page/Page column 32; 33
[4] Helvetica Chimica Acta, 1999, vol. 82, # 7, p. 981 - 1004
[5] Chemical Communications, 2012, vol. 48, # 5, p. 750 - 752
[6] Helvetica Chimica Acta, 2000, vol. 83, # 11, p. 2865 - 2883
[7] Patent: US2004/6229, 2004, A1, . Location in patent: Page/Page column 14
[8] Patent: US2003/232833, 2003, A1, . Location in patent: Page 13
[9] Patent: US2003/225281, 2003, A1, . Location in patent: Page 13
[10] European Journal of Organic Chemistry, 2000, # 3, p. 491 - 497
2.3.1. 1-((2,7-Dihydroxynaphthalen-8-yl)methyl)naphthalene-2,7-diol 3
In a 50 ml round bottom flask was dissolved 2,7-naphthalenediol (1 g, 6.25 mmol) in ethanol 96% (5 ml) and stirred at 45 °C. Hydrochloric acid 5% (30 ml) and formaldehyde 37% (0.3 g) were added to the mixture. After several minutes a white precipitate was formed. The precipitate was washed with water and dried under vacuum. Yield was 90%.
51%
With sodium carbonate In water at 20℃; for 0.5h;
50.6%
With sodium carbonate In water at 20 - 25℃; for 0.5h;
With air; copper(II) sulfate In chlorobenzene at 140℃; for 15h;
90%
With iron(III) chloride for 0.0666667h; microwave irradiation;
89%
With iron(III) chloride hexahydrate In ethanol; water at 25℃; for 24h;
82%
With iron(III) chloride hexahydrate In water; isopropyl alcohol at 40℃; for 1.5h; Inert atmosphere;
1 Example 1
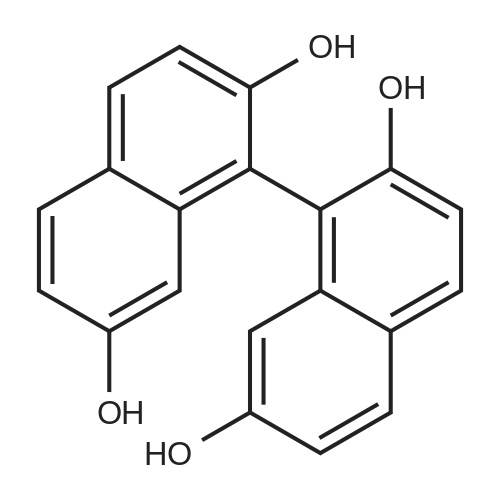
In a flask with a thermometer, a stirrer, and a reflux condenser, 139 g (0.5 moles) of iron(III) chloride hexahydrate and 1330 mL of water were charged under nitrogen gas purging, the inside of a reactor was replaced with nitrogen under stirring, and then a solution previously prepared by dissolving 82 g (0.5 moles) of naphthalene-2,7-diol in 190 mL of isopropyl alcohol was added to the reactor, followed by stirring at 40° C. for 30 minutes. A mixed solution of 139 g (0.5 moles) of iron(III) chloride hexahydrate, 664 mL of water, and 94 mL of isopropyl alcohol was added, and the resultant mixture was heated to 40° C. and then further stirred for 1 hout Then, 500 mL of ethyl acetate was added to the reaction solution and stirred for 10 minutes. The reaction solution was transferred to a separating flannel, an organiclayer was separated, and then an aqueous layer was extracted with ethyl acetate. The organic layers were combined and washed with saturated brine. Afier the solvent was distilled off under vacuum until the amount was about 200 mE, the solution was transferred to a SUS container provided with a thermometer, a stirrer, and Dean-Stark trap, and 5 L oftoluene was added. Then, ethyl acetate was evaporated by heating at a temperature lower than the boiling point of toluene, thereby replacing ethyl acetate as the solvent by toluene. The toluene solution was cooled to room temperature, and insoluble substances were filtered off with quantitative filter No. 5C manufactured by Advantec Co., Ltd. The solvent was distilled off from the filtrate under vacuum, and then the residue was dried at 1000 C. for 5 hours to produce 66 g (yield 82%) of a solid containing [1, 1’-binaphthalene] -2,2’,7,7’-tetraol as a main component. FIGS. 1 and 2 show a GPC chart and a MS spectrum chart, respectively, of the resultant compound. In the MS spectrum chart, besides [1,1’-binaphthalene]-2,2’,7, 7’-tetraol, small amounts of byproducts and naphthalene-2,7- diol used as the raw material were observed in the resultant compound. However, the purity determined by the GPC chart was 98%. In addition, it could be confirmed by differential scaiming calorimetry of the compound that the compound is an anhydride having a melting point of 218° C.
80%
With Cu(II) oxymetasilicate for 0.05h; microwave irradiation;
76%
With nitrosonium tetrafluoroborate; trifluoroacetic acid In dichloromethane at -15℃; for 1h; Sealed tube;
72%
With copper acetylacetonate for 0.0361111h; microwave irradiation;
72%
With dipotassium peroxodisulfate In trifluoroacetic acid at 20℃; for 16h; Sealed tube; Schlenk technique; regioselective reaction;
60%
With iron(III) chloride In 1,4-dioxane for 0.166667h; Heating;
60%
With aluminum oxide; Ru(OH)x; water; oxygen at 99.85℃; for 4h;
47%
With 3-methylbenzothiophene; dihydrogen peroxide; trifluoroacetic acid; trifluoroacetic anhydride In dichloromethane; water at 0 - 20℃; for 2h; Inert atmosphere;
40%
With air; Cu-exchange montmorillonite In chlorobenzene at 140℃; for 6h;
With water; iron(III) chloride Versetzen der Reaktionsloesung mit wss. HCl;
29 mg
With dipotassium peroxodisulfate; silver(I) acetate; copper diacetate In water at 20℃; for 12h;
8 Example 8:
1 mmol of 2,7-dihydroxynaphthalene was added to 4 ml of water-soluble (containing 2% Triton-X-100 surfactant), and 0.2 mmol of copper acetate was added thereto, 1.0mmol potassium persulfate, 1.0mmol silver acetate, reaction for 12 hours at room temperature, after the reaction is completed, a saturated NaCl aqueous solution is added to the reaction solution, and the mixture is extracted with dichloromethane. The organic layer is dried over anhydrous sodium sulfate, filtered, and evaporated to dryness under reduced pressure to obtain a crude compound. The crude compound was subjected to silica gel column chromatography, and a solution having a volume ratio of ethyl acetate and petroleum ether of 1: 5 was used as a mobile phase. The eluate with an Rf value of 0.3-0.5 was collected by TLC. The resulting eluate was collected, the solvent was removed under reduced pressure, and dried. 29 mg of a pure compound represented by the formula (II-8) was obtained.
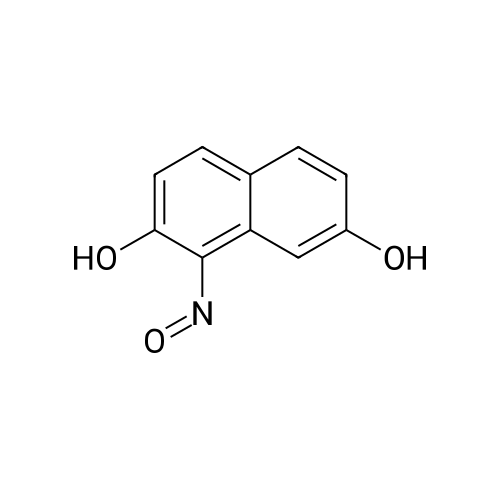
With sulfuric acid; sodium hydroxide; sodium nitrite In water at 0℃;
94%
With sulfuric acid; sodium hydroxide; sodium nitrite In water at 3℃; for 1.5h;
1
In a reaction flask, add 5.6 g of 2,7-naphthalenediol, 70 mL of an aqueous solution containing 2.8 g of sodium hydroxide, and stir well. After complete dissolution, the ice salt bath was cooled to 3 ° C and 2.5 g of Sodium nitrate, then slowly add 18g of sulfuric acid solution at a concentration of 40%, keep the temperature at 3 ° C; after the completion of the reaction to continue for 1.5h, get red brown sticky slurry; filter, washed with water to the filtrate is neutral, The filter cake was collected and dried naturally to give 6.2 g of 1-nitroso-2,7-naphthalenediphenol as a reddish brown solid, i.e., intermediate 3, yield 94%
92.1%
With sodium nitrite
88%
With acetic acid; sodium nitrite In water at 0℃; for 1.83333h;
4.2.3. General procedure for nitrosation
General procedure: To a stirred solution of 2-hydroxynaphthalene derivatives (1 equiv) in a 5:1 mixture of glacial acetic acid and water on an ice bath, an aqueous sodium nitrite (NaNO2, 1 equiv) was added dropwise. After 10 min, an additional aqueous NaNO2 (0.05 equiv) was added and the reaction mixture was further stirred at 0 °C for 2 h. The precipitate was collected by filtration, washed with dilute acetic acid and water, and dried in vacuo.
84%
With hydrogenchloride; sodium hydroxide; sodium nitrite In water 1a) 0 deg C, 3 h, b) 25 deg C, 12 h;
With hydrogenchloride; sodium nitrite
With sodium hydroxide; sodium nitrite Ansaeuern mit Salzsaeure;
With acetic acid; sodium nitrite
With sulfuric acid; sodium nitrite at 0℃; Yield given;
With sulfuric acid; sodium nitrite Yield given;
With sodium hydroxide; sodium nitrite
With sulfuric acid; sodium hydroxide; sodium nitrite
Stage #1: 2,7-Dihydroxynaphthalene With sodium hydroxide; sodium nitrite In water at -5℃;
Stage #2: With sulfuric acid In water for 1h;
2
A solution of 2,7-dihydroxynaphthalene (compound 4b, 1.60 g, 10 mmol) was completely dissolved in 21 mL of a 1.2 mol / L aqueous sodium hydroxide solution and the system was cooled to -5 ° C using an ice bath and kept at low temperature.NaNO2 (828 mg, 12 mmol) was added to the system to complete dissolution.Subsequently, 6 g of dilute sulfuric acid at a concentration of 45% was slowly added with stirring, and the mixture was stirred at a low temperature for 1 hour. After filtering, the filter cake was washed with water to neutral and vacuum dried to obtain an intermediate product (6b).The newly prepared N-propyl iodide (compound 5b, 3.29 g, 10 mmol) was then added to a flask containing 40 mL of absolute ethanol, triethylamine (3 mL) was added and heated at 70 ° C for 1 hour to give the intermediate product 5b ').While the reaction was carried out in the intermediate (5b), the intermediate product (6b) (compound 6b, 2.27 g, 12 mmol) was added to another flask containing 60 mL of absolute ethanol and heated at 70 ° C.After 1 hour, the solution in the flask containing the intermediate (6b) was added dropwise to another flask containing the intermediate (5a '), heating was continued, and the reaction was monitored by thin layer chromatography (TLC) until the reaction was completed.
4.2 g
With sulfuric acid; sodium nitrite In water at 0℃; for 1h;
S1 S1,Synthesis of 1-nitroso-2,7-dinaphthol 1
4g 2,7-dinaphthol is dissolved in 80mL of near boiling distilled water,Stir well and pour into the three-necked flask with the electric mixer.An aqueous solution of sodium nitrite (1.84 g of sodium nitrite dissolved in 40 mL of distilled water) was added under cooling and stirring, and after thoroughly mixing,Slowly add sulfuric acid solution (mixed sulfuric acid 1.44g and 20mL water mixed and cooled), control temperature below 0 °C, continue to stir for 1h after the addition is completed.A small amount of ammonium sulfate was added to remove excess nitrous acid, and the reaction was completed, followed by filtration under reduced pressure, and the solid was washed with distilled water and dried in vacuo to yield 4.20 g of purple compound.
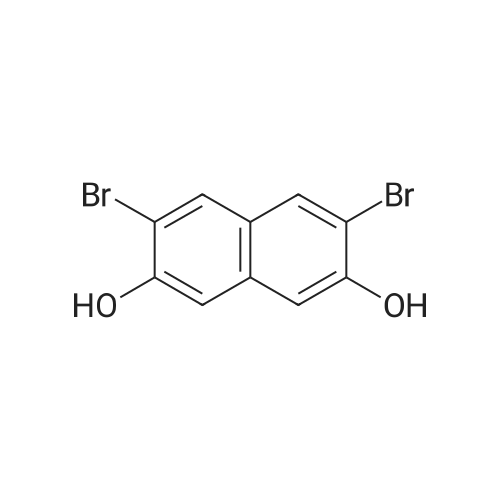
Bromine (5.5 mL, 109 mmol) dissolved in acetic acid (40 mL) was added dropwise over a 20 min period to a stirred solution of 2,7-dihydroxynaphthalene (S1, 5 g, 31 mmol) dissolved in acetic acid (115 mL). The mixture was diluted with water (3 mL) and brought to reflux. Upon discoloration of bromine, powdered tin (7.6 g, 56 mmol) was added drop by drop. The mixture was allowed to reflux for a further 6 h, then cool down to RT, and diluted with water (50 mL), the precipitated product was filtered as a colourless solid, which was recrystallized from AcOH to give 3 (9 g, 91%).
72%
With tin; water; bromine; acetic acid; for 4h;Inert atmosphere; Reflux;
Bromine (26.0 g, 0.162 mol) dissolved in acetic acid (50 mL) was added dropwise over a 20 min period to a stirred solution of 2,7-dihydroxynaphthalene (6.5 g, 0.046 mol) dissolved in acetic acid (150 mL). The mixture was diluted with water (20 mL) and brought to reflux. Upon discolouration of bromine, powdered tin (10 g, 0.084 mol) was added portionwise. The mixture was allowed to reflux for a further 4 h, allowed to cool to room temperature and diluted with water (100 mL), the precipitated product was filtered as a colourless solid (10.50 g, 72%), which was recrystallized from AcOH. Mp 188-190 C (lit. 30 mp 186-190 C). 1H NMR (300 MHz, DMSO-d6) delta 10.50 (s, 2H), 8.01 (s, 2H), 7.06 (s, 2H); 13C NMR (75 MHz, DMSO-d6) delta 152.5, 134.3, 131.0, 124.4, 110.1, 108.4. IR (ATR) (cm-1) 3500-3300 (b), 2866, 2745, 1615, 1588, 1525. HRMS (EI+, 70 eV); m/z: [M]+ calcd for C10H6Br2O2 315.8735, found 315.8741.
With N-chloro-succinimide In acetonitrile at 0 - 20℃; for 5h; Inert atmosphere;
1.A Synthetic Example 1 (Synthesis of 1,8-dichloronaphthalene-2,7-diol) ((A) Step)
Under a nitrogen atmosphere, 992 mg (6.20 mmol) of 2,7-dihydroxynaphthalene (Tokyo Chemical Industry Co., Ltd.) and 60 mL of acetonitrile (dehydrated grade) were added to a 200 mL three-necked flask. This solution was cooled to 0 °C. and 1.74 g (13.0 mmol) of N-chlorosuccinimide (Tokyo Chemical Industry) was added. The resulting mixture was stirred at room temperature for 5 hours, and the reaction was stopped by adding 60 mL of hydrochloric acid. The mixture was extracted with dichloromethane, and the organic phase was washed with brine and dried over anhydrous sodium sulfate. The residue was purified by silica gel column chromatography (developing solvent: dichloromethane) to obtain 1.06 g of 1,8-dichloronaphthalene-2,7-diol (yield 75%).
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione In nitromethane at 0 - 110℃;
83%
With bromine In acetic acid for 41h; Inert atmosphere; Reflux;
Synthesis of 1,3,6-tribromo-2,7-dihydroxynaphthalene
Synthesis of 1,3,6-tribromo-2,7-dihydroxynaphthaleneUnder a nitrogen atmosphere, 2,7-dihydroxynaphthalene (5 g, 31 mmol) was dissolved in acetic acid (150 ml). Note that the acetic acid was used as a solvent.Bromine (5.3 ml. 103 mmol) was dripped in the solution, and reaction was carried out under a reflux temperature for 41 hours.The reaction solution was cooled to a room temperature, and pure water (50 ml) was added thereto. A precipitated solid was separated and collected by filtering. This solid substance was rinsed by pure water, dried under a reduced pressure condition, thereby obtaining a white solid of 1,3,6-tribromo-2,7-dihydroxynaphthalene (10 g, yield: 83%).The reaction formula of the above-explained reaction is as follow. Various spectrum data of the obtained 1,3,6-tribromo-2,7-dihydroxynaphthalene are indicated below.1H-NMR (270 MHz, CDCl3) δ5.88 (s, 1H, OH), 6.24 (s, 1H, OH), 7.60 (s, 1H, ArH), 7.88 (s, 1H, ArH), 7.89 (s, 1H, ArH); EIMS (70 eV) m/z=396 (M+)
74%
With bromine In acetic acid for 2h; Heating;
With bromine; acetic acid at 20℃;
With bromine; acetic acid at 5 - 7℃; zuletzt bei 100grad;
With potassium carbonate; In acetonitrile; for 1h;Reflux;
General procedure: A mixture of 2-hydroxy-1-nitronaphthalene (500 mg,2.64 mmol), dimethyl sulfate (0.5 ml, 5.3 mmol), and K2CO3 (73.1 mg, 5.3 mmol) inacetonitrile (10 ml) was refluxed for 1hrs. The solvent was evaporated and the residue waspoured into water and extracted with ethyl acetate. The organic layer was washed with waterand brine and dried over anhydrous MgSO4 and concentrated in vacuo. The residue waspurified by column chromatography on silica gel with nhexane/ethyl acetate (15:1) as theeluent to obtain the product
30%
With sodium hydroxide; In dichloromethane; water; at 20℃; for 2h;
Step A: Sodium hydroxide (20 ml, 1M) and dimethyl sulfate (15.0 ml, 158 mmol) were added dropwise to a solution of 2,7-dihydroxynaphthalene (6.6 g, 40 mmol) in dichloromethane (100 ml) and water (60 ml). Additional sodium hydroxide (20 ml, 1M) and dimethyl sulfate (15.0 ml, 158 mmol) were added and the resulting mixture was stirred at room temperature for 2 hours. The two phases were separated and the aqueous layer was extracted with dichloromethane twice. The combined organic extracts were washed with 1 M HCl, dried over magnesium sulfate and concentrated under vacuum. The crude material was purified by column chromatography (5:1 heptane/ethyl acetate) to give 2-hydroxy-7-methoxynaphthalene (2.1 g, 30%, 100% AUC HPLC): 1H NMR (300 MHz, CDCl3) delta 7.66-7.70 (m, 2H), 6.94-7.08 (m, 4H), 4.90 (s, 1H), 3.92 (s, 3H).
With pyridine; at 0 - 20℃; for 3h;Inert atmosphere;
First, under a nitrogen atmosphere, 48.1g 2,7-naphthalendiol and 380ml of pyridine was added to the flask and cooled to 0 up by slowly dropwise addition of triflic anhydride 203.1g.Thereafter, to the reaction at 0 was stirred for 1 hour, then at room temperature the reaction was stirred for 2 hours.Then, the reaction mixture was added to water, extracted with toluene and the target component, then the use of the crude product on silica gel and the organic layer was concentrated under reduced pressure obtained was purified by column (columnpurification) (solvent: heptane / toluene = 6/1 (volume ratio )) to obtain a bis (trifluoromethanesulfonate) naphthalene-2,7-diyl ester 112.4 g (yield as a first intermediate compound: 88%).Which processes are shown in "Reaction 1."
88%
With pyridine; at 0 - 20℃; for 3h;Inert atmosphere;
First, 48.7 g of naphthalene-2,7-diol and 380 ml of pyridine were added to the flask under a nitrogen atmosphere. After cooling to 0 C, trifluoromethanesulfonic anhydride 203.1g was slowly added dropwise. Thereafter, the reaction solution was stirred at 0 C for 1 hour, and then the reaction solution was stirred at room temperature for 2 hours. Then, water was added to the reaction solution, the target component was extracted with toluene, and the crude product obtained by concentrating the organic layer under reduced pressure was purified with silica gel (solvent: heptane / toluene = 6/1 (volume ratio)),112.4g (Yield: 88%) of bis (trifluoromethanesulfonic acid) naphthalene-2,7-diyl ester as the first intermediate compound. The flow is shown in Reaction 1 below.
81%
With pyridine; In dichloromethane; at 0 - 20℃; for 12h;Inert atmosphere;
To an oven-dried round bottom flask equipped with a stirring bar a solution of 2,7-dihydroxynaphthalene (S4-1) (1.000 g, 6.24 mmol) in dichloromethane (30 ml) were added pyridine (2.1 ml, 26.06 mmol) and triflic anhydirde (2.3 ml, 13.70 mmol) at 0 oC under argon atmosphere. The mixture was stirred at room temperature for 12hours. To the reaction mixture was added 1M hydrochloric acid solution in water (25 ml) and extracted with diethyl ether (100 ml). The organic layer was washed with water (100 ml), dried over magnesium sulfate, filtered and concentrated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate : hexane = 1 : 10) as a white solid (S4) (2.140 g, 81 %). 1H NMR (400 MHz, CDCl3) : δ = 7.40 (dd, J = 8.8, 2.4 Hz, 2H), 7.73 (d, J = 2.4 Hz, 2 H), 7.92 (d, J = 9.2 Hz, 2H). ; 13C NMR (100MHz, CDCl3) : δ = 118.8, 119.5, 130.8, 131.3, 133.6, 148.3. ; HRMS m/z calc. C12H6F6O2S2Na [M+Na]+ 446.9408, found [M+Na]+ 446.9424; anal.CHN calcd. C12H6F6O2S2 C 34.0%, H 1.4%; found C 34.2%, H 1.2%.
79.5%
With pyridine; In dichloromethane; at 0 - 20℃; for 10h;Inert atmosphere;
1.6 g of 2,7-dihydroxynaphthalene was added to a dry round-bottomed double-necked flask containing 200 mL of dichloromethane as a reaction solvent, 20 mL of pyridine was added at 0C under nitrogen protection, and after stirring 8.6 mL of trifluoromethanesulfonic anhydride was added dropwise, stirred for 2 h, reacted at room temperature for 8 h, and the reaction progress was monitored by TLC dot plate. The aqueous phase was extracted with CH2Cl2. The organic phase was concentrated in vacuo. Purification by column chromatography gave the compound as a white solid in 79.5% yield
62%
With 2,6-dimethylpyridine; dmap; In tetrahydrofuran; dichloromethane; at -78 - 0℃; for 7.25h;Molecular sieve; Inert atmosphere;
A mixture of 2,7-dihydroxynaphthalene (3.00 g, 18.73 mmol), DMAP (0.42 g, 3.75 mmol) and 2,6-lutidine (4.41 g, 41.18 mmol) was suspended in anhydrous DCM (30 ml) and anhydrous THF (30 ml) in the presence of molecular sieves under an Ar2 atmosphere at -78 C. Tf2O (11.10 g, 39.33 mmol) was added dropwise to this stirred mixture over 15 minutes and the reaction was stirred at -78 C for 2 hours, then for 5 hours at 0 C. After that time, the reaction was carefully neutralised with saturated aqueous NaHCO3 solution (70 ml), extracted with DMC (6 x 120 ml), dried (MgSO4), filtered and the solvent removed in vacuo. The orange oil thus obtained was purified by flash column chromatography (petroleum ether:EtOAc , 95:5) to give compound 1 as a white solid (4.94 g, 62%); Rf 0.27 [5% EtOAc in petroleum ether]; mp 60-62 C; δH (CDCl3, 400 MHz) 7.92 (2H, d, J= 9.2 Hz, 2 x ArH), 7.73 (2H, d, J= 2.4 Hz, 2 x ArH), 7.40 (2H, dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 2 x ArH); δC (CDCl3, 100 MHz) 148.26 (2 x Ar-C), 133.62 (Ar-C), 131.27 (Ar-C), 130.80 (2 x Ar-CH), 121.13 (2 x Ar-CH), 119.48 (2 x Ar-CH), 118.77 (2 x CF3); HRMS m/z calc. C12H6F6O2S2Na [M+Na]+ 446.9408, found [M+Na]+ 446.9424; anal.CHN calcd. C12H6F6O2S2 C 34.0%, H 1.4%; found C 34.2%, H 1.2%. Spectroscopic data are in accordance with those reported in literature(Yao et al., 1998)
62%
With 2,6-dimethylpyridine; dmap; In tetrahydrofuran; dichloromethane; at -78 - 0℃; for 7.25h;Molecular sieve; Inert atmosphere;
Example 1 a: Synthesis Of Alkyne Building BlockProcedure for the synthesis of naphthalene-2,7-diyl bis(trifluoromethanesulfonate)DMAP, 2,6 lutidine, Tf02 2H6F606S2Molecular Weight: 424.2929 A mixture of 2,7-dihydroxynaphthalene (3.00 g, 18.73 mmol), DMAP (0.42 g, 3.75 mmol) and 2,6-lutidine (4.41 g, 41 .18 mmol) was suspended in anhydrous DCM (30 ml) and anhydrous THF (30 ml) in the presence of molecular sieves under an Ar2 atmosphere at -78C. Tf20 (1 1 .10 g, 39.33 mmol) was added dropwise to this stirred mixture over 15 minutes and the reaction was stirred at -78C for 2 hours, then for 5 hours at 0C. After that time, the reaction was carefully neutralised with saturated aqueous NaHC03 solution (70 ml), extracted with DMC (6 x 120 ml), dried (MgS04), filtered and the solvent removed in vacuo. The orange oil thus obtained was purified by flash column chromatography (petroleum ethenEtOAc , 95:5) to give compound 1 as a white solid (4.94 g, 62%); Rf 0.27 [5% EtOAc in petroleum ether]; mp 60-62 C; δΗ (CDCI3, 400 MHz) 7.92 (2H, d, J= 9.2 Hz, 2 x ArH), 7.73 (2H, d, J= 2.4 Hz, 2 x ArH), 7.40 (2H, dd, J1 = 8.8 Hz, J2 = 2.4 Hz, 2 x ArH); 5C (CDCI3, 100 MHz) 148.26 (2 x Ar-C), 133.62 (Ar-C), 131 .27 (Ar-C), 130.80 (2 x Ar-CH), 121 .13 (2 x Ar-CH), 1 19.48 (2 x Ar-CH), 1 18.77 (2 x CF3); HRMS m/z calc. C^HeFeC^Na [M+Na]+ 446.9408, found [M+Na]+ 446.9424; anal.CHN calcd. C12H6F6O2S2 C 34.0%, H 1 .4%; found C 34.2%, H 1 .2%. Spectroscopic data are in accordance with those reported in literature (Yao et al. , 1998).
With pyridine; In dichloromethane; at 0 - 20℃; for 2.5h;Inert atmosphere;
EXAMPLE 12 Preparation of 2-[(7-ethynyl-2-naphthalenyl)oxy]-N-(2-methoxy-l,l-dimethyletliyl)- butanamide (Compound 97)Step A: Preparation of l,r-(2,7-naphthalenediyl) bis(l,l,l-trifluoromethanesulfonate) To 200 mL of dichloromethane was added 2,7-dihydroxynaphthalene (5.4 g, 34 mmol) and pyridine (9.06 mL, 112 mmole) at room temperature. The mixture was cooled to 0 C under a nitrogen atomosphere with stirring using an ice/acetone bath. To this mixture at 0 C was then added trifluoromethanesulfonic anhydride (13.24 mL, 78.6 mmole) portionwise. After the addition, the mixture was stirred at ambient temperature for 2.5 hours. Dichloromethane (200 mL) and water (400 mL) were then added. The mixture was acicified with IN hydrochloric acid aqueous solution to pH 3 with stirring. The organic phase was separated, dried over MgS04 and concentrated under reduced pressure. The residue was purified by column chromatography (over silica gel with ethyl acetate and hexanes in volume ratios from 5 to 40 % as eluents) to give 14 g of the crude product. The crude product was then trituated with hexane (350 mL) and the solid was collected by filtration to give the title compound (3.5 g) as a solid. The solid which precipitated from the mother liquor was also collected by filtration to give another 424 mg of the title compound. The filtrate was the concentrated to about 100 mL in volume and the precipitate was collected by filtration to give another 8.1 g of the title compound..H NMR (CDCI3) δ 7.49 (d, 2H), 7.81 (s, 2H), 8.01 (d, 2H).
With pyridine; In dichloromethane; at 0 - 20℃;Cooling with acetone-dry ice; Inert atmosphere;
Step A: Preparation of 1 , 1 '-(2,7-naphthalenediyl) bis(l , 1 , 1 -trifluoromethanesulfonate) To 200 mL of dichloromethane was added 2,7-dihydroxynaphthalene (5.4 g, 34 mmol) and pyridine (9.06 mL, 112 mmole) at room temperature. The mixture was cooled to 0 C under a nitrogen atomosphere with stirring using an ice/acetone bath. To this mixture at 0 C was then added trifluoromethanesulfonic anhydride (13.24 mL, 78.6 mmole) portionwise. After the addition, the mixture was stirred at ambient temperature for 2.5 hours. Dichloromethane (200 mL) and water (400 mL) were then added. The mixture was acicified with IN hydrochloric acid aqueous solution to pH 3 with stirring. The organic phase was separated, dried over MgSC^ and concentrated under reduced pressure. The residue was purified by column chromatography (over silica gel with ethyl acetate and hexanes in volume ratios from 5 to 40 % as eluents) to give 14 g of the crude product. The crude product was then trituated with hexane (350 mL) and the solid was collected by filtration to give the title compound (3.5 g) as a solid. The solid which precipitated from the mother liquor was also collected by filtration to give another 424 mg of the title compound. The filtrate was the concentrated to about 100 mL in volume and the precipitate was collected by filtration to give another 8.1 g of the title compound..H MR (CDCI3) δ 7.49 (d, 2H), 7.81 (s, 2H), 8.01 (d, 2H).
112.4g
With pyridine; at 0 - 20℃; for 3h;Inert atmosphere;
First, under the nitrogen atmosphere, 2,7-naphthalene diol (48.1 g) and pyridine (380 ml) were added to a flask, cooled to 0C, and then slowly added dropwise with trifluoromethane sulfonic acid anhydride (203.1 g). After that, the reaction solution was stirred at 0C for 1 hour and at room temperature for 2 hours. Then, the reaction solution was added with water and the target component was extracted with toluene. The crude product obtained by concentrating the organic layer under reduced pressure was purified by silica gel column chromatography (solvent: heptane/toluene = 6/1 (volume ratio)) to give naphthalene-2,7-diyl bis(trifluoromethane sulfonate) (112.4 g) as a first intermediate compound. The scheme is shown in the following 'Reaction 1'.
With triethylamine; In dichloromethane; at 0 - 20℃;
To a 100 mL round bottom flask was added 2,7-dihydroxynaphthalene (0.801 g, 5.0 mmol), dichloromethane(50 mL), and triethylamine (2.09 mL, 1.518 g, 15 mmol). The flask was cooled to and stirred at 0C. To thisstirring flask, trifluoromethane sulfonic anhydride (1.68 mL, 2.821 g, 10 mmol) was added dropwise. Thesolution was allowed to slowly warm to room temperature overnight. Phase separation was accomplishedwith dichloromethane and 5% NH4OH (aq). The organic layer was separated and dried over MgSO4. Afterfiltration to remove the drying agent, solvents were removed via rotary evaporation to give 2.073 g of ayellow-brown powder.3 This crude naphthalene-2,7-diyl bis(trifluoromethanesulfonate) product was useddirectly in the next step without further purification.
Stage #1: 2,7-Dihydroxynaphthalene With sodium hydroxide In water at 60℃; for 0.5h;
Stage #2: With sulfuric acid; sodium nitrite In water at 0℃; for 3h;
1.1
2.5 g (62.4 mmol) of NaOH, 100 ml of water was added to a four-necked flask, stirred at 60 ° C, and then added10 g (62.4 mmol) of 2,7-dihydroxynaphthalene was stirred at 60 ° C for 30 min. After dissolving, 5.2 g (74.80 mmol) of NaNO2 was added, Mix 20min, ice bath cooling to 0 , at this temperature slowly dropping 8mL concentrated sulfuric acid and 15mL water mixture, dripping finished After the reaction 3h. After completion of the reaction, suction was carried out to obtain 11.33 g of a reddish brown solid, the yield was 96.2%.
90%
With sulfuric acid; sodium hydroxide; sodium nitrite at 0℃; for 1h;
50%
With 4-nitro-4-methyl-2,3,5,6-tetrabromo-2,5-cyclohexadien-1-one In diethyl ether for 2h; Ambient temperature;
With potassium permanganate In methanol; water at 20℃; for 12h;
2.3. The synthesis of DPD
The compound DPD has been synthesized based on previous literature[47,48]. In brief, 2,7-dihydroxynaphthalene (2.4 g, 15 mmol),methanol (60 mL), water (500 mL), and 0.05 M potassium permanganate(6 mL, 0.3 mmol) were mixed in 1 L flask. After stirring at roomtemperature for 12 h, the solution was adjusted to weak acidic through2 M HCl. The precipitation was washed with methylene chloride andethyl acetate to give a dark green solid (1.2 g, yield 50%). 1H NMR(500 MHz, DMSO-d6) δ 7.90 (br, 4H), 6.88 (br, 4H).
16%
With sodium hydroxide In methanol; water for 9h;
With potassium permanganate In methanol; water at 20℃; for 12h;
With 1H-imidazole In N,N-dimethyl-formamide at 0℃; for 2h;
36%
With 1H-imidazole In N,N-dimethyl-formamide at 0 - 20℃; for 2.5h;
33%
Stage #1: 2,7-Dihydroxynaphthalene; tert-butyldimethylsilyl chloride With 1H-imidazole; tetrakis(triphenylphosphine) palladium(0); potassium carbonate In ethanol; water; N,N-dimethyl-formamide; toluene at 90℃; for 4h; Schlenk technique; Inert atmosphere;
Stage #2: With boron tribromide In dichloromethane at -78 - 20℃;
General procedure (Scheme 5):
General procedure: To a Schlenk flask were added 2-bromo-3-methoxynaphthalene(1.0 equiv), aryl boronic acid (2.2 equiv), K2CO3 (3.0 equiv), Pd(PPh3)4 (2.5 mol%), and degassedEtOH/toluene/water (1/1/1) under Ar atmosphere. The mixture was heated at 90 °C until thecompletion of the reaction. Then the mixture was cooled to room temperature, and DCM was added.The mixture was washed with NaOH solution (20% wt), and the aqueous phase was extracted withDCM (2 × 20 mL). The combined organic phase was washed with brine (20 mL) and dried overanhydrous MgSO4. After removing the solvent, the residue was dissolved in anhydrous DCM. Thesolution was cooled to -78 °C, and BBr3 (1 M in DCM, 5.0 equiv) was added slowly by syringe. Thenthe mixture was warmed up to room temperature and stirred until the complete consumption of thestarting material. The mixture was poured into the ice water (50 mL) and extracted with DCM (3 × 50mL). The combined organic phase was washed with brine (100 mL) and dried over anhydrous Na2SO4. After removing the solvent, the residue was purified by silica gel chromatography to givethe desired product.
With 1H-imidazole In N,N-dimethyl-formamide
With 1H-imidazole In N,N-dimethyl-formamide for 2.5h; Cooling with ice; Inert atmosphere;
112.112-1
Step 112-1: TBSCl (21.2 g) was added in two portions to a solution of naphthalene-2,7-diol (25.0 g) and imidazole (10.6 g) in DMF (150 mL) under nitrogen atmosphere with ice-cooling. After stirring at the same temperature for 2.5 hours, the reaction solution was added to H2O, followed by extraction with Et2O twice. The combined organic layers were washed with brine, dried over MgSO4 and then concentrated under reduced pressure. The residue was purified by column chromatography (Chromatorex NH, mobile phase: EtOAc/hexane = 10/90 to 50/50; v/v) to obtain 7-[tert-butyl(dimethyl)silyl]oxy}-2-naphthol (23.2 g, colorless oil). 1H NMR (200 MHz, CDCl3, δ): 0.24 (s, 6H), 1.01 (s, 9H), 5.23 (s, 1H), 6.87-6.97 (m, 2H), 6.98-7.01 (m, 1H), 7.01-7.04 (m, 1H), 7.62 (d, J = 4.8 Hz, 1H), 7.66 (d, J = 4.8 Hz, 1H); ESI MS m/z 273, (M-H)-.
With triethylamine In ethyl acetate at 0 - 20℃; for 0.166667h; Green chemistry;
3. General Procedure for the Synthesis of Aryl Mesylates (Table 4)
General procedure: To a solution of hydroxyarene (25.0 mmol) in ethyl acetate (75 mL) at 0 °C was added triethylamine (5.06 g, 50.0 mmol) followed by MsCl (3.72 g, 32.5 mmol). After addition of MsCl, the ice-water bath was removed and the resulting thick slurry was vigorously stirred for 10 min. To the slurry was then added water (50 mL). The two-phase mixture was separated. The organic layer was washed with water (25 mL) and dried over anhydrous MgSO4. Removal of solvent under reduced pressure gave the corresponding pure aryl mesylate.
Stage #1: 2,7-Dihydroxynaphthalene With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 0.5h;
Stage #2: allyl bromide In N,N-dimethyl-formamide for 24h;
44%
Stage #1: 2,7-Dihydroxynaphthalene With potassium carbonate In acetone at 20℃; for 1h;
Stage #2: allyl bromide In acetone at 20℃; for 8h;
34%
Stage #1: 2,7-Dihydroxynaphthalene With potassium carbonate In acetone at 25℃; for 0.166667h;
Stage #2: allyl bromide In acetone
6 2.1.6 Synthesis of 7-(allyloxy)naphthalen-2-ol
2,7-Dihydroxynaphthalene (800 mg, 5.0 mmol) was dissolved in acetone (15 mL) followed by the addition of K2CO3 (759 mg, 5.5 mmol) at 25 °C. After stirring for 10 min, allyl bromide (475 μL, 5.5 mmol) was added dropwise to the solution mixture. The mixture was allowed to stir for an overnight. TLC indicated the disappearance of the starting material. The mixture was then filtered through a small pad of silica gel, which was further washed with dichloromethane. After that, the collected filtrate was concentrated and purified by column chromatography to give the product as colorless oil (346 mg, 1.7 mmol). Yield: 34%; Rf = 0.5 (EtOAc/n-hexanes, 1:6); 1H NMR (CDCl3, 400 MHz) δ 4.63 (d, J = 5.3 Hz, 2H), 5.13 (brs, 1H), 5.32 (dd, J = 0.9, 10.5 Hz, 1H), 5.47 (dd, J = 1.3, 17.3 Hz, 1H), 6.07-6.17 (m, 1H), 6.94 (dd, J = 2.4, 8.8 Hz, 1H), 6.98-7.04 (m, 3H), 7.66 (d, J = 8.8 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 68.9, 106.0, 108.9, 115.4, 116.7, 118.0, 124.5, 129.4, 129.7, 133.3, 136.0, 154.1, 157.3; HRMS (ESI) m/z Calc. for C13H12O2 [M+H]+ 201.0910. Found: 201.0913.
With sodium hydroxide; potassium carbonate In acetone
26 Preparation of 7-(2-propenyloxy)-2-naphthalenol
EXAMPLE 26 Preparation of 7-(2-propenyloxy)-2-naphthalenol A mixture of 36 g of 2,7-dihydroxynaphthalene and 40 g of anhydrous potassium carbonate in 250 ml of anhydrous acetone was stirred vigorously at room temperature for 2.5 hours. Allyl bromide (21.9 ml) was added and the mixture was stirred at 23° for 19 hours. The reaction mixture was filtered and the solid was washed well with acetone. The filtrate was concentrated in vacuo and the residue was acidified and extracted with methylene chloride. The extract was washed with three 200 ml of portions of 1N sodium hydroxide. The combined aqueous layer was acidified and extracted with methylene chloride. The dried (magnesium sulfate) extract was concentrated in vacuo and the residue was purified by chromatography on 250 g of silica gel. Elution with 10% ethyl acetate-toluene gave 17.7 g of somewhat impure product. Crystallization from etherhexane gave 7.9 g, mp 78°-80°, of 7-(2-propenyloxy)-2-naphthalenol.
[0709] To a mixture of naphthalene-2,7-diol (25 g, 156.08 mmol) and K2CO3 (32.3 g, 232.02 mmol) in acetone (300 ml) was added iodomethane (22.2 g, 156.41 mmol) dropwise with stirring at 0 C. The resulting solution was stirred overnight at room temperature. The solids were filtered off and the filtrate was concentrated under vacuum to give a residue, which was purified by silica gel column chromatography using 1%-10% ethyl acetate in petroleum ether to afford 7-methoxynaphthalen-2-ol as a light yellow solid (10 g, 37%). (ES, m/z): [M+H]+ 175.1; 1H NMR (400 MHz, DMSO-d6): delta 9.65 (s, 1H), 7.65 (d, J=8.8 Hz, 2H), 6.79 (dd, J=13.6, 1.6 Hz, 2H), 6.92-6.89 (m, 2H), 3.84 (s, 3H).
30%
General procedure: To a Schlenk flask were added 2-bromo-3-methoxynaphthalene(1.0 equiv), aryl boronic acid (2.2 equiv), K2CO3 (3.0 equiv), Pd(PPh3)4 (2.5 mol%), and degassedEtOH/toluene/water (1/1/1) under Ar atmosphere. The mixture was heated at 90 C until thecompletion of the reaction. Then the mixture was cooled to room temperature, and DCM was added.The mixture was washed with NaOH solution (20% wt), and the aqueous phase was extracted withDCM (2 × 20 mL). The combined organic phase was washed with brine (20 mL) and dried overanhydrous MgSO4. After removing the solvent, the residue was dissolved in anhydrous DCM. Thesolution was cooled to -78 C, and BBr3 (1 M in DCM, 5.0 equiv) was added slowly by syringe. Thenthe mixture was warmed up to room temperature and stirred until the complete consumption of thestarting material. The mixture was poured into the ice water (50 mL) and extracted with DCM (3 × 50mL). The combined organic phase was washed with brine (100 mL) and dried over anhydrous Na2SO4. After removing the solvent, the residue was purified by silica gel chromatography to givethe desired product.
Stage #1: 2,7-Dihydroxynaphthalene; benzyl bromide With tetrakis(triphenylphosphine) palladium(0); potassium carbonate In ethanol; water; toluene at 90℃; for 4h; Schlenk technique; Inert atmosphere;
Stage #2: With boron tribromide In dichloromethane at -78 - 20℃;
General procedure (Scheme 5):
General procedure: To a Schlenk flask were added 2-bromo-3-methoxynaphthalene(1.0 equiv), aryl boronic acid (2.2 equiv), K2CO3 (3.0 equiv), Pd(PPh3)4 (2.5 mol%), and degassedEtOH/toluene/water (1/1/1) under Ar atmosphere. The mixture was heated at 90 °C until thecompletion of the reaction. Then the mixture was cooled to room temperature, and DCM was added.The mixture was washed with NaOH solution (20% wt), and the aqueous phase was extracted withDCM (2 × 20 mL). The combined organic phase was washed with brine (20 mL) and dried overanhydrous MgSO4. After removing the solvent, the residue was dissolved in anhydrous DCM. Thesolution was cooled to -78 °C, and BBr3 (1 M in DCM, 5.0 equiv) was added slowly by syringe. Thenthe mixture was warmed up to room temperature and stirred until the complete consumption of thestarting material. The mixture was poured into the ice water (50 mL) and extracted with DCM (3 × 50mL). The combined organic phase was washed with brine (100 mL) and dried over anhydrous Na2SO4. After removing the solvent, the residue was purified by silica gel chromatography to givethe desired product.
With potassium carbonate In N,N-dimethyl-formamide at 35℃;
With potassium carbonate In N,N-dimethyl-formamide
With potassium carbonate In acetone at 20℃; for 22h; Heating / reflux;
4.1
EXAMPLE 4; This Example illustrates the preparation of 2-( 2-benzyloxynaphthyl-7-oxy)-2- methylthio-N-(2-methylpent-3-yn-2-yl) acetamide (Compound No. 4 of Table 24); Stage: 1 Preparation of 2-benzyloxynaphth-7-ol; A stirred solution of 2,7-dihydroxynaphthylene (4.8g) in acetone (50ml) containing anhydrous potassium carbonate (4.08g) and benzyl bromide (5.13gO was heated to refux for 4 hours , cooled to ambient temperature then stored for 18 hours. The mixture was filtered and the filtrate evaporated under reduced pressure to give a pale green solid that was fractionated by chromatography (silica; hexane/ ethyl acetate 95:5 to1:1 by volume) to give 2-benzyloxynaphth-7-ol, 1.63g, as a cream coloured solid, m.p.152-154°C.
With 5,10,15,20,22,24-hexahydro-5,5,10,15,15,20-hexamethyl-10α,20α-bis(4-nitrophenyl)-calix[4]pyrrole; caesium carbonate In acetonitrile at 40℃; regioselective reaction;
8,16-methano-16H-dinaphtho[2,1-d:1',2'-g][1,3]dioxocine-2,14-diol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
83%
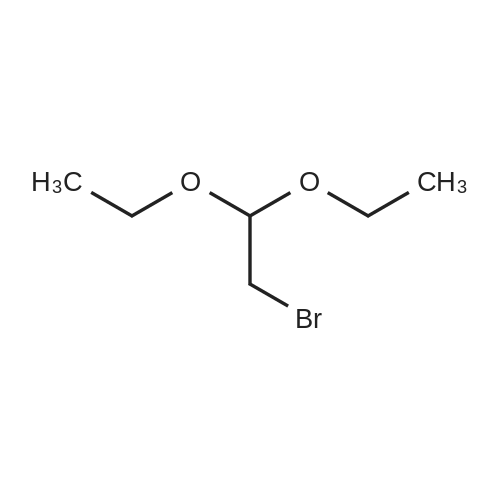
With polystyrene-bound p-toluenesulfonic acid In neat (no solvent) at 80℃; for 4h; Green chemistry; regioselective reaction;
General experimental procedure for the synthesis of 7
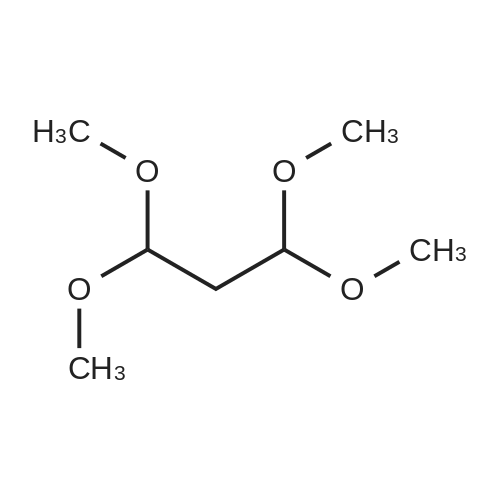
To a 25 mL round-bottomed flask was added 2,7-dihydroxynaphthalene 6 (1 mmol, 0.16 g), 1,1,3,3-tetramethoxypropane 2 (1equiv, 0.166 mL), and polystyrene-bound p-TSA (25 mg) (25 mg) and then capped. The reaction mixture was stirred at 80 °C under solvent-free conditions for 4 h. Finally, the completion of the reaction was confirmed by TLC and the reaction mixture was cooled to room temperature. Then, the reaction mixture was washed twice with ethyl acetate (2×5 mL) and filtered to recover the catalyst. The combined filtrate was evaporated under reduced pressure to obtain the crude product, which was further purified by recrystallization from a mixed solvent (EtOH/water, 5/1,v/v) to give the pure desired product.
With bromine; triphenylphosphine; In acetonitrile; at 0 - 250℃; for 1.5h;
A synthetic procedure was based on the literature method [12]. To a vigorously stirred mixture of triphenylphosphine (31.5g, 120mmol) in acetonitrile (50.0mL), bromine (19.2g, 120mmol) was added dropwise at 0C. The reaction mixture was allowed to reach room temperature, and 2,7-dihydroxynaphthalene 1 (16.0g, 100mmol) was added in one portion. The mixture was heated to 70C for 30min, after which the solvent was removed by rotary evaporation. The flask was equipped with a gas trap, and the black residue was heated to 250C for 1h. After cooling to room temperature, the mixture was dissolved in 200mL of dichloromethane and the viscous liquid was obtained after column chromatography (silica gel, petroleum ether/dichloromethane 1:1). The crude product was purified by column chromatography (silica gel, petroleum ether/dichloromethane 3:2) to give the compound 2 (18.8g, 84.3mmol, 84%) as a beige powder. 1H NMR (400MHz, CDCl3) delta (ppm): 5.09 (s, 1H, -OH), 7.06 (d, 1H, J 2.4Hz, naphthalene-H), 7.10 (dd, 1H, J 8.8, 2.8Hz, naphthalene-H), 7.24 (dd, 1H, J 8.8, 2.0Hz, naphthalene-H), 7.63 (d, 1H, J 8.8Hz, naphthalene-H), 7.72 (d, 1H, J 8.8Hz, naphthalene-H), 7.84 (d, 1H, J 1.2Hz, naphthalene-H). 13C NMR (100MHz, CDCl3) delta (ppm): 108.7, 118.1, 120.8, 127.0, 127.3, 128.3, 129.4, 129.9, 135.7, 154.0. HR-ESI-MS m/z: [M-H]- calcd. for C10H6BrO, 220.9602; found, 220.9601.
66%
With bromine; triphenylphosphine; In acetonitrile; at 70℃; for 1h;Schlenk technique;
To a stirring suspension of Triphenyl phosphine (31.5 g) in Acetonitrile (50 mL) in a 250 mL10 Schlenk-flaskwas added carefully Bromine (6.2 mL) at 0 00 with a syringe over 30 mm. The yellow solution was warmed to room temperature and 2,7-Dihydroxynaphthalene (16 g) was added in one portion. The reaction was refluxed at 70 00 for one hour. After cooling to room temperature, the solvent was removed under reduced pressure. The reaction flask was connected to a gas-washing bottle filled with a concentrated sodium hydroxide solution. The flask was heatedto 250 00 for two hours and the black residue dissolved in 100 mL Dichloromethane and purified via column chromatography (DCM:Pentan 1:1 to pure DCM). Product 101 was received as a beige powder (14.7 g, 66%)DC: Dichloromethane:Pentane, 1:1, R=0,21H-NMR: (300 MHz, ODd3) = 7.76 (d, 1 H), 7.63 (d, 1 H), 7.54 (d, 1 H), 7.32 (dd, 1 H), 7.03 (dd, 1H), 6.98 (d, 1H), 4.98 (b, 1H)
43%
Preparation 2 [0100] Preparation of 2-Bromo-7-methoxy-naphthalene. [CHEMMOL-00021] [0101] Preparation of 7-Bromo-2-naphthalenol. [0102] Triphenyl phosphine (89.7 g, 0.342 mol) and acetonitrile (350 mL) were combined in a 1-L flask under N2 atmosphere. The mixture was cooled to 10 C. Bromine (17.6 mL, 0.342 mol) was added dropwise over 10 minutes. The cooling bath was removed and 2,7-dihydroxynaphthalene (50.0 g, 0.312 mol) was added along with 350 mL of CH3CN rinse. The resulting yellow tan mixture was heated at reflux for 3 hours. Acetonitrile was distilled off under a water aspirator pressure over 2 hours, resulting in a grayish white solid. The solid was heated to 280 C. over 30 minutes giving a black liquid. The liquid was heated to 310 C. over 20 minutes and the temperature was maintained at 310 C. for an additional 15 minutes until gas evolution ceased. The black mixture was cooled to room temperature. Chromatography yielded 34.5 g of the intermediate title compound as an off-white solid which was 87% pure by HPLC (43% yield).
43%
Triphenyl phosphine (89.7 g, 0.342 mol) and acetonitrile (350 mL) were combined in a 1-L flask under N2 atmosphere. The mixture was cooled to 10 C. Bromine (17.6 mL, 0.342 mol) was added dropwise over 10 minutes. The cooling bath was removed and 2,7-dihydroxynaphthalene (50.0 g, 0.312 mol) was added along with 350 mL of CH3CN rinse. The resulting yellow tan mixture was heated at reflux for 3 hours. Acetonitrile was distilled off using a water aspirator over 2 hours resulting in a grayish white solid. The solid was heated to 280 C. over 30 minutes giving a black liquid. The liquid was heated to 310 C. over 20 minutes and the temperature was maintained at 310 C. for an additional 15 minutes until gas evolution ceased. The black mixture was cooled to room temperature. Chromatography yielded 34.5 g of the intermediate title compound as an off-white solid which was 87% pure by HPLC (43% yield).
43%
Triphenyl phosphine (89.7 g, 0.342 mol) and acetonitrile (350 mL) were combined in a 1-L flask under N2 atmosphere. The mixture was cooled to 10 C. Bromine (17.6 mL, 0.342 mol) was added dropwise over 10 minutes. The cooling bath was removed and 2,7-dihydroxynaphthalene (50.0 g, 0.312 mol) was added along with 350 mL of CH3CN rinse. The resulting yellow tan mixture was heated at reflux for 3 hours. Acetonitrile was distilled off using a water aspirator over 2 hours resulting in a grayish white solid. The solid was heated to 280 C. over 30 minutes giving a black liquid. The liquid was heated to 310 C. over 20 minutes and the temperature was maintained at 310 C. for an additional 15 minutes until gas evolution ceased. The black mixture was cooled to room temperature. Chromatography yielded 34.5 g of the intermediate title compound as an off-white solid which was 87% pure by HPLC (43% yield).
With sulfur(IV) oxide In water monomer for 0.5h; Irradiation;
66.7%
With sodium hydrogen sulphite In methanol at 60℃; for 2.5h; Autoclave; Inert atmosphere;
60%
With disodium metabisulfite In water monomer at 150℃; for 8h; Sealed tube;
1.1 EXAMPLE 1 Synthesis of Compounds of Chemical Formula 2 and Chemical Formula 1 (0055) (1) Synthesis of 7-(dimethylamino)naphthalen-2-ol (compound 5 of Scheme 1)
EXAMPLE 1 Synthesis of Compounds of Chemical Formula 2 and Chemical Formula 1 (0055) (1) Synthesis of 7-(dimethylamino)naphthalen-2-ol (compound 5 of Scheme 1) (0056) The present inventors performed a synthesis of 7-(dimethylamino)naphthalen-2-ol. (0057) 8 mL of water (H2O) and 10.5 mL of a dimethylamine solution (40% in H2O, 93.5 mmol) were added in a sealed-tube containing 3 g of a compound 4 (18.7 mmol, Sigma-aldrich, D116408) which was a synthesis starting material and 7.11 g of sodium metabisulphite (Na2S2O5, 37.4 mmol), and the sealed-tube was blocked. The mixture was stirred for 8 hours at 150° C. using a silicon oil container. Next, after a temperature of the reactant was cooled to room temperature (25° C.), the container was opened, 100 mL of dichloromethane, 100 mL of water, and 30 mL of saturated salt water were added, and then an organic layer was extracted by using a separating funnel. The organic layer was dried by 5 g of anhydrous sodium sulfate (Na2SO4), and concentrated by using an aspirator (at 25° C. and 20 to 500 mmHg). A light-brown solid compound obtained thus was separated (eluent: 20% EtOAc/Hexane) by using a column chromatography method (6 cm of diameter, 15 cm of height) using a silica gel (Merck-silicagel 60, 230-400 mesh) to obtain 2.10 g of a white solid compound 5 (60%). A product was analyzed by using a thin layer chromatography (TLC) (silical gel 60F-254 glass plate, Merck) to have an elution value of Rf=0.25 (20% EtOAc/Hexane-eluting once). 1H NMR (CDCl3, 300 MHz, 293K): δ 7.66-7.59(m, 2H), 7.05-7.02(m, 1H), 6.96-6.95(d, 1H), 6.85-6.82(m, 1H), 6.78-6.77(d, 1H), 5.10(s, 1H), 3.05(s, 6H). 13C NMR (CDCl3, 75 MHz, 293K): δ153.88, 149.17, 136.24, 129.39, 128.61, 122.44, 114.22, 113.80, 108.02, 105.32, 40.91. HRMS: m/z calcd. for C12H13NO 187.0997 found 187.0999.
60%
With disodium metabisulfite In water monomer at 150℃; for 8h; Sealed tube;
1.1 (1) Synthesis of 7-(dimethylamino)naphthalen-2-ol (compound 5 in Scheme 1)
(1) Synthesis of 7-(dimethylamino)naphthalen-2-ol (compound 5 in Scheme 1) The present inventors performed synthesis of 7-(dimethylamino)naphthalen-2-ol. Water (H2O, 8 mL) and an aqueous dimethylamine solution (40% in H2O, 10.5 mL, 93.5 mmol) were put into a sealed tube containing compound 4 (3 g, 18.7 mmol, Sigma-aldrich, D116408), which was a starting material for synthesis, and sodium metabisulfite (Na2S2O5, 7.11 g, 37.4 mmol), and the tube was sealed. The resulting mixture was stirred at 150° C. for 8 hours in a silicone oil tube. Thereafter, a reaction product was cooled to room temperature (25° C.), the tube was opened, and dichloromethane (100 mL), water (100 mL), and saturated brine (30 mL) were put into the tube to extract an organic layer through a separatory funnel. The organic layer was dried on anhydrous sodium sulfate (Na2SO4, 5 g), and concentrated using an aspirator (25° C., 20 to 500 mmHg). The light brown solid compound obtained thus was separated (developing solution: 20% EtOAc/hexane) through column chromatography (diameter: 6 cm, and height: 15 cm) using silica gel (Merck-silicagel 60, 230-400 mesh) to obtain a white solid compound 5 (2.10 g, 60%). The product had a developing value of Rf=0.25 (20% EtOAc/hexane-developing once), as analyzed through thin layer chromatography (TLC, silica gel 60E-254 glass plate, Merck). 1H NMR (CDCl3, 300 MHz, 293K): δ 7.66-7.59 (m, 2H), 7.05-7.02 (m, 1H), 6.96-6.95 (d, 1H), 6.85-6.82 (m, 1H), 6.78-6.77 (d, 1H), 5.10 (s, 1H), 3.05 (s, 6H). 13C NMR (CDCl3, 75 MHz, 293K): δ 53.88, 149.17, 136.24, 129.39, 128.61, 122.44, 114.22, 113.80, 108.02, 105.32, 40.91. HRMS: m/z calcd. for C12H13NO 187.0997 found 187.0999.
60%
With disodium metabisulfite In water monomer at 140℃; for 5h; Microwave irradiation;
60%
With disodium metabisulfite In water monomer at 150℃; for 3h; Inert atmosphere;
52%
With disodium metabisulfite In water monomer at 150℃; for 2h; Inert atmosphere;
With disodium metabisulfite In water monomer at 150℃; for 8h;
1.1.1-4.1.1 1.1 XD00
2,7-dihydroxynaphthol (0.3 g, 18.7 mmol), dimethylamine (40% in water, 1.05 mL, 9.35 mmol), sodium metabisulfite (0.711 g, 3.74 mmol),2 mL of deionized water was sequentially added to the Shrek bottle, reacted at 150 ° C for 8 h, and after cooling to room temperature, the pH of the reaction system was adjusted to about 4 with hydrochloric acid, and then dried by DCM extraction.After purification by column chromatography, the intermediate product white solid XD00 is obtained.
In acetonitrile at 20℃; for 0.333333h; Inert atmosphere;
In acetonitrile for 0.25h;
12,92-Dihydroxo-1,9(1,4),5(1,5)-trinaphthalina-2,4,6,8-tetraoxa-11,14-diaza-3,7-di(diethylamidate)thiophosphacyclopentadecaphanes (21, 22)
General procedure: b. To 1 mmol (0.247 g) of HETA in 1 mL of acetonitrile the solution of 0.5 mmol (0.08 g) of 2,7-dihydroxynaphthalene in 2 mL of acetonitrile was added at stirring. After 15 min to the reaction mixture the suspension of 0.5 mmol of compound 13 or 14 in 5 mL of acetonitrile was introduced and kept with stirring for 1 day. Then, to the solution 1 mmol (0.032 g) of elemental sulfur was added. On the next day the solution was filtered, the solvent removed in vacuum, the residue chromatographed on a column eluting the products by the system hexane-ethylacetate, 1 : 1. The obtained products 21 and 22 were dried in vacuum for 1 h (60°C, 1 Torr).
With 2-Picolinic acid; potassium phosphate; copper(l) iodide In dimethyl sulfoxide at 100℃; for 24h;
72%
With 2-Picolinic acid; potassium phosphate; copper(l) iodide In dimethyl sulfoxide at 90℃; for 24h; Inert atmosphere;
Synthesis of 2,2'-[naphthalene-2,7-diylbis(oxy)]dipyridine 2
A mixtureof 2,7-dihydroxynaphthalene (1.60 g, 10 mmol), 2-bromopyridine(3.16 g, 20 mmol), potassium phosphate (8.48 g, 40 mmol), copper(i)iodide (1.90 g, 10 mmol) and picolinic acid (0.25 g, 2 mmol) in dryDMSO (50 ml) was heated to 90 °C for 24 h in argon atmosphere. Thesolvent was removed under reduced pressure, ethyl acetate (50 ml) wasadded to the crude residue, and this was refluxed for 1 h. The solid wasfiltered off and the filter cake was washed with hot ethyl acetate (20 ml).The organic filtrate was concentrated under reduced pressure and the residue was purified by recrystallization from heptane to provide a whitesolid product (2.26 g, 7.2 mmol, 72% yield).
30%
With potassium carbonate In N,N-dimethyl-formamide for 72h; Heating;
EXAMPLE (II-1) STR36 12.5 g (0.05 mol) of 3,4,5-trichloro-benzotrifluoride are added slowly to a stirred mixture, heated to 60 C., of 8.0 g (0.05 mol) of 2,7-dihydroxy-naphthalene, 4.2 g (0.075 mol) of potassium hydroxide powder and 100 ml of dimethyl sulphoxide, and stirring of the reaction mixture is continued for approximately 3 hours at 60 C. After the mixture has cooled to approximately 20 C., it is diluted with water and methylene chloride and filtered. The organic phase is separated from the filtrate, washed with water, dried using sodium sulphate and filtered. The solvent is distilled off from the filtrate under a waterpump vacuum, the residue is stirred with petroleum ether, and the product which is obtained in this process in the form of crystals is isolated by filtering off with suction. 4.9 g (26% of theory) of 7-(2,6-dichloro-4-trifluoromethyl-phenoxy)-2-naphthol of melting point 98 C. are obtained.
STR73 12.5 g (0.05 mol) of 3,4,5-trichloro-benzotrifluoride are added slowly and with stirring to a mixture of 8.0 g (0.05 mol) of 2,7-dihydroxynaphthalene, 4.2 g (0.075 mol) of pulverulent potassium hydroxide and 100 ml of dimethyl sulphoxide, which has been warmed to 60 C., and the reaction mixture is then stirred for a further approx. 3 hours at 60 C. After the mixture has been cooled to approximately 20 C., it is diluted with water and methylene chloride, and filtered. The organic phase is separated off from the filtrate, washed with water, dried using sodium sulphate and filtered. The solvent is removed from the filtrate by distillation in a water-pump vacuum, the residue is stirred with petroleum ether and the product which is thus obtained in the form of crystals is isolated by filtering with suction. 4.9 g (26% of theory) of 7-(2,6-dichloro-4-trifluoromethylphenoxy)-2-naphthol of melting point 98 C. are obtained. In analogy to Example (II-1) the starting substances of the formula (II) listed below in Table 2 may be prepared. STR74
With dmap; trifluoromethanesulfonic acid anhydride; In tetrahydrofuran; dichloromethane;
Step 1 Trifluoro-methanesulfonic acid 7-(trifluoro-methanesulfonyloxy)-naphthalene-2-yl ester To a cold (-78 C.) mixture of naphthalene-2-7-diol (5.4 g, 33.8 mmol), 2,6-lutidine (8.0 g) and DMAP (824 mg) in THF (60 mL) and CH2Cl2 (60 mL) was added trifluoromethanesulfonic anhydride (20 g). The mixture was stirred at -78 C. for 1.5 h. and then warmed to 0 C. Aqueous NaHCO3 was added, and the mixture was extracted with CH2Cl2. The combined organic extracts were washed with brine, dried (anhyd. MgSO4) and concentrated in vacuo. The residue was purified by chromatography on silica gel and the product was isolated by recrystallization in EtOAc/Hexane to give 6 g (42%) of the title compound.
With sulfuric acid at 25 - 27℃; for 24h; Inert atmosphere; Cooling with ice;
1 Example 1: Synthesis of 8-hydroxy-4-methyl-naphthopyran-2-one (1a)
10.0 g (62.5 mmol) of 1,5-naphthalenediol were addedAnd 19.4 mL (77.5 mmol) of ethyl acetoacetate were placed in a 250 mL three-necked flask,Stir for half an hour under the protection of nitrogen (mechanical stirring).80 mL of 80% concentrated sulfuric acid was slowly added dropwise to the above reaction apparatus under an ice bath,Remove the ice bath after dropping,Stir at room temperature (25-27 ° C) for about 24 hours,The reaction was monitored by TLC (ethyl acetate: petroleum ether = 1: 1, v / v)After determining the raw material,Remove the reaction device,The reaction solution was poured into 500mL ice water,Stand overnight,A yellow solid precipitation.filter,The filter cake is then washed until neutral,drying.then,It was transferred to a flask,While stirring while slowly adding 10% sodium hydroxide solution,After the solid is dissolved,Filtered while still hotWashed,Collect the filtrate;After cooling to room temperature, concentrated hydrochloric acid was added dropwise to the filtrate until pH = 1-2.At this point there will be a lot of yellow solid filtrate appears,filter,Washed to neutral,Get crude after drying.The crude product is recrystallized from glacial acetic acid,Available pure (1a) 12.0g,Yield 85.0%.
75%
With poly(4-vinylpyridinium)perchlorate In neat (no solvent) at 20℃; for 0.416667h; Irradiation;
2.5 General procedure for the preparation of coumarins
General procedure: In a 25 mL batch reactor equipped with a distillation condenser the mixture of phenols (1 mmol), β-keto esters (2 mmol) and P(4-VPH)ClO4 (50 mg) was stirred and irradiated under ultrasonic irradiation (with a frequency of 35 kHz and a nominal power 200 W) at ambient temperature for the time mentioned in the Table 1. Ethyl acetoacetate was taken 2 equiv. for the proper solubility of phenol and ease of proper stirring of the reaction mixture. After completion of the reaction (monitored by TLC), ethanol was added to the reaction mixture and the catalyst was recovered by filtration. The filtrate was concentrated in vacuum, and the crude product was washed with water, dried and slowly re-crystallized in ethanol or ethanol-water system. The melting point, IR, 1H NMR and mass spectroscopic techniques were used to analyze the products and compared with the authentic samples.
With trifluorormethanesulfonic acid In dichloromethane at 20℃; for 432h; regioselective reaction;
95%
With trifluorormethanesulfonic acid In dichloromethane at 20℃; for 432h; regioselective reaction;
4.1.1. General procedure for the synthesis of 3-diethoxyphosphorylchromen-2-ones 12a-e
General procedure: To a solution of corresponding phenol 11a-c (11 mmol) or naphthol 11d,e (11 mmol) in CH2Cl2 (50 mL) trifluoromethanesulfonic acid (3.00 g, 20 mmol) or methanesulfonic acid (1.92 g, 20 mmol) and acrylate 10 (2.66 g, 10 mmol) were added and the resulting mixture was stirred at room temperature for the period of time given in Table 4. Next, saturated aqueous NaHCO3 solution was added (100 mL). Extraction with CH2Cl2 (3 × 30 mL), drying (MgSO4) and evaporation of the solvent gave a crude product, which was purified by crystallization from Et2O.
With ammonium chloride at 120℃; for 0.166667h; Neat (no solvent);
91%
With boron tri(hydrogen sulphate) at 120℃; for 0.333333h; Green chemistry;
General procedure for the preparation of tetrahydrobenzo[a]xanthen-11-one derivatives (6a-6h)
General procedure: A mixture of aldehyde (1 mmol), 2-naphthols (1 mmol), dimedone (1.1 mmol) and boron sulphonic acid (0.10 mmol) was stirred at 120 °C for the appropriate time indicated in Table 2. The progress of reactions was monitored by TLC (eluent:EtOAc/n-hexane, 1:5). After completion of the reaction, the reaction mixture was cooled to room temperature, the crude product was heated in ethanol (30 mL), and the catalyst was removed by filtration. The pure product was obtained by cooling of the filtrate.
90%
With guanidine hydrochloride at 80℃; for 0.333333h;
89%
With toluene-4-sulfonic acid; 1-butyl-3-methylimidazolium Tetrafluoroborate at 50℃; for 1.5h;
88%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.266667h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
85%
With N-methyl-2-oxopyrrolidin-1-ium dihydrogen phosphate at 80℃; for 0.333333h; Ionic liquid;
With guanidine hydrochloride at 80℃; for 0.333333h;
91%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.166667h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
90%
With ammonium chloride at 120℃; for 0.25h; Neat (no solvent);
89%
With boron tri(hydrogen sulphate) at 120℃; for 0.25h; Green chemistry;
General procedure for the preparation of tetrahydrobenzo[a]xanthen-11-one derivatives (6a-6h)
General procedure: A mixture of aldehyde (1 mmol), 2-naphthols (1 mmol), dimedone (1.1 mmol) and boron sulphonic acid (0.10 mmol) was stirred at 120 °C for the appropriate time indicated in Table 2. The progress of reactions was monitored by TLC (eluent:EtOAc/n-hexane, 1:5). After completion of the reaction, the reaction mixture was cooled to room temperature, the crude product was heated in ethanol (30 mL), and the catalyst was removed by filtration. The pure product was obtained by cooling of the filtrate.
With ammonium chloride at 120℃; for 0.166667h; Neat (no solvent);
90%
With zirconium oxide chloride octahydrate In water at 55℃; for 8h; Green chemistry;
89%
With guanidine hydrochloride at 80℃; for 0.25h;
87%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.233333h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
EXAMPLE 21 Preparation of N-( 1 -cyano- 1 -methylethyl)-2- [(7-ethoxy-2-naphthalenyl)oxy]butanamide (Compound 75) Step A: Preparation of methyl 2-[(7-hydroxy-2-naphthalenyl)oxy]butanoate To a solution of naphthalene-2,7-diol (5.0 g, 31 mmol) in acetone (200 mL) was added cesium carbonate (10.1 g, 31 mmol) at room temperature under a nitrogen atmosphere with stirring. After the addition, the mixture was stirred at room temperature for 3 min and then methyl-2-bromobutyrate (5.6 g, 31 mmol) was added. The mixture was stirred at room temperature overnight and then filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (over silica gel with ethyl acetate and hexanes in volume ratios from 10 to 80 % as eluents) to give the title compound (3.9 g) as a solid. in NMR (CDCI3) delta 7.62-7.70 (m, 2H), 6.85-7.05 (m, 4H), 5.85 (s, 1H), 4.68-4.78 (m, 1H),3.75 (s, 3H), 2.00-2.10 (m, 2H), 1.08-1.15 (t, 3H).
To a solution of naphthalene-2,7-diol (5.0 g, 31 mmol) in acetone (200 mL) was added cesium carbonate (10.1 g, 31 mmol) at room temperature under a nitrogen atmosphere with stirring. After the addition, the mixture was stirred at room temperature for 3 min and then methyl-2-bromobutyrate (5.6 g, 31 mmol) was added. The mixture was stirred at room temperature overnight and then filtered to remove solids. The filtrate was concentrated under reduced pressure and the residue was purified by column chromatography (over silica gel with ethyl acetate and hexanes in volume ratios from 10 to 80 % as eluents) to give the title compound (3.9 g) as a solid. in NMR (CDCI3) delta 7.62-7.70 (m, 2H), 6.85-7.05 (m, 4H), 5.85 (s, 1H), 4.68-4.78 (m, 1H),3.75 (s, 3H), 2.00-2.10 (m, 2H), 1.08-1.15 (t, 3H).
With toluene-4-sulfonic acid; 1-butyl-3-methylimidazolium Tetrafluoroborate at 50℃; for 2h;
90%
With N-methyl-2-oxopyrrolidin-1-ium dihydrogen phosphate at 80℃; for 0.333333h; Ionic liquid;
89%
With guanidine hydrochloride at 80℃; for 0.25h;
88%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.166667h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
With guanidine hydrochloride at 80℃; for 0.333333h;
92%
With toluene-4-sulfonic acid; 1-butyl-3-methylimidazolium Tetrafluoroborate at 50℃; for 1.5h;
90%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.2h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
85%
With alum at 60℃; for 1.25h; Green chemistry;
85%
With N-methyl-2-oxopyrrolidin-1-ium dihydrogen phosphate at 80℃; for 0.333333h; Ionic liquid;
With tetrabutylammomium bromide; sodium hydroxide at 80℃; for 36h;
60%
With potassium carbonate In acetone at 60℃; for 12h;
2, 7-Bis-(2-bromo-ethoxy)-naphthalene (2)
The intermediate 2 was synthesized by the reaction of 2, 7-dihydroxy naphthalene (1 gm, 6.25 mmol) with 1, 2-dibromo ethane (2.5 eqv.) in presence of K2CO3 (1.8 gm) in dry acetone for 12h at 600C (Scheme 2).Then acetone was evaporated and extract by EtOAc, dried over anhydrous Na2SO4 and evaporated under reduced pressure, then purified by coloumn chromatography (100-200 silica gel, by 6% EtOAc in petroleum ether as elluent).Yield 60% and characterized by 1H NMR spectroscopy (see Supporting Information). Mp 92°C, 1H-NMR (400 MHz, CDCl3): δH: 7.68 (d, 2H, j=9.76), 7.04 (d, 2H, j=3.03), 7.03 (d, 2H, j=2.44), 4.39 (t, 4H, j=12.8), 3.70(t, 2H, j=12.84).
48%
With potassium carbonate In acetone for 12h; Reflux;
With LACTIC ACID In neat (no solvent) at 70℃; for 0.666667h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With LACTIC ACID In neat (no solvent) at 70℃; for 0.833333h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With LACTIC ACID In neat (no solvent) at 70℃; for 0.75h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With LACTIC ACID In neat (no solvent) at 70℃; for 0.75h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With LACTIC ACID In neat (no solvent) at 70℃; for 0.833333h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With LACTIC ACID In neat (no solvent) at 70℃; for 0.666667h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With LACTIC ACID In neat (no solvent) at 70℃; for 0.833333h; Green chemistry;
Preparation of 2-hydroxy-12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano-[2,3-d]pyrimidine-9,11-(10H)-dione and 12-aryl-8,12-dihydro-8,10-dimethyl-9H-naphtho[1',2':5,6]pyrano[2,3-d]pyrimidine-9,11-(10H)diones. Gereral Procedure.
General procedure: A mixture of aromatic aldehyde 1 (1.0 mmol), 2,7-dihydroxynaphthalene (weight) or β-naphthol 2 (weight, 1.0 mmol), 1,3-dimethylbarbituric acid 3 (weight, 1.0 mmol), and lactic acid (weight) at the optimum temperature (70 °C or 100 °C), was stirred with out solvent in an oil bath. Completion of reaction was determined by TLC (solvent system). The crude reaction mixture was cooled to RT, the solid formed were suspended in ethanol(5 ml) and collected. The crude solid was recrystallized from ethanol. The structures of the compounds were confirmed by their IR, 1H NMR and 13C NMR spectra and found to be identical with those reported in the literature. Correct combustion analyses were obtained for previously un-reported compounds 4h and 4i.
With formic acid In neat (no solvent) at 80℃; for 0.333333h; Green chemistry;
General procedure: A mixture of heteroaryl amine(2.0 mmol), terephthaldehyde (1.0 mmol) and naphthol (2.0mmol) in the presence of formic acid (0.1 mmol) as catalystwas magnetically stirred on a preheated oil bath at 80 ° C forthe appropriate amount of time as indicated in (Table 1). Theprogress of the reaction was monitored by thin-layer chromatography(TLC). After completion, the reaction mixture wascooled to room temperature and EtOH (5 mL) was addeduntil solid products precipitated. The precipitate was filtered,washed with cold ethanol and dried. The crude product wasstirred for 5 min in boiling EtOH and the resulting whiteprecipitate was filtered. The obtained products 7 were foundto be pure upon TLC examination (Ethyl acetate/ n-Hexane:1/2).
With formic acid; In neat (no solvent); at 80℃; for 0.25h;Green chemistry;
General procedure: A mixture of heteroaryl amine(2.0 mmol), terephthaldehyde (1.0 mmol) and naphthol (2.0mmol) in the presence of formic acid (0.1 mmol) as catalystwas magnetically stirred on a preheated oil bath at 80 ° C forthe appropriate amount of time as indicated in (Table 1). Theprogress of the reaction was monitored by thin-layer chromatography(TLC). After completion, the reaction mixture wascooled to room temperature and EtOH (5 mL) was addeduntil solid products precipitated. The precipitate was filtered,washed with cold ethanol and dried. The crude product wasstirred for 5 min in boiling EtOH and the resulting whiteprecipitate was filtered. The obtained products 7 were foundto be pure upon TLC examination (Ethyl acetate/ n-Hexane:1/2).
With potassium carbonate In acetonitrile at 20℃; for 1.5h; Inert atmosphere;
(S)-3-Cyclohexyl-2-(7-hydroxynaphthalen-2-yloxy)-propionic acid methyl ester (7)
General procedure: A solution of 6 (1.68 g, 5.28 mmol) in dry acetonitrile (8 mL) was added to a mixture of 2,7-dihydroxynaphthalene (4.20 g, 26.2 mmol) and K2CO3 (1.80 g) in dry acetonitrile (17 mL) under argon atmosphere. The mixture was stirred for 1.5 h at room temperature and partitioned between CH2Cl2 (70 mL) and H2O (40 mL). The organic layer was washed twice with H2O (10 mL each) and once with saturated aq NaHCO3 (20 mL). Drying over Na2SO4 and removal of the solvent in vacuo furnished a crude residue which was purified by flash chromatography (cyclohexane/EtOAc 5:1) to give 7 as a colorless solid (1.49 g, 4.54 mmol, 86%).
With fluorosulfonyl fluoride; triethylamine In dichloromethane at 20℃; for 12h;
75%
Stage #1: 2,7-Dihydroxynaphthalene With triethylamine In acetonitrile at 20℃; for 0.166667h;
Stage #2: With 1-(fluorosulfuryl)-2,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate In acetonitrile for 1h;
75%
Stage #1: 2,7-Dihydroxynaphthalene With triethylamine In acetonitrile at 20℃; for 0.166667h;
Stage #2: With 1-(fluorosulfuryl)-2,3-dimethyl-1H-imidazol-3-ium trifluoromethanesulfonate In acetonitrile for 1h; Inert atmosphere;
18 Example 18 Preparation of 2,7-naphthyl-disulfonyl fluoride
Triethylamine (440 μL, 3.17 mmol) was added to a solution of 2,7-dihydroxynaphthalene [Compound 40] (169 mg, 1.04 mmol) in acetonitrile (1 mL) at room temperature and stirred for 10 min.A solution of 1-(fluorosulfonyl)-2-3-dimethyl-1H-imidazole trifluoromethanesulfonate [Compound 4] (840 mg, 2.56 mmol) in acetonitrile (1 mL) was added to the system in one portion and under argon Under protection 1h,TLC (petroleum ether: ethyl acetate = 10:1, product Rf = 0.54) followed the reaction,Column chromatography purification (silica gel 300-400 mesh,Petroleum ether: ethyl acetate = 10:1),Obtained white solid 2,7-naphthyl-disulfonyl fluoride [compound 41] (253 mg, 75%)(scheme 18).
With fluorosulfonyl fluoride; triethylamine In dichloromethane
With ammonium hydroxide; In neat (no solvent); at 120℃; for 0.166667h;
General procedure: A mixture of 2,7-naphthalenediol (1 mmol), aldehyde (2 mmol), carboxylic acid (1.2 mmol), and aqueous ammonia (0.2 mL, 25 %) was stirred under solvent-free conditions at 120 C for an appropriate time (indicated in Table 2). The progress of the reaction was monitored by TLC. After completion of the reactions, saturated aqueous NaCl (20 mL) was added, the suspension was stirred for 30 min, and the precipitate was isolated by filtration, washed with water, and air-dried. The crude products were washed with ethyl acetate-n-hexane, (20 mL, 1:4) to afford the pure products.
With ammonium hydroxide; In neat (no solvent); at 120℃; for 0.25h;
General procedure: A mixture of 2,7-naphthalenediol (1 mmol), aldehyde (2 mmol), carboxylic acid (1.2 mmol), and aqueous ammonia (0.2 mL, 25 %) was stirred under solvent-free conditions at 120 C for an appropriate time (indicated in Table 2). The progress of the reaction was monitored by TLC. After completion of the reactions, saturated aqueous NaCl (20 mL) was added, the suspension was stirred for 30 min, and the precipitate was isolated by filtration, washed with water, and air-dried. The crude products were washed with ethyl acetate-n-hexane, (20 mL, 1:4) to afford the pure products.
12-(2-hydroxy-3-methoxyphenyl)-9,9-dimethyl-2-hydroxy-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With guanidine hydrochloride at 80℃; for 0.5h;
89%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.166667h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
12-(5-bromo-2-hydroxyphenyl)-9,9-dimethyl-2-hydroxy-8,9,10,12-tetrahydrobenzo[a]xanthen-11-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With Zr-MCM-41 In neat (no solvent) at 80℃; for 0.2h; Green chemistry;
General Procedure for the Synthesis of Xanthenones (4, 5)
General procedure: Zr-MCM-41 (10 mol%) was added to a mixture of aldehyde (1 mmol), 5,5-dimethyl-1,3-cyclohexanedione (1 mmol), and naphthol (1 mmol) and heated at 80 °C for the appropriate amount of time as indicated in Table 2. The progress of the reaction was monitored by thin-layer chromatography (TLC). After completion, ethyl acetate (10 mL) was added and centrifugated until catalyst was separated. Then, ethyl acetate was evaporated under vacuum to give the crude product. The crude product was recrystallized from EtOH to yield pure xanthene-11-one derivatives.
88%
With guanidine hydrochloride at 80℃; for 0.416667h;
N2,N7-bis(4-hexylphenyl)naphthalene-2,7-diamine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Example 1-1 Preparation of N2,N7-bis(4-hexylphenyl)naphthalene-2,7-diamine 2,7-Dihydroxynaphthalene (354 mg, 2.21 mmol), I2 (24 mg, 0.09 mmol) and 4-n-hexylaniline (1 g, 5.64 mmol) were mixed and heated at 190 C. for 8 hours. After the reaction was completed, the reaction mixture was cooled to room temperature and purified using a silica gel column (eluent: EtOAc/n-Hex=1/10) to obtain the target compound (430 mg, yield: 40.6%) (compound 1a). 1H NMR (400 MHz, CDCl3): delta 7.602-7.580 (d, 2H), 7.167-7.162 (d, 2H), 7.128-7.072 (m, 8H), 6.980-6.953 (dd, 2H), 5.731 (s, 2H), 2.586-2.547 (t, 4H), 1.623-1.586 (m, 4H), 1.319 (m, 12H), 0.907-0.873 (t, 6H).
Stage #1: zinc(II) cyanide; 2,7-Dihydroxynaphthalene With hydrogenchloride In diethyl ether at 0 - 5℃;
Stage #2: With water In diethyl ether; ethanol Cooling with ice; Heating;
2,7-Dihydroxynaphthalene-1-carbaldehyde (2)
Anhydrous Zn(CN)2 (5.5g, 46.87 mmol) was taken in a three-necked round-bottomed flask and dry Et2O (120 mL) was added. The flask was then set-up on a magnetic stirrer maintaining the temperature between 0-5 °C under anhydrous conditions. 2,7-Dihydroxynaphthalene(5 g, 31.25 mmol) was then introduced into the flask along with dry Et2O (100 mL). Next, a stream of dry HCl gas was passed through the solution for ca. 4-5 h with continuous stirring. The solution gradually acquired a yellow color due to the intermediate product ‘aldimine hydrochloride’. Further HCl gas was passed through the solution for 2-3 h. After the total consumption of 2,7-dihydroxynaphthalene (monitored by TLC), the entire mixture was poured slowly into ice cold H2O. EtOH (30 %) (100 mL) was added and then the suspension was heated on a water bath for a further 15-20 min until all the aldimine had dissolved. The solution was cooled on ice and a solid started separating slowly. The precipitated thick greenish solid was filtered using a Büchner funnel and washed several times with H2O. The obtained crude product was air-dried overnight and was crystallized by using absolute EtOH to afford a yellowish green crystal solid of the aldehyde derivative. Yield: 4.6 g (78 %); mp 158 °C (Lit.8 159-160 °C). IR (KBr): 3209, 2923, 1618, 1580, 1518, 1403, 1319, 1225, 761 cm-1. 1H NMR (300 MHz, CDCl3 + DMSO-d6): δ = 12.56 (s, 1 H, OH, D2O exch), 10.60 (s, 1 H, H-C=O), 9.80 (s, 1 H, OH), 7.817 (d, J = 9.0 Hz, 1 H,C-4), 7.812 (d, J = 9.3 Hz, 1 H, C-8), 7.55 (d, J = 8.7 Hz, 1 H, C-5), 6.90 (dd, J = 8.7 Hz, 2.1 Hz, 1 H, C-6), 6.79 (d, J = 9.0 Hz, 1 H, C-3). 13C NMR (75 MHz, CDCl3 + DMSO-d6): δ = 193.1, 165.1, 158.9, 138.9, 134.9, 130.9, 122.3, 116.2, 114.9, 110.9, 103.0. MS (EI, 70 eV): m/z (%) = 51 (13), 77 (28), 103 (20), 131 (43), 160 (65),188 (45).Anal. Calcd for C11H8O3: C, 70.21; H, 4.28. Found: C, 70.32; H, 4.14.
With potassium permanganate In tetrahydrofuran; water at 20℃; for 48h;
1 Synthesis of perylenequinone
To 2, 7-dihydroxynaphthalene (0.50 g, 3.12 mmol) was added 50 mL of THF,Stirring until completely dissolved, adding 84.4 mL of water, 15.6 mL (0.25 eqv) of a 0.05 mol / L potassium permanganate solution,The reaction was carried out at room temperature for 2 days to obtain a reaction mixture.To the reaction mixture was added 50mL 2mol / L dilute hydrochloric acid,Adjust pH to pH = 6-7.Standing,Filter, pointsDo not rinse the filter cake three times with ethyl acetate and ether,457 mg of crude product was obtained,Re-crystallization of ethanol 30mL again,filter,In a vacuum oven at 50 drying,435 mg of violet black solid (perylenequinone) was obtained.The reaction yield was 88.7%.The spectral test results of the prepared products refer to the relevant drawings,among them,
1-(4,4-diethoxybutyl)-3-(naphthalen-1-yl)urea[ No CAS ]
2-(2,7-dihydroxynaphthalen-1-yl)-N-(naphthalen-1-yl)pyrrolidine-1-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
88%
With trifluoroacetic acid In chloroform at 20℃; for 12h;
General method for the synthesis of pyrrolidines 8
General procedure: To 0.30 g 1-(4,4-diethoxybutyl)-3-(naphthalen-1-yl)urea(6, 0.91 mmol) dissolved in 10 cm3 of dry chloroform 0.91 mmol of appropriate phenol and 0.10 g trifluoroacetic acid (0.91 mmol) were added. The mixture was stirred at room temperature for 12 h. The precipitate formed was filtered off, washed with 10 cm3 diethyl ether, and dried in vacuum (0.01 torr, 12 h, r.t.) to give target compound 8.
12-(2,4-difluorophenyl)-2-hydroxy-9,9-dimethyl-9,10-dihyro-8H-benzo[a]xanthen-11(12H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
89%
In neat (no solvent); at 60℃; for 0.383333h;Green chemistry;
General procedure: The mixtureof naphthalenediol (1a-b, 1 mmol), fluoro substituted benzaldehyde(2a-e, 1 mmol) and 1,3-dicarbonyl compound (3a-b,1 mmol) was stirred in a round bottom flask at 60 C under catalyst and solvent-free conditions for 20 min. After completionof the reaction (TLC) resulted solid was dissolved in THF, filtered and evaporated the solvent using rotary evaporator. After recrystallization by ethanol we obtained the pure products as white solids (4a-j).
In neat (no solvent); at 60℃; for 0.3h;Green chemistry;
General procedure: The mixtureof naphthalenediol (1a-b, 1 mmol), fluoro substituted benzaldehyde(2a-e, 1 mmol) and 1,3-dicarbonyl compound (3a-b,1 mmol) was stirred in a round bottom flask at 60 C under catalyst and solvent-free conditions for 20 min. After completionof the reaction (TLC) resulted solid was dissolved in THF, filtered and evaporated the solvent using rotary evaporator. After recrystallization by ethanol we obtained the pure products as white solids (4a-j).
Stage #1: 4,4'-diaminostilbene-2,2'-disulfonic acid With sodium hydroxide In water at 4 - 5℃;
Stage #2: With sodium nitrite at 4 - 5℃; for 1.5h;
Stage #3: 2,7-Dihydroxynaphthalene With sodium carbonate In water at 8 - 10℃; for 3h;
2.2. Synthesis of dye 1
1.95 g of 95% purity 4,40-diaminostilbene-2,20-disulfonic acid(0.005 mol) was suspended in 20 mL distilled water. 1.1 mL of 30%NaOH aqueous solution (0.010 mol) were added dropwise and themixture was cooled down to 4-5 °C. The sodium salt of the 4,40-diaminostilbene-2,20-disulfonic acid was treated with 3 mL(0.030 mol) of 32% HCl. The bis-diazotization of the resulting suspensionwas performed using 0.71 g of 98% purity solid NaNO2(0.010 mol) at 4-5 °C and the pH value of about 1. After the fast addition of NaNO2, the mixture was maintained under stirring for1.5 h to complete the diazotization reaction. The nitrous acid excess was neutralized by urea. 1.77 g of 98% purity 2,7-dihydroxynaphthalene (0.011 mol) used as a coupling component, was dissolved in a 10% NaOH aqueous solution and was directly introduced to the above-obtained suspension of the bis-diazonium salt. By the periodic direct introduction of 10% Na2CO3 aqueous solution, the pH of the reaction mixture was maintained around 8-8.5 and the temperature was kept at low values of 8-10 °C. Thecoupling reaction was controlled by a conventional method [18].Thereafter, for the completion of the reaction, the mixture was maintained under stirring for 3 h. The separation of crude dye 1 was made by filtration. The resulted precipitate was washed with methanol. After drying, dye 1 was purified by recrystallization from dioxane: pyridine 90:10 (v:v) mixture to give the pure product.The obtained dye 1 (3.06 g, 81%) Rf 0.73 (C4H8O/NH3/MeOH5:3:2); 1H NMR (500 MHz, (CD3)2SO), δ/ppm; 6.72 (d, J 9.27 Hz,2H, H59,H60), 6.97 (dd, J 8.52, 2.39 Hz, 2H, H61,H62), 7.65 (d,J 8.66 Hz, 2H, H55, H56), 7.83 (d, J 9.16 Hz, 2H, H51,H52)7.84-7.89 (m, 6H, H53,H54,H57,H58,H63, H64), 8.27 (s, 2H, H65,H66), 8.31 (s, 2H, H67, H68); 10.33 (s, 2H, H69,H70), 16.02 (s, 2H,H71,H72); 13C NMR (125 MHz, (CD3)2SO), δ/ppm; 126.7 (C1,C2),139.68 (C3,C4), 130.7 (C5,C6), 120.1 (C7,C8), 115.9 (C9,C10), 119.36(C11,C12), 105.17 (C13,C14), 116.9 (C15,C16), 122.37 (C17,C18),129.26 (C19,C20), 134.8 (C21,C22), 155.49 (C23,C24), 143.69(C25,C26), 158.54 (C27,C28), 167.95 (C29,C30), 147.13 (C31,C32), 127.79 (C33,C34); IR (KBr): ~v 3429(sv), 1331 (dv) cm-1 (OH),1214(sv) cm-1 (C-O),1136 (s) cm-1 (νasSO3-), 1075(sv) cm-1 (νsSO3); UV/Vis(water): λmax (lgε) 560 nm (4.45); HRMS (ESI): m/z calcd forC34H24N4O10S2: 711.0876 [M - H]-; found: 711.0875.
With 2,3,6,7-tetramethoxy-9(10H)-anthracenone; caesium carbonate In dimethyl sulfoxide at 25℃; for 18h; Irradiation; Sealed tube; regioselective reaction;
General Procedure for Carboxylation of Arenes - GP2c (glovebox)
General procedure: To a dry microwave vial (5 mL) equipped with stirring bar, was added the arene (if solid) (0.1 mmol) and2,3,6,7-tetramethoxyanthracen-9(10H)-one (6.3 mg, 0.02 mmol, 20 mol %). The vials were sealed andtransferred to a glovebox after the vial had evacuated and refilled with N2 (×3) within the antechamber viasyringe. Cs2CO3 (98 mg, 3 equiv.) and DMSO (1 mL) was added and the vial was sealed with a Supelcoaluminium crimp seal with septum (PTFE/butyl). The vials were removed from the glovebox and the arene(0.1 mmol) (if liquid) was added via syringe. The reaction mixture then had gaseous CO2 (22 cm3) added viaa gastight Hamilton syringe, into the head space of the vial. The vial was then irradiated from two sidesby two kessil lamps (vials approximately 6 cm away from the light source) with fans placed in front of thevials for cooling (temperatures measured via IR ranged between 25-30 °C). After 18 hrs. the irradiation wasstopped and the vials were decapped quenched with aq. HCl (1 mL, 0.3M) and monitored by LCMS and 1HNMR.Reactions were filtered of any solids and purified directly via prep-HPLC.
With 1,2-diamino-benzene In dichloromethane at 40℃; for 60h; Inert atmosphere; Molecular sieve;
4. General Procedure for the Synthesis of 2H-Benzo[h]Chromene 3
General procedure: Dry CH2Cl2 (1.0 mL) was added to a mixture of o-phenylenediamine (IX, 0.04 mmol),enal 1 (0.30 mmol) and arylol 2 (0.20 mmol) under Ar. The reaction was stirred at 40 Cuntil the completion of 2 monitored by TLC. Then, the mixture was applied to columnchromatography directly and eluted with ethyl ether and hexane (9/1) to give product 3.
(E)-1-((4-iodo-2,6-dimethylphenyl)diazenyl)naphthalene-2,7-diol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
87%
Stage #1: 4-iodo-2,6-dimethylaniline With hydrogenchloride; potassium carbonate; sodium nitrite In water at 0℃; for 1h;
Stage #2: 2,7-Dihydroxynaphthalene With sodium hydroxide In water at 21℃; for 3h;
(E)-1-((4-Iodo-2,6-dimethylphenyl)diazenyl)naphthalene-2,7-diol (S2)
This reaction was not performed under inert conditions.4-Iodo-2,5-dimethylanilin (2.47 g, 10 mmol, 1.00 equiv) and K2CO3 (1.26 g, 10.00 mmol, 1.00 equiv)were dissolved in water (10 mL). NaNO2 (680 mg, 10.0 mmol, 1.00 equiv) dissolved in water (10 mL) was added dropwise. The mixture was cooled to 0 °C under constant stirring and HCl (4 M, 10 mL) wasadded dropwise. The mixture was stirred for 1h and was then added dropwise to a mixture of 2,7-dihydroxynaphthalene (1.60 g, 10.0 mmol, 1.00 equiv) and 15 wt% NaOH solution (10 mL). The reactionwas stirred for a further 3 h while warming up to 21 °C. The mixture was diluted with HCl (1 M, 50 mL).The precipitate was removed by filtration and washed with water (2 × 10 mL). The residue wascrystallized from ethanol/water (5:1) at -21 °C to yield S2 as a dark red solid (3.49 g, 8.70 mmol, 87%).
General procedure: In a container (internal capacity: 1 L) equipped with a stirrer, a condenser tube, and a burette, 24 g (150 mmol) of 2,7-dihydroxynaphthalene (a reagent manufactured by Sigma-Aldrich), 25.4 g (71 mmol) of <strong>[2631-77-8]3,5-diiodosalicylaldehyde</strong> (a reagent manufactured by Tokyo Chemical Industry Co., Ltd.), and 200 mL of 1-methoxy-2-propanol were charged, and 1.3 g (14 mmol) of methanesulfonic acid (a reagent manufactured by Kanto Chemical Co., Inc.) was added to prepare a reaction solution. This reaction solution was stirred at 90 C. for 5 hours and allowed to react. After the reaction finished, 1.7 L of pure water was added to the reaction solution, extraction with ethyl acetate was performed, followed by concentration, to obtain a solution. The obtained solution was separated and purified by column chromatography to obtain 9.2 g of the objective compound (X-27N35IB) represented by the following formula (purity: 98.7% and yield: 20%). As a result of measuring the molecular weight of the obtained compound (X-27N35IB) by the above method, it was 658. Also, since the following peaks were found by performing the 1H-NMR measurement under the above measurement conditions, the compound was confirmed to have a chemical structure of the following formula (X-27N35IB). 1H-NMR (d6-DMSO): δ (ppm) 10.4 (1H, -OH), 9.8 (1H, -OH), 9.5 (1H, -OH), 6.7-8.0 (12H, Ph), 6.2 (1H, Methine)






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping