* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1983, # 10, p. 563 - 564
[2] Tetrahedron, 1990, vol. 46, # 8, p. 3093 - 3100
[3] Journal of Fluorine Chemistry, 1983, vol. 23, p. 487
[4] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
[5] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
[6] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
[7] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
[8] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
[9] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
2
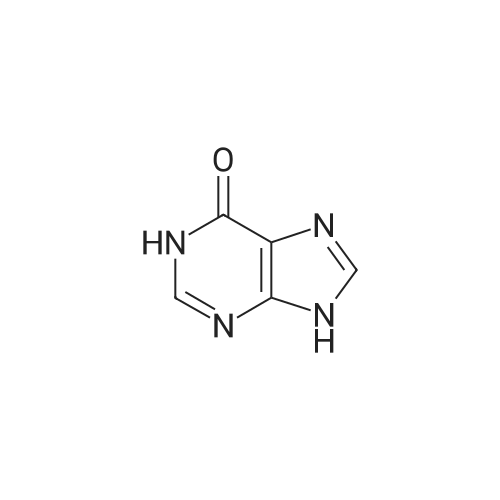
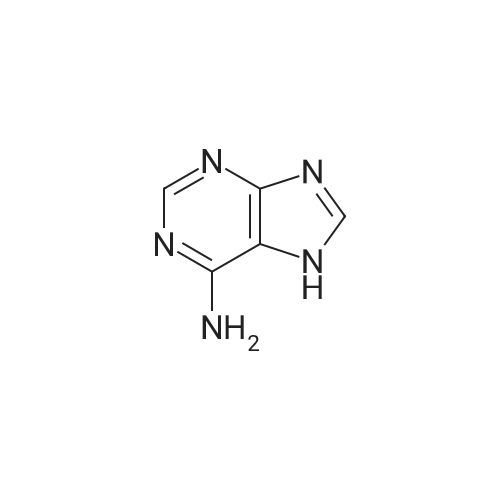
[ 58-96-8 ]
[ 316-46-1 ]
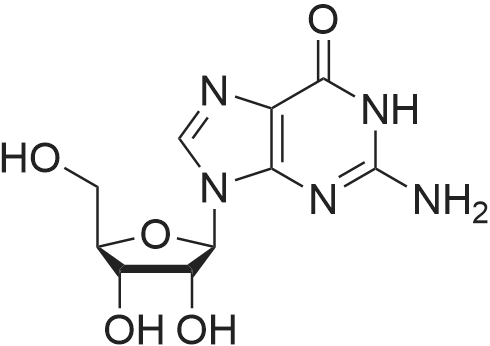
[ 119068-03-0 ]
[ 119003-30-4 ]
[ 119003-30-4 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
[2] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
3
[ 58-96-8 ]
[ 316-46-1 ]
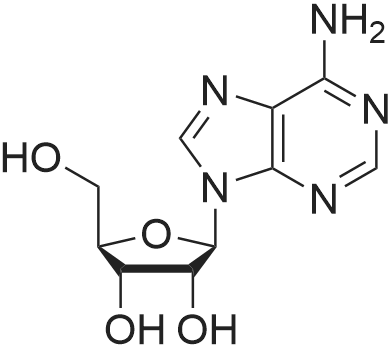
[ 119068-04-1 ]
[ 119003-30-4 ]
[ 119003-30-4 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
4
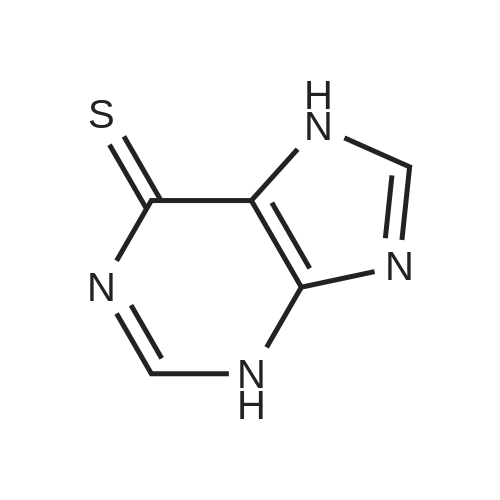
[ 78948-09-1 ]
[ 58-96-8 ]
[ 316-46-1 ]
[ 119068-03-0 ]
[ 119003-30-4 ]
[ 119003-30-4 ]
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1988, p. 2547 - 2554
5
[ 58-96-8 ]
[ 2880-89-9 ]
Reference:
[1] Synthesis, 2009, # 23, p. 3957 - 3962
[2] Journal of Organic Chemistry, 1990, vol. 55, # 16, p. 4928 - 4933
[3] Canadian Journal of Chemistry, 1994, vol. 72, # 9, p. 2005 - 2010
[4] Nucleosides, Nucleotides and Nucleic Acids, 2009, vol. 28, # 9, p. 821 - 834
[5] Journal of Biological Chemistry, 1951, vol. 190, p. 95,96
[6] Chemical Communications, 2012, vol. 48, # 45, p. 5587 - 5589
[7] European Journal of Medicinal Chemistry, 2013, vol. 65, p. 249 - 255
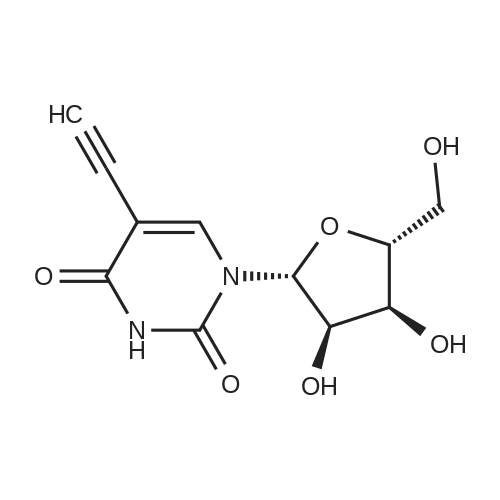
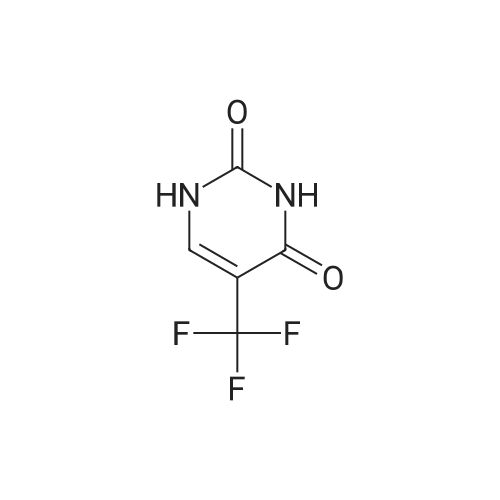
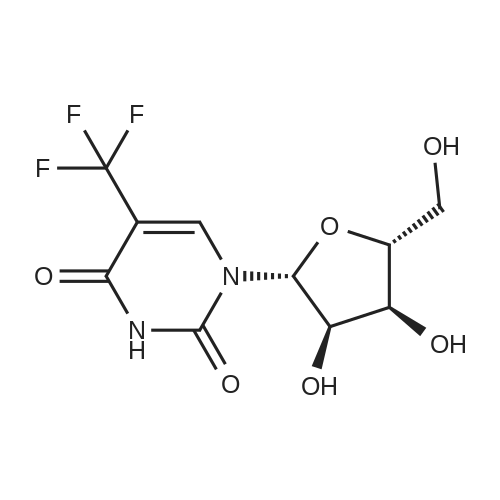
6
[ 58-96-8 ]
[ 957-75-5 ]
Yield
Reaction Conditions
Operation in experiment
79%
With sodium azide; bromoisocyanuric acid monosodium salt In water; acetonitrile at 20℃; for 0.5 h;
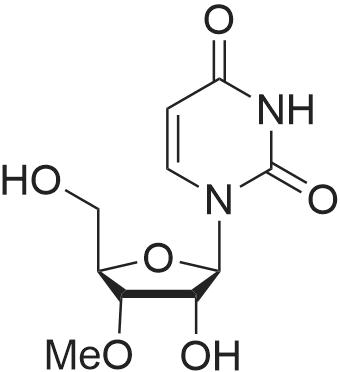
General procedure: 2'-O-Methyluridine (5, 0.103 g, 0.4 mmol) was dissolved in aqueous acetonitrile solution(H2O:CH3CN 1:9, 5 mL) under stirring. NaN3 (0.104 g, 1.6 mmol) was added, followed by addition of SMBI (0.101 g, 0.44 mmol) at r.t. and the mixture was stirred. Progress of the reaction was followedby TLC. On completion of the reaction after 1.5 h, the reaction mixture was filtered, evaporated todryness under reduced pressure and coevaporated with acetonitrile (2 × 2 mL). The crude reactionmixture was purified by column chromatography (4percent–6percent MeOH in DCM, v/v) to afford bromonucleoside 6 (0.117 g, 93percent) in pure form as a white solid
75%
With 1,3-dibromo-5,5-dimethylimidazolidine-2,4-dione In N,N-dimethyl-formamide at 25℃; for 0.33 h;
Typical procedure for the bromination of unprotected nucleosides: DBH (323 mg, 1.13 mmol) was added to a stirred solution of 1d (500 mg, 2.05 mmol) in DMF (5 mL). The resulting pale-yellow solution was stirred at room temperature for 20 minutes or until TLC showed absence of starting material and formation of less polar product. Volatiles were evaporated and the residue was coevaporated with MeCN. The resulting pale solid was crystallized from hot acetone to give 2d (500 mg, 75percent) as colorless crystals with data as reported.14
22 g
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile) In N,N-dimethyl-formamide at 80℃; for 4 h;
To a solution of uridine 146 (20.0 g, 82mmol) and N BS (21 .7 g, 0.12 mol) in anhydrous dimethylfomamide was added AIBN (0.1 eq) in anhydrous dimethylfomamide, then the solution was stirred at 80 °C for 4 h. Saturated sodium thiosulfate solution (20 mL) was added. After evaporation of the solvent, the residue was precipitation with methanol to give 22.0 g compound 147 as light yellow solid.
22.0 g
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile) In N,N-dimethyl-formamide at 80℃; for 4 h;
Synthesis of Compound 147: To a solution of uridine 146 (20.0 g, 82mmol) and NBS (21 .7 g,0.12 mol) in anhydrous dimethylfomamide was added AIBN (0.1 eq) in anhydrous dimethylfomamide,then the solution was stirred at 80 °C for 4 h. Saturated sodium thiosulfate solution (20 mL) was added. After evaporation of the solvent, the residue was precipitation with methanol to give 22.0 g compound 147 as light yellow solid.
22 g
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile) In N,N-dimethyl-formamide at 80℃; for 4 h;
Synthesis of Compound 1 47: To a solution of uridine 146 (20.0 g, 82mmol) and NBS (21 .7 g, 2 mol) in anhydrous dimethylfomamide was added AIBN (0.1 eq) in anhydrous dimethylfomamide, n the solution was stirred at 80 °C for 4 h. Saturated sodium thiosulfate solution (20 mL) was ded. Afte ompound 1
22 g
With N-Bromosuccinimide; 2,2'-azobis(isobutyronitrile) In N,N-dimethyl-formamide at 80℃; for 4 h;
Synthesis of Compound 147: To a solution of uridine 146 (20.0 g, 82mmol) and NBS (21.7 g, 0.12 mol) in anhydrous dimethylfomamide was added AIBN (0.1 eq) in anhydrous dimethylfomamide, then the solution was stirred at 80 °C for 4 h. Saturated sodium thiosulfate solution (20 mL) was added. After evaporation of the solvent, the residue was precipitation with methanol to give 22.0 g compound 147 as light yellow solid.
Reference:
[1] Canadian Journal of Chemistry, 1994, vol. 72, # 9, p. 2005 - 2010
[2] Synthesis, 2009, # 23, p. 3957 - 3962
[3] Journal of Organic Chemistry, 1990, vol. 55, # 16, p. 4928 - 4933
[4] Organic and Biomolecular Chemistry, 2008, vol. 6, # 16, p. 2884 - 2891
[5] Nucleosides, Nucleotides and Nucleic Acids, 2009, vol. 28, # 9, p. 821 - 834
[6] Molecules, 2013, vol. 18, # 10, p. 12740 - 12750
[7] Tetrahedron Letters, 2012, vol. 53, # 26, p. 3333 - 3336
[8] Chemistry - A European Journal, 2009, vol. 15, # 27, p. 6619 - 6625
[9] Recueil: Journal of the Royal Netherlands Chemical Society, 1981, vol. 100, # 7/8, p. 267 - 271
[10] Patent: WO2014/93924, 2014, A1, . Location in patent: Page/Page column 288; 289
[11] Journal of Inorganic Biochemistry, 2015, vol. 146, p. 61 - 68
[12] Patent: WO2015/196130, 2015, A2, . Location in patent: Page/Page column 621
[13] Patent: WO2015/196128, 2015, A2, . Location in patent: Page/Page column 629
[14] Patent: EP2918275, 2015, A1, . Location in patent: Paragraph 2169; 2170
[15] Patent: EP2918275, 2016, B1, . Location in patent: Paragraph 2169; 2170
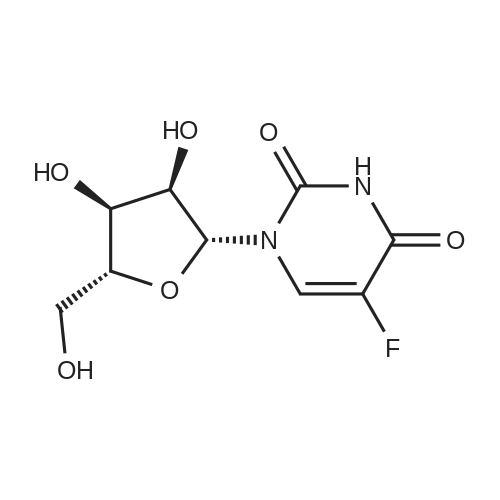
With iodine; nitric acid In chloroform at 80℃; for 5 h;
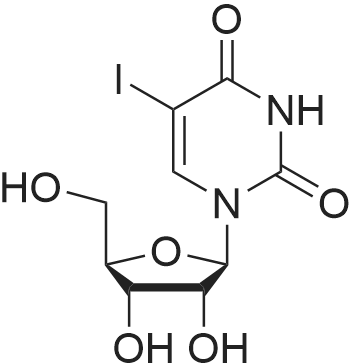
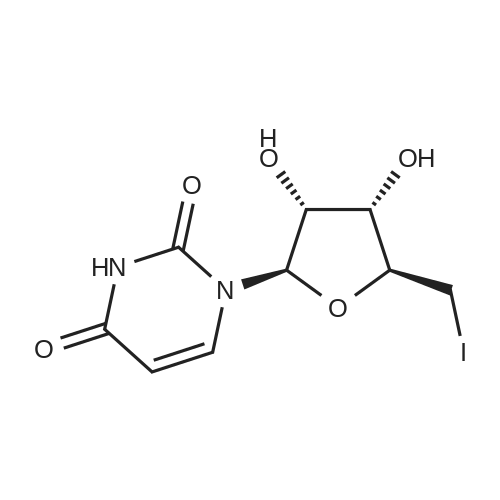
In a round bottom flask, uridine (1 g, 4.1 mmol) and iodine powder (1 .15 g, 4.5 mmol) were dissolved in a mixture of CHCI3 (55 ml) and 1 M HN03 (10 ml). The reaction was heated at reflux (80°C) for 5h. Reaction progress was monitored by TLC (30 percent methanol in chloroform, Rf: 0.60; RfsM: 0.45). Upon cooling of the reaction mixture to 4°C, crystals of the title compound formed as colourless needles. The precipitate was collected by filtration and dried under vacuum overnight to provide 1.39 g (92 percent) of 5-iodouridine 1 ; δΗ (DMSO-d6, 300 MHz) 3.49 - 3.71 (2H, m, H-5'), 3.84 - 3.88 (1 H, m, H-4'), 3.98 (1 H, t, J = 5.0 Hz, H-3'), 4.02 (1 H, t, J = 5.0 Hz, H-2'), 5.08 (1 H, d, J = 5.3 Hz, OH-3'), 5.27 (1 H, t, J = 4.7 Hz, OH-5'), 5.43 (1 H, d, J = 5.4 Hz, OH- 2'), 5.71 (1 H, d, J = 4.6 Hz, H-1 '), 8.48 (1 H, s, H-6) 1 1 .69 (1 H, s, NH); 5C (DMSO-d6, 300MHz) 61 .2 (C-5'), 70.3 (C-3'), 70.9 (C-5), 75.0 (C-2'), 85.7 (C-4'), 89.5 (C-1 '), 146.0 (C-6), 152.5 (C-2), 162.9 (C-4). m/z (ESI) 388.0000 [M+NH4]+, requires 388.0000.
78%
With silver(II) sulfate; iodine In methanol at 20℃; Inert atmosphere
Iodine (0.508 g) in 2.0 mL of methanol was added dropwise to a suspension of silver sulfate (0.624 g) and uridine (0.488 g) in 10 mL of methanol. The reaction mixture was stirred at room temperature until the orange color disappeared. The precipitate was removed by filtration. The filtrate was concentrated and recrystallized from methanol, affording a pale yellow solid (78percent). 1HNMR (CD3OD) δ 8.511 (1H, d, J = 3.2 Hz), 5.776 (1H, t, J = 1.6 Hz), 4.085(2H, m), 3.938 (1H, m), 3.814 (1H, d, J = 2.4 Hz), 3.784 (1H, d, J = 2.8 Hz), 3.674 (1H, d, J = 2.4 Hz), 2.644 (1H, d, J = 2.8 Hz); HRMS [M+1] calcd 370.9662, found: 370.9665.
72.2%
at 110℃; for 4 h;
Take uridine (13.06g, 53.50mmol) was dissolved in 252ml of dilute nitric acid, heated to 110 ,Was added (10.09g, 39.92mmol) elemental iodine, the reaction was monitored by thin layer chromatography case, exhibitionEluent, CH2Cl2: CH3OH = 7: 1, about 4 hours after the reaction raw material point disappears, and the reactionComplete, the reaction was stopped. After leaving to cool and extracted 3 times with 30ml petroleum ether, collecting the lower solution,Upper solution was extracted with 30ml of deionized water, lower solutions were twice extracted. Standing whiteThe solid precipitated, placed in the upper refrigerator overnight, a lot of white solid was obtained, that is, uridine 5-I-(14.30g, 38.64mmol), the yield was 72.22percent
40%
With iodine; iodic acid; acetic acid In tetrachloromethane; water at 40℃; for 2 h;
General procedure: The suspension of nucleosides 2a,b (19 mmol) in water (5.7 mL) was treated with HIO3(9.7 mmol, 1.7 g), AcOH (15.2 mL) and a solution of iodine (11.22 mmol, 2.85 g) inCCl4 (3.8 mL). The resulting mixture was stirred at 40C for 2 h until the starting materialwas consumed or some by-product was formed (monitored by HPLC). After that,water (20 mL) was added. The reaction mixture was cooled to 4C and filtered. The precipitatewas washed with water (2 £ 10 mL). The combined solutions were diluted withwater (250 ml) and extracted with benzene (3 £ 150 mL). The aqueous layer was evaporatedunder reduced pressure. The product was purified by RPC in a linear gradient ofEtOH in water (0–30percent) to give the product 3a,b.
Reference:
[1] Bulletin of the Korean Chemical Society, 2018, vol. 39, # 9, p. 1054 - 1057
[2] Synthetic Communications, 1990, vol. 20, # 21, p. 3391 - 3394
[3] Canadian Journal of Chemistry, 1994, vol. 72, # 9, p. 2005 - 2010
[4] Patent: WO2011/51733, 2011, A2, . Location in patent: Page/Page column 44
[5] Synthesis, 2009, # 23, p. 3957 - 3962
[6] Canadian Journal of Chemistry, 1982, vol. 60, p. 554 - 557
[7] Journal of Organic Chemistry, 1990, vol. 55, # 16, p. 4928 - 4933
[8] Nucleosides, Nucleotides and Nucleic Acids, 2009, vol. 28, # 9, p. 821 - 834
[9] Chemical Communications, 2017, vol. 53, # 68, p. 9406 - 9409
[10] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 5, p. 1511 - 1518
[11] Patent: CN105566304, 2016, A, . Location in patent: Paragraph 0040; 0057; 0058; 0059; 0060
[12] Organic Preparations and Procedures International, 2018, vol. 50, # 3, p. 332 - 342
[13] Antiviral Research, 2012, vol. 94, # 1, p. 35 - 43
[14] Organic and Biomolecular Chemistry, 2015, vol. 13, # 15, p. 4506 - 4513
[15] Organic Preparations and Procedures International, 2018, vol. 50, # 3, p. 332 - 342
10
[ 64-17-5 ]
[ 58-96-8 ]
[ 1024-99-3 ]
Reference:
[1] Recueil: Journal of the Royal Netherlands Chemical Society, 1981, vol. 100, # 7/8, p. 267 - 271
11
[ 58-96-8 ]
[ 606-02-0 ]
[ 58-97-9 ]
Reference:
[1] Organic Process Research and Development, 2014, vol. 18, # 11, p. 1575 - 1581
12
[ 58-96-8 ]
[ 58-97-9 ]
Reference:
[1] Organic Process Research and Development, 2014, vol. 18, # 11, p. 1575 - 1581
[2] Chemistry of Natural Compounds, 1989, vol. 25, # 5, p. 626 - 627[3] Khimiya Prirodnykh Soedinenii, 1989, vol. 25, # 5, p. 732 - 733
[4] Journal of the Chemical Society, 1940, p. 746,748
[5] Journal of the Chemical Society, 1957, p. 3798
[6] Biochimica et Biophysica Acta, 1958, vol. 28, p. 376,379
[7] Journal of Biological Chemistry, 1957, vol. 227, p. 329,333
[8] Biochimica et Biophysica Acta, 1958, vol. 28, p. 376,379
[9] Acta Chemica Scandinavica (1947-1973), 1957, vol. 11, p. 17,20
[10] Chemical and Pharmaceutical Bulletin, 1995, vol. 43, # 2, p. 210 - 215
[11] Journal of Organic Chemistry, 2005, vol. 70, # 3, p. 1100 - 1103
[12] Antimicrobial Agents and Chemotherapy, 2007, vol. 51, # 2, p. 503 - 509
[13] Antiviral Research, 2010, vol. 85, # 3, p. 470 - 481
[14] Advanced Synthesis and Catalysis, 2014, vol. 356, # 2-3, p. 563 - 570
13
[ 58-96-8 ]
[ 58-97-9 ]
[ 15548-52-4 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1980, vol. 45, # 10, p. 2830 - 2838
14
[ 58-96-8 ]
[ 58-97-9 ]
[ 63-39-8 ]
Reference:
[1] Nucleosides, Nucleotides and Nucleic Acids, 2010, vol. 29, # 3, p. 245 - 256
15
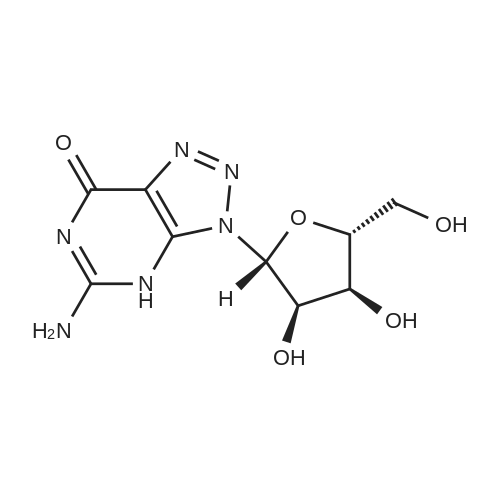
[ 477247-27-1 ]
[ 58-97-9 ]
[ 70580-90-4 ]
[ 58-96-8 ]
[ 479352-21-1 ]
Reference:
[1] Journal of the American Chemical Society, 2002, vol. 124, # 48, p. 14364 - 14372
16
[ 24514-44-1 ]
[ 63-39-8 ]
[ 58-96-8 ]
[ 58-97-9 ]
[ 66-22-8 ]
[ 58-98-0 ]
[ 302917-20-0 ]
Reference:
[1] Journal of the American Chemical Society, 2011, vol. 133, # 36, p. 14452 - 14459
17
[ 19140-04-6 ]
[ 58-97-9 ]
[ 58-96-8 ]
[ 15548-52-4 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 1980, vol. 45, # 10, p. 2830 - 2838
[2] Collection of Czechoslovak Chemical Communications, 1980, vol. 45, # 10, p. 2830 - 2838
Reference:
[1] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 20, p. 6171 - 6180
31
[ 5451-40-1 ]
[ 58-96-8 ]
[ 13276-52-3 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2015, vol. 357, # 6, p. 1237 - 1244
32
[ 68-12-2 ]
[ 58-96-8 ]
[ 871254-61-4 ]
Yield
Reaction Conditions
Operation in experiment
96.6%
Stage #1: at 5℃; for 0.5 h; Stage #2: at 5℃; for 2 h;
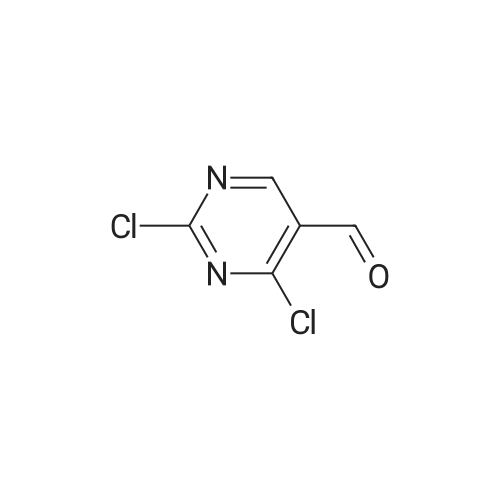
In the reaction flask, the solvent DMF (560 mL) was added, the temperature was lowered to 5 ° C, and phosphorus oxychloride (153 g) was slowly added dropwise, and the reaction temperature of the mixed system was controlled not to exceed 5 ° C, and the phosphorus oxychloride was added dropwise. Add boric acid (30.9g), stir for 30min, then add uridine 112g (1mol) in solid form to the above mixed system, control the reaction temperature not to exceed 5 ° C, stir for 2h, slowly warm to room temperature and continue to stir the reaction until The raw materials disappeared. Then, 560 mL of phosphorus oxychloride was added to the reaction system and the temperature was raised to reflux for 6 hours, then phosphorus oxychloride was recovered, and the residue was poured into a rapidly stirred ice water mixture to precipitate 2,4-dichloro-5-pyrimidine formaldehyde solid. 170 g, yield 96.6percent, purity 99.4percent.
Reference:
[1] Patent: CN108467368, 2018, A, . Location in patent: Paragraph 0010; 0011; 0012; 0013
With triethylamine In 1,4-dioxane at 25℃; for 36h;
100%
With pyridine at 20℃; for 16h;
100%
With triethylamine In N,N-dimethyl-formamide at 20℃;
99%
With pyridine Cooling with ice;
98%
With pyridine at 20℃; Cooling with ice;
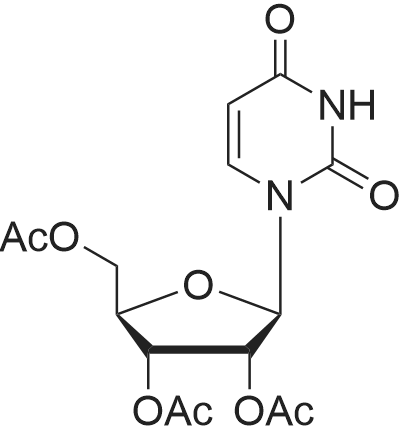
General Procedure for Acetylation of 2a, 2b, and 9 to Afford 7, 11, and 10 (Scheme 3and Scheme 4)
General procedure: Compound 2a, 2b or 9 (10 mmol) was co-evaporated with pyridine (3 £ 10 mL) to drynessunder reduced pressure. The dry residue was suspended in pyridine (50 mL). Aftercooling of the suspension in an ice bath, acetic anhydride (50 mmol, 4.6 mL for 2a and9; 60 mmol, 5.7 mL for 2b) was added dropwise with vigorous stirring. The ice bath wasremoved and the reaction mixture was well stirred overnight at r.t. After completion ofthe reaction, water (20 mL) was added to quench the acetic anhydride
96%
With dmap; triethylamine In acetonitrile at 20℃;
95%
With pyridine; dmap at 20℃;
95%
With dmap In pyridine at 4 - 20℃; for 24h; Inert atmosphere;
94%
With pyridine; dmap at 25℃; for 4h;
94%
With lithium perchlorate for 10h; Heating;
93%
With dmap In acetonitrile for 1h; Ambient temperature;
93%
at 100℃; for 1.5h;
93%
With dmap; 1-ethylene glycol monomethyl ether-3-methylimidazolium methanesulfonate at 20℃; for 0.416667h;
92%
In pyridine at 60℃; for 3h;
92%
Stage #1: acetic anhydride With molybdenium(VI) dioxodichloride In dichloromethane at 20℃; for 0.5h;
Stage #2: uridine In dichloromethane at 20℃; for 107h;
92%
In neat (no solvent) Molecular sieve; Microwave irradiation; Green chemistry;
Optimized MW-assisted peracetylation
General procedure: The substrate belonging to one of the subset reported in Table 1(NTC, TC, CP, DGNP) (0.1 mmol) was left to react under MWheating (Synthos 3000, Anton Paar) with dry acetic anhydride(1 mL, 10 mmol) in a 3 mL vial (Rotor 64MG5 ), equipped witha magnetic stirrer in the presence of molecular sieves(10 % w/w). The microwave, equipped with IR sensor forexternal temperature control (IR limit calculated as follows:Tinternal= 1.214 × TIR), has been set with the power programsprovided for its subset as described in Table 1. At the end of thereaction, the mixture was filtered, diluted with ethanol (2 mL)and left under vigorous stirring for 30 minutes at 50 °C. Themixture was then evaporated under reduced pressure and asmall amount of a saturated solution of sodium bicarbonate(3.8 mL, 10 mmol NaHCO3) was added. After the evolution ofCO2, the precipitation of the peracetylated product was observed. The products were separated by simple decantation. Forcompounds which do not precipitate upon addition of NaHCO3,an extraction with AcOEt was needed. The organic phase, afterdrying with Na2SO4, filtration and evaporation, gave the reaction crude.
90%
With vanadyl triflate In dichloromethane at 20℃; for 96h;
89%
With iron(III) sulfate at 20℃; for 7h;
88%
With pyridine at 60℃; for 3h;
84%
In pyridine at 20℃; Inert atmosphere;
4.3 5-Chlorouridine (9)
Uridine (6) (3.0g, 12.3mmol) was coevaporated with dry pyridine (2×10mL) and dissolved in dry pyridine (20mL). Acetic anhydride (4.1mL, 43.5mmol) was added and the mixture was stirred at room temperature overnight under argon. Work-up afforded the acetylated uridine intermediate 7 (3.81g, 10.3mmol, 84% yield) as a white foam [ESI for C15H18N2O9 (M+): 258.8 (2,3,5-tri-O-acetylribose+H+, 37.5%), 370.7 (M+H+, 13%), 392.9 (M+Na+, 100%)].
79%
With pyridine at 60℃; for 3h;
51 Synthesis of 5-carbanoylmethyl uridine (03601013036):
To a solution of uridine 146 (1 .0 g, 4.0 mmol) in 20 mL of pyridine was added 2 m L (2.16 g, 21 .0 mmol) of acetic anhydride. The resulting reaction mixture was heated to 60 °C for 3 h, and the TLC indicated its completion. The reaction mixture was concentrated, and the residue was purified by flash chromatography on a silica gel column using dichloromethane-methanol (80:1 ) as eluent giving 1 .2 g desired product 1 55 in 79% yield.
79%
With pyridine at 60℃; for 3h;
51 Synthesis of 2’,3’,5’-tri-O-acetyluridine (155).
Synthesis of 2’,3’,5’-tri-O-acetyluridine (155). To a solution of uridine 146 (1.0 g, 4.0 mmol) in20 mL of pyridine was added 2 mL (2.16 g, 21.0 mmol) of acetic an hydride. The resulting reactionmixture was heated to 60 CC for 3 h, and the TLC indicated its completion. The reaction mixture was concentrated, and the residue was purified by flash chromatography on a silica gel column using dichloromethane-methanol (80:1) as eluent giving 1 .2 g desired product 155 in 79% yield.
79%
With pyridine at 60℃; for 3h;
44; 51
yn es s o , , - r - -ace y ur ne . o a so u on o ur ne . g, . mmo n mL of pyridine was added 2 mL (2.1 6 g, 21 .0 mmol) of acetic anhydride. The resulting reaction xture was heated to 60 °C for 3 h, and the TLC indicated its completion. The reaction mixture was ncentrated, and the residue was purified by flash chromatography on a silica gel column using hloromethane-methanol (80:1 ) as eluent giving 1 .2 g desired product 155 in 79% yield.
With sodium acetate
In pyridine
With pyridine at 20℃; for 1h;
With pyridine for 4h; Ambient temperature;
With pyridine
With pyridine at 60℃;
With dmap; triethylamine In acetonitrile at 20℃; for 1h;
In pyridine
In 1,4-dioxane
With pyridine at 0℃; for 5h;
With pyridine for 5h;
1
Example 1: Triacetyl Uridine RG2133 The investigational drug used in the Examples below was RG2133 (2',3',5'-tri-O- acetyluridine). RG2133 was produced under cGMP conditions from uridine via exhaustive acetylation and purified by repeated precipitation after residual acetic anhydride is removed by distillation. The purified drug substance was dried under reduced pressure and sieved to obtain a uniform solid.
With cytidine deaminase enzyme; In aq. phosphate buffer; at 37℃; for 0.0833333h;pH 7.0;Enzymatic reaction;
Comparative Example 1 : Deamination of cytidine (compound 1 ) to uridine A 100 mM solution of the cytidine (495 muIota) in 100 mM phosphate buffer at pH 7 was mixed with 50 L of cytidine deaminase enzyme solution containing >300 AU in phosphate buffer. The reaction was performed at 37C during 5 minutes and stopped with HCI. Then, the crude reaction was filtered through a 10 KDa membrane, and a portion was diluted and analyzed by HPLC under UV-DAD (ultraviolet-diode array detection). Product identification was performed by comparison to a standard sample. Uridine was obtained in quantitative yield (>99%).
With toluene-4-sulfonic acid In acetone for 1h; Reflux;
100%
With toluene-4-sulfonic acid In acetone for 4h; Reflux;
99%
With toluene-4-sulfonic acid In acetone for 1h; Reflux;
96%
With toluene-4-sulfonic acid In acetone at 60℃; for 24h; Inert atmosphere;
96%
With toluene-4-sulfonic acid In acetone at 60℃; for 24h;
2.2.1 Synthesis of 2′',3′-O-isopropylideneuridine
To a stirred solution of uridine (3, 10.0g, 41.0mmol) and p-toluenesulfonic acid (0.779g, 4.10mmol) in acetone (100mL) was added 2,2-dimethoxypropane (6.40g, 61.4mmol) under argon. The mixture was stirred at 60°C for 24h before being concentrated under reduced pressure. The crude material was purified by flash chromatography (1:9 v/v MeOH/DCM) to yield the title compound (11.2g, 96%) as a white foam; mp. 169°C; 1H NMR (500MHz, CD3OD): δ 7.78 (d, J=8.1Hz, 1H), 5.86 (d, J=2.8Hz, 1H), 5.50 (d, J=8.0Hz, 1H) 4.96 (dd, J=2.8, 6.3Hz, 1H), 4.63 (dd, J=3.4, 6.3Hz, 1H), 4.22 (m, 1H), 3.75 (dd, J=3.6, 11.9Hz, 1H), 3.72 (dd, J=4.5, 11.9Hz, 1H), 1.54 (s, 3H), 1.30 (s, 3H); 13C NMR (100MHz, CD3OD): δ 166.8, 153.1, 144.5, 113.1, 101.6, 92.5, 88.5, 85.6, 82.4, 62.1, 26.7, 25.5. Data are in agreement with that previously reported [39].
95%
With toluene-4-sulfonic acid In acetonitrile for 3h; Inert atmosphere; Reflux;
94%
With toluene-4-sulfonic acid In acetonitrile for 3h; Reflux;
94%
With toluene-4-sulfonic acid In acetonitrile for 3h; Reflux;
4.2. Preparation of 20,30-O-Isopropylidene Uridine (9)
Uridine 8 (500 mg, 2 mmol) was dissolved in acetonitrile (30 mL). 2,2-dimethoxypropane (2 mL,16 mmol) and p-toluenesulfonic acid (15 mg) were added and the mixture was refluxed for 3 h.The reaction mixture was concentrated under vacuum to give a brown residue. The residue waspurified by flash chromatography (100% DCM to 100% acetone in a stepwise gradient) to give thedesired compound 9. [47] (534 mg, 94% yield). 1H-NMR (CD3OD, 500 MHz) (ppm) 7.86 (d, J = 8.1 Hz,1H), 5.89 (d, J = 3.0 Hz, 1H), 5.71 (d, J = 8.1 Hz, 1H), 4.93 (dd, J = 6.6, 3.2 Hz, 1H), 4.84 (dd, J = 6.5,3.4 Hz, 1H), 4.23 (m, 1H), 3.79 (dd, J = 12.0, 3.6 Hz, 1H), 3.73 (dd, J = 12.2, 3.8 Hz, 1H), 1.57 (s, 3H),1.38 (s, 3H). 13C-NMR-DEPTQ (CD3OD, 126 MHz) (ppm) 164.8, 150.6, 142.4, 113.7, 101.2, 92.7, 86.9,84.4, 80.8, 61.6, 26.1, 24.1. MS (ES+) m/z 285.1 ([M + H]+, 100%), 569.2 ([2M + H]+, 67%), HRMS (ES+)m/z calc. for C12H17N2O6 [M + H]+ 285.1081, found 285.1075.
94%
With stannous chloride In acetonitrile at 20℃; for 1h;
93%
With toluene-4-sulfonic acid In acetone at 20℃; for 2h;
91%
With sulfuric acid In acetone at 0 - 55℃; for 8.5h; Industrial scale;
90%
With toluene-4-sulfonic acid In N,N-dimethyl-formamide
90%
With toluene-4-sulfonic acid In acetone at 20℃; for 1.5h; Inert atmosphere;
88%
With toluene-4-sulfonic acid In acetone at 20℃; Inert atmosphere;
2',3'-O-Isopropylideneuridine (4).
To a stirred solution of uridine (3.65 g, 15 mmol) in anhydrous acetone (100 mL) was added 2,2-dimethoxypropane (10.05 mL, 82 mmol) and p-toluenesulfonic acid monohydrate (0.15 g, 0.75 mmol). The reaction mixture was stirred overnight at room temperature. The solvent was removed under reduced pressure, and the residue was dissolved in EtOAc, washed with 5% NaHCO3, brine, and dried over sodium sulfate anhydrous. The solvent was removed under vacuum, and the crude was purified by silica gel column chromatography (3:1 ratio of EtOAc to hexanes) to afford compound 4 (3.75 g, 88%).
85%
With toluene-4-sulfonic acid at 20℃; for 3h;
82%
With toluene-4-sulfonic acid In acetone at 20℃; Inert atmosphere;
Synthesis of 2′,3′-O-isopropylideneuridine (1)
Anhydrous acetone (100 mL) was added to a mixture ofuridine (3.6 g, 15 mmol), 2,2-dimethoxypropane (10.05 mL,82 mmol) and p-toluenesulfonic acid monohydrate (0.45 g,2.5 mmol). The reaction mixture was stirred under nitrogenatmosphere at room temperature overnight. The solvent wasremoved under reduced pressure and the crude was redissolvedin EtOAc, washed with 5% NaHCO3, brine, anddried over anhydrous Na2SO4. The mixture was then concentratedunder reduced pressure and purified by CombiflashRf+ chromatography (50% ethyl acetate in hexanes)to afford pure compound 1 (82%, 3.53 grams). 1H-NMR(500 MHz, DMSO-d6): δ 11.37 (s, 1H, NH), 7.78 (d, 1H,J = 7.8 Hz, H-6), 5.82 (d, 1H, J = 3.0 Hz, H-1′), 5.63 (d,1H, J = 7.8 Hz, H-5), 5.14 (t, 1H, J = 4.9 Hz, OH-5′), 4.88(dd, 1H, J = 6.4 Hz, H-2′), 4.73 (dd, 1H, J = 3.4 Hz, H-3′),4.06 (q, 1H, J = 7.8 Hz, H-4′), 3.60-3.52 (m, 2H, H-5′),1.47 (s, 3H, CH3), 1.28 (s, 3H, CH3); 13C NMR (125 MHz,DMSO-d6): δ 163.6, 150.8, 142.4, 113.4, 102.2, 91.6, 87.0,84.1, 80.9, 61.7 27.5, 25.6.
81%
With toluene-4-sulfonic acid In water monomer; acetone at 60℃; for 2h; Inert atmosphere;
81%
With toluene-4-sulfonic acid In acetone at 60℃; for 2h; Inert atmosphere;
80%
With toluene-4-sulfonic acid In acetone at 20℃;
79%
79%
With toluene-4-sulfonic acid In acetone for 24h; Reflux; Inert atmosphere;
General procedure for 2',3'-O-isopropylidenenucleosides
General procedure: To a suspension of N6-benzoyladenosine, uridine, or N2-isobutyrylguanosine (15.70 mmol) in anhydrous acetone (100 mL), p-toluenesulfonic acid monohydrate (0.30 g, 1.57 mmol) and 2,2-dimethyoxypropane (17.3mL, 141.19 mmol) were added. After 24 h reflux,the mixture was filtered to remove any unreacted starting material and the filtrate was concentrated under vacuum. The residue was dissolved in EtOAc, washed with5% NaHCO3, brine, and was dried over sodium sulfate anhydrous. The crude mixture was washed with anhydrous diethyl ether and filtered to afford pure compound1, 7, or 13, respectively.
73%
With toluene-4-sulfonic acid In acetone
65%
With toluene-4-sulfonic acid In acetone at 0℃; for 1h; Reflux; Inert atmosphere;
61%
With toluene-4-sulfonic acid In N,N-dimethyl-formamide at 20℃; for 18h;
To a solution of uridine (700 mg, 2.87 mmol) in dry N,N-dimethylformamide (70 ml) were added 2,2-dimethoxypropane (7 ml, 57 mmol) and p-toluenesulfonic acid monohydrate (70 mg, 0.37 mmol) at room temperature. After stirring for 18 h, the reaction was quenched by adding saturated NaHCO3 solution to the reaction mixture until the pH became 7.0. The reaction mixture was diluted with distilled water and loaded onto an HP-20 column and the column was washed with distilled water. The column was then eluted with MeOH and the eluent was concentrated under reduced pressure to give 2,3-O-isopropylidene-1-uracil-β-D-ribofuranoside (3, 500 mg, 61% yield). Spectroscopic data were in good agreement with reported values.
58%
With DOWEX (50WX8) ion exchange resin (H+) In acetone
56%
With toluene-4-sulfonic acid In N,N-dimethyl-formamide at 40℃; for 3h; Molecular sieve;
24.8%
With toluene-4-sulfonic acid In N,N-dimethyl-formamide at 40℃; for 1.5h; Inert atmosphere; Molecular sieve;
With toluene-4-sulfonic acid
With toluene-4-sulfonic acid In tetrahydrofuran
Stage #1: 2,2-dimethoxy-propane; uridine With toluene-4-sulfonic acid In water monomer; N,N-dimethyl-formamide at 45℃; for 2h; Molecular sieve;
Stage #2: With Amberlyst A-21 resin In water monomer; N,N-dimethyl-formamide at 20℃; for 0.333333h;
45 g
With sulfuric acid In acetone Inert atmosphere;
8
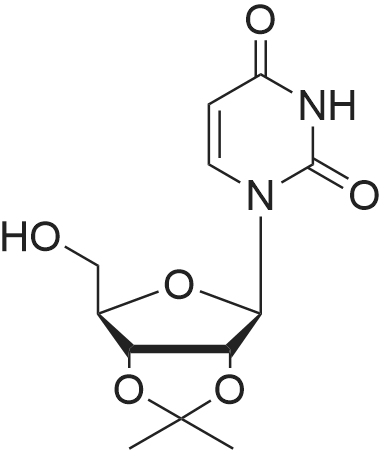
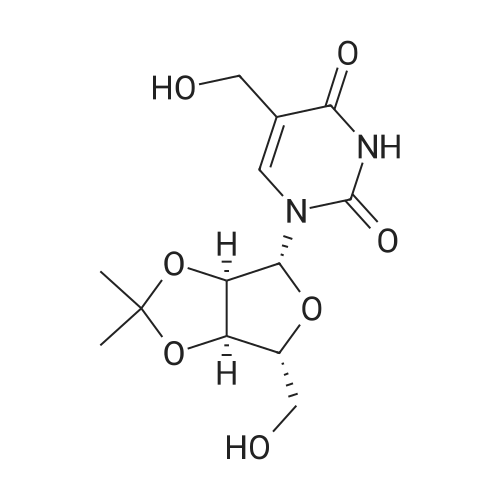
A 2L 3-neck RBF was charged with l-[(3R,4S,5R)-3,4-dihydroxy-5- (hydroxymethyl)tetrahydrofuran-2-yl]pyrimidine-2,4-dione (61.4g, 251.43 mmol) and acetone (1400 mL). The resulting slurry was stirred at RT and sulfuric acid (2 mL was added. Stirring was continued ovemite. The clear colorless solution was quenched/adjusted to basic pH with 100 mL of trimethylamine. The crude solution was concentrated under reduced pressure to yield a pale yellow oil. The residue was dissolved in 600 mL of EtOAc and washed with water x 2, bicarb x 2, water, brine x 2 and dried over sodium sulfate. The colorless solution was concentrated under reduced pressure to yield l-[(3aR,6R,6aR)-6-(hydroxymethyl)-2,2-dimethyl- 3a,4,6,6a-tetrahydrofuro[3,4-d][l,3]dioxol-4-yl]pyrimidine-2,4-dione (45 g) as a white solid.
With toluene-4-sulfonic acid In N,N-dimethyl-formamide
Stage #1: 2,2-dimethoxy-propane; uridine In acetone for 0.166667h;
Stage #2: With sulfuric acid In acetone at 20℃; for 0.5h;
1.1 Step 1: Synthesis of compound S2
Dissolve uridine (5g, 20.5mmol) in acetone (150mL), slowly drip 2,2-dimethoxypropane (12.6mL, 102.5mmol), stir for 10min, slowly drip into concentrated sulfuric acid (1.5mL, 26.7mmol), after stirring at room temperature for 30 minutes, slowly adding sodium bicarbonate solution (2.5 g sodium bicarbonate dissolved in 20 mL of water) to quench, stirring for 10 minutes, the solvent was removed under reduced pressure,Extract with ethyl acetate (50 mL), wash with saturated brine, and dry with anhydrous sodium sulfate,After filtration and concentration, the crude compound S2 (5.5 g, 95%) was obtained, which was directly used in the next step.
With sulfuric acid; triethylamine In acetone at 0 - 57℃;
1
A 100 L cylindrical vessel was charged with uridine (11.66 kg), acetone (70 L), 2,2- dimethoxypropane (1.05 equiv), and sulfuric acid (0.01 equiv). The reaction mixture was heated at 50°C-57°C until the reaction was deemed complete. Triethylamine (0.04 equiv) was added, followed by seed, and the slurry was cooled to 0°C-5°C. The crystalline solid was collected and washed with MTBE to afford Intermediate 1.
With pyridine; at 20℃; for 16.0h;Cooling with ice;
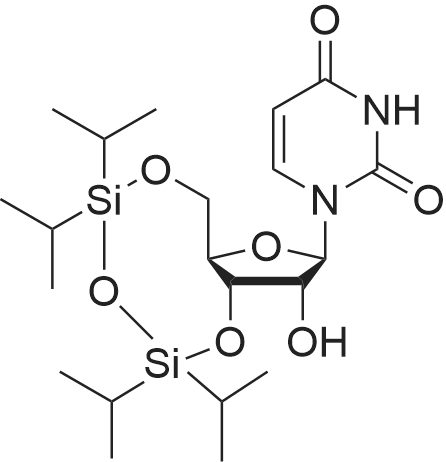
To an ice cooled solution of 82-1 (50 g, 204.9 mmol) in dry Py (400 mL) was added TIPDSCl (70.78 g, 225.4 mmol) dropwise. The mixture was stirred at R.T. for 16 h, and then concentrated at low pressure. The residue was purified by chromatography using 20% EA in PE to generate 82-2 (111.5 g, 100%) as a white solid.
100%
With pyridine; at 20℃; for 16.0h;Cooling with ice;
To an ice cooled solution of 226-1 (50 g, 204.9 mmol) in dry Py (400 mL) was added TIPDSCl (70.78 g,225.4 mmol) dropwise. The mixture was stirred at RT for 16 h, and then concentrated at low pressure. Theresidue was purified by chromatography using 20% EA in PE to generate 226-2 (111.5 g, 100%) as a white solid
100%
With pyridine; at 20℃; for 16.0h;
To an ice cooled solution of 226-1 (50 g, 204.9 mmol) in dry pyridine (400 mE) was added TIPDSC1 (70.78 g, 225.4 mmol) dropwise. The mixture was stirred at RT for 16 h, and then concentrated at low pressure. The residue was purified by chromatography using 20% EA in PE to generate 226-2 (111.5 g, 100%) as a white solid.
100%
With pyridine; at 20℃; for 16.0h;
To an ice cooled solution of 226-1 (50 g, 204.9 mmol) in dry pyridine (400 mL) was added TIPDSCl (70.78 g, 225.4 mmol) dropwise. The mixture was stirred at RT for 16 h, and then concentrated at low pressure. The residue was purified by chromatography using 20% EA in PE to generate 226-2 (111.5 g, 100%) as a white solid.
92%
With pyridine; at 20℃; for 16.0h;Inert atmosphere;
In a 100ml two-necked bottle, add 40ml of pyridine and 2.4g (10mmol) of uridine, under nitrogen protection,3.2g (10mmol) 1,3 dichloro-1,1,3,3-tetraisopropyl disiloxane was added dropwise,Then the reaction was stirred at room temperature for 16h. After the reaction was detected by TLC, the pyridine was removed by spin-drying, 50 ml of dichloromethane was added, washed three times with water (30 ml × 3), dried over anhydrous sodium sulfate, and the solvent was spin-dried. Column chromatography gave 4.5 g of the compound of formula II with a yield of 92%.
91%
With pyridine; at 0℃;
A solution of uridine (2, 9.8 g, 40 mmol) in dry pyridine (50 mL) was treated with 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (TIPDSCl, 12.6 mL, 40.4 mmol) and stirred at 0 C overnight. The reaction mixture was diluted with EtOAc (100 mL) and washed with 1 M HCl (50 mL x 3). The combined aqueous phase was extracted with EtOAc (50 mL x 3). All the organic layers were combined, dried over anhydrous Na2SO4, concentrated, and then purified through silica gel column chromatography [EtOAc/petroleum ether (PE) 25:75] to give compound 3 (17.7 g, 91%) as a colorless solid: mp 75-76 C; 1H NMR (400 MHz, CDCl3) delta 9.88 (br, s, 1H), 7.80 (d, J = 8.4 Hz, 1H), 5.75 (s, 1H), 5.71 (dd, J = 8.0, 2.0 Hz, 1H), 4.32-4.29 (m, 1H), 4.24-4.16 (m, 3H), 4.00 (dd, J = 13.2, 2.4 Hz, 1H), 1.11-1.00 (m, 28H); 13C NMR (100 MHz, CDCl3) delta 163.7, 150.3, 139.9, 102.0, 91.0, 81.9, 75.1, 68.6, 60.1, 17.5, 17.4, 17.3, 17.2, 17.04, 16.97, 16.9, 16.8, 13.4, 13.0, 12.9, 12.4; HRMS (m/z) [M + Na]+ calcd for C21H38N2NaO7Si2, 509.2110, found 509.2101.
90%
With pyridine; at 20℃; for 48.0h;Inert atmosphere;
In a 100 ml two-necked flask, 40 ml of pyridine and 2.4 g (10 mmol) of uridine were separately added under nitrogen protection conditions.3.2 g (10 mmol) of 1,3 dichloro-1,1,3,3-tetraisopropyldisiloxane was added dropwise, followed by stirring at room temperature for 2 days.After the TLC reaction was completed, the pyridine was removed by spin-drying, 50 ml of dichloromethane was added, and the mixture was washed three times with water (30 ml x 3).After drying over anhydrous magnesium sulfate, the solvent was evaporated to dryness, and then applied4.4 g of 3',5'-O-(1,1,3,3-tetraisopropyl-1,3-disiloxane)uridine were obtained in a yield of 90%.
90%
With pyridine; at 20℃; for 48.0h;Inert atmosphere;
In a 100 ml two-necked flask, 40 ml of pyridine and 2.4 g (10 mmol) of uridine were separately added under nitrogen protection conditions.3.2 g (10 mmol) of 1,3 dichloro-1,1,3,3-tetraisopropyldisiloxane was added dropwise.The reaction was then stirred at room temperature for 2 days. After the TLC reaction was completed, the pyridine was removed by spin-drying, and 50 ml of dichloromethane was added.After washing three times with water (30 ml x 3), dried over anhydrous magnesium sulfate, the solvent was evaporated to dryness.Get 4.4g3',5'-O-(1,1,3,3-tetraisopropyl-1,3-disiloxane) uridine,The yield was 90%.
85%
at 20℃;Inert atmosphere; Cooling with ice;
Step 1: Compound P16-2 [0236] To an ice-cold solution of P16-1 (2.0 g, 7.6 mmol) in anhydrous pyridine (20 mL) was added 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (2.40 g, 7.6 mmol) under N2. The reaction mixture was stirred at room temperature overnight. The solvent was removed under vacuum. The resulting residue was diluted with ethyl acetate (100 mL), and washed with saturated NaHCO3 and brine. The organic layer was separated, dried over anhydrous Na2SO4, and filtered. The filtrate was concentrated in vacuo to give a residue. The residue was purified by silica gel column chromatography (DCM/MeOH=100/1 to 50/1) to give P16-2 as a white solid (3.2 g, 85%).
85%
With pyridine; dmap; at 0 - 20℃; for 18.0h;Inert atmosphere;
General procedure: A solution of uridine (1.68 g, 6.87 mmol) in distilled pyridine (20.5 mL) was prepared and cooled to 0 C in an ice-water bath,before the addition of the 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane(2.3 mL, 7.2 mmol). The reaction mixture was stirred overnight and subsequently quenched by the addition of distilled H2O(21 mL). The solvents were removed under reduced pressure and the residue obtained was partitioned between EtOAc (70 mL) and distilled H2O (2 mL). The organic fraction was dried over anhydrous Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel, using a gradient of EtOAc-hexane (10-50%) for elution. Product 10 (2.80 g) was obtained as a white foam, yield: 85%. 1H and 13C NMR spectra obtained correlated well to those reportedby Matsuda and co-workers.60 1H NMR (300 MHz, CDCl3): d 9.64(br s, 1H), 7.77 (d, J = 8.1 Hz, 1H), 5.75 (d, J = 2.1 Hz, 1H), 5.73-5.69 (m, 1H), 4.32 (dd, J = 4.7, 8.8 Hz, 1H), 4.25-4.14 (m, 3H),4.00 (dd, J = 2.5, 13.2 Hz, 1H), 3.62 (br s, 1H), 1.10-1.02 (m, 28H);13C NMR (50 MHz, CDCl3): d 163.5, 150.2, 139.9, 102.0, 90.9, 81.8,75.1, 68.7, 60.1, 17.4, 17.33, 17.25, 17.2, 17.0, 16.91, 16.88, 16.8,13.3, 12.93, 12.88, 12.4; HRMS (ESI) m/z calculated for C21H39N2O7-Si2 (M+H)+ 487.22903, found 487.22848.
85%
at 0 - 20℃; for 3.25h;Inert atmosphere;
A solution of uridine (lO.Og, 40.9 mmol, 1.0 eq.) under argon atmosphere, at 0 C was added l,3-dichloro-l,l,3,3-tetraisopropyldisiloxane (14.4 mL, 45.0 mmol, 1.1 eq.) drop wise via syringe over a 15 min period. The reaction mixture was warmed to room temperature and further stirred for 3 hours. The reaction was quenched with MeOH (lOmL) and the solvent was evaporated. The viscous residue was dissolved in DCM (400 mL) and washed with sat. NaC03H, brine, and dried over MgS04. The solvent was evaporated and coevaporated with toluene to remove traces of pyridine. The product was purified by silica gel column chromatography, eluding with hexane:ethylacetate (3:2) to provide 17 (17.0 g, 85 % y ield) as white foam.
85%
With pyridine; at 0 - 20℃; for 3.15h;Inert atmosphere;
A solution of uridine (lO.Og, 40.9 mmol, 1.0 eq.) in dry pyridine under argon atmosphere, at 0 C was added l,3-dichloro-l,l,3,3-tetraisopropyldisiloxane (14.4 mL, 45.0 mmol, 1.1 eq.) drop wise via syringe over a 15 min period. The reaction mixture was warmed to room temperature and further stirred for 3 hours. The reaction was quenched with MeOH (lOmL) and the solvent was evaporated. The viscous residue was dissolved in DCM (400 mL) and washed with sat. NaC03H, brine, and dried over MgS04. The solvent was evaporated and coevaporated with toluene to remove traces of pyridine. The product was purified by silica gel column chromatography, eluding with hexane:ethylacetate (3:2) to provide 17 (17.0 g, 85 % yield) as white foam
76%
In pyridine; at 20℃; for 17.0h;
Uridine (15, 100.0 g, 409.5 mmol) was co-evaporated to dryness with anhydrous pyridine (600 mL) and re-suspended in anhydrous pyridine (700 mL). To this stirred fine suspension was added l,3-dichloro-l,l,3,3-tetraisopropyldisiloxane (135.7 g, 482.5 mmol) over 60 min at ambient temperature. After stirring the fine suspension for 17 h at ambient temperature, the reaction was quenched by adding methanol (20 mL) and then concentrated under reduced pressure. The residue was partitioned between ethyl acetate (1.5 L) and water (2 L). The organic layer was further washed with 5% hydrochloric acid (2 x 1 L), brine (500 mL), dried over solid sodium sulfate (50 g), filtered and concentrated under reduced pressure to the crude product, ca 250 g. The residue was subjected to a filtration column using silica gel (1.75 kg) and a gradient of ethyl acetate in hexanes 20-65%. The pure product fractions as judged by a homogenous TLC (Rf 0.55 in 1 : 1 hexanes-ethyl acetate) were combined and concentrated under reduced pressure and dried (40 C, 0.2 mm Hg, 24 h) to afford 145.5 g (76%) of 16 as a white foam solid. An additional fraction (35 g) of slightly impure 16 was also collected. 1H NMR (DMSO-J6) delta (ppm) 11.35 (s, IH, NH), 7.66 (d, IH, J= 7.6 Hz, H-6), 5.57 (d, 1Eta, J= 4.8 Hz, 2'-OH), 5.50-5.49 (m, 2Eta, V-H and HS), 4.14-4.18 (m, 3H, 2', 3', 4'-H), 3.97-3.87 (m, 2Eta, 5'-Ha and Hb), 1.02- 0.95 (m, 28Eta, CH(CH3)2).
75%
With pyridine; at 20℃; for 14.0h;
Uridine 31a (10.0 g, 41 mmol) and anhydrous pyridine (50 mL) were co-evaporated to dryness and resuspended in anhydrous pyridine (60 mL). <strong>[69304-37-6]1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane</strong> (13.5 g, 48.3 mmol) was added to the stirred suspension at ambient temperature. After the fine suspension was stirred at ambient temperature for 14 h, methanol (3 mL) was added and quenched and concentrated under reduced pressure. The residue was partitioned between ethyl acetate (150 ml) and water (200 ml). The organic layer was further washed with 5% hydrochloric acid (2X 100 mL), brine (50 mL), dried over solid sodium sulfate, filtered and concentrated under reduced pressure to crude product. Column chromatography (20-50% ethyl acetate-hexane) carried out. Obtained a white foam solid 32a, 14.5g (75%)
70%
With pyridine; for 16.0h;Inert atmosphere;
To a solution of uridine (2.44 g, 10.0 mmol) in pyridine (50 mL) 1,1,3,3-tetraisopropyl-1,3-dichloro-disiloxane (3.84 mL, 12.0 mmol) was added dropwise with stirring under Ar. Stirring was continued for 16 h. The reaction mixture was diluted with CH2Cl2 and washed with sat. aq. NaHCO3 and brine. The aqueous phases were reextracted with CH2Cl2. The organic layers were combined, dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography to give the title compound (3.44 g, 7.07 mmol, 70 % yield). 1H NMR (400 MHz, CDCl3) delta 8.80 (s, 1H), 7.63 (d, J = 8.2 Hz, 1H), 5.67 (s, 1H), 5.63 (d, J = 8.1 Hz, 1H), 4.30 (dd, J = 8.6, 4.9 Hz, 1H), 4.15-4.11 (m, 2H), 4.06-4.03 (m, 1H), 3.94 (dd, J = 13.1, 2.9 Hz, 1H), 1.03-0.95 (m, 28H).
With Geobacillus kaustophilus ATCC 8005 entrapped in agarose matrix supplemented with bentonite; In aq. phosphate buffer; at 60℃; for 6h;pH 7.0;Green chemistry;
Ribavirin biosynthesis was assayed using 5 × 109 colony-forming units (CFU) in 1 mL sodium phosphate buffer (30 mM, pH7.0) containing equimolar concentrations of uridine (Urd) and TCA(2.5 mM). Reactions were performed at 60C and 200 rpm in aperiod of 16 h.
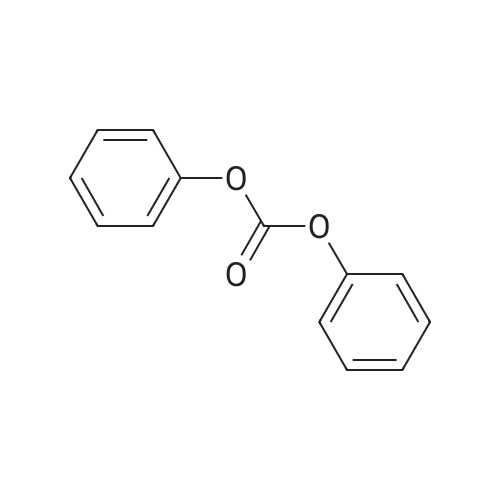
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N,N,N,N,N-hexamethylphosphoric triamide at 150℃; for 0.333333h;
92%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide for 4h;
90%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 80℃; for 3h;
90%
With bis(phenyl) carbonate; sodium bicarbonate In N,N-dimethyl-formamide at 150℃; for 0.333333h;
89%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 110℃; for 2.5h;
84%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 110 - 120℃; for 4.5h;
80%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 140℃; for 1.66667h;
80%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 140℃;
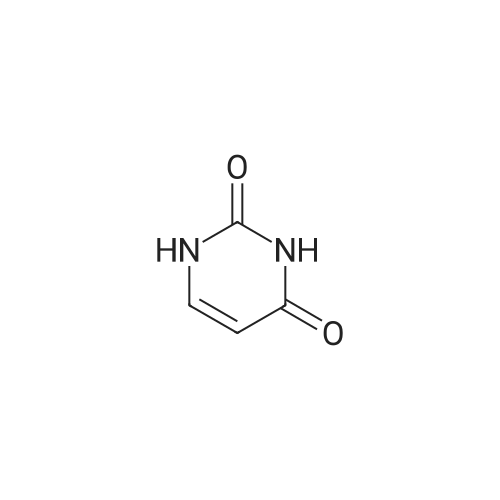
2 Synthesis of 2,2'-cyclouridine 2
Example 2 Synthesis of 2,2'-cyclouridine 2 500 mg (2.05 mmol) of uridine 1, 570 mg of (2.65 mmol, 1.3 gram-equivalent (eq)) of diphenyl carbonate, and 10 mg (0.12 mmol, 0.059 eq) of sodium bicarbonate are dissolved in 1 mL of dimethyl formamide and heated to 140° C. After the starting materials are completely consumed, the solution is cooled down to room temperature to form white precipitates. The white precipitates are washed by ice formic acid. Residual part of the solution is removed at reduced pressure. After collection, a white powder product (2,2'-cyclouridine) 2 is obtained. Its weight and yield are 370 mg and 80%, respectively. Mp: 250˜253° C. [lit.30, 244˜246° C., lit.31, 238˜244° C.; commercial authentic sample: 248˜251° C.]. Anal. C9H10N2O5, calcd: C, 47.79, H, 4.46, N, 12.39. found: C, 47.41; H, 4.44; N, 12.41; MW: 226.2, ESI+Q-TOF MS, M=226.1 (m/z), [M+H]+=227.1, [M+Na]+=249.0, [2M+H]+=453.2, [2M+Na]+=475.2; 1H-NMR (500 MHz, CD3OD): δ 3.45 (dd, J5'a,4'=4.0, J5'a,5'b=12.0 Hz, 1H, H5'a), 3.49 (dd, J5'b,4'=4.0, J5'b,5'a=12.0 Hz, 1H, H5'b), 4.23 (dd, J4',5'=4.0, J4',5'b=4.0 Hz, 1H, H4'), 4.54 (s, 1H, H3'), 5.28 (d, J2',1'=6.0 Hz, 1H, H2'), 6.05 (d, J5,6=7.5 Hz, 1H, H5), 6.37 (d, J1',2'=6.0 Hz, 1H, H1'), 7.82 (d, J6,5=7.5 Hz, 1H, H6).
79%
With bis(phenyl) carbonate In N,N-dimethyl-formamide
75%
With bis(phenyl) carbonate; sodium hydrogencarbonate; uridine In N,N-dimethyl-formamide at 120℃; for 4h;
74.6%
With 4-hydroxymethyl-1,3-dioxolan-2-one; sodium carbonate at 100℃; for 2h;
1
EXAMPLE No. 1; . Synthesis of cyclouridine; 220 grams of Uridine (MW 244.202) were reacted in 660 ml of glycerolcarbonate, in the presence of 3.3 g of Sodium Carbonate, for 2 hours at 100°C, and the product synthesis was monitored using HPLC (see the diagram below) [Show Image] At the end of the reaction the reaction mixture (containing crystals in suspension) formed was concentrated under vacuum at 60°C to remove the excess solvent. The residue was then taken up with methanol resulting in further crystal formation. After cooling at 4°C overnight, the crystalline solid was separated by filtration and dried thus obtaining 152g of Cyclouridine (MW 226.187) with a purity measured by HPLC of 99.9%. The yield (w/w) was thus of 74.6%. The reaction has a duration of two hours and can be carried out at a lower temperature than the one used in the known methods. Its processing according to the method of the present invention is simpler than that of the known methods, since it is possible, in addition to the temperature, to also reduce the reaction time, another essential parameter for an industrial process.
74%
Stage #1: uridine With bis(phenyl) carbonate In N,N-dimethyl-formamide at 80℃;
Stage #2: With sodium hydrogencarbonate In N,N-dimethyl-formamide at 115℃;
74%
Stage #1: uridine With bis(phenyl) carbonate In N,N-dimethyl-d<SUB>6</SUB>-formamide at 85℃; Inert atmosphere;
Stage #2: With sodium hydrogencarbonate In N,N-dimethyl-formamide at 120℃; for 4h;
72%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 120℃; for 4h;
71%
With bis(phenyl) carbonate In N,N-dimethyl-formamide at 80℃;
56%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 130℃; for 0.5h;
45.6%
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 100℃; for 14h;
1 Synthesis of Compound 5:
Synthesis of Compound 5: Compound 4 (10.0g, 41mmol) and diphenyl carbonate (10.0g, 49mmol) was dissolved in dimethylformamide (10mL), was added sodium bicarbonate (0.35g, 4.1mmol),The reaction was stirred at 100 deg.] C for 14 hours.The reaction was cooled to room temperature, poured into ice water (400mL) with methylene chloride (3 × 200mL)And the combined organic phases were dried over anhydrous sodium sulfate, and concentrated under reduced pressure to give a viscous mixture of methylene chloride and the solid was washed with acetonitrile three times to give a white solid compound 5 (4.2g, 45.6%)).
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N,N,N,N,N-hexamethylphosphoric triamide at 150℃; for 0.333333h; Yield given;
With diphenyl sulfite; sodium hydrogencarbonate In N,N,N,N,N,N-hexamethylphosphoric triamide at 150℃; Yield given;
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 100℃; for 4h;
11.31 g
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl acetamide at 100℃; for 5h;
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 150℃;
Multi-step reaction with 2 steps
1: 85 percent / thionyl chloride / acetonitrile / 4.5 h / 5 °C
2: 94 percent / sodium acetate / dimethylformamide / 4 h / 85 °C
With bis(phenyl) carbonate at 120℃; Inert atmosphere;
With bis(phenyl) carbonate; sodium bicarbonate; N,N-dimethyl acetamide at 100℃; for 5h;
Preparation of 2,2'-Anhydro-1-β-D-arabinofuranosyluracil
Preparation of 2,2'-Anhydro-1-β-D-arabinofuranosyluracil. Uridine (12.21g, 50mmol), diphenyl carbonate (11.79g, 55mmol), sodium hydrogen carbonate (0.21g, 2.5mmol) and dry DMA (10ml) were heated together, with stirring, at 100°C. After 5h, the products were cooled to room temperature, and diethyl ether (100ml) was added with stirring. After 2 hours, the colourless precipitate (11.70g) was collected by filtration and was washed with ether (2 x 50ml). The sole nucleoside constituent of the precipitated material was identified as 2,2'-anhydro-1-β-D-arabinofuranosyluracil (calculated quantitative yield, 11.31g) by comparison with authentic material.
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide at 80℃; for 2h;
Multi-step reaction with 2 steps
1.1: pyridine / 18 h / 0 - 5 °C
1.2: 21 h / 20 °C
2.1: ammonia / methanol / 3 h / 0 - 20 °C
With bis(phenyl) carbonate; sodium hydrogencarbonate; phosphoramide at 150℃;
1 Example 1 Synthesis of Compound VIII '
A solution of 24.4 g of uridine IX '(Uridine), 32.1 g of diphenyl carbonate (1.5 eq), 8.4 g of sodium bicarbonate, 250 mL of phosphoric acid triamide was added to a 500 mL three-necked flask. Heated to 150 ° C reaction, HPLC to control the reaction is complete, then cooled to room temperature, add water, extracted with chloroform, combined extract, anhydrous sodium sulfate drying, filtration. The filtrate enters the next step.
Multi-step reaction with 2 steps
1: thionyl chloride / acetonitrile / 2 h / 5 - 70 °C
2: sodium acetate / N,N-dimethyl-formamide / 1 h / 85 °C
With bis(phenyl) carbonate; sodium hydrogencarbonate In N,N-dimethyl-formamide
With iodine; nitric acid; In chloroform; at 80℃; for 5h;
In a round bottom flask, uridine (1 g, 4.1 mmol) and iodine powder (1 .15 g, 4.5 mmol) were dissolved in a mixture of CHCI3 (55 ml) and 1 M HN03 (10 ml). The reaction was heated at reflux (80C) for 5h. Reaction progress was monitored by TLC (30 % methanol in chloroform, Rf: 0.60; RfsM: 0.45). Upon cooling of the reaction mixture to 4C, crystals of the title compound formed as colourless needles. The precipitate was collected by filtration and dried under vacuum overnight to provide 1.39 g (92 %) of 5-iodouridine 1 ; deltaEta (DMSO-d6, 300 MHz) 3.49 - 3.71 (2H, m, H-5'), 3.84 - 3.88 (1 H, m, H-4'), 3.98 (1 H, t, J = 5.0 Hz, H-3'), 4.02 (1 H, t, J = 5.0 Hz, H-2'), 5.08 (1 H, d, J = 5.3 Hz, OH-3'), 5.27 (1 H, t, J = 4.7 Hz, OH-5'), 5.43 (1 H, d, J = 5.4 Hz, OH- 2'), 5.71 (1 H, d, J = 4.6 Hz, H-1 '), 8.48 (1 H, s, H-6) 1 1 .69 (1 H, s, NH); 5C (DMSO-d6, 300MHz) 61 .2 (C-5'), 70.3 (C-3'), 70.9 (C-5), 75.0 (C-2'), 85.7 (C-4'), 89.5 (C-1 '), 146.0 (C-6), 152.5 (C-2), 162.9 (C-4). m/z (ESI) 388.0000 [M+NH4]+, requires 388.0000.
78%
With silver(II) sulfate; iodine; In methanol; at 20℃;Inert atmosphere;
Iodine (0.508 g) in 2.0 mL of methanol was added dropwise to a suspension of silver sulfate (0.624 g) and uridine (0.488 g) in 10 mL of methanol. The reaction mixture was stirred at room temperature until the orange color disappeared. The precipitate was removed by filtration. The filtrate was concentrated and recrystallized from methanol, affording a pale yellow solid (78%). 1HNMR (CD3OD) delta 8.511 (1H, d, J = 3.2 Hz), 5.776 (1H, t, J = 1.6 Hz), 4.085(2H, m), 3.938 (1H, m), 3.814 (1H, d, J = 2.4 Hz), 3.784 (1H, d, J = 2.8 Hz), 3.674 (1H, d, J = 2.4 Hz), 2.644 (1H, d, J = 2.8 Hz); HRMS [M+1] calcd 370.9662, found: 370.9665.
72.2%
With iodine; nitric acid; at 110℃; for 4h;
Take uridine (13.06g, 53.50mmol) was dissolved in 252ml of dilute nitric acid, heated to 110 ,Was added (10.09g, 39.92mmol) elemental iodine, the reaction was monitored by thin layer chromatography case, exhibitionEluent, CH2Cl2: CH3OH = 7: 1, about 4 hours after the reaction raw material point disappears, and the reactionComplete, the reaction was stopped. After leaving to cool and extracted 3 times with 30ml petroleum ether, collecting the lower solution,Upper solution was extracted with 30ml of deionized water, lower solutions were twice extracted. Standing whiteThe solid precipitated, placed in the upper refrigerator overnight, a lot of white solid was obtained, that is, uridine 5-I-(14.30g, 38.64mmol), the yield was 72.22%
72%
With iodine; silver sulfate; In methanol; at 20℃; for 0.5h;
Iodine (2.0mmol, 1eq) in 2mL of methanol was added to a suspension of silver sulfate (2.0mmol, 1eq) and uridine (2.0mmol, 1eq) in 10mL of methanol. The reaction was stirred at room temp until the color disappeared (~30min). The precipitate was removed by filtration over celite. The filtrate was concentrated, giving a pale yellow solid as product, which was used immediately in the next reaction.
40%
With iodine; iodic acid; acetic acid; In tetrachloromethane; water; at 40℃; for 2h;
General procedure: The suspension of nucleosides 2a,b (19 mmol) in water (5.7 mL) was treated with HIO3(9.7 mmol, 1.7 g), AcOH (15.2 mL) and a solution of iodine (11.22 mmol, 2.85 g) inCCl4 (3.8 mL). The resulting mixture was stirred at 40C for 2 h until the starting materialwas consumed or some by-product was formed (monitored by HPLC). After that,water (20 mL) was added. The reaction mixture was cooled to 4C and filtered. The precipitatewas washed with water (2 £ 10 mL). The combined solutions were diluted withwater (250 ml) and extracted with benzene (3 £ 150 mL). The aqueous layer was evaporatedunder reduced pressure. The product was purified by RPC in a linear gradient ofEtOH in water (0-30%) to give the product 3a,b.
2-Acetylamino-3-{3-[1-((2R,3R,4S,5R)-3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-2,4-dioxo-1,2,3,4-tetrahydro-pyrimidin-5-yl]-4-hydroxy-phenyl}-N-ethyl-propionamide[ No CAS ]
Multi-step reaction with 5 steps
1: triethylamine; acetonitrile / 1 h / Ambient temperature; other reagent
2: POCl3 / acetonitrile / 0.25 h / 0 °C
3: acetonitrile / 3 h / 0 °C
4: 1.62 g / acetic acid; methanol / 5 h / Ambient temperature
5: 97 percent / ethanol / 1.5 h / Ambient temperature
Multi-step reaction with 3 steps
1: pyridine
2: P2S5
Example 1 : Synthesis of fluorescent nucleoside analogsExtending upon the established synthesis of the 2'-deoxy ribose nucleoside analogs 1 and 2 (Fig. 2), a first set of novel furano and pyrrolo-nucleosides with modification in the ribose portion was prepared. Fluorescent analogues of the prodrugs ddT, ddC, and d4T were prepared as outlined in Fig. 6. Briefly, reduction of uridine (21) led to 2',3'-didehydro-2',3'-dideoxy (23) and 2',3'-dideoxy (24) uridine analogs, which subsequently were converted to the corresponding furano-pyrimidines 14 and 15. Both compounds can be prepared in large quantities and good yields by this synthesis.
With methanol; water; triethylamine at 71℃; Microwave irradiation;
Multi-step reaction with 3 steps
1: triethylamine / tetrahydrofuran / 17 h / Ambient temperature
2: 78 percent / conc. NH4OH / methanol
3: 95 percent / triphenylmethyl fluoroborate / acetonitrile; H2O / 1 h / Ambient temperature
With keratinase from Doratomyces microsporus Hydrolysis; Enzymatic reaction;
With ammonia In methanol at 20℃;
1 4.4. Preparation of uridines 6a-e
General procedure: A mixture of commercially available β-d-ribose tetraacetate (6.28 mmol) with pyrimidine base (8.8 mmol), 30.8 mL of acetonitrile, hexamethyldisilazane (10.9 mmol, 1.24 equiv), saccharine (0.4 mmol, 0.046 equiv) and trimethylsilyl trifluoromethane-sulfonate (8.8 mmol, 1.4 equiv) was taken in a Erlenmeyer flask. The Erlenmeyer flask was placed in a microwave oven and irradiated under at low power (100 W) for 3 min. The reaction mixture was cooled at room temperature, neutralized with aqueous sodium bicarbonate, and extracted with CH2Cl2. The organic extract was dried over anhydrous sodium sulfate, filtered and evaporated to dryness. The residue was purified by column chromatography eluting with AcOEt/hexane 7:3, resulting in the desired nucleosides 5a-e, in 75-82% yields. Finally, the protected nucleosides 5a-e (1 mmol) were treated with ammonia/MeOH (saturated at 0 °C, 41.8 mL) overnight at room temperature. The solvent was evaporated under reduced pressure to give compounds 6a-e, in 85-92% yields. Chemical and physical properties of the ribofuranosyl nucleosides were in agreement with previous data.31-35
Uridine (20; 30.0 kg), TPP (46.8 kg) and imidazole (12.2 kg) were slurried in THF (267 kg). A solution of iodine (33.2 kg) in THF (87 kg) was added slowly to the slurry while the reaction temperature was maintained below 28 C. The reaction mixture was stirred overnight (ca. 18 h) at about 25 C. to achieve complete conversion. The reaction mixture was quenched with a small amount (2.3 L) of water. The reaction mixture was distilled under moderate vacuum while adding isopropanol (maximum internal temperature: 50 C.) till IPA content (by gc) of the distillate was greater than 87% (v/v). The resulting slurry was cooled to room temperature (ca. 22 C.) and aged overnight. The precipitated product was filtered and washed with isopropanol (2×50 kg) and dried at about 50 C. under vacuum with a slow nitrogen stream to afford 21 (36.5 kg; 83.9% theory.).
83.9%
Uridine (IVG ; 30.0 kg), TPP (46.8 kg) and imidazole (12.2 kg) were slurried in THF (267 kg). A solution of iodine (33.2 kg) in THF (87 kg) was added slowly to the slurry while the reaction temperature was maintained below 28 C. The reaction mixture was stirred overnight (ca. 18 h) at about 25 C to achieve complete conversion. The reaction mixture was quenched with a small amount (2.3 L) of water. The reaction mixture was distilled under moderate vacuum while adding isopropanol (maximum internal temperature: 50 C) till IPA content (by gc) of the distillate was greater than 87% (v/v). The resulting slurry was cooled to room temperature (ca. 22 C) and aged overnight. The precipitated product was filtered and washed with isopropanol (2 x 50 kg) and dried at about 50 C under vacuum with a slow nitrogen stream to afford IVh (36.5 kg; 83.9% theory. ).
83%
With pyridine; iodine; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 14h;
To a stirred suspension of compound 3-1 (30.5 g, 125 mmol), PPh3 (39.3 g, 150 mmol) and pyridine (100 mL) in anhydrous THF (200 mL) was added dropwise a solution of I2 (38.1 g, 150 mmol) in THF (100 mL) at 0 C. The mixture was warmed to R.T. and stirred for 14 hours. The precipitate was removed by filtration, and the filtrate was concentrated. The residue was dissolved in EA and washed with saturated Na2S2O3 aqueous solution and then brine. The organic layer was dried over Na2SO4 and concentrated. The residue was purified on a silica gel column (DCM/MeOH=100:1 to 20:1) to afford compound 3-2 as a white solid (36.5 g, 83%).
79%
With pyridine; 1H-imidazole; iodine; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 14h;
To a stirred suspension of 23-1 (20.0 g, 81.3 mmol), imidazole (15.9 g, 234.0 mmol), PPh3 (53.5 g, 203.3 mmol) and pyridine (90 mL) in anhydrous THF (100 mL) was added a solution of I2 (41.3 g, 162.6 mmol) in THF (150 mL) dropwise at 0 C. The mixture was slowly warmed to R.T. and stirred for 14 h. The reaction was quenched with sat. aq. Na2S2O3 (150 mL) and extracted with THF/EA (1/1) (100 mL×3). The organic layer was dried over Na2SO4, and concentrated at a low pressure. The residue was recrystallized from EtOH to afford pure 23-2 (23 g, 79%) as a white solid.
79%
With pyridine; 1H-imidazole; iodine; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃;
To a stirred suspension of 12-1 (20.0 g, 81.3 mmol), imidazole (15.9 g, 234.0 mmol), PPh3 (53.5 g, 203.3 mmol) and pyridine (90 mL) in anhydrous THF (100 mL) was added a solution of I2 (41.3 g, 162.6 mmol) in THF (150 mL) dropwise at 0 C. The mixture was slowly warmed to RT and stirred for 14 h. The reaction was quenched with sat. aq. Na2S2O3 (150 mL) and extracted with THF/EA (1/1) (100 mL×3). The organic layer was dried over Na2SO4, and concentrated at a low pressure. The residue was recrystallized from EtOH to afford pure 12-2 (23 g, 79%) as a white solid
79%
With pyridine; 1H-imidazole; iodine; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 14h;
To a stirred suspension of 12-1 (20.0 g, 81.3 mmol), imidazole (15.9 g, 234.0 mmol), PPh3 (53.5 g, 203.3 mmol) and pyridine (90 mE) in anhydrous THF (100 mE) was added a solution of 12 (41.3 g, 162.6 mmol) in THF (150 mE) dropwise at 0 C. The mixture was slowly warmed to RT and stirred for 14 h. The reaction was quenched with sat. aq. Na2S2O3 (150 mE) and extracted with THF/EA (1/1) (100 mEx3). The organic layer was dried over Na2SO4, and concentrated at a low pressure. The residue was recrystallized from EtOR to afford pure 12-2 (23 g, 79%) as a white solid.
79%
With 1H-imidazole; iodine; triphenylphosphine; In tetrahydrofuran; pyridine; at 0 - 20℃; for 14h;
To a stirred suspension of 12-1 (20.0 g, 81.3 mmol), imidazole (15.9 g, 234.0 mmol), PPh3 (53.5 g, 203.3 mmol) and pyridine (90 mL) in anhydrous THF (100 mL) was added a solution of I2 (41.3 g, 162.6 mmol) in THF (150 mL) dropwise at 0 C. The mixture was slowly warmed to RT and stirred for 14 h. The reaction was quenched with sat. aq. Na2S2O3 (150 mL) and extracted with THF/EA (1/1) (100 mL×3). The organic layer was dried over Na2SO4, and concentrated at a low pressure. The residue was recrystallized from EtOH to afford pure 12-2 (23 g, 79%) as a white solid.
With iodine; triphenylphosphine; In 1,4-dioxane; at 20℃;Inert atmosphere; Alkaline conditions;
Uridine (1 mmol) was suspended in dioxane (4 mL) followed by the addition of pyridine (2 mmol), PPh3 (1.5 mmol), and iodine (1.5 mmol) under an argon atmosphere. The mixture was stirrd at room temperature overnight. The reaction mixture was quenched with methanol and saturated aqueous Na2S203and was then evaporated to dryness to provide crude compound 10 , which was used directly in the next step.
Novozym 435; In tert-Amyl alcohol; at 60℃; for 68h;Enzymatic reaction; Molecular sieve;Conversion of starting material;
Example 4: Synthesis of acyl uridine with different acyl donors The synthesisof uridine esters (acyl uridines) was performed with different acyl donors. 1 g uridine (4 mmol) was esterified with 8 mmol of 3-phenylpropionic acid, octadecanoic diacid, octadecanoic diacid or azelaic acid in 20 ml 2-methyl-2-butanol with 0.2 g Novozym435 as biocatalyst. Reactions were performed in shaked flasks at60 C for 68 hrs with addition of 2 g of molecular sieves. Conversion was calculated on the amount of acyl donor consumed.
55
[ 24057-28-1 ]
[ 58-96-8 ]
2',3'-Methoxymethylideneuridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
With pyridine; trimethyl orthoformate; In tetrahydrofuran; water;
2',3'-Methoxymethylideneuridine Uridine (50 gm., 0.205M) was added to a 1 liter round-bottomed flask under an nitrogen atmosphere. Dry freshly distilled tetrahydrofuran (500 mL), pyridinium p-toluene sulphonate (5 gm., 20 mM) were added to the reaction mixture. Trimethyl orthoformate (109 gm., 1.03M) was then added slowly via an addition funnel. The reaction mixture was left to stir for 18 hr. at ambient temperature during which time the reaction became homogeneous. Water (18 gm., 1M) was added and the reaction stirred for a further 0.5 hr. after which time pyridine (20 mL) was added. The reaction was stirred at ambient temperature for another 18 hr. and the solvents then removed in vacuo. The resultant white solid was flash chromatographed on silica to give the desired product as a white solid (40 gm. 68%). M.p. 188-190 C. (lit. 189-190 C.).
Stage #1: bis(phenyl) carbonate; uridine In N,N-dimethyl-formamide at 80℃; for 1h; Inert atmosphere;
Stage #2: With sodium hydrogencarbonate at 115℃; for 4h;
With sodium hydrogencarbonate In N,N-dimethyl-formamide at 100℃; for 4h; Inert atmosphere;
Diphenyl carbonate (4.82 g, 22.54 mmol) was dissolved in anhydrous DMF (10 mL). uridine (5.0 g, 20.49 mmol) and NaHCO3 (86 mg, 1.02 mmol) were added. The suspension was heated to 100 °C, for 4 hours. The suspension was cooled to room temperature and diethyl ether (50 mL) was added and stirred for 30 min at room temperature. The light brown suspension was filtrated and the solid was washed with diethyl ether (2 × 30 mL) and dried under vacuum overnight. The crude nucleoside [16] (13, 5.21 g, 20.49 mmol), NaN3 (5.33 g, 81.97 mmol), and 15-crown-5-ether (0.63 g, 2.86 mmol) were dissolved in anhydrous DMF (20 mL). The suspension was heated to 150 °C for 20 min, the suspension changed from colorless to dark brown. Benzoic acid (2.75 g, 22.54 mmol) was added and the suspension was heated to 150 °C for 2 hours and then cooled to room temperature. The reaction mixture was diluted with H2O (30 mL) and extracted with CHCl3 (2 × 30 mL). The chloroform layer was extracted with H2O (30 mL). The water phase was evaporated to dryness affording a yellow foam. The resulting residue was purified by column chromatography (0-12% CH3OH in CH2Cl2) to afford 14 (3.58 g, 65%) as a white foam, and the data were found correct.[17]
Stage #1: 5'-CGGCUXUUAACCGA-3', X=2-deoxouridine With 1U P1 nuclease In aq. buffer at 37℃; for 16h; Enzymatic reaction;
Stage #2: With 2U alkaline phosphatase In aq. buffer at 37℃; for 1h; Enzymatic reaction;
Enzymatic digestion
Hydrolysis of desulfured S2URNA (1) was performed as follows: 20 µg of RNA suspended in 25 mM Tris pH 7.0 was digested with 1U P1 nuclease at 37 °C for 16 h. Then 2U alkaline phosphatase (Calf Intestinal, CIP) and 1x CIP buffer were added and the reaction mixture was incubated at 37 °C for 1 h. The hydrolysate was applied to RP-HPLC column (Kinetex 5µ C18 100A 250 x 4.60 mm) equilibrated with buffer A: 0.1 M CH3COONH4/H2O, and eluted at 1 mL/min with buffer A and then with buffer B: 0.1M CH3COONH4/H2O + 40% CH3CN, with gradient conditions as follows: 0-20 min, linear gradient with 100 % buffer A, 20-60 min linear gradient hold at 100 % buffer B. Each ribonucleoside was identified with the diode array UV detector.
51 Example 51.
To a solution of uridine 146 (1.0 g, 4.0 mmol) in 20 mL of pyridine was added 2 mL (2.16 g, 21.0 mmol) of acetic anhydride. The resulting reaction mixture was heated to 60 °C for 3 h, and the TLC indicated its completion. The reaction mixture was concentrated, and the residue was purified by flash chromatography on a silica gel column using dichloromethane-methanol (80:1) as eluent giving 1.2 g desired product 155 in 79% yield.
General Nucleoside Acetylation Protocol
General procedure: Nucleoside/nucleotide (2; 100 mM) and N-acetyl imidazole (1a;10 equiv) were dissolved in water (pH 8; adjusted with 4 MNaOH). The solution was incubated at r.t. for 4 h, and NMR spectra were periodically acquired. The product was purified byreverse-phase (C18) flash coumn chromatography (eluted at pH4 with 100 mM NH4HCO2/MeCN = 98:2 to 80:20). The fractions containing 5 were lyophilised to yield a white powder.
With purine nucleoside phosphorylase; uridine phosphorylase; In aq. phosphate buffer; dimethyl sulfoxide; at 80.0℃; for 5h;Enzymatic reaction;
One ml catalyst (cell lysate) with transglycosylationactivity of about 12 units/ml cell lysate was added to 150 ml of a solution kept thermostatically at 80 C andhaving the following composition- 15mM Uridine/2?-Deoxyuridine,- 5mM 6-Mercaptopurine, and- 30 mM potassium phosphate buffer, pH 7.[0099] The assays were performed in 10% (v/v) of DMSO as co-solvent using the conditions described above. After5 hours at 80C, the reaction mixture was filtered by centrifugation at 2000 3g for 30 min, at 4C, through an Amiconultra-4 Centrifugal Filter Devices (Millipore, Bedford, MA) with a 3000-Da cut-off, and the filtrate was recovered. Thebioconversion yield of the reaction was higher than 50%. The resulting <strong>[50-44-2]6-<strong>[50-44-2]mercaptopurine</strong></strong> nucleosides (<strong>[50-44-2]6-<strong>[50-44-2]mercaptopurine</strong></strong>riboside and <strong>[50-44-2]6-<strong>[50-44-2]mercaptopurine</strong></strong> deoxyriboside) were analyzed by HPLC.
With purine nucleoside phosphorylase; uridine phosphorylase; In aq. phosphate buffer; at 60℃; for 1.5h;Enzymatic reaction;
Transglycosylation reaction was carried out at analytical scale in the followingconditions: 250 ml of cell lysates (equivalent to 14 units of each UPase and PNPase enzymatic activities) was added to10 ml of a solution having the following composition: 4 mM 1-beta-D-ribofuranosyluracil (uridine nucleoside), 4 mM <strong>[73-24-5]adenine</strong>base, 30 mM potassium phosphate buffer pH 7, thermostatically controlled at 60C. After 1.5 hours at 60C, the reactionwas stopped by diluting the mixture 1:5 and cooling in ice. The percentage of bioconversion of <strong>[73-24-5]adenine</strong> base to 9-beta-Dribofuranosyl<strong>[73-24-5]adenine</strong>(adenosine nucleoside) was determined by analyzing an aliquot of the reaction mixture by highperformance liquid chromatography (HPLC) with the use of Kromasil 100-5C18 (Akzo Nobel) column of a size of 250 x4.6mm and eluted with a 4% methanol-water solution. The transglycosylation catalytic activity was expressed as units· ml -1 (mmoles ofAra-A formed in 1.5 hours · ml -1 of mixture of cell lysates) or in units · g -1 of moist resin (mmoles ofD-ribofuranosyl<strong>[73-24-5]adenine</strong> formed in 1.5 hours · ml -1 of cell lysate) and was calculated relative to a standard D-ribofuranosyl<strong>[73-24-5]adenine</strong>solution eluted by HPLC in the same conditions. Under these conditions, about 55 percent of adenosinenucleoside was formed (approximately 9 units per ml of cell lysate).
With purine nucleoside phosphorylase; uridine phosphorylase; In aq. phosphate buffer; dimethyl sulfoxide; at 80℃; for 3h;pH 7.0;Enzymatic reaction;
One ml catalyst (cell lysate) with transglycosylation activity of about 12 units/ml cell lysate were added to 150ml of a solution kept thermostatically between 80C, and having the following composition:- 7.5mM uridine/2?-Deoxyuridine,- 2.5mM Trifluoromethyluracil, and- 20 mM potassium phosphate buffer, pH 7.[0089] After 3 hour at 80 C, the reaction mixture was filtered by centrifugation at 20003g for 30 min, at 4C, throughan Amicon ultra-4 Centrifugal Filter Devices (Millipore, Bedford, MA) with a 3000-Da cut-off, and the filtrate was recovered.The bioconversion yield of the reaction was higher than 60%. The resulting trifluoromethyluracil nucleosides (trifluoromethyluridine or 2?-deoxytrifluoromethyluridine) were analyzed by HPLC.
1,3-dichloro-1,1,3,3-tetrakis(2-methylethyl)disiloxane[ No CAS ]
[ 58-96-8 ]
[ 69304-38-7 ]
Yield
Reaction Conditions
Operation in experiment
With pyridine at 20℃; Cooling with ice; Inert atmosphere;
Robust preparation of intermediates 8 and 9
General procedure: Uridine (5; 20.00g; 82mmol) was azeotroped with pyridine (approx. 50ml) and then dissolved in anhydrous pyridine (180ml). 1,3-Dichloro-1,1,3,3-tetraisopropyldisiloxane (30ml; 82ml) was added dropwise to this solution, while cooled in an ice bath, under an atmosphere of nitrogen. The resulting reaction mixture was allowed to warm to room temperature overnight and patitioned between EtOAc and 10% aq. HCl. The organic phase was separated, washed with a further portion of 10% aq. HCl, sat. aq. sodium bicarbonate, brine, dried (MgSO4). The volatiles were removed under reduced pressure to provide the 1′,3′-diprotected uridine intermediate (6; 39.9g; 100%). LCMS: MH+, 487.48. This material was used in the next step (below) without purification. Sodium bicarbonate (20.67g; 246mmol) was dissolved in water (80ml) and added to a stirred solution of the crude alcohol (6; 39.9g; 82mmol), TEMPO (1.92g; 12.3mmol) and potassium bromide (1.46g; 12.3mmol) in dichloromethane (120ml). The resulting mixture was cooled in an ice bath and stirred vigorously before the dropwise addition of sodium hypochlorite (189ml of a 5.75% aq. solution; 176mmol). The reaction was stirred until LCMS indicated that the reaction was complete (approx. 1h.) then partitioned between 10% aq. sodium thiosulfate and dichloromethane. The organic phase was separated, washed with an additional portion of sodium thiosulfate, brine, dried (MgSO4), and the volatiles removed under reduced pressure to give the crude ketone (7; 33.0g; 83%) that co-exists with its hydrate form, consistent with a previous observation in a similar system.11 LCMS: MH+, 485.50 and (M+H2O+H)+, 503.27. This material was used in the next step (below) without purification. n-Butyllithium (77ml of a 1.6M solution in hexane; 124mmol) was added dropwise to a stirred solution of trimethylsilylacetylene (12.16g; 124mmol) in anhydrous THF (125ml) at -78°C, under an atmosphere of nitrogen. When the addition was complete, the solution was stirred for a further 30min. and the crude ketone (7; 20.00g; 41.3mmol) in THF (40ml) was added. After stirring for a further 3h., the reaction was quenched by the addition of sat. aq. ammonium chloride followed by EtOAc. The organic layer was separated, dried (MgSO4) and the volatiles removed under reduced pressure. The residue was purified by silica gel column chromatography using 0 to 20% EtOAc in hexanes as eluent to give (2‘-S)-intermediate (9; 16.60g; 69.0%).
In the reaction flask, the solvent DMF (560 mL) was added, the temperature was lowered to 5 C, and phosphorus oxychloride (153 g) was slowly added dropwise, and the reaction temperature of the mixed system was controlled not to exceed 5 C, and the phosphorus oxychloride was added dropwise. Add boric acid (30.9g), stir for 30min, then add uridine 112g (1mol) in solid form to the above mixed system, control the reaction temperature not to exceed 5 C, stir for 2h, slowly warm to room temperature and continue to stir the reaction until The raw materials disappeared. Then, 560 mL of phosphorus oxychloride was added to the reaction system and the temperature was raised to reflux for 6 hours, then phosphorus oxychloride was recovered, and the residue was poured into a rapidly stirred ice water mixture to precipitate 2,4-dichloro-5-pyrimidine formaldehyde solid. 170 g, yield 96.6%, purity 99.4%.
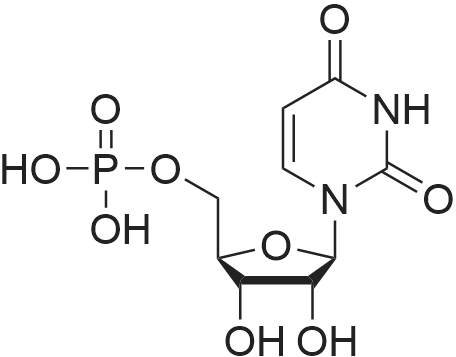
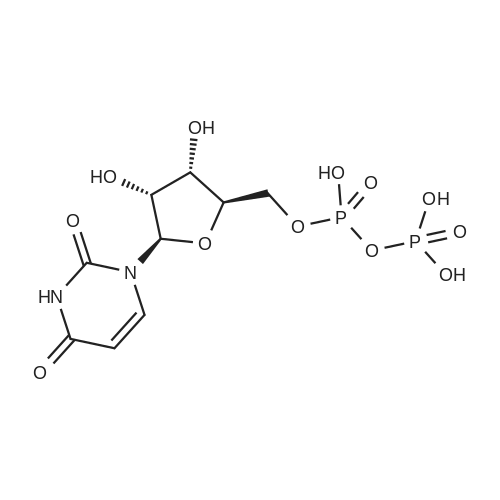
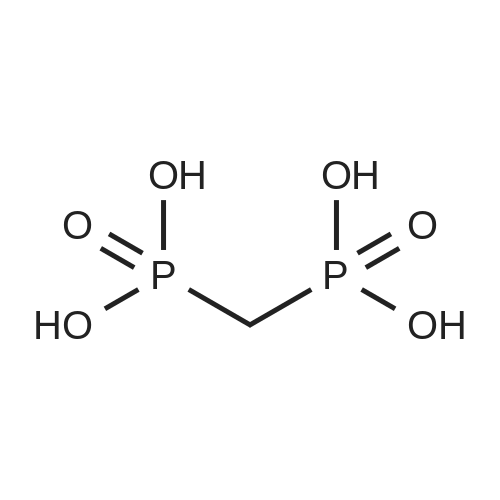
uridine-5'-O-[(phosphonomethyl)phosphonic acid] ditriethylamine salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
5%
Stage #1: Methylenediphosphonic acid; uridine With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20℃;
Stage #2: triethylamine carbonate In water; N,N-dimethyl-formamide at 20℃; for 0.5h;
uridine-5’-O-[(phosphonoethyl)phosphonic acid] tritriethylene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
8%
Stage #1: Methylenediphosphonic acid; uridine With dicyclohexyl-carbodiimide In N,N-dimethyl-formamide at 20℃;
Stage #2: triethylamine carbonate In N,N-dimethyl-formamide at 20℃; for 0.5h; Cooling;
1 General procedure A for the synthesis of nucleotides
General procedure: To a solution of DCC (3 eq.) and nucleoside in DMF (2 mL) methylene diphosphonic acid (1.5 eq.) was added at rt and the mixture was allowed to stir at rt for 6-24 h. Samples were withdrawn at 3 - 12 h interval for LC-MS to check the disappearance of nucleosides and to monitor the formation of the desired nucleotide. On the disappearance of a nucleoside, 10 mL of cold TEAC-solution was added. The mixture was stirred at rt for 30 min followed by filtration and lyophilization of the aqueous solution. The mixture of nucleotide and dinucleotide was separated by ion-exchange chromatography on Source 15Q. Fractions containing the product were pooled and evaporated to dryness. The compound was then purified by RP-HPLC using a gradient of 10 mM triethylammonium acetate buffer - CHbCN from 80:20 to 20:80 in 40 min, suitable fractions were pooled and lyophilized to obtain the final product as glassy solid
With potassium phosphate; at 40℃; for 6.0h;pH 7.5;Enzymatic reaction;Equilibrium constant;
General procedure: To produce dihalogenated nucleoside analogues, base concentrations of 5 mM and sugar donorconcentrations of 22 to 30.5 mM were applied in a reaction volume of 50 mL. Thermostable pyrimidineand purine nucleoside phosphorylase PyNP 02 and PNP 02 were added in a concentration of 0.1 mgmL1 each. Enzymatic reactions were performed in 0.5 mM potassium phosphate buer at pH 7.5using a reaction temperature of 40 C. Reactions were stirred at 100 rpm. Regular samples were takento monitor enzymatic reactions by HPLC as described above. The reactions were stopped by enzyme removal after 2 h and 6 h for deoxyriboside and ribosidenucleosides, respectively. Protein was removed by filtration using centrifugal filter units with a cut-oof 30 kDa (Millipore, Darmstadt, Germany) at room temperature. Filtrate was stored at 4 C untilpurification by semi-preparative HPLC.Analytical HPLC was performed to determine product quantity after each step. Product loss perstep was determined based on the product quantity of step 1 (e.g., after enzymatic synthesis) and theproduct quantity of the following step (e.g., after protein depletion and HPLC).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping