* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
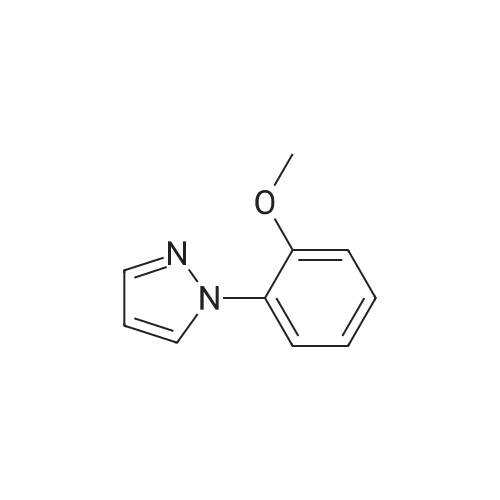
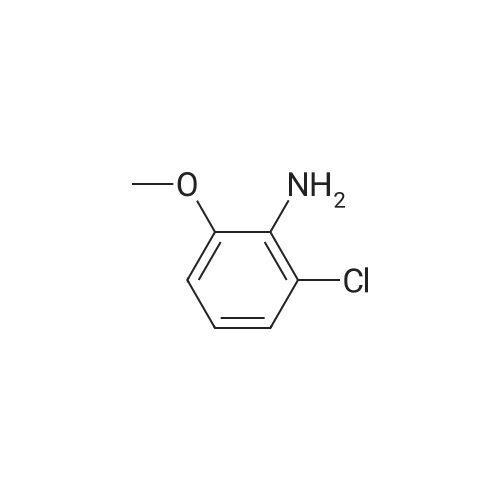
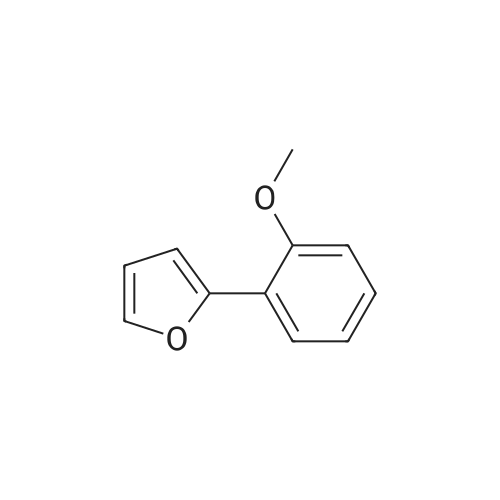
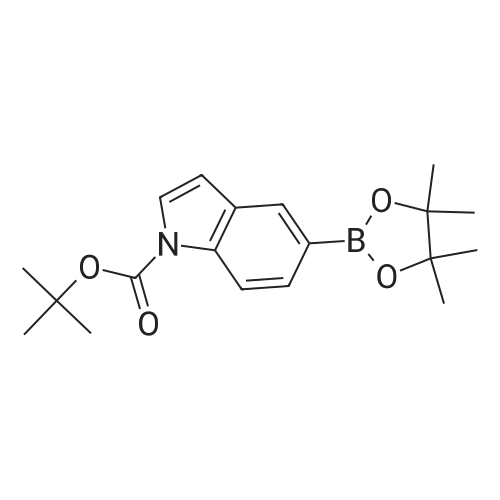
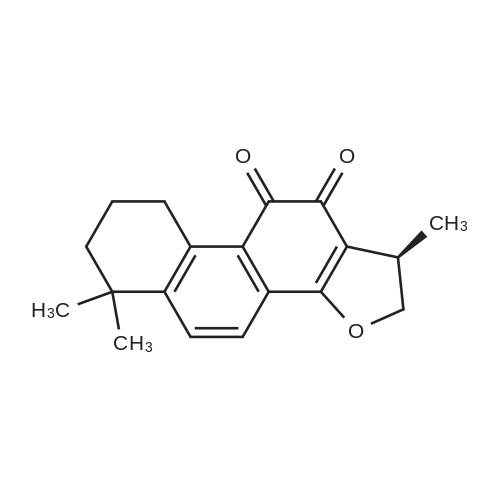
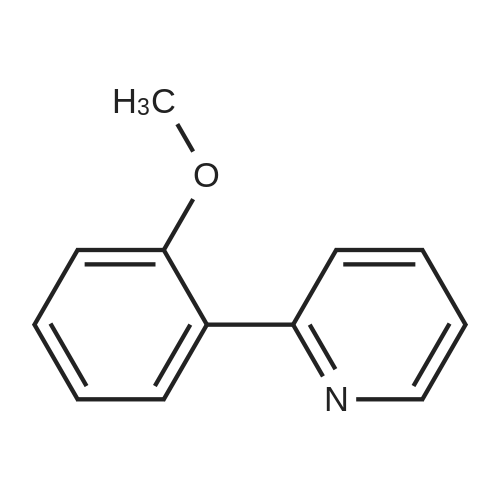
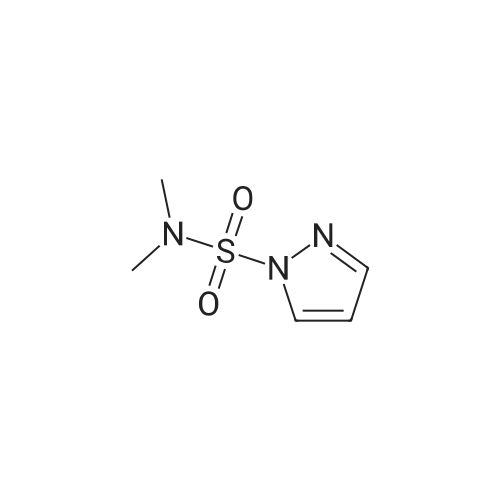
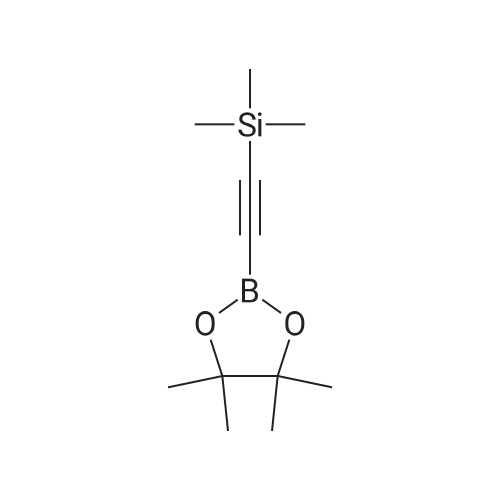
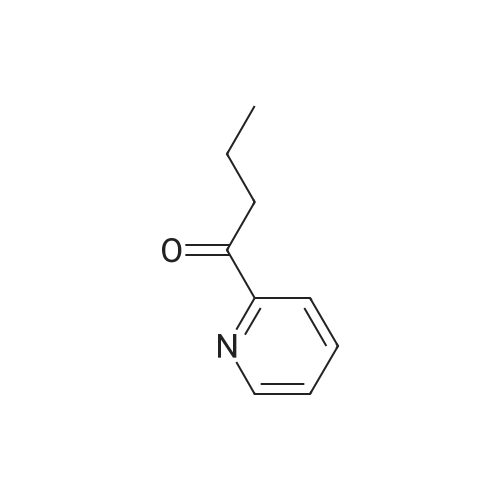
With copper diacetate; sodium hydroxide; 3-(diphenylphosphino)propionic acid In 1,4-dioxane at 120℃; for 36 h; Sealed tube
General procedure: Cu(OAc)2 (0.03mmol), L2 (0.06mmol), aryl idione or bromide (0.5mmol), 1H-pyrazole (0.75mmol), NaOH (1mmol), and 1,4-dioxane (1mL) was added into a 5mL tube, then sealed. The mixture was stirred at 100°C for certain time. After cooling to room temperature, the mixture was quenched with 10mL H2O and extracted with EtOAc (3×20mL). The combined EtOAc extracts were dried with anhydrous Na2SO4 and filtrated and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with PE/EtOAc, as the eluent, to afford the desired products.
Reference:
[1] Advanced Synthesis and Catalysis, 2007, vol. 349, # 17-18, p. 2673 - 2676
[2] Chinese Chemical Letters, 2014, vol. 25, # 5, p. 775 - 778
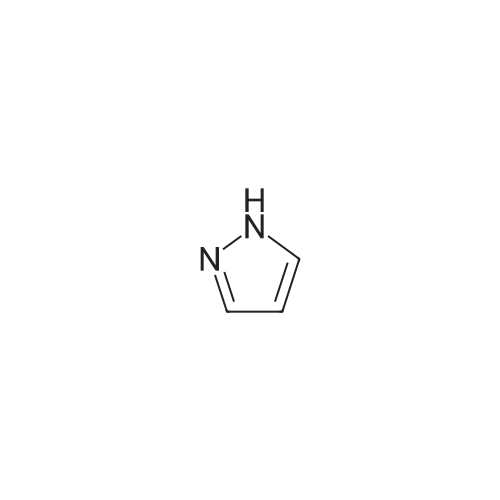
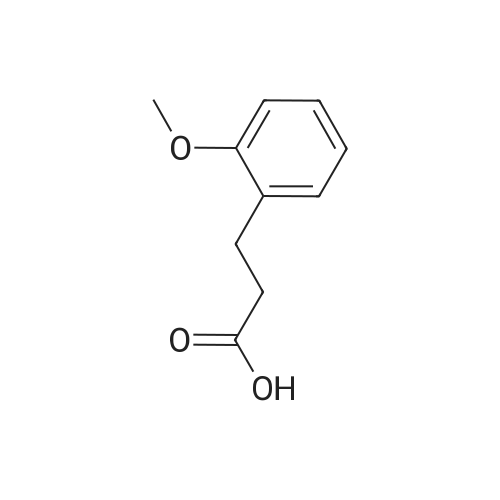
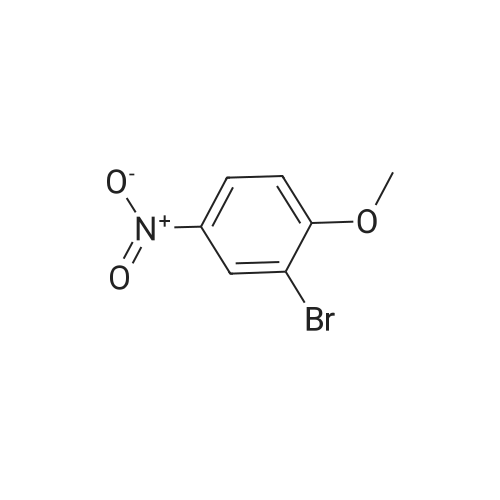
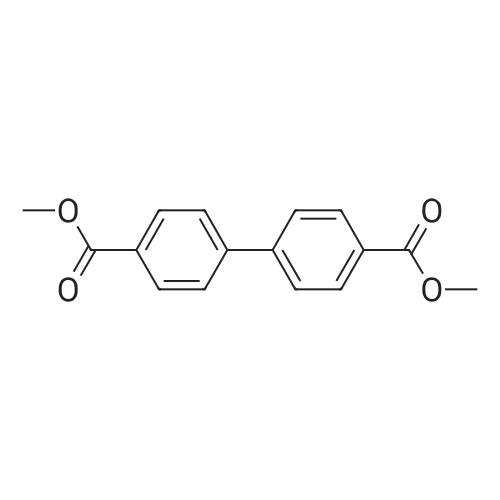
2
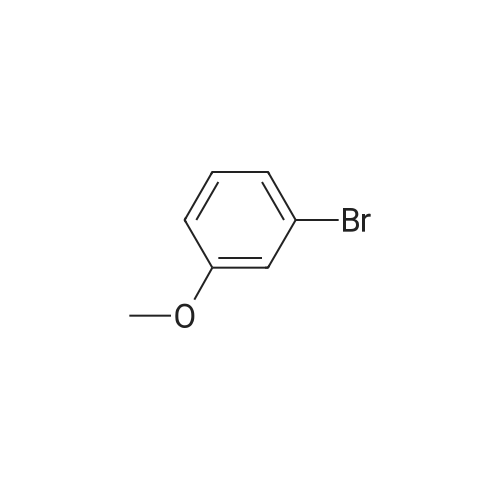
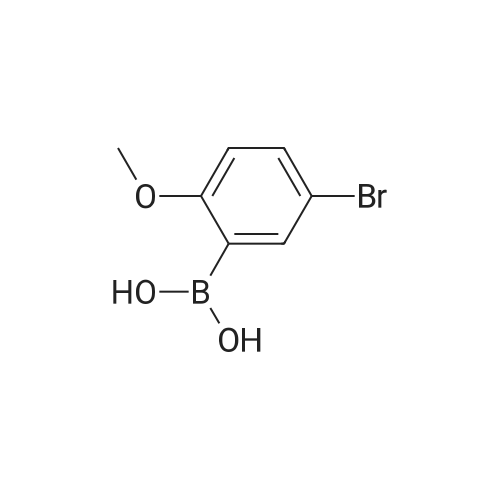
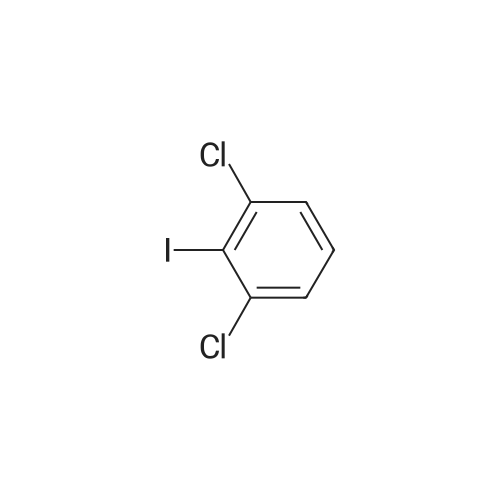
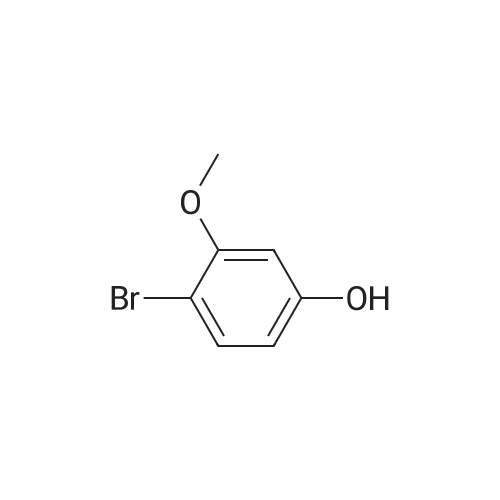
[ 578-57-4 ]
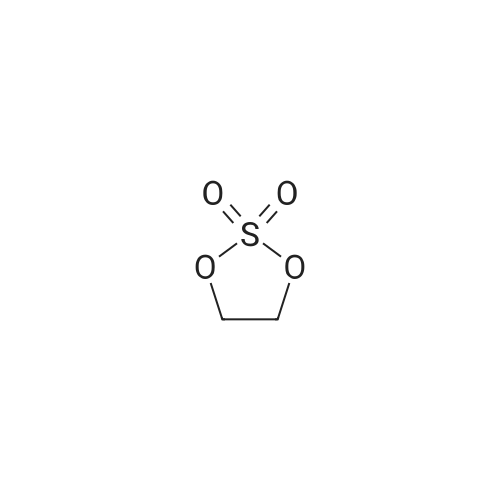
[ 6850-57-3 ]
Yield
Reaction Conditions
Operation in experiment
19%
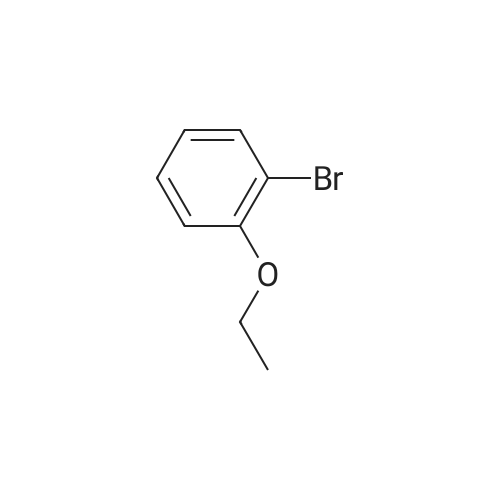
Stage #1: With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; water; caesium carbonate In 1,4-dioxane at 95℃; Stage #2: With hydrazine In 1,4-dioxane; methanol at 20℃; for 1 h; Heating / reflux
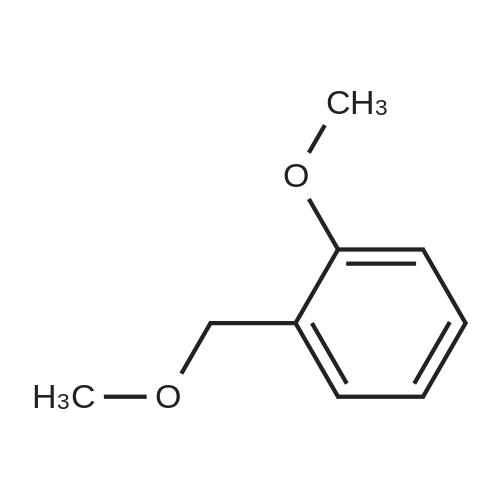
To the mixture of 2-bromoanisole (0.020 ml, 0.16 mmol) and 1,4-dioxane (2 ml), water (0.2 ml), cesium carbonate (0.32 g, 0.97 mmol), sodium 1,3-dioxo-1,3-dihydro-isoindole-2-ylmethyl trifluoroborate (81 mg, 0.32 mmol), palladium(II) acetate (3.6 mg, 0.016 mmol), and 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl (13 mg, 0.032 mmol) were added, and then the obtained reaction mixture was stirred at 95° C. (an outer temperature) overnight. The reaction mixture was cooled to room temperature, and then hydrazine hydrate (0.039 ml, 0.81 mmol) and methanol (2 ml) were added thereto, followed by heating to reflux for 1 hour. The reaction mixture was cooled to room temperature, and then water and ethyl acetate were added to the reaction mixture, followed by filtration with celite. The organic layer of the filtrate was separated and washed with saline. The solvents were evaporated under reduced pressure from the organic layer, and then the obtained residue was purified with NH-silica gel column chromatography (heptane:ethyl acetate=1:4), thereby obtaining the entitled compound (4.1 mg, 19percent) as the mixture with 2'-(dicyclohexyl-phosphinoyl)-2,6-dimethoxy-biphenyl (6.2 mg). 1H-NMR Spectrum (CDCl3) δ(ppm): 3.81(2H, s), 3.85(3H, s), 6.87(1H, d, J=8.1 Hz), 6.91(1H, dt, J=1.1, 7.3 Hz), 7.20-7.25(2H, m)
Reference:
[1] Chemistry - A European Journal, 2017, vol. 23, # 3, p. 563 - 567
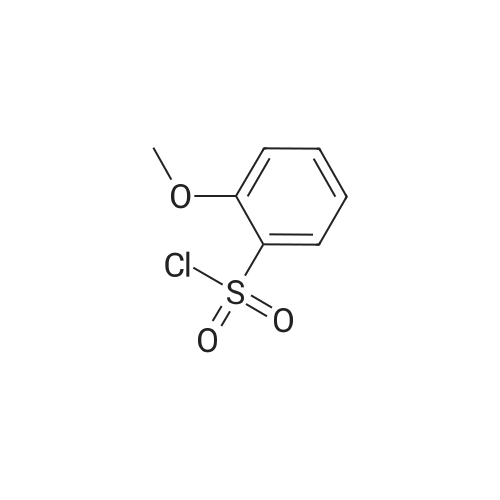
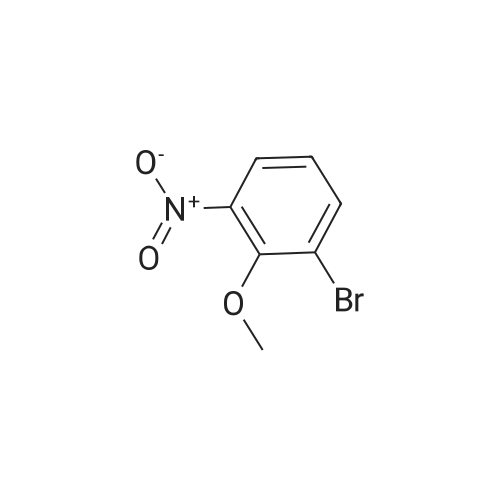
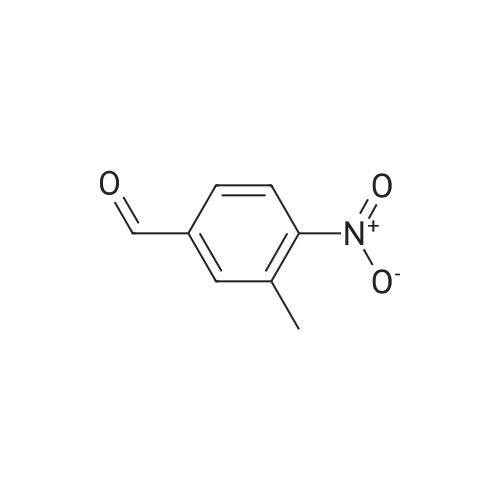
4
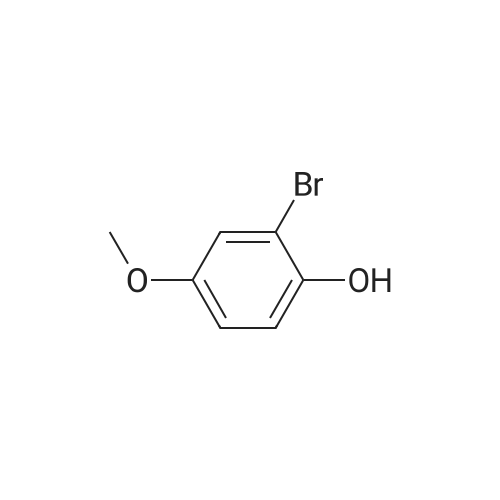
[ 578-57-4 ]
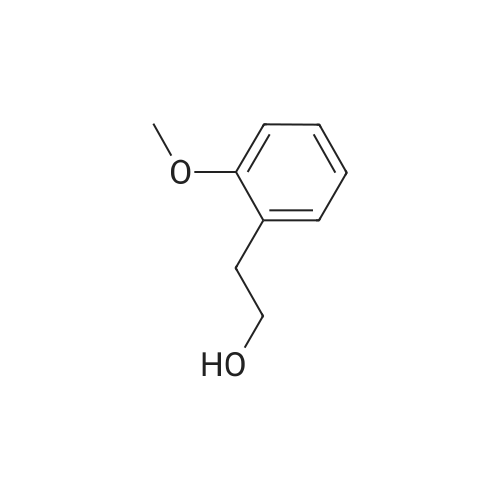
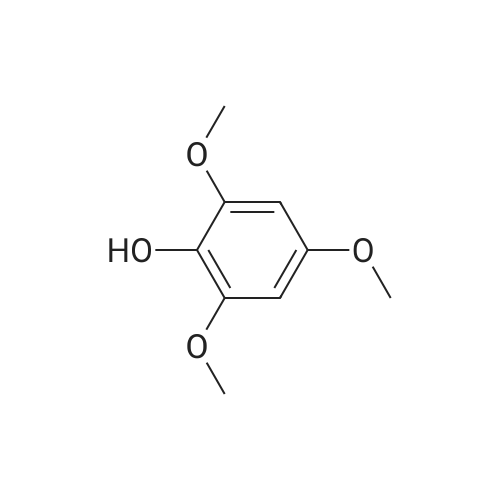
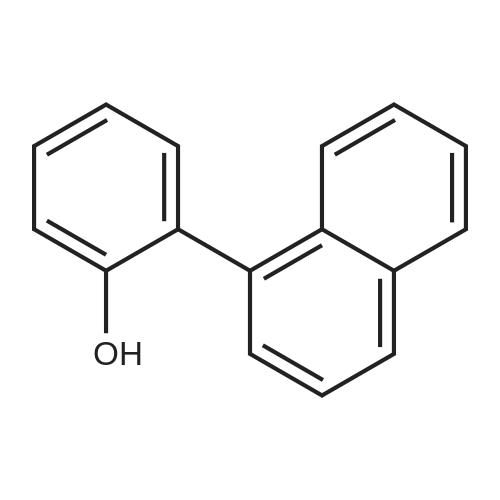
[ 2398-37-0 ]
[ 591-20-8 ]
Reference:
[1] Patent: US4447660, 1984, A,
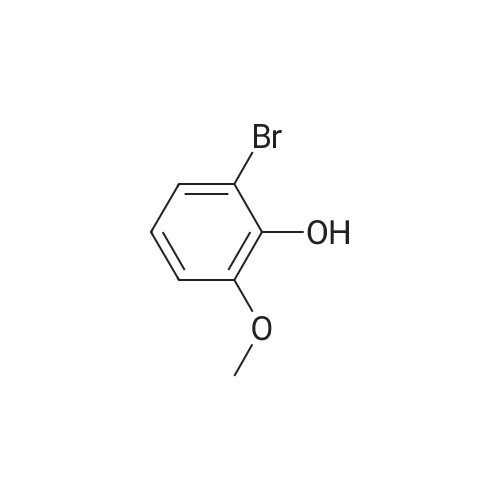
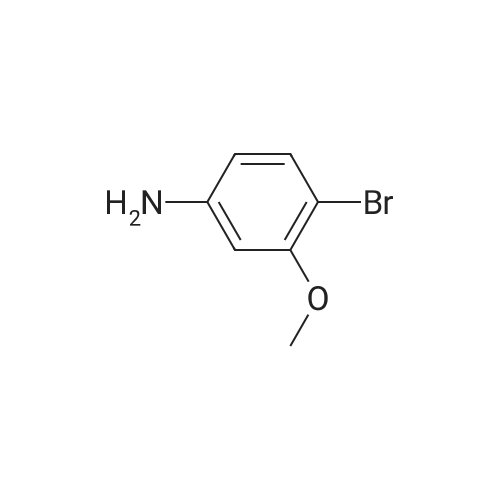
5
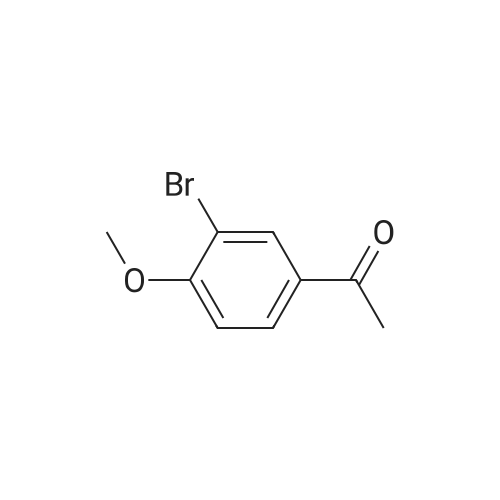
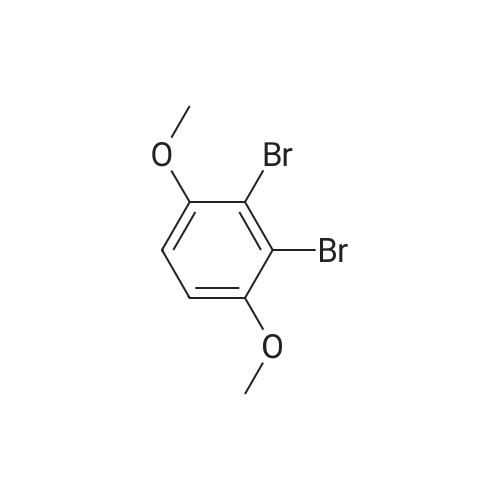
[ 1072-53-3 ]
[ 578-57-4 ]
[ 7417-18-7 ]
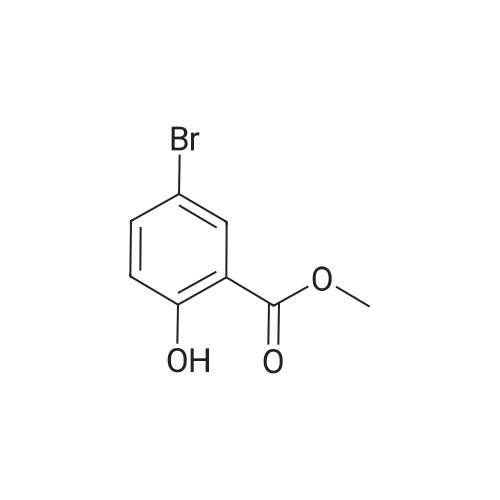
Reference:
[1] Synthesis, 2008, # 11, p. 1793 - 1797
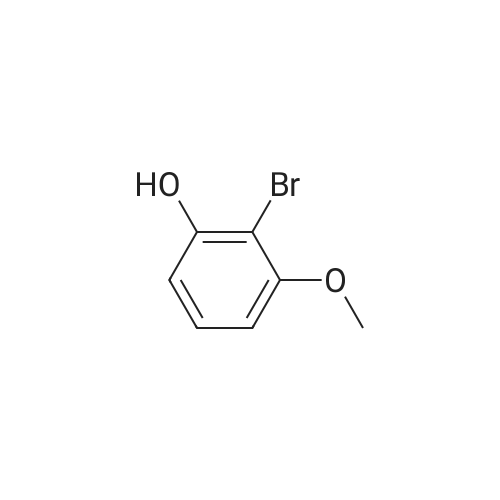
6
[ 578-57-4 ]
[ 75-36-5 ]
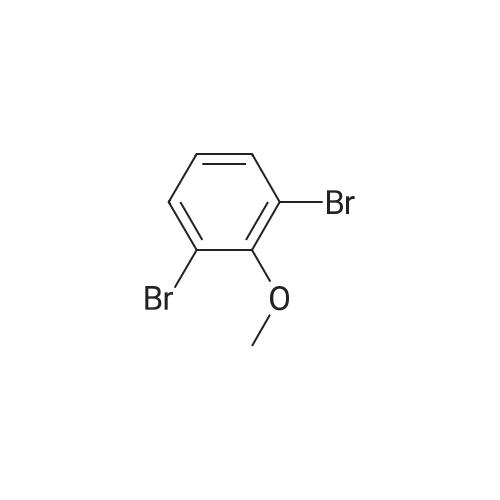
[ 35310-75-9 ]
Yield
Reaction Conditions
Operation in experiment
12.1%
With o-tetrachloroquinone; (η5,η5-(C5H4)2SiMe2)Mo2(CO)6 In 1,2-dichloro-ethane at 80℃; for 24 h; Inert atmosphere; Schlenk technique
General procedure: Under an argon atmosphere, the monobridged bis(cyclopentadienyl)molybdenum carbonyl complex (0.1 mmol) and o-chloranil (0.098 g, 0.4 mmol) were mixed with 1,2-dichloroethane (3.5 mL)in a 25 mL round-bottom flask at room temperature with magneticstirring. The solution was immediately darkened. After the mixturehad been stirred for 40 min at room temperature, aromatic compoundsand acylation reagents were added by syringe. The reactionmixture was heated at 80 °C in an oil bath for 24 h. After cooling toroom temperature, the solvent was removed by rotary evaporation,and the residue was purified by Al2O3 column chromatography,eluting with petroleum ether developed colorless liquid thatafforded the corresponding white solid product. The catalyst wasrecovered and washed with petroleum ether, then reused forfurther cycles. The course of the reaction was monitored using anAgilent 6820 gas chromatograph.
Reference:
[1] Marine Drugs, 2013, vol. 11, # 5, p. 1427 - 1439
[2] Polyhedron, 2018, vol. 147, p. 75 - 79
[3] Comptes Rendus Hebdomadaires des Seances de l'Academie des Sciences, 1947, vol. 224, p. 1363
7
[ 578-57-4 ]
[ 108-24-7 ]
[ 35310-75-9 ]
Reference:
[1] Tetrahedron Letters, 2002, vol. 43, # 36, p. 6331 - 6333
[2] Bioorganic and Medicinal Chemistry, 2010, vol. 18, # 2, p. 971 - 977
[3] Journal of Organic Chemistry, 1993, vol. 58, # 6, p. 1385 - 1392
Reference:
[1] Journal of the American Chemical Society, 1934, vol. 56, p. 1787,1790
13
[ 578-57-4 ]
[ 5197-28-4 ]
Reference:
[1] Bulletin de la Societe Chimique de France, 1897, vol. <3> 17, p. 115
[2] Chemische Berichte, 1896, vol. 29, p. 2598[3] Chemische Berichte, 1899, vol. 32, p. 161 Anm.
14
[ 578-57-4 ]
[ 7697-37-2 ]
[ 64-19-7 ]
[ 5197-28-4 ]
Reference:
[1] Chemische Berichte, 1896, vol. 29, p. 2598[2] Chemische Berichte, 1899, vol. 32, p. 161 Anm.
15
[ 578-57-4 ]
[ 6322-83-4 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2015, vol. 106, p. 50 - 59
16
[ 578-57-4 ]
[ 7439-89-6 ]
[ 19056-40-7 ]
Reference:
[1] Patent: US2003/158179, 2003, A1,
17
[ 578-57-4 ]
[ 4731-65-1 ]
Reference:
[1] Journal of the American Chemical Society, 1975, vol. 97, # 7, p. 1787 - 1794
[2] Dalton Transactions, 2012, vol. 41, # 15, p. 4552 - 4557
[3] Patent: US8309779, 2012, B2, . Location in patent: Page/Page column 18
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 4, p. 1180 - 1183
21
[ 5720-06-9 ]
[ 578-57-4 ]
[ 21702-84-1 ]
[ 89694-45-1 ]
Reference:
[1] Journal of Organic Chemistry, 2004, vol. 69, # 2, p. 566 - 569
22
[ 578-57-4 ]
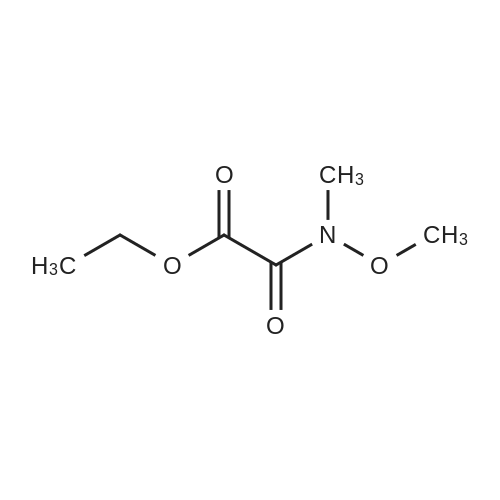
[ 23094-96-4 ]
Yield
Reaction Conditions
Operation in experiment
98%
With chlorosulfonic acid In dichloromethane at -5 - 20℃; for 1.5 h;
L-BROMO-2-METHOXY-BENZENE (1.87 g, 10.0 mmol) was dissolved in chloroform (5 mL) and the solution was cooled in an ice-salt bath to-5° C to 0° C. The cooled solution was carefully charged with CHLOROSULFONIC acid (2.0 mL, 30.0 mmol) over 30 minutes and the mixture was allowed to warm to room temperature over 1 hour. The mixture then was poured onto chopped ice and transferred to a separatory funnel. The aqueous layer was separated and extracted (X2). The combined organic layers were dried and concentrated to give 3-bromo-4-methoxyphenylsulfonyl chloride (2.80 g, 98percent). 3-Bromo-4-methoxyphenylsulfonyl chloride (2.80 g, 9.8 mmol) was dissolved in dichloromethane (30 mL) and the solution was treated with triethylamine (1.76 mL, 12.6 mmol), followed by the dropwise addition of t-butylamine (1.33 mL, 12.6 mmol). The mixture was allowed to stand for 2 hours and then was poured onto a mixture of 5percent citric acid solution and dichloromethane. The organic layer was separated and the aqueous layer was extracted with dichloromethane/ (X2). The combined organic layers were dried and concentrated. Crystallization of the crude solid from ethyl acetate/hexane gave 3-BROMO-N- tert-butyl-4-methoxybenzenesulfonamide (2.43 g, 77percent)
98%
With chlorosulfonic acid In chloroform at -7 - 20℃; for 1.5 - 1.83333 h;
o-Bromoanisole (1.87 g, 10. OMMOL) was dissolved in chloroform (5 mL) and the solution was cooled in an ice-salt bath to-5° C to 0° C. The solution was carefully charged with CHLOROSULFONIC acid (2.0 mL, 30.0 mmol) over 30 minutes and then allowed to warm to room temperature over 1 hour. The mixture was poured onto chopped ice and transferred to separatory funnel. The organic layer was separated and the aqueous layer was extracted, dried and concentrated to give 3-bromo-4-methoxyphenylsulfonyl chloride (2.80 g, 98percent).; REFERENCE 17 N-TERT-BUTYL-4-METHOXY-3- (4, 4,5, 5-TETRAMETHYL- [1, 3,2] DIOXABOROLAN-2-YL) - BENZENESULFONAMIDE [0214] A 500 mL 3-necked flask, equipped with thermometer, overhead stirrer and a 60 mL DROPPING FUNNEL, WAS CHARGED WITH 2-BROMOANISOLE (46.8 G, 0.25 MOL, 1.0 EQ. ) AND ANHYDROUS chloroform (250 mL). The flask was flushed with nitrogen and cooled with a brine-ice cool- bath to an internal temperature of-7°C and then chlorosulfonic acid (87.4 g, 0.75 mol, 3.0 EQ.) was added via dropping funnel over 1 hour while maintaining an internal temperature of less THAN-5°C. The reaction mixture was stirred for 50 minutes and then poured on to ice (500 g). The mixture was stirred until the ice melted and then the organic layer was separated and washed with water (2 x 200 mL). The combined aqueous layers were backwashed with chloroform (2 x 200 mL) and the combined organic phases were washed with brine and dried (MGS04). [0215] The organic phase was treated at room temperature with triethylamine (87 mL, 63 g, 0.625 MOL, 2.5 EQ. ) AND THEN TERT-BUTYLAMINE (34 ML, 24 G, 0.325 MOL, 1.3 EQ. ). THE REACTION mixture was stirred overnight, cooled in an ice-bath and poured into ice cold 2M hydrochloric acid (500 mL). The organic layer was separated and washed with 2M hydrochloric acid (2 x 250 mL) and brine, dried (MGSO4) and concentrated. Crystallization of the residue from chloroform-hexane gave 3-BROMO-N-TERT-BUTYL-4-METHOXY-BENZENESULFONAMIDE (37.5 g, 47percent) as brilliant white crystals. RP-HPLC (10-95S) RT = 3.92 MIN.APOS;H NMR (400 MHz, d6- DMSO): 8 7. 94 (1H, d, J = 2. 4 HZ), 7.77 (1H, dd, J = 8. 8,2. 4 HZ), 7.48 (1H, s), 7.25 (1H, d, 8.8 Hz), 3.92 (3H, s), 1.08 (9H, s). [0216] A mixture OF 3-BROMO-N-TERT-BUTYL-4-METHOXY-BENZENESULFONAMIDE (36.2 g, 112 mmol, 1.0 eq), bis (pinocalto) diborane (30.0 g, 117 mmol, 1.05 EQ.), potassium acetate (33.0 G, 336 MMOL, 3.0 EQ. ) AND PDCL2 (DPPF)-DCM (533 MG, 0.653 MMOL, 5.8 MOL percent, IN 115 ML of 1,4-dioxane) was heated at 100 °C under nitrogen and then 4,4, 5,5, 4', 4', 5', 5'- OCTAMETHYL- [2, 2APOS;] BI [ [1, 3, 2] DIOXABOROLANYL] (0.35 EQ. ) WAS ADDED TO THE MIXTURE. THE REACTION mixture was heated for 28 hours and then allowed to cool. The mixture was filtered of solids and concentrated. The residue was dissolved in ethyl acetate (500 mL) and the solution was washed with 5percent citric acid (3x 200 mL), saturated sodium bicarbonate (3 x 200 mL) and then brine, dried (MGSO4) and concentrated. Purification of product from the residue by silica-gel chromatography, eluting with 10-50percent EtOAc/Hex gave N-tert-butyl-4-methoxy-3- (4, 4,5, 5- TETRAMETHYL- [1, 3,2] DIOXABOROLAN-2-YL) -BENZENESULFONAMIDE (30 G, 72percent) AS A PALE-ORANGE solid. RP-HPLC (10-95S): RT = 3.17 min.'HNMR (400MHz, d6-DMSO) : 57. 95 (1H, d, J = 2.4 Hz), 7.85 (1H, dd, J = 8.8, 2.4 Hz), 7.36 (1H, s), 7.11 (1H, d, J = 8.8), 3.81 (3H, s), 1.29 (12H, s), 1.07 (9H, s); M/Z (LCMS-ESI): Q+ 310 (boronic acid+Na), 370 (M+H), 392 (M+Na); Q~ 354 (M-Me), 556 (boronic acid anhydride).
97%
With chlorosulfonic acid In dichloromethane at 0 - 20℃; Cooling with ice
A solution of 2-bromoanisole (5.00 g, 26.7 mmol) in CH2Cl2 (100 mL) was cooled on ice. Chlorosulfonic acid (9.3 g, 80 mmol) in CH2Cl2 (100 mL) was added dropwise at 0° C. The reaction mixture was allowed to reach room temperature overnight and was then added slowly to a stirred solution of brine. The organic phase was separated and washed with brine, dried over MgSO4, filtered and concentrated. The intermediate 3-bromo-4-methoxybenzenesulfonyl chloride was obtained in 97percent yield (7.33 g). 1H NMR (600 MHz, CDCl3) δ ppm 4.03 (s, 3H) 7.04 (d, J=8.85 Hz, 1H) 7.99 (dd, J=8.85, 2.44 Hz, 1H) 8.22 (d, J=2.44 Hz, 1H). MS (ESI+) m/z 249 [M-Cl]+.
Reference:
[1] Patent: WO2004/50637, 2004, A2, . Location in patent: Page 68-69
[2] Patent: WO2004/62661, 2004, A1, . Location in patent: Page/Page column 24-25; 37-38
[3] Patent: US2015/25068, 2015, A1, . Location in patent: Paragraph 0794
[4] Journal of the Chemical Society [Section] C: Organic, 1969, p. 1341 - 1345
[5] Bioorganic and Medicinal Chemistry Letters, 1997, vol. 7, # 4, p. 485 - 488
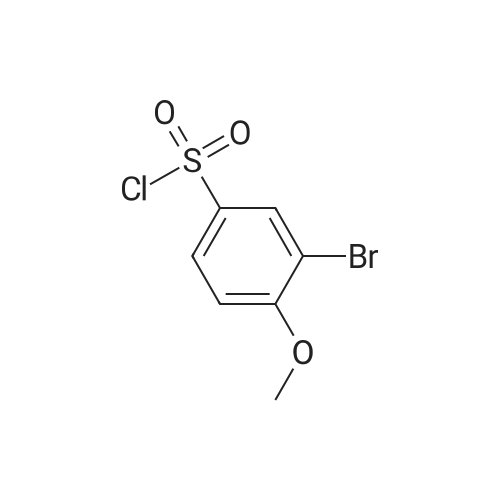
23
[ 578-57-4 ]
[ 116557-46-1 ]
Reference:
[1] Journal of the American Chemical Society, 1934, vol. 56, p. 1787,1790
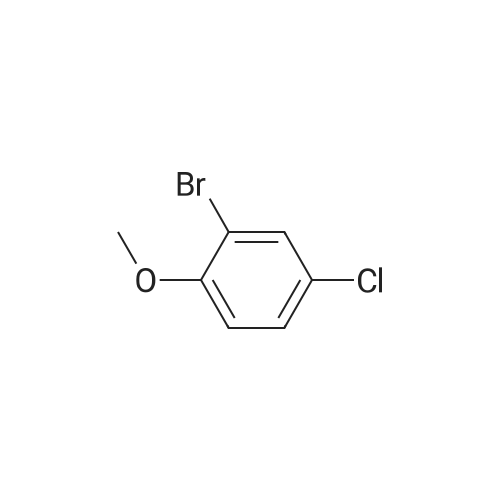
24
[ 578-57-4 ]
[ 60633-25-2 ]
Reference:
[1] Chemical Science, 2017, vol. 8, # 10, p. 7009 - 7013
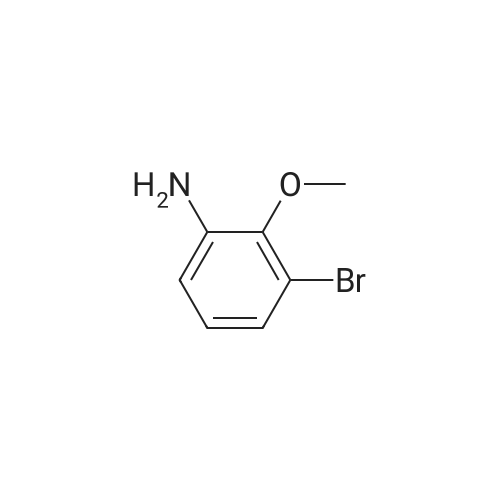
25
[ 578-57-4 ]
[ 172217-13-9 ]
[ 158966-62-2 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1995, vol. 43, # 8, p. 1287 - 1293
26
[ 578-57-4 ]
[ 182056-39-9 ]
Yield
Reaction Conditions
Operation in experiment
92%
With N-iodo-succinimide; [bis(trifluoromethanesulfonyl)imidate](triphenylphosphine)gold(I) In dichloromethane; toluene at 20℃; for 14 h;
General procedure: To a stirred solution of the substrate (1 mmol) in CH2Cl2 or (CH2Cl)2 (0.1 M) were added Ph3PAuNTf2 (0.025 mmol, 19 mg; complex Ph3PAuNTf2 toluene, 2:1) followed by N-iodosuccinimide (1.1 mmol, 248 mg). The resulting solution was stirred at r.t. or under reflux until complete conversion of the starting material. After removal of the solvent under reduced pressure, the crude material was purified by flash column chromatography using different gradients of hexanes and EtOAc to obtain the pure desired products.
Reference:
[1] Synlett, 2014, vol. 25, # 3, p. 399 - 402
[2] Synthetic Communications, 2007, vol. 37, # 8, p. 1259 - 1265
[3] Canadian Journal of Chemistry, 2005, vol. 83, # 10, p. 1808 - 1811
[4] Tetrahedron Letters, 2004, vol. 45, # 43, p. 8015 - 8018
27
[ 578-57-4 ]
[ 5197-28-4 ]
[ 98775-19-0 ]
Reference:
[1] Journal of the American Chemical Society, 1934, vol. 56, p. 1787,1790
With o-tetrachloroquinone; (eta5,eta5-(C5H4)2SiMe2)Mo2(CO)6; In 1,2-dichloro-ethane; at 80℃; for 24h;Inert atmosphere; Schlenk technique;
General procedure: Under an argon atmosphere, the monobridged bis(cyclopentadienyl)molybdenum carbonyl complex (0.1 mmol) and o-chloranil (0.098 g, 0.4 mmol) were mixed with 1,2-dichloroethane (3.5 mL)in a 25 mL round-bottom flask at room temperature with magneticstirring. The solution was immediately darkened. After the mixturehad been stirred for 40 min at room temperature, aromatic compoundsand acylation reagents were added by syringe. The reactionmixture was heated at 80 C in an oil bath for 24 h. After cooling toroom temperature, the solvent was removed by rotary evaporation,and the residue was purified by Al2O3 column chromatography,eluting with petroleum ether developed colorless liquid thatafforded the corresponding white solid product. The catalyst wasrecovered and washed with petroleum ether, then reused forfurther cycles. The course of the reaction was monitored using anAgilent 6820 gas chromatograph.
The preparation of tri(2-methoxyphenyl)phosphorus was carried out in the following manner. 91.1 g (3.75 mol) of magnesium pieces were slowly added to 95 mL (0.75 mol) of 2-bromo-anisol in 2 liters of THF. After intensive reaction, the reaction mixture was heated under reflux for 2 hours to obtain a Grignard reagent. The Grignard reagent was added dropwise to a solution of 17.5 mL (0.2 mol) of PCl3 in 2 liters of THF at -78° C. over 2 hours with stirring. After completion of the dropwise addition, the dry ice/acetone bath was removed, and the reaction material was warmed to room temperature. The reaction material was stirred overnight, and the solvent was removed in a vacuum. The phosphine product was used in a subsequent step without removal.
With chlorosulfonic acid; In dichloromethane; at -5 - 20℃; for 1.5h;
L-BROMO-2-METHOXY-BENZENE (1.87 g, 10.0 mmol) was dissolved in chloroform (5 mL) and the solution was cooled in an ice-salt bath to-5 C to 0 C. The cooled solution was carefully charged with CHLOROSULFONIC acid (2.0 mL, 30.0 mmol) over 30 minutes and the mixture was allowed to warm to room temperature over 1 hour. The mixture then was poured onto chopped ice and transferred to a separatory funnel. The aqueous layer was separated and extracted (X2). The combined organic layers were dried and concentrated to give 3-bromo-4-methoxyphenylsulfonyl chloride (2.80 g, 98%). 3-Bromo-4-methoxyphenylsulfonyl chloride (2.80 g, 9.8 mmol) was dissolved in dichloromethane (30 mL) and the solution was treated with triethylamine (1.76 mL, 12.6 mmol), followed by the dropwise addition of t-butylamine (1.33 mL, 12.6 mmol). The mixture was allowed to stand for 2 hours and then was poured onto a mixture of 5% citric acid solution and dichloromethane. The organic layer was separated and the aqueous layer was extracted with dichloromethane/ (X2). The combined organic layers were dried and concentrated. Crystallization of the crude solid from ethyl acetate/hexane gave 3-BROMO-N- tert-butyl-4-methoxybenzenesulfonamide (2.43 g, 77%)
98%
With chlorosulfonic acid; In chloroform; at -7 - 20℃; for 1.5 - 1.83333h;Product distribution / selectivity;
o-Bromoanisole (1.87 g, 10. OMMOL) was dissolved in chloroform (5 mL) and the solution was cooled in an ice-salt bath to-5 C to 0 C. The solution was carefully charged with CHLOROSULFONIC acid (2.0 mL, 30.0 mmol) over 30 minutes and then allowed to warm to room temperature over 1 hour. The mixture was poured onto chopped ice and transferred to separatory funnel. The organic layer was separated and the aqueous layer was extracted, dried and concentrated to give 3-bromo-4-methoxyphenylsulfonyl chloride (2.80 g, 98%).; REFERENCE 17 N-TERT-BUTYL-4-METHOXY-3- (4, 4,5, 5-TETRAMETHYL- [1, 3,2] DIOXABOROLAN-2-YL) - BENZENESULFONAMIDE [0214] A 500 mL 3-necked flask, equipped with thermometer, overhead stirrer and a 60 mL DROPPING FUNNEL, WAS CHARGED WITH 2-BROMOANISOLE (46.8 G, 0.25 MOL, 1.0 EQ. ) AND ANHYDROUS chloroform (250 mL). The flask was flushed with nitrogen and cooled with a brine-ice cool- bath to an internal temperature of-7C and then chlorosulfonic acid (87.4 g, 0.75 mol, 3.0 EQ.) was added via dropping funnel over 1 hour while maintaining an internal temperature of less THAN-5C. The reaction mixture was stirred for 50 minutes and then poured on to ice (500 g). The mixture was stirred until the ice melted and then the organic layer was separated and washed with water (2 x 200 mL). The combined aqueous layers were backwashed with chloroform (2 x 200 mL) and the combined organic phases were washed with brine and dried (MGS04). [0215] The organic phase was treated at room temperature with triethylamine (87 mL, 63 g, 0.625 MOL, 2.5 EQ. ) AND THEN TERT-BUTYLAMINE (34 ML, 24 G, 0.325 MOL, 1.3 EQ. ). THE REACTION mixture was stirred overnight, cooled in an ice-bath and poured into ice cold 2M hydrochloric acid (500 mL). The organic layer was separated and washed with 2M hydrochloric acid (2 x 250 mL) and brine, dried (MGSO4) and concentrated. Crystallization of the residue from chloroform-hexane gave 3-BROMO-N-TERT-BUTYL-4-METHOXY-BENZENESULFONAMIDE (37.5 g, 47%) as brilliant white crystals. RP-HPLC (10-95S) RT = 3.92 MIN.'H NMR (400 MHz, d6- DMSO): 8 7. 94 (1H, d, J = 2. 4 HZ), 7.77 (1H, dd, J = 8. 8,2. 4 HZ), 7.48 (1H, s), 7.25 (1H, d, 8.8 Hz), 3.92 (3H, s), 1.08 (9H, s). [0216] A mixture OF 3-BROMO-N-TERT-BUTYL-4-METHOXY-BENZENESULFONAMIDE (36.2 g, 112 mmol, 1.0 eq), bis (pinocalto) diborane (30.0 g, 117 mmol, 1.05 EQ.), potassium acetate (33.0 G, 336 MMOL, 3.0 EQ. ) AND PDCL2 (DPPF)-DCM (533 MG, 0.653 MMOL, 5.8 MOL %, IN 115 ML of 1,4-dioxane) was heated at 100 C under nitrogen and then 4,4, 5,5, 4', 4', 5', 5'- OCTAMETHYL- [2, 2'] BI [ [1, 3, 2] DIOXABOROLANYL] (0.35 EQ. ) WAS ADDED TO THE MIXTURE. THE REACTION mixture was heated for 28 hours and then allowed to cool. The mixture was filtered of solids and concentrated. The residue was dissolved in ethyl acetate (500 mL) and the solution was washed with 5% citric acid (3x 200 mL), saturated sodium bicarbonate (3 x 200 mL) and then brine, dried (MGSO4) and concentrated. Purification of product from the residue by silica-gel chromatography, eluting with 10-50% EtOAc/Hex gave N-tert-butyl-4-methoxy-3- (4, 4,5, 5- TETRAMETHYL- [1, 3,2] DIOXABOROLAN-2-YL) -BENZENESULFONAMIDE (30 G, 72%) AS A PALE-ORANGE solid. RP-HPLC (10-95S): RT = 3.17 min.'HNMR (400MHz, d6-DMSO) : 57. 95 (1H, d, J = 2.4 Hz), 7.85 (1H, dd, J = 8.8, 2.4 Hz), 7.36 (1H, s), 7.11 (1H, d, J = 8.8), 3.81 (3H, s), 1.29 (12H, s), 1.07 (9H, s); M/Z (LCMS-ESI): Q+ 310 (boronic acid+Na), 370 (M+H), 392 (M+Na); Q~ 354 (M-Me), 556 (boronic acid anhydride).
97%
With chlorosulfonic acid; In dichloromethane; at 0 - 20℃;Cooling with ice;
A solution of 2-bromoanisole (5.00 g, 26.7 mmol) in CH2Cl2 (100 mL) was cooled on ice. Chlorosulfonic acid (9.3 g, 80 mmol) in CH2Cl2 (100 mL) was added dropwise at 0 C. The reaction mixture was allowed to reach room temperature overnight and was then added slowly to a stirred solution of brine. The organic phase was separated and washed with brine, dried over MgSO4, filtered and concentrated. The intermediate 3-bromo-4-methoxybenzenesulfonyl chloride was obtained in 97% yield (7.33 g). 1H NMR (600 MHz, CDCl3) delta ppm 4.03 (s, 3H) 7.04 (d, J=8.85 Hz, 1H) 7.99 (dd, J=8.85, 2.44 Hz, 1H) 8.22 (d, J=2.44 Hz, 1H). MS (ESI+) m/z 249 [M-Cl]+.
With chlorosulfonic acid; In chloroform; at -10 - 20℃; for 1h;
10 g of 1-bromo-2-methoxybenzene was dissolved in chloroform (56 mL), and then chlorosulfuric acid (11 mL, 3 equivalents) was added thereto at -10 C., followed by stirring at room temperature for 1 hour. The formation of the product was confirmed by TLC, and then the reaction mixture was poured into distilled water under ice-cooling, followed by extraction using ethyl acetate. Thereafter, the organic phase was dried over anhydrous magnesium sulfate, followed by filtration, and thus obtaining as a crude product 14 g of the title compound by vacuum concentration of the filtrate.MS (ESI) m/z: 285 (M+H)+
With caesium carbonate; 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl;palladium diacetate; In 1,4-dioxane; at 20 - 100℃;Inert atmosphere;
Synthesis of Intermediate XLII. Nitrogen was purged through a stirred solution of arylbromide (XLI, 2.15 mmol) in 1,4-dioxane (20 ml) at room temperature for 30 minutes. BINAP (0.134 gm, 0.215 mmol), palladium acetate (0.0096 g, 0.043 mmol) and cesium carbonate (1.40 gm, 4.3 mmol) were added to the reaction mixture and the nitrogen purging was continued for another 20 minutes and finally diamine (XL, 2.15 mmol) was added and stirred at 100 C. overnight under nitrogen atmosphere. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated under vacuum. The residue was dissolved in water, extracted with ethyl acetate (3×50 ml). Combined organic extracts were washed with brine (20 ml), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude product was then purified by column chromatography (60-120 silica gel) using 20% ethyl acetate-hexane to afford compound XLII (40-60%).
General procedure: Nitrogen was purged through a stirred solution of arylbromide (XLI, 2.15 mmol) in 1,4-dioxane (20 ml) at room temperature for 30 minutes. BINAP (0.134 gm, 0.215 mmol), palladium acetate (0.0096 g, 0.043 mmol) and cesium carbonate (1.40 gm, 4.3 mmol) were added to the reaction mixture and the nitrogen purging was continued for another 20 minutes and finally diamine (XL, 2.15 mmol) was added and stirred at 100 C. overnight under nitrogen atmosphere. After completion of the reaction (monitored by TLC), the reaction mixture was concentrated under vacuum. The residue was dissolved in water, extracted with ethyl acetate (3×50 ml). Combined organic extracts were washed with brine (20 ml), dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure. The crude product was then purified by column chromatography (60-120 silica gel) using 20% ethyl acetate-hexane to afford compound XLII (40-60%).
With N-iodo-succinimide; [bis(trifluoromethanesulfonyl)imidate](triphenylphosphine)gold(I); In dichloromethane; toluene; at 20℃; for 14h;
General procedure: To a stirred solution of the substrate (1 mmol) in CH2Cl2 or (CH2Cl)2 (0.1 M) were added Ph3PAuNTf2 (0.025 mmol, 19 mg; complex Ph3PAuNTf2 toluene, 2:1) followed by N-iodosuccinimide (1.1 mmol, 248 mg). The resulting solution was stirred at r.t. or under reflux until complete conversion of the starting material. After removal of the solvent under reduced pressure, the crude material was purified by flash column chromatography using different gradients of hexanes and EtOAc to obtain the pure desired products.
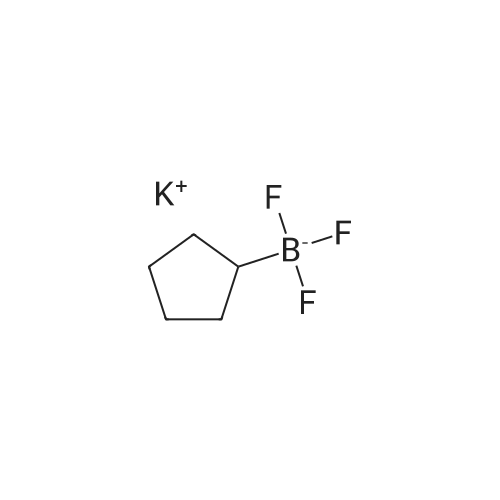
With caesium carbonate;dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; palladium diacetate; In 1,4-dioxane; water; at 100℃;
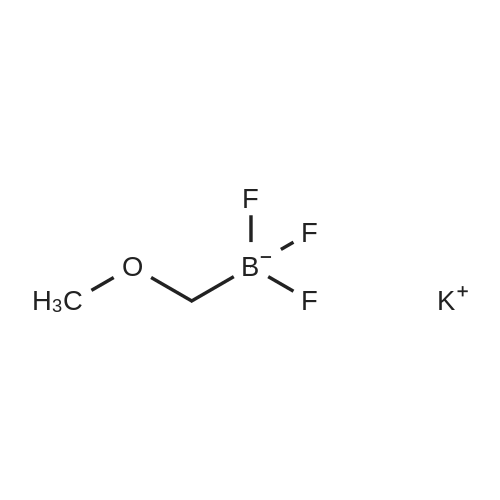
To a mixture of 2-bromoanisole (50 mg, 0.27 mmol) and 1,4-dioxane (2 ml) were added water (0.2 ml), cesium carbonate (0.26 g, 0.80 mmol), <strong>[910251-11-5]potassium methoxymethyl trifluoroborate</strong> (81 mg, 0.53 mmol), palladium (II) acetate (3.0 mg, 0.013 mmol) and 2-dicyclohexelphosphino-2',6'-dimethoxybiphenyl (11 mg, 0.027 mmol). Then, the reaction mixture was stirred at 100C (external temperature) overnight. After the reaction mixture was allowed to cool at room temperature, water and butane were added to the mixture, and the resultant was filtered using Celite. The organic layer was separated, and the organic layer was washed with an aqueous saturated sodium chloride solution. The organic layer was separated, and the solvent was distilled off under reduced pressure. The residue was purified by NH-silica gel column chromatography (heptane) to obtain the title compound (27 mg, 0.18 mmol, 66%). 1H-NMR Spectrum (CDCl3) delta (ppm) : 3.42 (3H, s), 3.84 (3H, s) , 4.51 (2H, s) , 6.87-6.89(1H, m) , 6.96(1H, td, J=7.4, 1.1Hz), 7.25-7.29(1H, m), 7.34-7.36(1H, m).
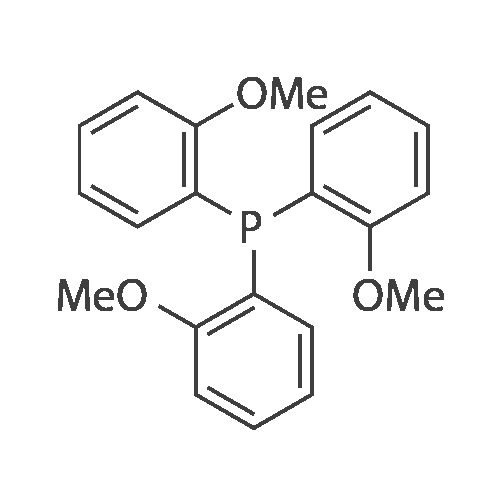
tris(o-methoxy-m-sulfonic acid)phosphine[ No CAS ]
[ 4731-65-1 ]
Yield
Reaction Conditions
Operation in experiment
With anhydrous phosphorus trichloride;
(a) Preparation of tris(o-methoxyphenyl)phosphine Tris(o-methoxyphenyl)phosphine is prepared in an analogous manner to that of J. Org. Chem. 43, 2941 et seq. (1978) for synthesis of tris(o-tolyl)phosphine by reaction of phosphorus trichloride with the Grignard reagent obtained from o-methoxybromobenzene.
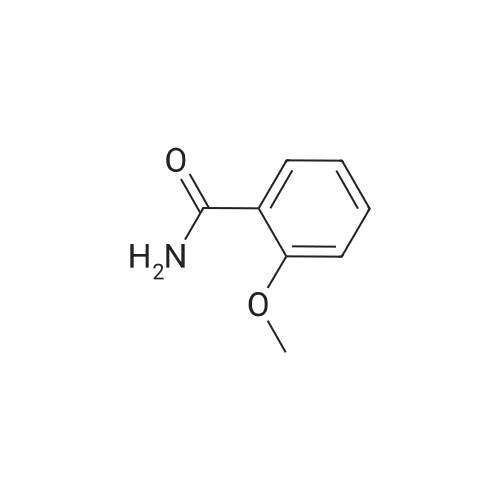
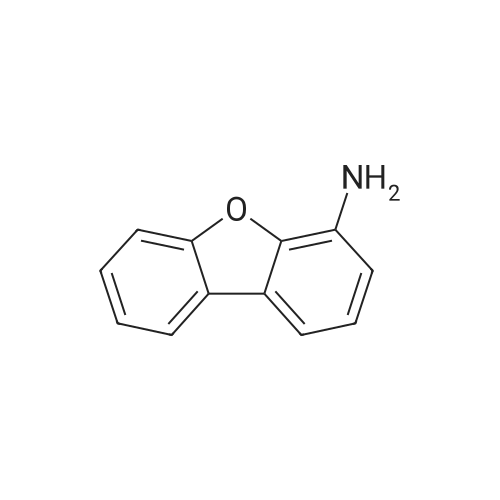
With copper diacetate; sodium hydroxide; 3-(diphenylphosphino)propionic acid; In 1,4-dioxane; at 120℃; for 36h;Sealed tube;
General procedure: Cu(OAc)2 (0.03mmol), L2 (0.06mmol), aryl idione or bromide (0.5mmol), 1H-pyrazole (0.75mmol), NaOH (1mmol), and 1,4-dioxane (1mL) was added into a 5mL tube, then sealed. The mixture was stirred at 100C for certain time. After cooling to room temperature, the mixture was quenched with 10mL H2O and extracted with EtOAc (3×20mL). The combined EtOAc extracts were dried with anhydrous Na2SO4 and filtrated and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel with PE/EtOAc, as the eluent, to afford the desired products.
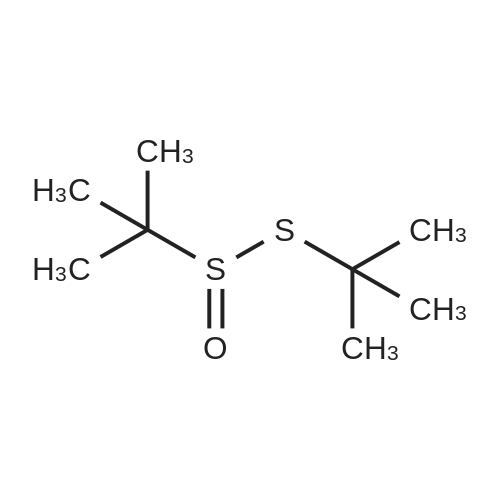
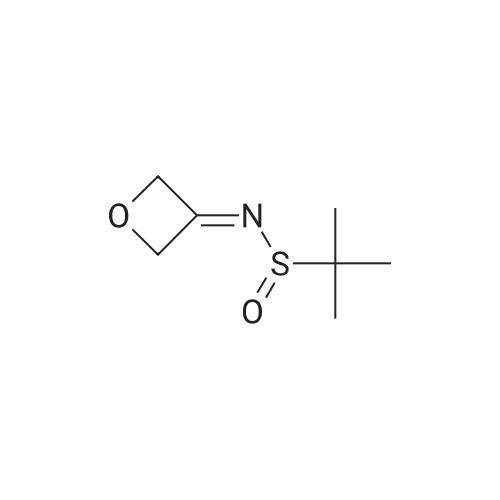
General procedure: Under an argon atmosphere, to a solution of substituted bromobenzene (2.2 mmol) in THF (10 mL) was added dropwise n-BuLi (1.0 mL, 2.5 mmol) at -78 C and the mixture was stirred for 30 min. A solution of <strong>[67734-35-4](R)-tert-butyl tert-butanethiosulfinate</strong> 6 (0.39 g, 2.0 mmol) in THF (2 mL) was then added dropwise to the reaction mixture at -78 C. The temperature was allowed to slowly warm to room temperature. The reaction mixture was stirred overnight at room temperature, and then evaporated under reduced pressure. Saturated NH4Cl (10 mL) was added, and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were washed with brine and dried over anhydrous Na2SO4, filtered, and evaporated under reduced pressure. The residue was purified by column chromatography with petroleum ether/EtOAc (4:1, V/V) to afford 7.13
With tris-(dibenzylideneacetone)dipalladium(0); (R)-(-)-1-[(SP)-2-(dicyclohexylphosphino)ferrocenyl]ethyldi-tert-butylphosphine; potassium tert-butylate; lithium chloride; In toluene; at 130℃; for 3h;Inert atmosphere;
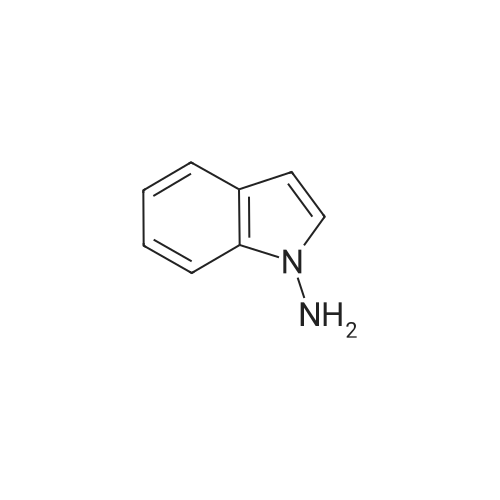
General procedure: A flame-dried resealable Schlenk tube was charged with Pd2(dba)3 (0.025 mmol, 2.5 mol %), Josiphos (0.05 mmol, 5 mol %), the solid reactant(s) (1.0 mmol of the <strong>[53406-38-5]1-aminoindole</strong>, 2.0 mmol of the aryl halide), LiCl (2.0 mmol) and KOtBu (1.4 mmol). The Schlenk tube was capped with a rubber septum, evacuated, and backfilled with argon; this evacuation/backfill sequence was repeated one additional time. The liquid reactant(s) and toluene (2 mL/mmol) were added through the septum. The septum was replaced with a teflon screwcap. The Schlenk tube was sealed, and the mixture was stirred at 130 C for 3 h. The resulting suspension was cooled to room temperature and filtered through a pad of celite eluting with ethyl acetate, and the inorganic salts were removed. The filtrate was concentrated and purification of the residue by silica gel column chromatography gave the desired product. All the compounds gave satisfactory spectroscopic data. Data for the selected compounds are given below:Compound 3a: Yield: 94%; TLC : Rf 0.39 (c-hexane/AcOEt 8:2). IR (neat): 3306, 1604, 1508, 1446, 1359, 1246, 1220, 1112, 1063, 832, 754 cm-1; 1H NMR (CDCl3, 300 MHz) : delta 7.55 (dd, 1H, J = 6.5, 2.3 Hz), 7.25-6.95 (m, 4H), 6.65 (d, 2H, J = 8.9 Hz), 6.42 (d, 1H, J = 3.3 Hz), 6.37 (d, 2H, J = 8.9 Hz), 6.32 (s, 1H), 3.62 (s, 3H).13C NMR (75 MHz, CDCl3) delta 154.5, 141.0, 135.9, 128.7, 126.6, 122.3, 121.1, 120.3, 114.7 (2C), 114.2 (2C), 109.5, 100.6, 55.6. m/z MS (ES+) 239.0 (M+H+).Compound 3i: Yield: 63%; TLC : Rf 0.40 (c-hexane/AcOEt 8:2). IR (neat): 3326, 1521, 1466, 1236, 1220, 1125, 1103, 832, 754 cm-1; 1H NMR (CDCl3, 300 MHz) : delta 7.79-7.61 (m, 1H), 7.33-7.10 (m, 4H), 6.99-6.83 (m, 2H), 6.55 (dd, 1H, J = 3.3, 0.7 Hz), 6.54, (br s, 1H), 6.46 (dd, 2H, J = 9.0, 4.4 Hz). 13C NMR (75 MHz, CDCl3) delta 159.4 (d, 1C, JC-F = 237.0 Hz), 143.4, 135.6, 128.5, 126.7, 122.5, 121.2, 120.4, 116.0 (d, 2C, JC-F = 23.5 Hz), 114.0 (d, 2C, JC-F = 8.3 Hz), 109.3, 100.9. m/z MS (ES+) 227.0 (M+H+).
With potassium fluoride; copper(l) iodide; palladium diacetate; catacxium A; In N,N-dimethyl-formamide; at 90℃; for 12h;Inert atmosphere;
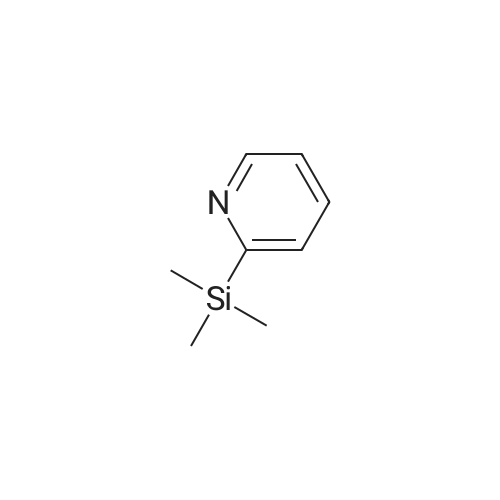
General procedure: 2-Trimethylsilylpyridine (1.2mmol), the aryl halide (1mmol), palladium acetate (0.10mmol), CataXCium A (0.2mmol), CuI (76mg, 0.4mmol) and KF (2.20mmol) were combined in reaction tubes in a Radleys green-house parallel synthesiser under a flow of nitrogen and degassed DMF (1ml) was added. The resulting suspensions were stirred at 90 oC under nitrogen for 12 hours. Reactions were analysed by LC-MS at 1mg/1ml in methanol to determine the yield.
With bis-triphenylphosphine-palladium(II) chloride; potassium acetate; In 1,4-dioxane; at 20 - 90℃; for 5h;Inert atmosphere;
To a stirred solution of 1-Bromo-2-methoxybenzene (3.0 g, 16.04 mmol) in 1,4-dioxane (30 mL) was added bis(pinacolato)diboron (4.06 g, 16.04 mmol), PdCl2(PPh3)2 (0.56 g, 0.802 mmol) and potassium acetate (3.14 g, 32.08 mmol) at room temperature, and the resulting mixture was stirred at 90 C under nitrogen for 5 h. The reaction mixture was diluted with ethyl acetate (50 mL) and filtered through celite. The filtrate was concentrated under vacuum and the residue was purified by column chromatography on silica gel (hexane) to give 23 [82] (3.0 g, 80.0 %) as an off-white solid, mp 76.5-77.8 C (ref. [82] 80-81 C): 1H NMR (400 MHz,CDCl3) delta 7.68 (dd, J = 7.2, 1.2 Hz, 1H), 7.39 (t, J = 7.2 Hz, 1H), 6.95 (t, J = 7.2 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 3.83 (s, 3H), 1.35 (s, 12H); IR (Neat) numax 2979.20, 1576.49, 1356.44, 1145.35 cm-1.
61%
With trans-bis(triphenylphosphine)palladium dichloride; potassium acetate; In 1,4-dioxane; at 130℃;
After adding and dissolving compound B-1(50g, 267mmol), bis(pinacolato)diboron(88g, 347mmol), bis(triphenylphosphine)palladium(II) dichloride(Pd(PPh3)Cl2)(9g, 13.35mmol), KOAc(65g, 667mmol) and 1,4-dioxane(1.3L) into a flask, the mixture was reacted at 130. After completing the reaction, the organic layer was extracted with ethyl acetate. The remaining moisture was removed from the obtained organic layer with magnesium sulfate. Then, the organic layer was dried. The remaining product was subjected to column chromatography to obtain compound1-1(38g, yield: 61%).
With 1H-imidazole; 1,1'-bis-(diphenylphosphino)ferrocene; palladium diacetate; ammonium chloride; N-ethyl-N,N-diisopropylamine; In 1,4-dioxane; at 90℃; for 3h;Sealed tube;
General procedure: To a stirred solution of aryl halide (Br/I) (1 mmol) in dry dioxane in a 25 mL sealed tube, was added Pd(OAc)2 (5 mol%), dppf (6 mol %), DIPEA (2 mmol), imidazole (0.25 mmol), ammonium chloride (2 mmol) and then Co2(CO)8 (0.3 mmol). The seal tube was closed immediately and stirred at 90 C for 3h. After the reaction time the reaction mixture was cooled to room temperature. The reaction mixture was filtered through celite pad and washed with dioxane, the filtrate was concentrated under reduced pressure and the residue obtained was purified by column chromatography.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In 1,4-dioxane; water; at 100℃;Inert atmosphere;
[00429] To a mixture of l-bromo-2-methoxy-benzene (5.1 g, 27.2 mmol), 2-bethoxypyridne-4-boronic acid (5.0 g, 32.7 mmol), H20 (30 mL) and K2C03 (7.5 g, 54 mmol) in dioxane (90 mL) was added Pd(dppf)Cl2 (222 mg, 0.27 mmol). The mixture was stirred at 100 °C under N2 for overnight, cooled to room temperture and evaporated under reduced pressure to dryness. The residue was diluted with EA (100 mL x2) and washed with water, dried over anhydrous Na2S04, filtered. The filtrate was evaporated in vacuum to residue, which was purified by silica gel chromatography (from PE to PE/EA = 10/1) to give (5.3 g, yield: 91 percent) of 2-methoxy-4-(2-methoxy-phenyl)-pyridine as yellow oil. MS: m/z 216.2 (M+ H+).
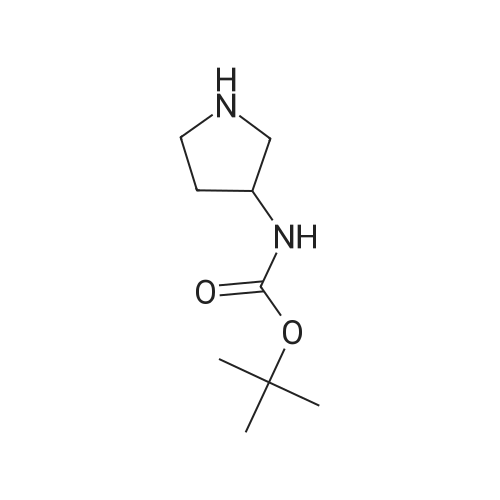
tert-butyl(1-(2-methoxyphenyl)pyrrolidin-3-yl)carbamate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With tris-(dibenzylideneacetone)dipalladium(0); 2,2'-bis-(diphenylphosphino)-1,1'-binaphthyl; In toluene; at 100℃; for 16.5h;Inert atmosphere;
[00578] Step 1 : To a solution of 2-bromo anisole (1.2 g, 6.45 mmol) in degassed toluene (20 mL) was added a suspension of Pd2(dba)2 (494 mg, 0.54 mmol), BINAP (1 g, 1.61 mmol) and tert-butyl pyrrolidin- 3-ylcarbamate (1 g, 5.37 mmol), and the mixture was stirred at 100 C for 16.5 h under argon. The reactant was portioned between water (30 mL) and EtOAc (20 mL). The organic layer was separated and the aqueous phase was extracted with EtOAc (20 mL x2). The combined organic layers were washed with brine (10 mL x 2) and concentrated to dryness. The crude product was purified by Combi flash (1406) (PE/EtOAc = 50/1 -20/1) to give tert-butyl (l-(2-methoxyphenyl)pyrrolidin-3-yl)carbamate (1.43 g, yield: 91%) as yellow oil. (1407) [00579] lH NMR (400 MHz, CDC13): delta = 6.91-6.84 (m, 4H), 6.73 (d, J= 6.4 Hz, 1H), 4.85 (brs, 1H), 4.30 (brs, 1H), 3.83 (s, 3H), 3.55-3.44 (m, 2H), 3.25-3.15 (m, 2H), 2.31-2.23 (m, 1H), 1.85-1.79 (m, 1H), 1.45 (s, 9H).
N-(2-methoxyphenyl)dibenzo[b,d]furan-4-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
The dibenzo [b, d] furan-4-amine 0.10molAnd sodium tert-butoxide 0.30 mol,Toluene 400 mL was added to the reaction flask,Stirred for 30 minutes,Nitrogen protection,Then 0.12 mol of 1-bromo-2-methoxybenzene was added,1.5 g of tris (dibenzylideneacetone) dipalladium,Finally, tri-tert-butylphosphine 4 g,And the temperature was raised to 100 C for 24 hours.After the reaction was completed, the reaction system was cooled,Water was added to terminate the reaction,filter,The filtrate was separated,Spin-dried toluene,A small amount of dichloromethane was added to dissolve the product after spinning,(Petroleum ether: dichloromethane = 3: 1 by volume)To give N- (2- methoxyphenyl) dibenzo [b, d] furan-4-amine (Intermediate D-1)(0.05mol, y = 50%).
4-(4-methoxyphenyl)-3-(4-ethoxyphenyl)sydnone[ No CAS ]
[ 159087-46-4 ]
1-(4-ethoxyphenyl)-4-(2-methoxyphenyl)-5-(4-methoxyphenyl)pyrazole[ No CAS ]
C18H18N2O2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
General procedure: A flask equipped with a reflux condenser was charged with pyrazole (1 eq.), and TBAF (10 eq. (1) M solution in THF) and heated at reflux under an inert atmosphere of nitrogen for 24 h. The mixture was allowed to cool to r.t., poured into water and extracted with ethyl acetate. The combined organic layers were dried over MgSO4 and concentrated in vacuo. Flash silica chromatography (gradient starting with 100% 40-60 petroleum ether and ending with 60% ethyl acetate in 40-60 petroleum ether) afforded the target desilated pyrazoles.
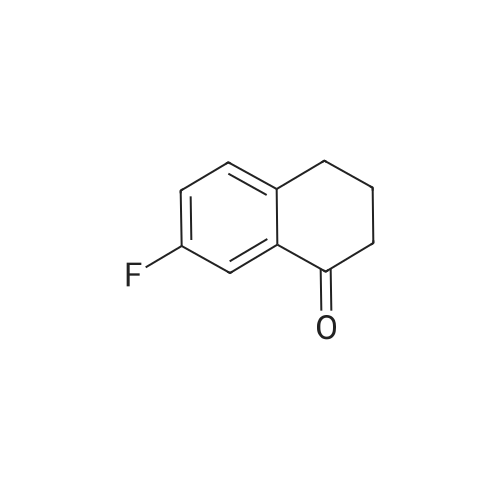
7-fluoro-2-(2-methoxyphenyl)-3,4-dihydronaphthalen-1(2H)-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
90%
With sodium hydroxide; bis(dibenzylideneacetone)-palladium(0); tri tert-butylphosphoniumtetrafluoroborate; In 1,4-dioxane; water; at 100℃; for 1h;Inert atmosphere; Microwave irradiation;
General procedure: In a microwave tube were added a suspension of Pd(dba)2 (5mol%), tBu3PHBF4 (10mol%), NaOH (2eq), aryl bromide (1.2eq) and alpha-tetralone (1.0eq) in a mixture of degassed dioxane/water (4:1, v/v, 2-4mL) and heated under Ar and microwave irradiation (100W of initial power, 100C, 60min, infrared probe). Then, the mixture was allowed to cool to rt, diluted in AcOEt, washed with saturated NH4Cl solution, dried over anhydrous MgSO4, filtered and concentrated under reduced pressure. The crude material was purified by silica gel column with n-hexane:AcOEt (95:5) as solvent.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping