* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With diphenyl phosphoryl azide; triethylamine In N,N-dimethyl-formamide at 20℃; for 3 h; Stage #2: With water In N,N-dimethyl-formamide at 100℃; for 1 h;
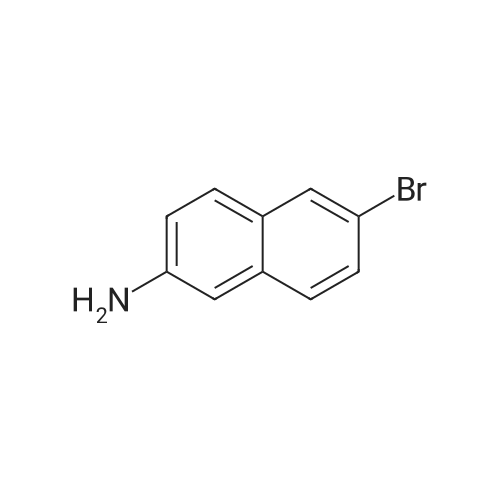
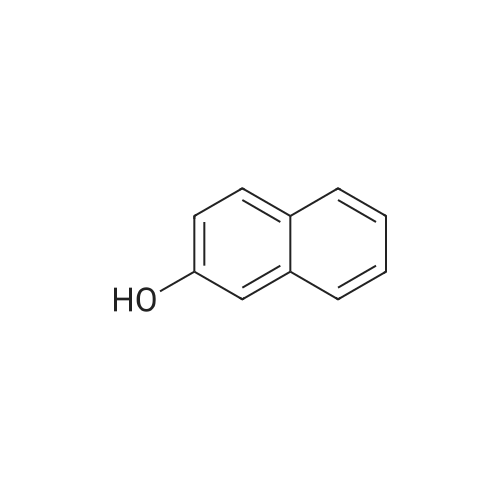
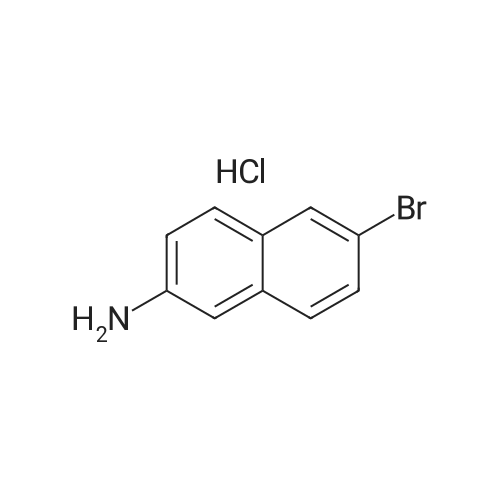
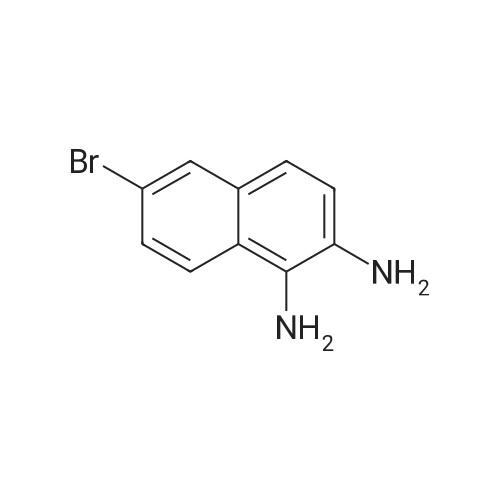
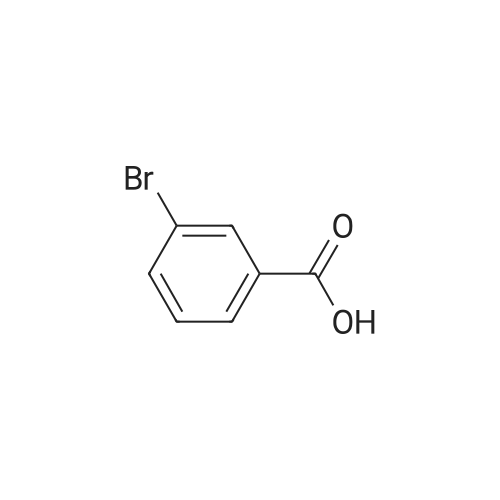
Part B. Preparation of 6-bromonaphthalen-2 -amine.; [00748] A solution of the product Part A (5.07g, 20.19mmol) and triethylamine (4.22mL, 3.07g, 30.3mmol) in dry DMF (155mL) was treated with the diphenylphosphoroyl azide (6.55mL, 8.34g, 30.3mmol) followed by stirring at room temperature for 3h. The solution was then treated with water (2OmL) followed by warming at 1000C for Ih. The solution was cooled and the flask fitted with a short- path distillation head and the DMF removed by distillation under high vacuum. The solid residue was dissolved in EtOAc and washed with saturated sodium bicarbonate solution. Filtered through celite and the filtrate was washed with water (3x) and then with brine. Dried over Na2SC^, filtered and concentrated under vacuum to give the title compound as a beige solid (4.48g, 100 percent).
100%
Stage #1: With diphenyl phosphoryl azide; triethylamine In N,N-dimethyl-formamide at 20℃; for 3 h; Stage #2: With water In N,N-dimethyl-formamide at 100℃; for 1 h;
[00490] Part B. Preparation of 6-bromonaphthalen-2-amine.; [00491] A solution of the product Part A (5.07g, 20.19mmol) and triethylamine (4.22mL, 3.07g, 30.3mmol) in dry DMF (155mL) was treated with the diphenylphosphoroyl azide (6.55mL, 8.34g, 30.3mmol) followed by stirring at room temperature for 3h. The solution was then treated with water (2OmL) followed by warming at 1000C for Ih. The solution was cooled and the flask fitted with a short- path distillation head and the DMF removed by distillation under high vacuum. The solid residue was dissolved in EtOAc and washed with saturated sodium bicarbonate solution. Filtered through celite and the filtrate was washed with water (3x) and then with brine. Dried over Na2SO4, filtered and concentrated under vacuum to give the title compound as a beige solid (4.48g, 100 percent).
100%
Stage #1: With diphenyl phosphoryl azide; triethylamine In N,N-dimethyl-formamide at 20℃; for 3 h; Stage #2: With water In N,N-dimethyl-formamide at 100℃; for 1 h;
Part 1. Preparation of 6-bromonaphthalen-2 -amine.[00328| A solution of the product Part H (5.07 g, 20.19 mmol) and triethylamine (4.22 mL, 3.07 g,30.3 mmol) in dry N.N-dimethylformamide (155mL) was treated with the diphenylphosphoroyl azide(6.55 mL, 8.34 g, 30.3 mmol) followed by stirring at room temperature for 3 hours. The solution was then treated with water (20 mL) followed by warming at 100 0C for 1 hour. The solution was cooled and the flask fitted with a short-path distillation head and the NN-dimethylformamide was removed by distillation under high vacuum. The solid residue was dissolved in ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was filtered through diatomaceous earth, and the filtrate was washed with water (3*) and then with brine. The organic layer was dried overNa2SO4, filtered and concentrated under vacuum to give the title compound as a beige solid (4.48 g,100 percent).
With copper(l) iodide; copper(l) chloride In N,N-dimethyl-formamide for 4 h; Inert atmosphere; Reflux
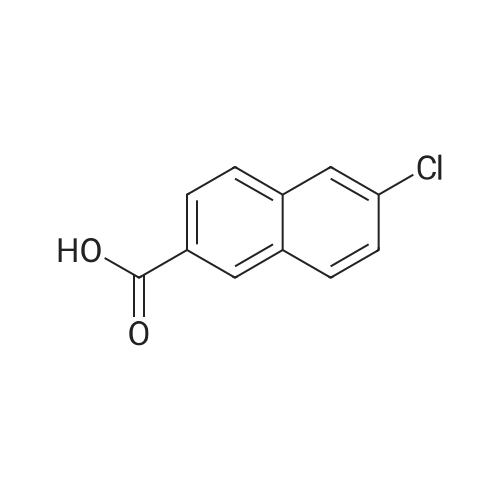
Example 32 Synthesis of Compound 32 6-chloro-2-naphthoic acid A suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCl (11.7 g, 114.64 mmol) and CuI (2.19 g, 11.50 mmol) in degassed DMF (45 mL) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 mL) and the resulting mixture was extracted with EtOAc (2*500 mL). The combined organic layers were then washed with H2O (4*500 mL) followed by brine (1*500 mL), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 32 (2.2 g, 93percent) as an off-white solid. HPLC tR 6.47 min.
93%
With copper(l) iodide; copper(l) chloride In N,N-dimethyl-formamide for 4 h; Inert atmosphere; Darkness; Reflux
Example 66 - Synthesis of Compound 68 6-chloro-2-naphthoic acid686-chloro-2 -naphthoic acidA suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCI (1 1.7 g, 114.64 mmol) and CuI (2.19 g, 1 1.50 mmol) in degassed DMF (45 ml.) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 ml.) and the resulting mixture was extracted with EtOAc (2 x 500 mL). The combined organic layers were then washed with H2O (4 x 500 mL) followed by brine (1 x 500 mL), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 68 (2.2 g, 93percent) as a off-white solid. HPLC fR 6.47 min.
93%
With copper(l) iodide; copper(l) chloride In N,N-dimethyl-formamide for 4 h; Reflux; Inert atmosphere; Darkness
A suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCI (1 1.7 g, 114.64 mmol) and CuI (2.19 g, 1 1.50 mmol) in degassed DMF (45 ml.) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 ml.) and the resulting mixture was extracted with EtOAc (2 x 500 ml_). The combined organic layers were then washed with H2O (4 x 500 ml.) followed by brine (1 x 500 ml_), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 70 (2.2 g, 93percent) as a off-white solid. HPLC fe 6.47 min
93%
With copper(l) iodide; copper(l) chloride In N,N-dimethyl-formamide for 4 h; Inert atmosphere; Darkness; Reflux
A suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCl (11.7 g, 114.64 mmol) and CuI (2.19 g, 11.50 mmol) in degassed DMF (45 mL) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 mL) and the resulting mixture was extracted with EtOAc (2*500 mL). The combined organic layers were then washed with H2O (4*500 mL) followed by brine (1*500 mL), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 68 (2.2 g, 93percent) as a off-white solid. HPLC tR 6.47 min.
75%
With CuI In N,N-dimethyl-formamide
Step 1. To a solution of 6-bromo-2-naphthoic acid (4.4 g, 17.5 mmol) in 50 mL anhydrous DMF were added CuCl (8.7 g, 87.5 mmol) and CuI (0.2 g). The slurry was refluxed for 1 hour. At room temperature it was diluted with 300 mL EtOAc and stirred for 2 hours. It was filtered through celite. The filtrate was evaporated in vacuuo to afford 6-chloro-2-naphthoic acid (2.7 g, 75percent). ES-MS: (M+H)+207.
Stage #1: With lithium hydroxide monohydrate In tetrahydrofuran; water Stage #2: With hydrogenchloride In tetrahydrofuran; water
General procedure: Compound 14 (4.58 g, 20 mmol) in 1:1 THF/H2O (200 mL) was treated with LiOH.H2O (4.2 g, 100 mmol), stirred overnight, and concentrated to a thick slurry and then treated with 2M HCl to pH = 3 and the precipitate was filtered, washed with water and dried under vacuum to provide 3.8 g (88percent) of 15 as a white powder
84%
With potassium hydroxide In methanol at 50℃; for 8 h;
A suspension of methyl 6-bromo-2-naphthoate (3b) (2.7 g, 10.0 mmol) and potassium hydroxide (1.1 g, 20.0 mmol) in methanol (50 mL) was vigorously stirred at 50 °C. The reaction mixture becomes homogeneous after the consumption of the initial compound 3b. After 8 h, the solvent was evaporated under reduced pressure (ca 2/3 vol.), water (1500 mL) was added and the unreacted ester extracted with ethyl acetate. The aqueous solution was acidified with 10percent H2SO4 to pH 3 and, after extraction with ethyl acetate (3 .x. 200 mL) and drying on anhydrous sodium sulfate, removal of the solvent afforded the pure acid 3a (2.1 g, 8.4 mmol, 84percent yield). Mp 290-294 °C (decomp.); HRMS (ESI+): m/z [M+1]+ Calcd for C11H8BrO2: 252.08591. Found: 252.08582. 1H NMR (DMSO-d6) δ = 7.72 (1 H, dd, J = 8.5 and 1.8 Hz, 7-naphthyl H), 7.99 (1 H, d, J = 8.6 Hz, 4-naphthyl H), 8.02 (1 H, dd, J = 8.6 and 1.4 Hz, 8-naphthyl H), 8.09 (1 H, d, J = 8.8 Hz, 3-naphthyl H), 8.30 (1 H, d, J = 1.6 Hz, 5-naphthyl H), 8.62 (1 H, s, 1-naphthyl H), 13.15 (1H, br. s, COOH); 13C NMR (125.76 MHz, DMSO-d6) δ = 121.93, 126.50, 127.61, 128.81, 129.83, 130.01, 130.64, 130.90, 131.63, 136.11, 167.30.
80%
Stage #1: With water; lithium hydroxide In tetrahydrofuran; ethanol at 50℃; for 16 h; Stage #2: With hydrogenchloride In water
Example 176-BROMONAPHTHALENE-2-CARBOXYLIC ACID [(R)-1-(4-METHANESULFONYLAMINO-3-METHYLPHENYL)ETHYL]AMIDE 17A) 6-BROMONAPHTHALENE-2-CARBOXYLIC ACID To a stirred solution of 6-bromonaphthalene-2-carboxylic acid methyl ester (2 g, 8 mmol) in tetrahydrofuran (66 mL) and ethanol (22 mL) was added a solution of lithium hydroxide (542 mg, 22 mmol) in water (22 mL). The reaction was stirred at 50° C. for 16 hours. After cooling, the organic solvents were removed by evaporation, and the aqueous residue was diluted with water (100 mL) then washed with EtOAc (2.x.50 mL). The aqueous layer was acidified using 1N HCl and the products were extracted with EtOAc (3.x.50 mL). The combined organics were washed with brine (100 mL), dried (MgSO4), filtered and concentrated. Trituration with DCM gave the title compound (1.594 g, 80percent) as an off-white solid.1H NMR (400 MHz, DMSO-d6) δ 7.74 (dd, 1H, J=8.7 Hz, 1.9 Hz), 7.99-8.04 (m, 2H), 8.10 (d, 1H, J=8.8 Hz), 8.32 (s, 1H), 8.64 (s, 1H).
70%
Stage #1: With lithium hydroxide; water In tetrahydrofuran at 20℃; for 48 h; Stage #2: With hydrogenchloride; water In tetrahydrofuran at 0℃;
Part A. Preparation of 6-bromo-2 -naphthoic acid.; [00746] A solution of methyl 6-bromo-2-naphthoate (7.7Og, 29.0mmol) in 2:1 THF:water (15OmL) was treated with lithium hydroxide hydrate (2.44g, 58.1mmol) followed by stirring at room temperature for 48h. Concentrated under vacuum, diluted with water and cooled to O0C. Acidified to pH3 with 4N HCl. Solids were collected by filtration, dissolved in toluene-EtOAc (ca. 2L) and washed with brine. Dried over Na2SO4, filtered and concentrated under vacuum. Brown solid was triturated with ether, collected by filtration, and dried under vacuum to give the title compound as a nearly white solid (5.07g, 70percent).
70%
Stage #1: With lithium hydroxide; water In tetrahydrofuran at 20℃; for 48 h; Stage #2: With hydrogenchloride In water at 0℃;
[00487] Example 1. Preparation ofN-(6-(3-tert-butyl-5-(2,4-dioxotetrahydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IA-LO-2.9).; [00488] Part A. Preparation of 6-bromo-2 -naphthoic acid.; [00489] A solution of methyl 6-bromo-2-naphthoate (7.7Og, 29.0mmol) in 2:1 THF:water (15OmL) was treated with lithium hydroxide hydrate (2.44g, 58.1mmol) followed by stirring at room temperature for 48h. Concentrated under vacuum, diluted with water and cooled to O0C. Acidified to pH3 with 4N HCl. Solids were collected by filtration, dissolved in toluene-EtOAc (ca. 2L) and washed with brine. Dried over Na2SC^, filtered and concentrated under vacuum. Brown solid was triturated with ether, collected by filtration, and dried under vacuum to give the title compound as a nearly white solid (5.07g, 70percent).
70%
Stage #1: With lithium hydroxide monohydrate In tetrahydrofuran; water at 20℃; for 48 h; Stage #2: With hydrogenchloride In tetrahydrofuran; water
Part H. Preparation of 6-bromo-2 -naphthoic acid.[00326] A solution of methyl 6-bromo-2-naphthoate (7.7Og, 29.0mmol) in 2: 1 tetrahydrofuran: water(150 mL) was treated with lithium hydroxide hydrate (2.44 g, 58.1 mmol) followed by stirring at room temperature for 48 hours. The mixture was concentrated under vacuum, diluted with water and cooled to 0 0C. The mixture was acidified to pH3 with 4 N HCl. Solids were collected by filtration, dissolved in toluene-ethyl acetate (ca. 2 L) and washed with brine. The organic layer was dried overNa2SO+, filtered and concentrated under vacuum. The brown solid was triturated with ether, collected by filtration, and dried under vacuum to give the title compound as a nearly white solid (5.07g, 70percent).
56%
With potassium hydroxide In methanol at 50℃; for 48 h;
Potassium hydroxide (127 mg, 2.26 mmol) was added to a suspension of methyl 6-bromo-2-naphthoate (200 mg, 0.75 mmol) in methanol (50 mL) and the mixture heated at 50 °C for 48 h. The solvent was evaporated and the residue diluted with water (30 mL), acidified with HCl (1 M) and the extracted with ethyl acetate (30 mL * 2). The combined organic layers were dried with anhydrous magnesium sulfate and the solvent evaporated under reduced pressure. The crude product was purified via recrystallization using ethyl acetate to give the product as white crystals (105 mg, yield 56percent). 1H NMR matched that previously reported. 1H NMR (400 MHz, CD3OD): δ 8.58 (s, 1H; Ar-H), 8.14 (s, 1H; Ar-H), 8.06 (dd, J = 1.8, 8.7 Hz, 1H; Ar-H), 7.92 (d, J = 8.7 Hz, 1H; Ar-H), 7.87 (d, J = 8.7 Hz, 1H; Ar-H), 7.64 (dd, J = 1.8, 8.7 Hz, 1H; Ar-H).
Reference:
[1] Organic and Biomolecular Chemistry, 2015, vol. 13, # 27, p. 7477 - 7486
[2] Organic and Biomolecular Chemistry, 2015, vol. 13, # 27, p. 7477 - 7486
[3] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 4, p. 1557 - 1568
[4] Bioorganic Chemistry, 2011, vol. 39, # 4, p. 151 - 158
[5] Patent: US2012/88746, 2012, A1, . Location in patent: Page/Page column 47-48
[6] Patent: WO2009/39127, 2009, A1, . Location in patent: Page/Page column 174
[7] Patent: WO2009/39134, 2009, A1, . Location in patent: Page/Page column 103
[8] Patent: WO2010/111437, 2010, A1, . Location in patent: Page/Page column 73-74
[9] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 16, p. 4355 - 4367
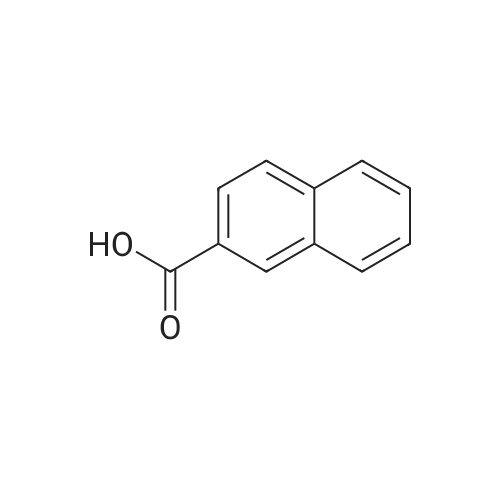
6
[ 1590-25-6 ]
[ 5773-80-8 ]
Yield
Reaction Conditions
Operation in experiment
70%
With sodium hypochlorite; sodium hydroxide In 1,4-dioxane; water at 70℃; for 4 h;
1-(6-bromo-naphthalen-2-yl)ethaneone (0.22 g, 0.88 mmol) obtained in Preparation Example 100 was dissolvedin 3 mL of 1,4-dioxane. NaOH (0.353 g, 8.8 mmol) dissolved in 3 mL of water and 9-11percent NaOCl solution (1.67mL, 2.64 mmol) were added thereto, and the mixture was heated to 70°C and stirred for 4 hours. After addition of NaHSO3aqueous solution and water, the reaction solution was extracted with ether. 1N HCl was added thereto, and the organiclayer was separated, dried with MgSO4 and purified by column chromatography to obtain the title compound (0.156 g,70percent).1H-NMR (CDCl3) δ 8.56 (1H, s), 8.05 (2H, m), 7.79 (2H, m), 7.58 (1H, m).
88%
With sodium hydroxide; sodium hypochlorite In 1,4-dioxane; water
6-bromo-2-naphthalenecarboxylic acid (Compound M) To a solution of sodium hypochlorite (62 ml, 5.25percent in water (w/w), 3.6 g, 48.18 mmol) and sodium hydroxide (6.4 g, 160.6 mmol) in 50 ml of water was added a solution of 2-acetyl-6-bromonaphthalene (Compound L) 4 g, (16.06 mmol) in 50 ml of 1,4-dioxane. The yellow solution was heated to 70° C. in an oil bath for 2 hours, cooled to ambient temperature, and extracted with ethyl ether (2*50 ml). The aqueous layers were diluted with NaHSO3 solution (until KI indicator solution remained colorless) and then acidified (pH <2) with 1N sulfuric acid to give a white precipitate. The mixture was extracted with ethyl ether, and the combined organic phase washed with saturated aqueous NaCl, dried (MgSO4) and concentrated to give 3.54 g (88percent) of the title compound as a solid. 1H NMR (DMSO-d6): δ 8.63 (1H, br s), 8.32 (1H, d, J=2.0 Hz), 8.10 (1H, d, J=8.8 Hz), 8.00-8.05 (2H, m), 7.74 (1H, dd, J=2.0, 8.8 Hz).
88%
With sodium hydroxide; sodium hypochlorite In 1,4-dioxane; sulfuric acid; water
6-bromo-2-naphthalenecarboxylic acid (Compound M) To a solution of sodium hypochlorite (62 ml, 5.25percent in water (w/w), 3.6 g, 48.18 mmol) and sodium hydroxide (6.4 g, 160.6 mmol) in 50 ml of water was added a solution of 2-acetyl-6-bromonaphthalene (Compound L) 4 g, (16.06 mmol) in 50 ml of 1,4-dioxane. The yellow solution was heated to 70° C. in an oil bath for 2 hours, cooled to ambient temperature, and extracted with ethyl ether (2*50 ml). The aqueous layers were diluted with NaHSO3 solution (until KI indicator solution remained colorless) and then acidified (pH<2) with IN sulfuric acid to give a white precipitate. The mixture was extracted with ethyl ether, and the combined organic phase washed with saturated aqueous NaCl, dried (MgSO4) and concentrated to give 3.54 g (88percent) of the title compound as a solid. 1H NMR (DMSO-d6): δ 8.63 (1H, br s), 8.32 (1H, d, J=2.0 Hz), 8.10 (1H, d, J=8.8 Hz), 8.00-8.05 (2H, m), 7.74 (1H, dd, J=2.0, 8.8 Hz).
Reference:
[1] Patent: EP3239143, 2017, A2, . Location in patent: Paragraph 0306
[2] Patent: US5877207, 1999, A,
[3] Patent: US5958954, 1999, A,
7
[ 580-13-2 ]
[ 1590-25-6 ]
[ 75-36-5 ]
[ 5773-80-8 ]
Reference:
[1] Patent: US4454341, 1984, A,
8
[ 37796-78-4 ]
[ 5773-80-8 ]
Yield
Reaction Conditions
Operation in experiment
8.29 g
at 110℃; for 4.5 h;
A glass container of 500 ml, provided with a reflux condenser, a gas injecting tube, an exhaust tube, a temperature measurement tube, and an electromagnetic stirrer, was set up with 210 g (3.5 mol) of acetic acid, 1.66 g (6.66 mmol) of cobalt acetate tetrahydrate, 1.65 g (6.73 mmol) of manganese acetate tetrahydrate, 1.06 g (8.91 mmol) of potassium bromide, 10.0 g (45 2 mmol) of 6-bromo-2-methylnaphthalene, and 5.2 g (51.0 mmol) of acetic acid anhydride. An oxidation reaction for producing BNA was performed by stirring the mixture in the glass container during 4.5 hours under ordinary pressure while keeping the inner temperature of the glass container at 110 degrees C. and injecting pure oxygen with a flow rate of 0.2 litters/minute into the glass container. [0043] After completing the reaction and cooling the reaction liquid to 30 degrees C., the cooled liquid was filtrated to obtain a precipitate as a residue. The obtained residue was dried under reduced pressure of 5 mmHg and 40 degrees C. until being of constant mass. A crude BNA product was thus obtained with a purity of 99.2percent and a weight of 8.29 g.
6-bromo-2-naphthol crystals. Finally, the crude crystals were reintroduced to a mass fraction of 66.7percent with a mass of 2160 Kg Of the acetic acid solution in the re-dissolved, dissolved and then static 1. 5h, filtration, washing, After drying, a recrystallization of 6-bromo-2-naphthoic acid was obtained in a yield of 95. 3percent pure Degrees 99. 55percent, single miscellaneous 0. 45percent
Reference:
[1] Patent: CN106542971, 2017, A, . Location in patent: Paragraph 0035
11
[ 135-19-3 ]
[ 5773-80-8 ]
Reference:
[1] Patent: CN106542971, 2017, A,
12
[ 580-13-2 ]
[ 5773-80-8 ]
Reference:
[1] Patent: EP3239143, 2017, A2,
13
[ 1590-25-6 ]
[ 5773-80-8 ]
[ 32405-50-8 ]
Reference:
[1] Indian Journal of Chemistry, Section A: Inorganic, Physical, Theoretical & Analytical, 1980, vol. 19, # 7, p. 646 - 649
14
[ 67-56-1 ]
[ 5773-80-8 ]
[ 33626-98-1 ]
Yield
Reaction Conditions
Operation in experiment
100%
Reflux
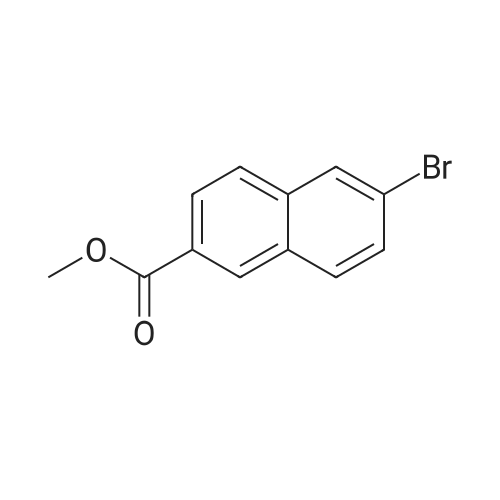
6-Bromo-2-naphthalenecarboxylic acid (2.4996 g, 10.0 mmol) was added50mL dried egg-shaped flask, add 20mL of anhydrous methanol to dissolve,Then 1 mL of concentrated sulfuric acid was slowly added dropwise and the system was refluxed overnight.TLC tracking until the conversion of raw materials is completed, stop heating,After cooling to room temperature, saturated aqueous sodium carbonate solution was added to quench the reaction.The reaction system was adjusted to neutrality, extracted with ethyl acetate,The organic phase is washed three times with anhydrous sodium sulfate.Concentration under reduced pressure afforded S7 (2.63 g, 100percent yield) as a white solid.
93%
Reflux
To a solution of 6-bromo-2-naphthoic acid (2.65 g, 10 mmol) in methanol 30 mL,concentrated sulfuric acid 1 mL was added drop wise. The reaction mixture was refluxed for 2 h, then added water 150 mL. The foliated crystal was collected thought filtration. Yield: 93percent;
26.4 g
for 7 h; Reflux
Reactor to 6-bromo-2-naphthalene carboxylic acid (T-1, manufactured by Tokyo Kasei Kogyo (Ltd.)) (25g, 99.6mmol), concentrated sulfuric acid (5.4ml, 99.6mmol), and methanol (80ml) It was placed. The mixture was stirred with heating under reflux for 7 hours. After cooling to room temperature, water was added to the reaction mixture and extracted with dichloromethane. Aqueous organic layers were washed with sodium carbonate, washed with water, then saturated brine, and dried over anhydrous magnesium sulfate. By concentrating the solution under reduced pressure to give compound (T-2) (26.4g, 99.6mmol) and. Compound (T-2) was used in the next reaction without purification.
Reference:
[1] Tetrahedron, 2009, vol. 65, # 7, p. 1349 - 1360
[2] Journal of the American Chemical Society, 2008, vol. 130, # 50, p. 16836 - 16837
[3] Patent: CN107286150, 2017, A, . Location in patent: Paragraph 0153; 0154; 0155; 0156; 0157
[4] European Journal of Medicinal Chemistry, 2015, vol. 102, p. 277 - 287
[5] Patent: JP2016/37458, 2016, A, . Location in patent: Paragraph 0172; 0173
15
[ 5773-80-8 ]
[ 71590-31-3 ]
Yield
Reaction Conditions
Operation in experiment
91%
Stage #1: With diphenyl phosphoryl azide; triethylamine In <i>tert</i>-butyl alcohol for 15 h; Reflux Stage #2: With hydrogenchloride In methanol at -78 - 20℃; for 15 h;
A mixture of 6-bromo-2-naphthoic acid (80.3 g, 319 mmol), diphenylphosphoryL azide (71 mL, 352 mmol) and triethylamine (50 mL, 358 mmol) in fert-butanol (400 mL) was heated to reflux and allowed to stir at this temperature for 15 hours. The reaction mixture was then cooled to room temperature and poured over saturated aqueous NaHC03 solution (6Ψ0 mL) and stirred vigorouslyfor 30 minutes. The resulting suspension was filtered, washed with water (200 mL) and dried in vacuo at 65 °C. The resulting white solid was suspended in MeOH (500 mL) and cooled to -78 °C, then HCl gas was bubbled into the mixture until saturated. The reaction mixture was then allowed to stir at room -temperature for 15 hours, after which time the resulting solids were collected by filtration, then washed with- ice-cold MeOH (100 mL) to provide Compound Int-6a as an off-white solid (74.8 g, 91percent), which was used without further purification. l NM (DMSO-_3/4) 6 10.5-10.0 (br s, 3H), 8.23 (s, 1H), 7.99 (d, J= 9.0 Hz, 1H), 7.92 (d, J= 9.0 Hz, 1H), 7.84 (s, 1H), 7.68-7.65 (m, 1H), 7.56-7.51 (m, 1H . "LRMS: (M+2H)+ = 223.
91%
Stage #1: With diphenyl phosphoryl azide; triethylamine In <i>tert</i>-butyl alcohol for 15 h; Reflux Stage #2: With sodium hydrogencarbonate In tert-butyl methyl ether for 0.5 h; Stage #3: With hydrogenchloride In methanol at -78 - 20℃; for 15 h;
Preparation of Compound 1A1AA mixture of 6-bromo-2--naphthoic acid-(80.3 g, 319 mmol),diphenylphosphorylazide (71 mL, 352 mmol) and triethylamine (50 mL, 358 mmol) in fer/-butanol (400 mL) was heated to reflux for 15 hours then cooled to room temperature. The reaction mixture was poured over saturated NaHC03 (600 mL) and stirred vigorously for 30 minutes. The solids were filtered, washed with water (200 mL) and dried under vacuum at 65 °C. The resulting. white solid was suspended in MeOH (500 mL), then HCl(g) was bubbled into the mixture at -78 °C until saturated. The reaction mixture was stirred at room temperature for 15 hours, then the resulting solids were collected by filtration, and washed with ice-cold MeOH (100 mL) to provide Compound 1A as an off- white solid (74.8 g, 91percent). 1H NMR (DMSO-<4) δ 10.5-10.0 (br s, 3H), 8.23 (s, 1H), 7.99 (d, J= 9.0 Hz, 1H), 7.92 (d, J= 9.0 Hz, 1H), 7.84 (s, 1H), 7.68-7.65 (m, 1H), 7.56-7.51 (m, 1H). LRMS: (M+2H)+ = 223.
With dimethylsulfide borane complex In tetrahydrofuran at 0 - 20℃;
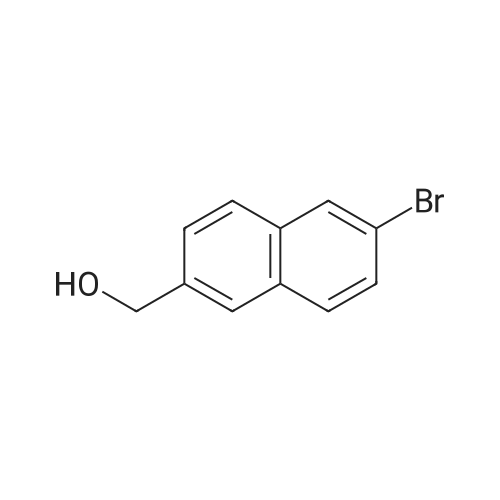
(6-bromo-2-naphthvπmethanol To a solution of 6-bromo-2-naphthoic acid (1g, 3.98mmol) in THF (20ml) at O0C was added 2M borane dimethyl sulfide in THF (0.53 ml, 5.97 mmol) portion wise. The mixture was warmed to RT and stirred overnight. The reaction was cooled to O0C and MeOH was added. The solution was concentrated under reduced pressure. The crude product was purified by column chromatography (EtOAc in heptane) to afford the title compound (0.4 g, 89percent). <n="33"/>LCMS: m/z 238 [M+H]\\
85%
With borane-THF In tetrahydrofuran at 0 - 25℃;
Into a 250-mL 3-necked round-bottom flask was placed 6-bromo-2-naphthoic acid (10 g, 39.83 mmol, 1.00 equiv), tetrahydrofuran (40 mL). This was followed by the addition of BH3THF (1 M) (80 mL, 2.00 equiv) dropwise with stirring at 0°C. The resulting solution was stirred overnight at 25°C. The reaction was then quenched by the addition of 100 mL of ice/water. The resulting solution was extracted with 2x200 mL of ethyl acetate and the organic layers were combined. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 8 g (85percent) of (6-bromonaphthalen-2-yl)methanol as a light yellow solid.
77%
With borane-THF In tetrahydrofuran at 0 - 20℃;
Preparation of {6- [5- (2-Phenoxy-ethylsulfanylmethyl)- [1, 3,4] oxadiazol-2-yl] -naphthalen- 2-yl}-methanol a) (6-Bromo-naphthalen-2-yl)-methanol A THF solution of 6-bromo-2-naphthoic acid (3.5 g, 13.94 mmol, 1 eq. ) was cooled to 0°C in an ice bath and then treated dropwise with borane-THF complex (1 M solution in THF, 16.7 mL, 16.7 mmol, 1.2 eq. ) via syringe. After addition was complete, the reaction was allowed to gradually warm to room temperature and stir at that temperature overnight. Several milliliters of water were added to the reaction to quench the remainder of the borane. The reaction was diluted with water and extracted with ethyl acetate. The organic layer was separated and then washed with aqueous 1 M NaOH and then brine. The organic layer was then collected, dried over MgSO4, filtered and the solvent removed in vacuo leaving (6-bromo-naphthalen-2-yl) -methanol (2.56 g, 77percent yield) as a white solid in the flask.
74%
With diisobutylaluminium hydride In tetrahydrofuran at 0 - 25℃; for 16 h;
To a solution of 6-bromo-2-naphthoic acid (1.0 g, 4.0 mmol) in TUF (20 mL) at 0°C was addedDIBAL-H (8.0 mE, 8.0 mmol). After addition, the reaction mixture was stirred at 25 °C for 16h. The rcaction was quenched by the addition of saturated aqueous NH4C1 (20 mE) arid thenextracted with ethyl acetate (3 x 30 mE). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography(petroleum ether: ethyl acetate 2:1) to give the title compound (0.7 g, 74 percent yield) as a white solid.
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 1, p. 267 - 273
[2] Patent: WO2009/130475, 2009, A1, . Location in patent: Page/Page column 31-32
[3] Patent: WO2016/40891, 2016, A2, . Location in patent: Paragraph 00335
[4] Patent: WO2005/40157, 2005, A2, . Location in patent: Page/Page column 83
[5] Patent: WO2016/112284, 2016, A1, . Location in patent: Page/Page column 132; 133
[6] Journal of Medicinal Chemistry, 1993, vol. 36, # 17, p. 2485 - 2493
[7] Patent: JP2016/37458, 2016, A,
[8] Patent: WO2018/118838, 2018, A1, . Location in patent: Paragraph 00249
[9] Patent: CN108530392, 2018, A, . Location in patent: Paragraph 0025; 0033; 0035
17
[ 459423-32-6 ]
[ 5773-80-8 ]
[ 106685-40-9 ]
Yield
Reaction Conditions
Operation in experiment
99%
Stage #1: With potassium carbonate In tetrahydrofuran; water for 8 h; Heating / reflux Stage #2: With hydrogenchloride; water In tetrahydrofuran for 1 h;
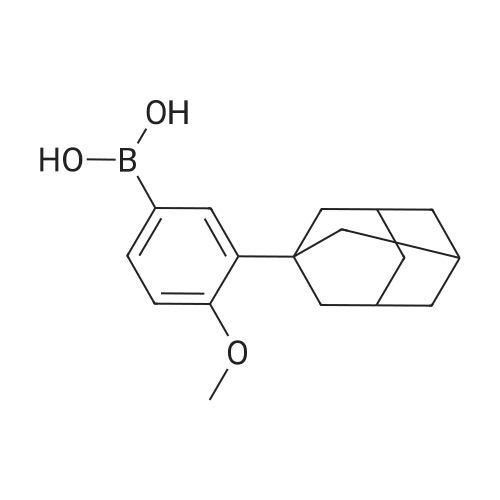
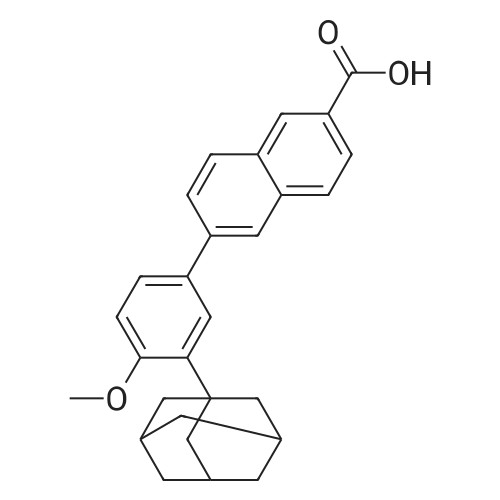
Example 3 :Preparation of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2- naphthoic acid (I) :20 ml (12 vol) of tetrahydrofuran, 2 g (7 mmol) of 3-adamantyl-4-methoxyphenylboronic acid (II), 1.65 g (6.6 mmol) of 6 -broτno-2 -naphthoic acid (III) and 20 mL of a 2 M aqueous potassium carbonate solution are introduced into a round-bottomed flask equipped with EPO <DP n="13"/>stirring and under a nitrogen stream. 0.7 g (5percent) of 10percent palladium on carbon (50percent wet; Keraeus type K-0218) is then introduced.The medium is heated under reflux for 8 hours. The catalyst is filtered on a cartridge, and then slowly poured over 30 ml of a 1 N aqueous hydrochloric acid solution.The medium is kept stirring for one hour. The precipitate is filtered, washed with water and then dried under reduced pressure. 2.06 g of 6- [3- (1- adamantyl) -4-methoxyphenyl] -2-naphthoic acid are obtained in the form of a white solid whose purity, determined by HPLC, is 99.9percent (yield = 79percent; m.p. 321°C) .
99%
Stage #1: With potassium hydroxide In tetrahydrofuran; water at 55℃; for 2 h; Heating / reflux Stage #2: With hydrogenchloride; water In tetrahydrofuran at 20℃;
Example 2 :Preparation of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2- naphthoic acid (I) :80 g (0.319 mol) of 6-bromo-2 -naphthoic acid, 95.7 g (0.335 mol, 1.05 eq) of 3 -adamantyl-4 -methoxyphenyl- boronic acid, 0.8 g of 5percent palladium on carbon (50percent wet,Degussa type E105CA/W) and 800 ml of tetrahydrofuran(10 vol) are introduced into a 4 litre reactor. The medium is heated to 55°C. 85 g (1.05 mol, 3.3 eq) of potassium hydroxide at 85percent are dissolved in 240 ml of water (3 vol) .The solution obtained is poured over the reaction medium. The addition is exothermic. The reaction medium reaches the reflux temperature. The reflux is EPO <DP n="12"/>maintained for about 2 hours .The reaction medium is filtered at about 35-400C on a cartridge and rinsed with 400 ml of a THF/water mixture (1/1) .The medium is cooled to 200C and 100 ml of HCl at 35percent in 600 ml of water are added. 6- [3- (1-adamantyl) -4- methoxyphenyl] -2-naphthoic acid precipitates. It is filtered and washed with 4 litres of water. The pH of the washings is about 6-7. The product is dried under vacuum at 1000C for 24 hours.131 g of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2 -naphtho- ic acid are obtained (crude yield = 99percent) .This crude material is dissolved in 15 to 22 volumes of THF under reflux. After filtration in the hot state, 15 to 22 volumes of heptane are added and the medium is cooled to about 50C for 1 to 2 hours.The 6- [3- (1-adamantyl) -4-methoxyphenyl] -2-naphthoic acid is filtered on sintered glass and it is rinsed with 1 to 2 volumes of heptane.108 g of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2-naphthoic acid are obtained in the form of a white solid whose purity, determined by HPLC, is 99.9percent (yield = 82percent; m.p. = 320-3220C) .
94.8%
Stage #1: With potassium carbonate In tetrahydrofuran; water for 2 - 4 h; Heating / reflux Stage #2: With hydrogenchloride; water In tetrahydrofuran for 1 h;
b) - Preparation of 6- [3- (1-adamantyl) -4-methoxy- phenyl] -2-naphthoic acid (I):20 mL of tetrahydrofuran (12 vol) , 2 g (7 mmol) of 3 -adamantyl-4-methoxyphenylboronic acid (II), 1.65 g (6.6 mmol) of 6-bromo-2 -naphthoic acid (III) and 20 mL of a 2 M aqueous potassium carbonate solution are introduced into a round-bottomed flask equipped with stirring and under a nitrogen stream. 15 mg (1percent) of palladium acetate and 46 mg (2percent) of 2- (dicyclohexyl- phosphino) biphenyl are then introduced. The medium is EPO <DP n="11"/>heated under reflux for 2 hours. Kinetic monitoring by HPLC indicates that the percent of 6- [3- (i-adamantyi) -4- methoxyphenyl] -2 -naphthoic acid formed is 94percent after one hour and 98percent after 2 h.After returning to room temperature, the catalyst is filtered on a cartridge, and then slowly poured over 30 ml of a 1 N aqueous hydrochloric acid solution.The medium is kept stirring for one hour. The precipitate is filtered, washed with water and then dried under reduced pressure. 2.68 g of 6-[3-(l- adamantyl) -4-methoxyphenyl] -2 -naphthoic acid are obtained in the form of a white solid whose purity, determined by HPLC, is 99.9percent (yield = 94.8percent; m.p. = 3210C) .The following melting points (m.p.) exist in the literature: m.p. = 319°-322°C (B. Charpentier et al . , J. Med. Chem., 1995, 38, 4993-5006) and m.p. = 325°-327°C (EP 0 199 636) .
Stage #1: With thionyl chloride; N,N-dimethyl-formamide In toluene at 45 - 50℃; for 1 h; Stage #2: With triethylamine In methanol; toluene at 10 - 25℃; for 1 h;
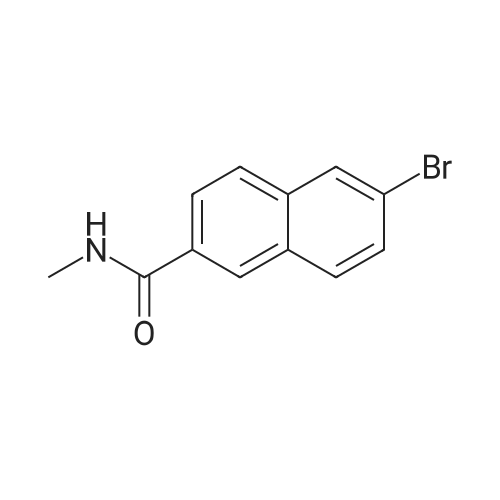
Reference Example 76-Bromo-2-naphthoic acid (10.1 g, 40.1 mmol) and N,N- dimethylformamide (4.75 g, 65.0 mmol) were added to toluene (80 mL) . To the reaction mixture was added dropwise thionyl chloride (5.7 g, 48.2 mmol) at 45 to 50°C, and the mixture was stirred for 1 hr, and allowed to cool to room temperature. The reaction mixture was added dropwise at 10 to 25°C to a solution prepared by adding triethylamine (11.4 g, 112.4 mmol) and 40percent methylamine methanol solution (8.1 g, 104.4 mmol) to toluene (80 rnL) , and the mixture was stirred at room temperature for 1 hr.. To the reaction mixture was added dropwise water (50 mL) , and the mixture was stirred at room temperature. The crystals were collected by filtration, and washed with a mixed solvent (25 mL) of methanol/water (2:8) to give wet crystals. The total amount of the wet crystals was added to N,N- dimethylacetamide (70 mL) , and dissolved with heating to 60°C. The reaction mixture was allowed to cool to room temperature, and water (140 mL) was added dropwise thereto. The crystals were collected by filtration, and washed with water (80 mL) to give wet crystals. The total amount of the wet crystals was suspended in ethyl acetate -(25 mL) with stirring at roomtemperature. The crystals were collected by filtration, and washed with ethyl acetate (5 mL) . The obtained wet crystals were dried under reduced pressure to give 6-bromo-N-methyl-2- naphthamide (9.4 g, 35.6 mmol). yield 89percent.3/4 NMR (500 MHz, DMSO-d6) δ 2.84 (d, J = 4.4 Hz, 3H) , 7.71 (dd, J = 8.8, 2.2 Hz, 1H) , 7.93 - 8.03 (m, 3H) , 8.28 (d, J = 1.9 Hz, 1H), 8.44 (s, 1H) , 8.62 (d, J = 4.1 Hz, 1H) ; HRMS (ESI) m/z Calcd for a Ci2HuNOBr [M+H]+: 264.0024, Found: 264.0019; Anal. Calcd for a Ci2Hi0NOBr: C, 54.57; H, 3.82; N, 5.30; Br, 30.25. Found: C, 54.56; H, 3.70; N, 5.34; Br, 30.23.
82%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine In tetrahydrofuran; N,N-dimethyl-formamide at 0 - 20℃; for 18 h; Inert atmosphere
Under an argon atmosphere, to a cooled (0 °C) solution of 8 (60.26 g, 240 mmol), EDCI*HCl (55.21 g, 288 mmol), HOBt*H2O (44.1 g, 288 mmol) and N,N-diisopropylethylamine ((i-Pr)2NEt) (37.23 g, 288 mmol) in anhydrous N,N-dimethylformamide (DMF) (960 mL) was added dropwise to a solution of MeNH2 (2 M solution in THF; 192 mL, 384 mmol) and the whole was stirred at room temperature for 18 h. After dilution with water, the precipitate was filtered off, washed with H2O and i-Pr2O and dried under the reduced pressure to give 9a (60.6 g, 82percent) as a colorless powder. 1H NMR (CDCl3 + CD3OD) δ: 3.04 (3H, s), 7.60 (1H, dd, J = 1.8 Hz, 8.6 Hz), 7.78 (2H, d, J = 8.6 Hz), 7.85 (1H, dd, J = 1.8 Hz, 8.6 Hz), 8.03 (1H, d, J = 1.8 Hz), 8.25 (1H, s). IR (KBr): 3274, 1638, 1622, 1559, 1495, 1408, 1316, 1159 cm-1. Anal. Calcd for C12H10NOBr.0.1H2O: C, 54.20; H, 3.87; N, 5.27; Br, 30.25. Found: C, 54.03; H, 3.72; N, 5.24.
80%
Stage #1: With thionyl chloride In DMF (N,N-dimethyl-formamide); ethyl acetate at 30 - 65℃; for 0.5 h; Stage #2: With triethylamine In methanol; DMF (N,N-dimethyl-formamide); water; ethyl acetate at 25℃; for 3 h;
4 Liters of ethyl acetate and 25 ML of DMF were added to 500 g (1.99 mol) of 6-bromo-2-naphthoic acid. 188 ML (2.61 mol, 1.3eq) of thionyl chloride was added dropwise at 30°C or lower.. The mixture was stirred at 65°C for 30 minutes.. After cooled to 25°C, a mixture of 408 ML (3.93 mol, 2eq) of a 40percent solution of methylamine in methanol and 558 ML (4.01 mol, 2eq) of triethylamine was added dropwise at 25°C or lower.. The mixture was stirred at 25°C for 3 hours. 2.5 Liters of water was added dropwise at 25°C or lower.. Crystals were filtered, and washed successively with 1.25 liters of a mixed solution of methanol/water=1/4.. Vacuum drying (50°C) to a constant weight afforded 422 g of 6-bromo-N-methyl-2-naphthamide (yield 80percent).1H NMR (CDCl3+CD3OD): δ 3.04 (3H, s), 7.60 (1H, dd, J=8.6, 1.8 Hz), 7.78 (2H, d, J=8.6 Hz), 7.85 (1H, dd, J=8.6, 1.8 Hz), 8.03 (1H, d, J=1.8 Hz), 8.25 (1H, s).
Reference:
[1] Patent: WO2012/173280, 2012, A1, . Location in patent: Page/Page column 32-33
[2] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 21, p. 6356 - 6374
[3] Patent: EP1471056, 2004, A1, . Location in patent: Page 32
With dimethylsulfide borane complex; In tetrahydrofuran; at 0 - 20℃;
(6-bromo-2-naphthv?methanol To a solution of 6-bromo-2-naphthoic acid (1g, 3.98mmol) in THF (20ml) at O0C was added 2M borane dimethyl sulfide in THF (0.53 ml, 5.97 mmol) portion wise. The mixture was warmed to RT and stirred overnight. The reaction was cooled to O0C and MeOH was added. The solution was concentrated under reduced pressure. The crude product was purified by column chromatography (EtOAc in heptane) to afford the title compound (0.4 g, 89%). <n="33"/>LCMS: m/z 238 [M+H]
85%
With borane-THF; In tetrahydrofuran; at 0 - 25℃;
Into a 250-mL 3-necked round-bottom flask was placed 6-bromo-2-naphthoic acid (10 g, 39.83 mmol, 1.00 equiv), tetrahydrofuran (40 mL). This was followed by the addition of BH3THF (1 M) (80 mL, 2.00 equiv) dropwise with stirring at 0C. The resulting solution was stirred overnight at 25C. The reaction was then quenched by the addition of 100 mL of ice/water. The resulting solution was extracted with 2x200 mL of ethyl acetate and the organic layers were combined. The mixture was dried over anhydrous sodium sulfate and concentrated under vacuum. This resulted in 8 g (85%) of (6-bromonaphthalen-2-yl)methanol as a light yellow solid.
77%
With borane-THF; In tetrahydrofuran; at 0 - 20℃;
Preparation of {6- [5- (2-Phenoxy-ethylsulfanylmethyl)- [1, 3,4] oxadiazol-2-yl] -naphthalen- 2-yl}-methanol a) (6-Bromo-naphthalen-2-yl)-methanol A THF solution of 6-bromo-2-naphthoic acid (3.5 g, 13.94 mmol, 1 eq. ) was cooled to 0C in an ice bath and then treated dropwise with borane-THF complex (1 M solution in THF, 16.7 mL, 16.7 mmol, 1.2 eq. ) via syringe. After addition was complete, the reaction was allowed to gradually warm to room temperature and stir at that temperature overnight. Several milliliters of water were added to the reaction to quench the remainder of the borane. The reaction was diluted with water and extracted with ethyl acetate. The organic layer was separated and then washed with aqueous 1 M NaOH and then brine. The organic layer was then collected, dried over MgSO4, filtered and the solvent removed in vacuo leaving (6-bromo-naphthalen-2-yl) -methanol (2.56 g, 77% yield) as a white solid in the flask.
74%
With diisobutylaluminium hydride; In tetrahydrofuran; at 0 - 25℃; for 16h;
To a solution of 6-bromo-2-naphthoic acid (1.0 g, 4.0 mmol) in TUF (20 mL) at 0C was addedDIBAL-H (8.0 mE, 8.0 mmol). After addition, the reaction mixture was stirred at 25 C for 16h. The rcaction was quenched by the addition of saturated aqueous NH4C1 (20 mE) arid thenextracted with ethyl acetate (3 x 30 mE). The combined organic layers were dried over Na2SO4 and concentrated under reduced pressure. The residue was purified by chromatography(petroleum ether: ethyl acetate 2:1) to give the title compound (0.7 g, 74 % yield) as a white solid.
63%
With lithium aluminium tetrahydride; In tetrahydrofuran; at 0 - 20℃; for 1h;
To a solution of 6-bromo-2-naphthoic acid (1.48 g, 5.9 mmol, 1 eq) dissolved in dry THF (75 mL) at 0C was added dropwise a lithium aluminium hydride solution (2.4 M, 5 mL, 12 mmol, 2 eq). The reaction was allowed to warm to RT, and after 1 h of stirring it was quenched by addition of H2O (0.5 mL), queous NaOH (10%, 1 mL) solution and H2O (3 mL), after which it was stirred for 16 h. The mixture was dried by addition of MgSO4, filtered and the filtrate was concentrated under reduced pressure. The residue was purified via flash-column-chromatography (SiO2, 10% to 40% EtOAc in pentane) to yield the product (0.872 g, 63%). 1H NMR (400 MHz, chloroform-d) delta 7.99 (s, 1H), 7.79 - 7.72 (m, 2H), 7.69 (d, J = 8.7 Hz, 1H), 7.55 (dd, J = 8.7, 1.9 Hz, 1H), 7.53 - 7.46 (m, 1H), 4.84 (s, 2H), 1.79 (s, 1H). 13C NMR (101 MHz, chloroform-d) delta 138.94, 134.07, 131.89, 129.90, 129.71, 129.67, 127.55, 126.29, 125.41, 119.95, 65.37.
With borane-THF; In tetrahydrofuran; at 0 - 20℃;
To a solution of compound B6-1 (2.51 g, 10 mmol) in THF (60 mL) wasadded a solution of BH3THF in THF (1 M, 15 mL) at 0 C. The reaction mixture was stirred at room temperature overnight, quenched with water (30 mL), and extracted with ethyl acetate (100 mL x 2). The combined organic extracts were washed with aqueous sodium hydroxide solution (1 N, 100 mL) and brine (100 mL), dried over anhydrous sodium sulfate, filtered, and concentrated to give Compound B6-2. LC-MS (ESI) m/z: 219 [M-OH] ?HNIVIR (CDC13, 400 IVIHz): 5 (ppm) 1.98 (s, 1H), 4.84 (s, 2H), 7.48-7.56 (m, 2H), 7.68-7.77 (m, 3H), 7.99 (s, 1H).
With diborane; In tetrahydrofuran; at 0 - 20℃; for 16h;Inert atmosphere;
6-bromonaphthalene-2-carboxylic acid (20 g, 80 mmol, 1.00 eq.) at 0 C under N2 protection.BH3 (1N in THF, 160 mL, 160 mmol, 2.00 eq.) was added dropwise to THF (60 mL).The resulting mixture was stirred at room temperature for 16 hours.It was then quenched with ice water and extracted three times with EtOAc.The combined organic layers were washed with brine.Dry over Na2SO4 and concentrate in vacuo.Obtained as a white solid(6-Bromonaphthalen-2-yl)methanol (18.1 g, crude) was used in the next step without further purification.
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane;
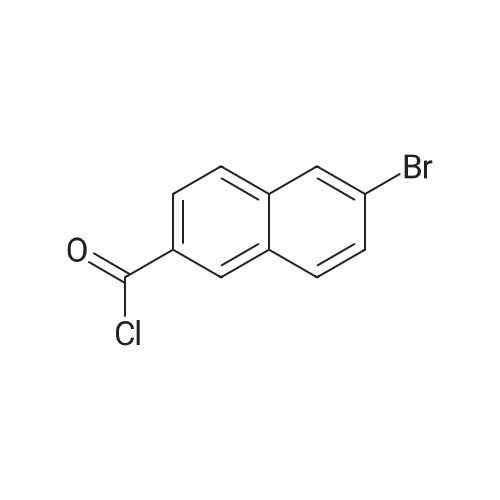
<strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (72.3 g, 282 mmol, 1.0 equiv.) was suspended in dichloromethane (600 mL) and DMF (catalytic, 5 drops) was added. Oxalylchloride (71.6 g, 564 mmol, 2.0 equiv.) was added portion wise during 1 hour. The reaction mixture was stirred overnight with a CaCl2 drying tube mouthed on the flask. Complete dissolution occurred. The reaction mixture was concentrated, dichloromethane (100 mL) was added and the solvent was evaporated again, yielding 6-bromo-2-naphthoyl chloride (76.1 g, 100 %) as an oil which was used as such in next step.
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; for 18h;
To a mixture of 6-bromo-naphthalene-2-carboxylic acid (1 g, 3.9 mmol, Lancaster) and CH2Cl2 (25 mL) was added oxalyl chloride (2.98 mL, 2 M solution in CH2Cl2, 5.8 mmol, Aldrich) and DMF (2 drops) with stirring at 0 C. The reaction mixture was stirred at room temperature for 18 h and the solvents were removed in vacuo to afford the title compound as a light-brown amorphous solid, which was used in the next step withouth purification. MS (ESI, pos. ion) m/z: 266 (M+1).
With thionyl chloride; at 80℃; for 1h;
6-Bromo-2-naphthoic acid (4.00 g, 15.9 mmol) was treated with 40 mL of thionyl chloride at 80 C. for 1 h. The mixture was concentrated in vacuo, and the resultant unpurified acid chloride (3 g, 11.1 mmol) was combined with 2,2'-azobisisobutyronitrile (731 mg, 4.45 mmol) in 25 mL of carbon tetrachloride and 15 mL of chlorobenzene. This mixture was added slowly via dropping funnel to a mixture of 2-mercaptopyridine-1-oxide sodium salt (1.99g, 13.7 mmol) and 4-(dimethylamino)pyridine (150 mg, 1.23 mmol) at 100 C. After the addition was complete, the mixture was stirred for an additional 4 h, cooled, and the solid by-product precipitate removed by filtration. The filtrate was concentrated in vacuo and the residue purified by flash column chromatography (SiO2, hexanes) to provide the title compound as a white solid. 1H NMR (500 MHz, CDCl3) delta: 8.02 (br s, 1H); 7.83 (d, J=1.6 Hz, 1H); 7.72 (d, J=8.7 Hz, 1H); 7.66 (d, J=8.7 Hz, 1H); 7.60 (dd, J=2.0 8.9 Hz, 1H); 7.47 (dd, J=2.1, 8.7 Hz, 1H).
With thionyl chloride; In tetrahydrofuran; N,N-dimethyl-formamide; toluene;
Reference Example 21 Production of 6-bromo-N,N-diisopropyl-2-naphthamide A suspension of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (100 g), thionyl chloride (37.7 mL) and DMF (0.5 mL) in THF (1000 mL) was stirred with heating at 60ØC for 90 min. The mixture was cooled to room temperature and the solvent was evaporated under reduced pressure. The obtained solid was dissolved in toluene and the solvent was evaporated to give a pale-yellow powder of 6-bromo-2-naphthoyl chloride.
With thionyl chloride; In tetrahydrofuran; N,N-dimethyl-formamide; toluene;
(i) Production of 6-bromo-N,N-diisopropyl-2-naphthamide A suspension of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (100 g), thionyl chloride (37.7 ml) and DMF (0.5 ml) in THF (1000 ml) was stirred at 60 C. with heating for 90 min. After cooling to room temperature, the solvent was evaporated under reduced pressure. The resulting solid was dissolved in toluene and the solvent was evaporated to give 6-bromo-2-naphthoyl chloride as a pale yellow powder.
With thionyl chloride; at 80℃; for 1h;
Step A 2-Bromo-6-chloronaphthalene.; 6-Bromo-2-naphthoic acid (4.00 g, 15.9 mmol) was treated with 40 mL of thionyl chloride at 80 C. for 1 h. The mixture was concentrated in vacuo, and the resultant unpurified acid chloride (3 g, 11.1 mmol) was combined with 2,2'-azobisisobutyronitrile (731 mg, 4.45 mmol) in 25 mL of carbon tetrachloride and 15 mL of chlorobenzene. This mixture was added slowly via dropping funnel to a mixture of 2-mercaptopyridine-1-oxide sodium salt (1.99 g, 13.7 mmol) and 4-(dimethylamino)pyridine (150 mg, 1.23 mmol) at 100 C. After the addition was complete, the mixture was stirred for an additional 4 h, cooled, and the solid by-product precipitate removed by filtration. The filtrate was concentrated in vacuo and the residue purified by flash column chromatography (SiO2, hexanes) to provide the title compound as a white solid. 1H NMR (500 MHz, CDCl3) delta: 8.02 (br s, 1H); 7.83 (d, J=1.6 Hz, 1H); 7.72 (d, J=8.7 Hz, 1H); 7.66 (d, J=8.7 Hz, 1H); 7.60 (dd, J=2.0, 8.9 Hz, 1H); 7.47 (dd, J=2.1, 8.7 Hz, 1H).
With thionyl chloride; In N,N-dimethyl-formamide; for 4h;Heating / reflux;
To <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (5.0 g, 20 mmol), 20 mL of thionyl chloride (SOCl ), and dimethylformaldehyde (DMF, 1 mL) were added, and the mixture was stirred under heating for 4 hours. An excessive amount of thionyl chloride (SOCl ) was distilled off in vacuum, and then to the reaction mixture, 20 mL of N- methylpyrrolidine (NMP), and N-phenyl-l,2-diaminobenzene(3.7 g, 20 mmol) were added, and the mixture was stirred at 160 0C for 12 hours. The mixture was cooled to ambient temperature, and then an excessive amount of water was added thereto to form a solid. The solid was filtered, washed with water and then ethanol, and dried to prepare a compound B-9 (6.2 g, yield 78 %).[132] MS [M+H]+ = 399
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 18 - 30℃; for 2h;
To methylene chloride solution including 6-bromo-2-naphthonic acid (0.42 g) and N,N-dimethylformamide (300 muL), oxalyl dichloride (2.6 ml) was added under stirring at room temperature (18 to 3O0C, the same shall apply hereinafter) followed by further stirring for 2 hours. Subsequently, the solvent was distilled off under the reduced pressure (760 to 20 mmHg, the same shall apply hereinafter). Resulting 6-bromo-2-naphthoyl chloride was dissolved in methylene chloride, added with pyridine (2.1 ml) and 2-ethyl-4-(l,l,1^3-3-3-heptafluoropropan-2-yl)-6-methylaniline (0.45 g), followed by stirring for 5 hours. After dilution with methylene chloride and water, the organic phase was separated and dried over magnesium sulfate. The drying agent (magnesium sulfate) was removed by filtration and the solvent was distilled off under the reduced pressure. The residue was separated and purified by column chromatography to obtain -bromo-N-beta-emyl-^U.l^^-heptafluoro (0.75 g, yield 84%). - 74 -1H-NMR (CDCl3) delta: 1.23 (3H, t), 2.35 (3H, s), 2.71 (2H, q), 7.39 (2H, s), 7.63-7.66 (2H, m), 7.80 (IH, d), 7.85 (IH, d), 7.96 (1 H, dd), 8.08 (IH, s), 8.40 (IH, s).
With thionyl chloride; In tetrahydrofuran; N,N-dimethyl-formamide; at 60℃; for 1.5h;
A solution of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> 32a (100 g, 398 mmol), thionylchloride (37.7 mL, 517 mmol) and DMF (0.5 mL) in THF (1000 mL) was stirred at 60 C for 90 min. After cooling to rt, the solvent and excess amount of thionylchloride was evaporated. The resulting residue was dissolved in dry THF (400 mL) and the solution was added dropwise to a cooled (0 C) solution of diisopropylamine (112 mL, 799 mmol) and triethylamine (112 mL, 804 mmol) in THF (800 mL). After being stirred at rt for 1 h, about 50% of the solvent was evaporated. Then the mixture was diluted with AcOEt and the organic phase was washed with H2O, 1 N NaOH solution and brine, dried over MgSO4 and evaporated. The residue was washed with i-Pr2O to give 32b (117 g, 88%) as a colorless crystal.
With thionyl chloride; at 70℃; for 16h;
Example 2: Synthesis of 6-{4-<1-hydroxyethyl)pyridin-3-yl)-2-naphthonitrile Step 1 : 6-bromo-2-naphthamide (2a) <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (2 g, 7.97 mmol) was stirred in thionyl chloride (13.28 ml) at 70 C for 16h. Solvent was evaporated, and the residue was dissolved in CH2CI2 and concentrated again. To the acid chloride intermediate was added ammonia (7N in MeOH, 13.66 ml, 96 mmol) and the mixture was stirred at room temperature for 3h. The reaction mixture was concentrated, diluted in ethyl acetate and filtered. The solid was rinsed with ethyl acetate and then dried. The title compound 2a (1.802 g, 90%) was isolated as beige solid. LC-MS (M+1) 251.9, t = 1.34 min.
With thionyl chloride; N,N-dimethyl-formamide; In tetrahydrofuran; at 60℃; for 1.5h;
A solution of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> 8 (100 g, 398 mmol), thionylchloride (SOCl2) (37.7 mL, 517 mmol) and DMF (0.5 mL) in THF (1000 mL) was stirred at 60 C for 90 min. After cooling to room temperature, the solvent and excess thionylchloride were evaporated. The resulting residue was dissolved in dry THF (400 mL) and the solution was added dropwise to a cooled (0 C) solution of diisopropylamine (112 mL, 799 mmol) and Et3N (112 mL, 804 mmol) in THF (800 mL). After stirring at room temperature for 1 h, about half the amount of solvent was evaporated. The mixture was then diluted with AcOEt and the organic phase was washed with H2O, 1 N NaOH solution and brine, dried over MgSO4 and evaporated. The residue was washed with i-Pr2O to give 9e (117 g, 88%) as a colorless crystal.
With thionyl chloride; at 70℃; for 16h;
<strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (2 g, 7.97 mmol) was stirred in thionyl chloride (13.28 ml) at 70 0C for 16h. After concentration in vacuo, the residue was rediiuted in CH2Ci7 and concentrated again. To the acid chloride intermediate was added ammonia in MeOH (7 M, 13.66 ml, 96 mmol) and the mixture was stirred at room temperature for 3h. The mixture was then concentrated. The residue was taken up in AcOEt, filtered, rinsed with AcOEt and then dried over MgSO4, filtered and concentrated in vacuo. The title compound (1.802 g. 90%) was isolated as a beige solid. LC-MS (M+1) 251.9, t = 1.34 min.
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; for 12.08h;Inert atmosphere;
Intermediate 2 (Scheme 1); 6-bromo-N-methyl-N-(methyloxy)-2-naphthalenecarboxamide:To a solution of <strong>[5773-80-8]6-bromo-2-naphthalenecarboxylic acid</strong> (lg, 3.98 mmol) indichloromethane (20 ml) under nitrogen at 0C was added a solution of oxalyl chloride (1.74 ml, 19.91 mmol) for 5 min. Drop of DMF was added and the reaction mixture was stirred at RT for 12h. The solution become clear and solvent was concentrated in vacuo. The yellowish solid was co-evaporated twice with toluene to provide slightly reddish powder and was preceded to the next step without further purification.
With thionyl chloride; triethylamine; for 1h;Reflux;
General procedure: Amides 7a-e were prepared from the corresponding commercially available carboxylic acids (1 mmol) 6a-e treated with an excess of SOCl2 (7 mmol) in presence of Et3N (0.1 mmol) under reflux for 1 h. The corresponding acylchloride was added to a mixture (45 mL) of NH4OH (28%), H2O and CH2Cl2 (1:1:1). The mixture was stirred at room temperature for 4 h. Insoluble amides 7a-d were separated by filtration while the amide 7e was obtained removing the solvent under vacuo. The crude was washed with H2O (20 mL) and extracted with AcOEt (3 × 20 mL).
With thionyl chloride; In N,N-dimethyl-formamide; for 4h;Reflux;
<strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (5.0g, 20ml) in thionyl chloride (SOCl2, 20mL) and dimethylformamide (DMF,1mL) was refluxed with stirring for 4 hours. After removal of excessthionyl chloride (SOCl2) in a vacuum distillation, N-methylpyrrolidine (NMP,20 mL) was added to the reaction mixture, N-phenyl-1,2-diaminobenzene (3.7g, 20mmol) was stirred at 160C for 12 hours. The resulting mixture was cooled to room temperature, excess of water was added to form a solid. The resulting mixture was filtered, washed with water and ethanol and dried to obtain compound B-18 (6.2g, yield: 78%).
With thionyl chloride; N,N-dimethyl-formamide; In dichloromethane; for 0.5h;Inert atmosphere; Reflux;
6-Bromo-2-naphthalenecarboxylic acid (251.1 mg, 1.0 mmol) was added to 25 mLDry egg-shaped bottle, the replacement of nitrogen three times to make the system in a nitrogen atmosphere,Add 5mL of freshly distilled methylene chloride to dissolve,Thionyl chloride (0.36 mL, 5.0 mmol) was then slowly added dropwise,After adding dropwise catalytic amount of ultra-dry N, N- dimethylamide to promote the reaction.Heated to reflux after 30 minutes to stop heating,Spin dry the solvent pump pumping half an hour directly cast one step reaction.The second step was N, O-dimethylhydroxylamine hydrochloride (107.3 mg, 1.1 mmol) andTriethylamine (0.42 mL, 3.0 mmol) was added to a 25 mL dry egg-shaped flask,5mL dichloromethane was added to completely dissolve the starting material,The system was cooled to 0 C in an ice-water bath,Under the protection of nitrogen, the new acid chloride prepared in the previous step was slowly added dropwise to the system,After the dropwise addition and holding at 0 C for 10 minutes, then gradually returned to room temperature,Stir for 1 hour. TLC tracking until the completion of the conversion of raw materials, add water to quench the reaction, The product was extracted with dichloromethane.The organic phase was washed three times with water and dried over sodium sulfate. Concentrated under reduced pressure,The crude product was purified by column chromatography (dichloromethane ? dichloromethane:Methanol = 100: 1) to afford S1 as a white solid (235 mg, 80% over two steps 235).
With thionyl chloride; In chloroform; at 60℃;Cooling with ice;
Under ice-cooling, <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (5.0 g, 19.91 mmol) and thionyl chloride (14.2 g, 119.48 mmol) were added to a certain amount of chloroform (30 ml).The reaction was stirred at 60 C. for 14-16 hours. After the reaction was completed, chloroform was evaporated under reduced pressure to give 6-bromo-2-naphthoyl chloride (Compound 1) as a white solid.
With thionyl chloride; at 60℃; for 16h;
6-Bromonaphthalene-2-carboxylic acid (25.1 g) was suspended in thionyl chloride (200 mL), stirred at 60C for 16 hours and evaporated under vacuum. Solid was dissolved in dichloromethane (50 mL) and evaporated under vacuum, giving 6-bromonaphthalene-2-carbonyl chloride (27.0 g, crude) as a white solid.
With copper(l) iodide; copper(l) chloride; In N,N-dimethyl-formamide; for 4h;Inert atmosphere; Reflux;
Example 32 Synthesis of Compound 32 6-chloro-2-naphthoic acid A suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCl (11.7 g, 114.64 mmol) and CuI (2.19 g, 11.50 mmol) in degassed DMF (45 mL) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 mL) and the resulting mixture was extracted with EtOAc (2*500 mL). The combined organic layers were then washed with H2O (4*500 mL) followed by brine (1*500 mL), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 32 (2.2 g, 93%) as an off-white solid. HPLC tR 6.47 min.
93%
With copper(l) iodide; copper(l) chloride; In N,N-dimethyl-formamide; for 4h;Inert atmosphere; Darkness; Reflux;
Example 66 - Synthesis of Compound 68 6-chloro-2-naphthoic acid686-chloro-2 -naphthoic acidA suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCI (1 1.7 g, 114.64 mmol) and CuI (2.19 g, 1 1.50 mmol) in degassed DMF (45 ml.) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 ml.) and the resulting mixture was extracted with EtOAc (2 x 500 mL). The combined organic layers were then washed with H2O (4 x 500 mL) followed by brine (1 x 500 mL), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 68 (2.2 g, 93%) as a off-white solid. HPLC fR 6.47 min.
93%
With copper(l) iodide; copper(l) chloride; In N,N-dimethyl-formamide; for 4h;Reflux; Inert atmosphere; Darkness;
A suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCI (1 1.7 g, 114.64 mmol) and CuI (2.19 g, 1 1.50 mmol) in degassed DMF (45 ml.) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 ml.) and the resulting mixture was extracted with EtOAc (2 x 500 ml_). The combined organic layers were then washed with H2O (4 x 500 ml.) followed by brine (1 x 500 ml_), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 70 (2.2 g, 93%) as a off-white solid. HPLC fe 6.47 min
93%
With copper(l) iodide; copper(l) chloride; In N,N-dimethyl-formamide; for 4h;Inert atmosphere; Darkness; Reflux;
A suspension of 6-bromo-2-naphthoic acid (3.0 g, 11.47 mmol), CuCl (11.7 g, 114.64 mmol) and CuI (2.19 g, 11.50 mmol) in degassed DMF (45 mL) was heated to reflux under argon in dark for 4 hrs. After cooling to room temperature, the solution was decanted into H2O (200 mL) and the resulting mixture was extracted with EtOAc (2*500 mL). The combined organic layers were then washed with H2O (4*500 mL) followed by brine (1*500 mL), dried over MgSO4, filtered and concentrated under reduced pressure to dryness. The residue was trituated with CH3CN and the solid obtained was then re-crystallized from EtOAc to give the pure product 68 (2.2 g, 93%) as a off-white solid. HPLC tR 6.47 min.
75%
With CuI;copper(l) chloride; In N,N-dimethyl-formamide;
Step 1. To a solution of 6-bromo-2-naphthoic acid (4.4 g, 17.5 mmol) in 50 mL anhydrous DMF were added CuCl (8.7 g, 87.5 mmol) and CuI (0.2 g). The slurry was refluxed for 1 hour. At room temperature it was diluted with 300 mL EtOAc and stirred for 2 hours. It was filtered through celite. The filtrate was evaporated in vacuuo to afford 6-chloro-2-naphthoic acid (2.7 g, 75%). ES-MS: (M+H)+207.
With copper(l) iodide; copper(l) chloride; In N,N-dimethyl-formamide; at 60℃; for 5h;Inert atmosphere; Darkness; Reflux;
6-Bromo-2-naphthoic acid (9.00 g, 35.85 mmol) and DMF (140 mL) were combined, stirred, and degassed with subsurface N2 for 10 minutes before adding CuCl (35.49 g, 358.50 mmol) and Cul (6.96 g, 36.57 mmol). The reaction vessel was wrapped in aluminum foil and refluxed for 5 h under a nitrogen atmosphere. The mixture was allowed to cool to 60 C then filtered through celite. The filtrate was diluted with EtOAc and 1 M HCl. The organic phases were washed once more with 1 M HCl, water (2X) and concentrated in vacuo. The resulting crude solid was suspended in acetonitrile, stirred for 1 hr, and filtered to afford 6-chloro-2-naphthoic acid as a white solid.
With copper(l) iodide; copper(l) chloride; In N,N-dimethyl-formamide; at 150℃; for 3h;Inert atmosphere;
A mixture of 6-bromo-2-naphthoic acid (Compound 106A) (3.00 g, 11.47 mmol), Cul (2.19 g, 11.50 mmol), and CuCl (11.70 g, 114.64 mmol) in DMF (20 mL) was stirred at 150 C under nitrogen for 3 hours. The mixture was cooled down to room temperature and diluted with an aqueous HC1 solution (3 M, 100 mL). A solid was formed, filtered, and washed with water (100 mL). It was suspended in THF (100 mL), filtered, and washed with THF (100 mL). The combined filtrates were concentrated under reduced pressure. The residue was suspended in an aqueous HC1 solution (3 M, 100 mL), stirred at room temperature for 10 minutes, and filtered. The cake was washed with water (50 mL) and CH3CN (50 mL) and dried under vacuum to yield Compound 106B. LC-MS (ESI) m/z: 205 [M-H] ; 1H-NMR (DMSO-r/r,, 400 MHz): d (ppm) 7.61-7.64 (m, 1H), 7.99-8.05 (m, 2H), 8.15-8.19 (m, 2H), 8.65 (s, 1H), 13.18 (s, 1H).
Part B. Preparation of 6-bromonaphthalen-2 -amine.; [00748] A solution of the product Part A (5.07g, 20.19mmol) and triethylamine (4.22mL, 3.07g, 30.3mmol) in dry DMF (155mL) was treated with the diphenylphosphoroyl azide (6.55mL, 8.34g, 30.3mmol) followed by stirring at room temperature for 3h. The solution was then treated with water (2OmL) followed by warming at 1000C for Ih. The solution was cooled and the flask fitted with a short- path distillation head and the DMF removed by distillation under high vacuum. The solid residue was dissolved in EtOAc and washed with saturated sodium bicarbonate solution. Filtered through celite and the filtrate was washed with water (3x) and then with brine. Dried over Na2SC^, filtered and concentrated under vacuum to give the title compound as a beige solid (4.48g, 100 percent).
100%
[00490] Part B. Preparation of 6-bromonaphthalen-2-amine.; [00491] A solution of the product Part A (5.07g, 20.19mmol) and triethylamine (4.22mL, 3.07g, 30.3mmol) in dry DMF (155mL) was treated with the diphenylphosphoroyl azide (6.55mL, 8.34g, 30.3mmol) followed by stirring at room temperature for 3h. The solution was then treated with water (2OmL) followed by warming at 1000C for Ih. The solution was cooled and the flask fitted with a short- path distillation head and the DMF removed by distillation under high vacuum. The solid residue was dissolved in EtOAc and washed with saturated sodium bicarbonate solution. Filtered through celite and the filtrate was washed with water (3x) and then with brine. Dried over Na2SO4, filtered and concentrated under vacuum to give the title compound as a beige solid (4.48g, 100 percent).
100%
Part 1. Preparation of 6-bromonaphthalen-2 -amine.[00328| A solution of the product Part H (5.07 g, 20.19 mmol) and triethylamine (4.22 mL, 3.07 g,30.3 mmol) in dry N.N-dimethylformamide (155mL) was treated with the diphenylphosphoroyl azide(6.55 mL, 8.34 g, 30.3 mmol) followed by stirring at room temperature for 3 hours. The solution was then treated with water (20 mL) followed by warming at 100 0C for 1 hour. The solution was cooled and the flask fitted with a short-path distillation head and the NN-dimethylformamide was removed by distillation under high vacuum. The solid residue was dissolved in ethyl acetate and washed with saturated sodium bicarbonate solution. The organic layer was filtered through diatomaceous earth, and the filtrate was washed with water (3*) and then with brine. The organic layer was dried overNa2SO4, filtered and concentrated under vacuum to give the title compound as a beige solid (4.48 g,100 percent).
With diphenyl phosphoryl azide; triethylamine; In water; N,N-dimethyl-formamide; at 25 - 100℃; for 5h;
[000705j To a solution of Compound 89A (3.77 g, 15 mmol) and TEA (2.27 g, 22.5 mmol) in DMF (100 mL) was added DPPA (5.47 g, 22.5 mmol). The mixture was stirred at 25 °C for 3 h. Water (30 mL) was added, and the mixture was heated at 100 °C for 2 h. DMF was removed by distillation under vacuum. The residue was dissolved in ethyl acetate (300 mL) and saturated sodium bicarbonate (200 mL), and filtrated through celite. The organic phase was separated, washed with water (200 mL) and brine (200 mL), dried over anhydrous sodium sulfate, and evaporated to furnish Compound 89B. LC-MS (ESI) mlz: 222 [M+H] ?H-NMR (DMSO-d6, 400 MHz) 5 Qpm) 5.55 (s, 2H), 6.83 (s, 1H), 6.99 (d, J= 8.8 Hz, 1H), 7.36 (dd, J= 8.8, 1.6 Hz, 1H), 7.47 (d, J= 8.4 Hz, 1H), 7.59 (d, J= 8.8 Hz, 1H), 7.87 (s, 1H).
N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; for 18h;Product distribution / selectivity;
(a) 6-Bromo-naphthalene-2-carbonyl chloride. To a mixture of 6-bromo-naphthalene-2-carboxylic acid (1 g, 3.9 mmol, Lancaster) and CH2Cl2 (25 mL) was added oxalyl chloride (2.98 mL, 2 M solution in CH2Cl2, 5.8 mmol, Aldrich) and DMF (2 drops) with stirring at 0 C. The reaction mixture was stirred at room temperature for 18 h and the solvents were removed in vacuo to afford the title compound as a light-brown amorphous solid, which was used in the next step withouth purification. MS (ESI, pos. ion) m/z: 266 (M+1).
With sodium hypochlorite; sodium hydroxide; In 1,4-dioxane; water; at 70℃; for 4h;
1-(6-bromo-naphthalen-2-yl)ethaneone (0.22 g, 0.88 mmol) obtained in Preparation Example 100 was dissolvedin 3 mL of 1,4-dioxane. NaOH (0.353 g, 8.8 mmol) dissolved in 3 mL of water and 9-11% NaOCl solution (1.67mL, 2.64 mmol) were added thereto, and the mixture was heated to 70C and stirred for 4 hours. After addition of NaHSO3aqueous solution and water, the reaction solution was extracted with ether. 1N HCl was added thereto, and the organiclayer was separated, dried with MgSO4 and purified by column chromatography to obtain the title compound (0.156 g,70%).1H-NMR (CDCl3) delta 8.56 (1H, s), 8.05 (2H, m), 7.79 (2H, m), 7.58 (1H, m).
3.54 g (88%)
With sodium hydroxide; sodium hypochlorite; In 1,4-dioxane; water;
6-bromo-2-naphthalenecarboxylic acid (Compound M) To a solution of sodium hypochlorite (62 ml, 5.25% in water (w/w), 3.6 g, 48.18 mmol) and sodium hydroxide (6.4 g, 160.6 mmol) in 50 ml of water was added a solution of 2-acetyl-6-bromonaphthalene (Compound L) 4 g, (16.06 mmol) in 50 ml of 1,4-dioxane. The yellow solution was heated to 70 C. in an oil bath for 2 hours, cooled to ambient temperature, and extracted with ethyl ether (2*50 ml). The aqueous layers were diluted with NaHSO3 solution (until KI indicator solution remained colorless) and then acidified (pH <2) with 1N sulfuric acid to give a white precipitate. The mixture was extracted with ethyl ether, and the combined organic phase washed with saturated aqueous NaCl, dried (MgSO4) and concentrated to give 3.54 g (88%) of the title compound as a solid. 1H NMR (DMSO-d6): delta 8.63 (1H, br s), 8.32 (1H, d, J=2.0 Hz), 8.10 (1H, d, J=8.8 Hz), 8.00-8.05 (2H, m), 7.74 (1H, dd, J=2.0, 8.8 Hz).
3.54 g (88%)
With sodium hydroxide; sodium hypochlorite; In 1,4-dioxane; sulfuric acid; water;
6-bromo-2-naphthalenecarboxylic acid (Compound M) To a solution of sodium hypochlorite (62 ml, 5.25% in water (w/w), 3.6 g, 48.18 mmol) and sodium hydroxide (6.4 g, 160.6 mmol) in 50 ml of water was added a solution of 2-acetyl-6-bromonaphthalene (Compound L) 4 g, (16.06 mmol) in 50 ml of 1,4-dioxane. The yellow solution was heated to 70 C. in an oil bath for 2 hours, cooled to ambient temperature, and extracted with ethyl ether (2*50 ml). The aqueous layers were diluted with NaHSO3 solution (until KI indicator solution remained colorless) and then acidified (pH<2) with IN sulfuric acid to give a white precipitate. The mixture was extracted with ethyl ether, and the combined organic phase washed with saturated aqueous NaCl, dried (MgSO4) and concentrated to give 3.54 g (88%) of the title compound as a solid. 1H NMR (DMSO-d6): delta 8.63 (1H, br s), 8.32 (1H, d, J=2.0 Hz), 8.10 (1H, d, J=8.8 Hz), 8.00-8.05 (2H, m), 7.74 (1H, dd, J=2.0, 8.8 Hz).
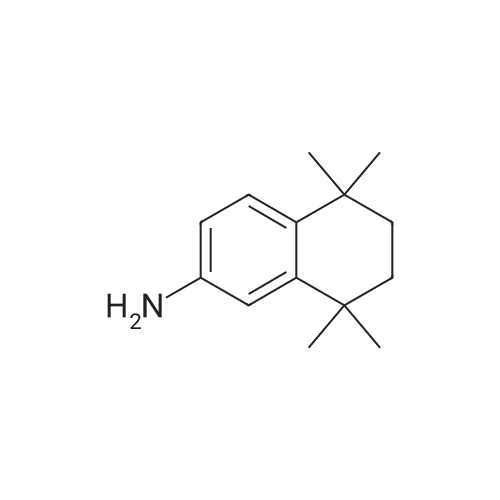
methyl 6-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthylamino)naphthoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; In 1,4-dioxane; sulfuric acid;
(a) Preparation of methyl 6-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthylamino)naphthoate: 4.04 g (19.9 mmol) of <strong>[92050-16-3]5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthylamine</strong>, 5 g (19.9 mmol) of 6-bromo-2-naphthoic acid, 3.3 ml (30 mmol) of N-methyl-morpholine, 2.9 g (19.9 mmol) of Cu2 O and 60 ml of dioxane were introduced in succession into a round-bottomed flask. The mixture was heated at reflux for 24 hours, the reaction medium was poured into 60 ml of 5N hydrochloric acid, and the precipitate was filtered off, washed with water and dried. The solid was purified by chromatography on a silica column eluted with ethyl acetate and, after evaporation of the solvents, 1.2 g of the expected acid was recovered, which was converted into the methyl ester by reacting same with 50 ml of methyl alcohol in the presence of 100 mul of concentrated sulfuric acid. 720 mg (10%) of methyl 6-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthylamino)-naphthoate were obtained, in the form of a chestnut-brown oil.
Example 3 :Preparation of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2- naphthoic acid (I) :20 ml (12 vol) of tetrahydrofuran, 2 g (7 mmol) of 3-adamantyl-4-methoxyphenylboronic acid (II), 1.65 g (6.6 mmol) of 6 -brotauno-2 -naphthoic acid (III) and 20 mL of a 2 M aqueous potassium carbonate solution are introduced into a round-bottomed flask equipped with EPO <DP n="13"/>stirring and under a nitrogen stream. 0.7 g (5%) of 10% palladium on carbon (50% wet; Keraeus type K-0218) is then introduced.The medium is heated under reflux for 8 hours. The catalyst is filtered on a cartridge, and then slowly poured over 30 ml of a 1 N aqueous hydrochloric acid solution.The medium is kept stirring for one hour. The precipitate is filtered, washed with water and then dried under reduced pressure. 2.06 g of 6- [3- (1- adamantyl) -4-methoxyphenyl] -2-naphthoic acid are obtained in the form of a white solid whose purity, determined by HPLC, is 99.9% (yield = 79%; m.p. 321C) .
99%
Example 2 :Preparation of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2- naphthoic acid (I) :80 g (0.319 mol) of 6-bromo-2 -naphthoic acid, 95.7 g (0.335 mol, 1.05 eq) of 3 -adamantyl-4 -methoxyphenyl- boronic acid, 0.8 g of 5% palladium on carbon (50% wet,Degussa type E105CA/W) and 800 ml of tetrahydrofuran(10 vol) are introduced into a 4 litre reactor. The medium is heated to 55C. 85 g (1.05 mol, 3.3 eq) of potassium hydroxide at 85% are dissolved in 240 ml of water (3 vol) .The solution obtained is poured over the reaction medium. The addition is exothermic. The reaction medium reaches the reflux temperature. The reflux is EPO <DP n="12"/>maintained for about 2 hours .The reaction medium is filtered at about 35-400C on a cartridge and rinsed with 400 ml of a THF/water mixture (1/1) .The medium is cooled to 200C and 100 ml of HCl at 35% in 600 ml of water are added. 6- [3- (1-adamantyl) -4- methoxyphenyl] -2-naphthoic acid precipitates. It is filtered and washed with 4 litres of water. The pH of the washings is about 6-7. The product is dried under vacuum at 1000C for 24 hours.131 g of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2 -naphtho- ic acid are obtained (crude yield = 99%) .This crude material is dissolved in 15 to 22 volumes of THF under reflux. After filtration in the hot state, 15 to 22 volumes of heptane are added and the medium is cooled to about 50C for 1 to 2 hours.The 6- [3- (1-adamantyl) -4-methoxyphenyl] -2-naphthoic acid is filtered on sintered glass and it is rinsed with 1 to 2 volumes of heptane.108 g of 6- [3- (1-adamantyl) -4-methoxyphenyl] -2-naphthoic acid are obtained in the form of a white solid whose purity, determined by HPLC, is 99.9% (yield = 82%; m.p. = 320-3220C) .
94.8%
b) - Preparation of 6- [3- (1-adamantyl) -4-methoxy- phenyl] -2-naphthoic acid (I):20 mL of tetrahydrofuran (12 vol) , 2 g (7 mmol) of 3 -adamantyl-4-methoxyphenylboronic acid (II), 1.65 g (6.6 mmol) of 6-bromo-2 -naphthoic acid (III) and 20 mL of a 2 M aqueous potassium carbonate solution are introduced into a round-bottomed flask equipped with stirring and under a nitrogen stream. 15 mg (1%) of palladium acetate and 46 mg (2%) of 2- (dicyclohexyl- phosphino) biphenyl are then introduced. The medium is EPO <DP n="11"/>heated under reflux for 2 hours. Kinetic monitoring by HPLC indicates that the % of 6- [3- (i-adamantyi) -4- methoxyphenyl] -2 -naphthoic acid formed is 94% after one hour and 98% after 2 h.After returning to room temperature, the catalyst is filtered on a cartridge, and then slowly poured over 30 ml of a 1 N aqueous hydrochloric acid solution.The medium is kept stirring for one hour. The precipitate is filtered, washed with water and then dried under reduced pressure. 2.68 g of 6-[3-(l- adamantyl) -4-methoxyphenyl] -2 -naphthoic acid are obtained in the form of a white solid whose purity, determined by HPLC, is 99.9% (yield = 94.8%; m.p. = 3210C) .The following melting points (m.p.) exist in the literature: m.p. = 319-322C (B. Charpentier et al . , J. Med. Chem., 1995, 38, 4993-5006) and m.p. = 325-327C (EP 0 199 636) .
With hydrogenchloride;AlCl3; In hexane; nitrobenzene;
6-Bromo-2-naphthoic acid. A solution of 14.7 (70.9 mmol) of 2-bromonaphthalene and 6.40 g (81.5 mmol) of acetyl chloride in 55 mL of nitrobenzene was added dropwise to a vigorously stirred solution of 10.4 g (78 mmol) of anhydrous AlCl3 in 25 mL of nitrobenzene at 25-30 C. over a period of 1.3 h. After being stirred for 1 h more at 25 C., the reaction mixture was poured onto 200 g of ice and 30 mL of concentrated HCl. The product was extracted with Et2 O (300, 200, and 100 mL). The extracts were washed with brine and saturated NaHCO3, dried (MgSO4), and concentrated at reduced pressure to afford a brownish oil. Short-path distillation (45-55 C./25 mm) was used to remove nitrobenzene. The crystalline residue was dissolved in hot hexane (300 ml), filtered, and concentrated at reduced pressure to give 16.3 g (92% yield) of crude <strong>[1590-25-6]2-acetyl-6-bromonaphthalene</strong>. Two crystallizations from hexane afforded 5.3 g (21% yield) of pure product, which had only one spot by TLC (10% EtOAc/hexane) Rf 0.22, mp 99-101.5 C.; IR (mull) 1670, 1623, 1365, 1355, 1265, 1225, 1175, 880, 820, 800 cm-1; 1 H NMR (CDCl3) delta 2.71 (s, 3, CH3 CO), 7.5-8.2 (m, 4, 3,4,7,8-ArH), 8.03 (s, 1, 5-ArH), 8.44 (s, 1, 1-ArH).
N-[(6-bromo-2-naphthalenyl)carbonyl]-N-methylglycine methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
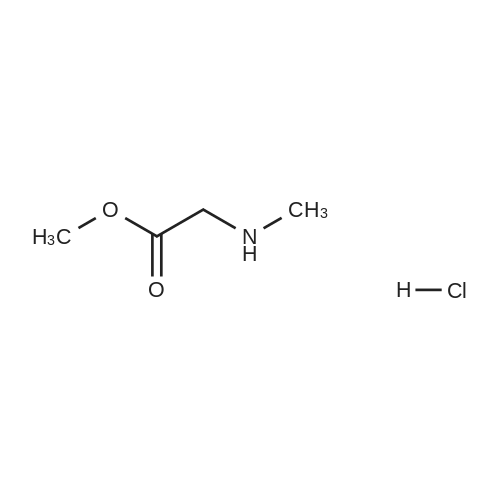
With N-ethylmorpholine;; In pyridine; N-methyl-acetamide; thionyl chloride; water; ethyl acetate; N,N-dimethyl-formamide;
A suspension of 6-bromo-2-naphthalenecarboxylic acid (5.37 g, 21.4 mmoles, a starting material of formula II) in thionyl chloride (54 ml) containing 5 drops of dimethylformamide (DMF) was refluxed for 30 min. The mixture was evaporated to dryness under reduced pressure. The residue was dissolved in dry pyridine (54 ml). <strong>[637-96-7]N-Methylglycine hydrochloride</strong> (2.8 g, 20.2 mmoles), a starting material of formula III, was added to the solution. The resulting mixture was stirred at 20-22 C. for 2 hr and then refluxed for 3 hr. The mixture was evaporated to dryness. The residue was dissolved in ethyl acetate (600 ml) and water (100 ml). After shaking the mixture and separating the two layers, the aqueous layer was extracted with more ethyl acetate. The combined organic extracts were washed with 2 N aqueous HCl, brine and water, dried (MgSO4) and evaporated to dryness. The residue (5.6 g) was crystallized from ethanol to give 3.4 g of the title compound; mp 103-105 C.; NMR (CDCl3) delta 2.75 (s, 3H), 3.08 (s, 3H), 4.25 (s, 2H), 7.30-8.20 (m, 6H); IR (CHCl3) 1738, 1630, 1580 cm-1; UVlambdamax (EtOH) 280 nm (epsilon6,980), 273 (6,720), 232 (64,740); Anal Calcd: C, 53.59% H, 4.20% N, 4.17%; Found: C, 53.41% H, 4.29% N, 4.27%. Procedure B for preparing the title compound A stirred mixture of the starting material of formula II, 6-bromo-2-naphthalenecarboxylic acid (12.8 g, 52 mmoles), and 1-hydroxybenzotrizole (HOBt, 7.0 g, 52 mmoles) in DMF (200 ml) was cooled to 0 C. N,N-dichlohexylcarbodiimide (DCC, 10.6 g, 52 mmoles) in DMF (30 ml) was added to the mixture. The resulting mixture was stirred at 0 C. for 30 min and at 20 C. for 1 hr and then cooled again to 0 C. N-Methylglycine methyl ester hydrochloride (7.25 g, 52 mmoles), followed by N-ethylmorpholine (6.7 ml, 52 mmoles), were added to the cooled mixture. The mixture was stirred for 30 min at 0 C. and then for 18 hr at 20 C. Thereafter, the mixture was filtered and concentrated to dryness under reduced pressure. The residue was subjected to chromatography on 325 g of silica gel using ethyl acetate-hexane (1:1) as the eluant. The pure fractions were pooled to yield 10.5 g of product which was recrystallized from ethyl acetate to give the title compound, identical to the product of procedure A of this example.
With sodium hydroxide; sodium hypochlorite; In (2S)-N-methyl-1-phenylpropan-2-amine hydrate; water;
EXAMPLE 1 N-[(6-Bromo-2-naphthalenyl)carbonyl]-N-methylglycine Methyl Ester (IV, R1 and R3 =CH3 and R2 =6-Br) The title compound can be prepared by two procedures designated as procedure A and procedure B below. The starting material of formula II used in each of these procedures is prepared as follows: A sodium hypochlorite solution was prepared by introducing chlorine gas (11.2 g) into a solution of NaOH (15.3 g) in 200 ml of ice water. Solid <strong>[1590-25-6](6-bromo-2-naphthalenyl)ethanone</strong> [9.5 g, 38.1 mmoles, prepared according to the procedure of R. B. Girdler et al., J. Chem. Soc. (C), 518 (1966)] was added to the stirred sodium hypochlorite solution at 0 C. and then the mixture was heated on a steam bath for 1 hr. The precipitate was removed by filtration. Sodium metabisulfite (5 g) was added to the cooled (0 C.) filtrate. The mixture was adjusted to pH 5 with concentrated HCl. The precipitate was collected and dried. The collected precipitate was crystallized from boiling ethanol by the addition of water to afford 5.5 g (two crops) of 6-bromo-2-naphthalenecarboxylic acid; mp 288-290 C.; NMR (CDCl3) delta 7.68 (2d, J=8 Hz, J2 =2 Hz, 1H), 7.97 (m, 2H), 8.05 (d, J=8 Hz, 1H), 8.25 (d, H), 8.58 (d, 1H), IR (CHCl3), 2800, 1680, 1625, 1567 cm-1; UVlambdamax (EtOH) 333 nm (epsilon1080), 323 (880), 317 (1020), 286 (9040), 237 (54,350). Procedure A for preparing the title compound:
6-Bromo-2-naphthalenecarboxylic acid (2.4996 g, 10.0 mmol) was added50mL dried egg-shaped flask, add 20mL of anhydrous methanol to dissolve,Then 1 mL of concentrated sulfuric acid was slowly added dropwise and the system was refluxed overnight.TLC tracking until the conversion of raw materials is completed, stop heating,After cooling to room temperature, saturated aqueous sodium carbonate solution was added to quench the reaction.The reaction system was adjusted to neutrality, extracted with ethyl acetate,The organic phase is washed three times with anhydrous sodium sulfate.Concentration under reduced pressure afforded S7 (2.63 g, 100% yield) as a white solid.
93%
With sulfuric acid;Reflux;
To a solution of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (2.65 g, 10 mmol) in methanol 30 mL,concentrated sulfuric acid 1 mL was added drop wise. The reaction mixture was refluxed for 2 h, then added water 150 mL. The foliated crystal was collected thought filtration. Yield: 93%;
26.4 g
With sulfuric acid; for 7h;Reflux;
Reactor to 6-bromo-2-naphthalene carboxylic acid (T-1, manufactured by Tokyo Kasei Kogyo (Ltd.)) (25g, 99.6mmol), concentrated sulfuric acid (5.4ml, 99.6mmol), and methanol (80ml) It was placed. The mixture was stirred with heating under reflux for 7 hours. After cooling to room temperature, water was added to the reaction mixture and extracted with dichloromethane. Aqueous organic layers were washed with sodium carbonate, washed with water, then saturated brine, and dried over anhydrous magnesium sulfate. By concentrating the solution under reduced pressure to give compound (T-2) (26.4g, 99.6mmol) and. Compound (T-2) was used in the next reaction without purification.
General procedure: Compound 14 (4.58 g, 20 mmol) in 1:1 THF/H2O (200 mL) was treated with LiOH.H2O (4.2 g, 100 mmol), stirred overnight, and concentrated to a thick slurry and then treated with 2M HCl to pH = 3 and the precipitate was filtered, washed with water and dried under vacuum to provide 3.8 g (88%) of 15 as a white powder
84%
With potassium hydroxide; In methanol; at 50℃; for 8h;
A suspension of methyl 6-bromo-2-naphthoate (3b) (2.7 g, 10.0 mmol) and potassium hydroxide (1.1 g, 20.0 mmol) in methanol (50 mL) was vigorously stirred at 50 C. The reaction mixture becomes homogeneous after the consumption of the initial compound 3b. After 8 h, the solvent was evaporated under reduced pressure (ca 2/3 vol.), water (1500 mL) was added and the unreacted ester extracted with ethyl acetate. The aqueous solution was acidified with 10% H2SO4 to pH 3 and, after extraction with ethyl acetate (3 × 200 mL) and drying on anhydrous sodium sulfate, removal of the solvent afforded the pure acid 3a (2.1 g, 8.4 mmol, 84% yield). Mp 290-294 C (decomp.); HRMS (ESI+): m/z [M+1]+ Calcd for C11H8BrO2: 252.08591. Found: 252.08582. 1H NMR (DMSO-d6) delta = 7.72 (1 H, dd, J = 8.5 and 1.8 Hz, 7-naphthyl H), 7.99 (1 H, d, J = 8.6 Hz, 4-naphthyl H), 8.02 (1 H, dd, J = 8.6 and 1.4 Hz, 8-naphthyl H), 8.09 (1 H, d, J = 8.8 Hz, 3-naphthyl H), 8.30 (1 H, d, J = 1.6 Hz, 5-naphthyl H), 8.62 (1 H, s, 1-naphthyl H), 13.15 (1H, br. s, COOH); 13C NMR (125.76 MHz, DMSO-d6) delta = 121.93, 126.50, 127.61, 128.81, 129.83, 130.01, 130.64, 130.90, 131.63, 136.11, 167.30.
80%
Example 176-BROMONAPHTHALENE-2-CARBOXYLIC ACID [(R)-1-(4-METHANESULFONYLAMINO-3-METHYLPHENYL)ETHYL]AMIDE 17A) 6-BROMONAPHTHALENE-2-CARBOXYLIC ACID To a stirred solution of 6-bromonaphthalene-2-carboxylic acid methyl ester (2 g, 8 mmol) in tetrahydrofuran (66 mL) and ethanol (22 mL) was added a solution of lithium hydroxide (542 mg, 22 mmol) in water (22 mL). The reaction was stirred at 50 C. for 16 hours. After cooling, the organic solvents were removed by evaporation, and the aqueous residue was diluted with water (100 mL) then washed with EtOAc (2×50 mL). The aqueous layer was acidified using 1N HCl and the products were extracted with EtOAc (3×50 mL). The combined organics were washed with brine (100 mL), dried (MgSO4), filtered and concentrated. Trituration with DCM gave the title compound (1.594 g, 80%) as an off-white solid.1H NMR (400 MHz, DMSO-d6) delta 7.74 (dd, 1H, J=8.7 Hz, 1.9 Hz), 7.99-8.04 (m, 2H), 8.10 (d, 1H, J=8.8 Hz), 8.32 (s, 1H), 8.64 (s, 1H).
70%
Part A. Preparation of 6-bromo-2 -naphthoic acid.; [00746] A solution of methyl 6-bromo-2-naphthoate (7.7Og, 29.0mmol) in 2:1 THF:water (15OmL) was treated with lithium hydroxide hydrate (2.44g, 58.1mmol) followed by stirring at room temperature for 48h. Concentrated under vacuum, diluted with water and cooled to O0C. Acidified to pH3 with 4N HCl. Solids were collected by filtration, dissolved in toluene-EtOAc (ca. 2L) and washed with brine. Dried over Na2SO4, filtered and concentrated under vacuum. Brown solid was triturated with ether, collected by filtration, and dried under vacuum to give the title compound as a nearly white solid (5.07g, 70%).
70%
[00487] Example 1. Preparation ofN-(6-(3-tert-butyl-5-(2,4-dioxotetrahydropyrimidin-l(2H)-yl)-2- methoxyphenyl)naphthalen-2-yl)methanesulfonamide (compound IA-LO-2.9).; [00488] Part A. Preparation of 6-bromo-2 -naphthoic acid.; [00489] A solution of methyl 6-bromo-2-naphthoate (7.7Og, 29.0mmol) in 2:1 THF:water (15OmL) was treated with lithium hydroxide hydrate (2.44g, 58.1mmol) followed by stirring at room temperature for 48h. Concentrated under vacuum, diluted with water and cooled to O0C. Acidified to pH3 with 4N HCl. Solids were collected by filtration, dissolved in toluene-EtOAc (ca. 2L) and washed with brine. Dried over Na2SC^, filtered and concentrated under vacuum. Brown solid was triturated with ether, collected by filtration, and dried under vacuum to give the title compound as a nearly white solid (5.07g, 70%).
70%
Part H. Preparation of 6-bromo-2 -naphthoic acid.[00326] A solution of methyl 6-bromo-2-naphthoate (7.7Og, 29.0mmol) in 2: 1 tetrahydrofuran: water(150 mL) was treated with lithium hydroxide hydrate (2.44 g, 58.1 mmol) followed by stirring at room temperature for 48 hours. The mixture was concentrated under vacuum, diluted with water and cooled to 0 0C. The mixture was acidified to pH3 with 4 N HCl. Solids were collected by filtration, dissolved in toluene-ethyl acetate (ca. 2 L) and washed with brine. The organic layer was dried overNa2SO+, filtered and concentrated under vacuum. The brown solid was triturated with ether, collected by filtration, and dried under vacuum to give the title compound as a nearly white solid (5.07g, 70%).
56%
With potassium hydroxide; In methanol; at 50℃; for 48h;
Potassium hydroxide (127 mg, 2.26 mmol) was added to a suspension of methyl 6-bromo-2-naphthoate (200 mg, 0.75 mmol) in methanol (50 mL) and the mixture heated at 50 C for 48 h. The solvent was evaporated and the residue diluted with water (30 mL), acidified with HCl (1 M) and the extracted with ethyl acetate (30 mL * 2). The combined organic layers were dried with anhydrous magnesium sulfate and the solvent evaporated under reduced pressure. The crude product was purified via recrystallization using ethyl acetate to give the product as white crystals (105 mg, yield 56%). 1H NMR matched that previously reported. 1H NMR (400 MHz, CD3OD): delta 8.58 (s, 1H; Ar-H), 8.14 (s, 1H; Ar-H), 8.06 (dd, J = 1.8, 8.7 Hz, 1H; Ar-H), 7.92 (d, J = 8.7 Hz, 1H; Ar-H), 7.87 (d, J = 8.7 Hz, 1H; Ar-H), 7.64 (dd, J = 1.8, 8.7 Hz, 1H; Ar-H).
Reference Example 76-Bromo-2-naphthoic acid (10.1 g, 40.1 mmol) and N,N- dimethylformamide (4.75 g, 65.0 mmol) were added to toluene (80 mL) . To the reaction mixture was added dropwise thionyl chloride (5.7 g, 48.2 mmol) at 45 to 50C, and the mixture was stirred for 1 hr, and allowed to cool to room temperature. The reaction mixture was added dropwise at 10 to 25C to a solution prepared by adding triethylamine (11.4 g, 112.4 mmol) and 40% methylamine methanol solution (8.1 g, 104.4 mmol) to toluene (80 rnL) , and the mixture was stirred at room temperature for 1 hr.. To the reaction mixture was added dropwise water (50 mL) , and the mixture was stirred at room temperature. The crystals were collected by filtration, and washed with a mixed solvent (25 mL) of methanol/water (2:8) to give wet crystals. The total amount of the wet crystals was added to N,N- dimethylacetamide (70 mL) , and dissolved with heating to 60C. The reaction mixture was allowed to cool to room temperature, and water (140 mL) was added dropwise thereto. The crystals were collected by filtration, and washed with water (80 mL) to give wet crystals. The total amount of the wet crystals was suspended in ethyl acetate -(25 mL) with stirring at roomtemperature. The crystals were collected by filtration, and washed with ethyl acetate (5 mL) . The obtained wet crystals were dried under reduced pressure to give 6-bromo-N-methyl-2- naphthamide (9.4 g, 35.6 mmol). yield 89%.¾ NMR (500 MHz, DMSO-d6) delta 2.84 (d, J = 4.4 Hz, 3H) , 7.71 (dd, J = 8.8, 2.2 Hz, 1H) , 7.93 - 8.03 (m, 3H) , 8.28 (d, J = 1.9 Hz, 1H), 8.44 (s, 1H) , 8.62 (d, J = 4.1 Hz, 1H) ; HRMS (ESI) m/z Calcd for a Ci2HuNOBr [M+H]+: 264.0024, Found: 264.0019; Anal. Calcd for a Ci2Hi0NOBr: C, 54.57; H, 3.82; N, 5.30; Br, 30.25. Found: C, 54.56; H, 3.70; N, 5.34; Br, 30.23.
82%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; N,N-dimethyl-formamide; at 0 - 20℃; for 18h;Inert atmosphere;
Under an argon atmosphere, to a cooled (0 C) solution of 8 (60.26 g, 240 mmol), EDCI·HCl (55.21 g, 288 mmol), HOBt·H2O (44.1 g, 288 mmol) and N,N-diisopropylethylamine ((i-Pr)2NEt) (37.23 g, 288 mmol) in anhydrous N,N-dimethylformamide (DMF) (960 mL) was added dropwise to a solution of MeNH2 (2 M solution in THF; 192 mL, 384 mmol) and the whole was stirred at room temperature for 18 h. After dilution with water, the precipitate was filtered off, washed with H2O and i-Pr2O and dried under the reduced pressure to give 9a (60.6 g, 82%) as a colorless powder. 1H NMR (CDCl3 + CD3OD) delta: 3.04 (3H, s), 7.60 (1H, dd, J = 1.8 Hz, 8.6 Hz), 7.78 (2H, d, J = 8.6 Hz), 7.85 (1H, dd, J = 1.8 Hz, 8.6 Hz), 8.03 (1H, d, J = 1.8 Hz), 8.25 (1H, s). IR (KBr): 3274, 1638, 1622, 1559, 1495, 1408, 1316, 1159 cm-1. Anal. Calcd for C12H10NOBr.0.1H2O: C, 54.20; H, 3.87; N, 5.27; Br, 30.25. Found: C, 54.03; H, 3.72; N, 5.24.
80%
4 Liters of ethyl acetate and 25 ML of DMF were added to 500 g (1.99 mol) of 6-bromo-2-naphthoic acid. 188 ML (2.61 mol, 1.3eq) of thionyl chloride was added dropwise at 30C or lower.. The mixture was stirred at 65C for 30 minutes.. After cooled to 25C, a mixture of 408 ML (3.93 mol, 2eq) of a 40% solution of methylamine in methanol and 558 ML (4.01 mol, 2eq) of triethylamine was added dropwise at 25C or lower.. The mixture was stirred at 25C for 3 hours. 2.5 Liters of water was added dropwise at 25C or lower.. Crystals were filtered, and washed successively with 1.25 liters of a mixed solution of methanol/water=1/4.. Vacuum drying (50C) to a constant weight afforded 422 g of 6-bromo-N-methyl-2-naphthamide (yield 80%).1H NMR (CDCl3+CD3OD): delta 3.04 (3H, s), 7.60 (1H, dd, J=8.6, 1.8 Hz), 7.78 (2H, d, J=8.6 Hz), 7.85 (1H, dd, J=8.6, 1.8 Hz), 8.03 (1H, d, J=1.8 Hz), 8.25 (1H, s).
To <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (5.0 g, 20 mmol), were added 20 mL of thionyl chloride (SOCl ), and dimethylformamide (DMF, 1 mL), and the mixture was heated under stirring for 4 hours. An excessive amount of thionyl chloride (SOCl ) is distilled off in vacuo, and to the reaction mixture, were added 20 mL of N-methylpyrrolidine (NMP), and N-phenyl-l,2-diamino benzene (3.7 g, 20 mmol). The mixture was stirred at 160C for 12 hours, and cooled to normal temperature, and an excessive amount of water was added thereto to form a solid. The solid was filtered, washed with water and then ethanol, and dried to prepare a compound L-I (6.2 g, yield 78 %).[216] MS [M+H]+ = 399
78%
Thionyl chloride (SOC12) (20 mL) and dimethylformamide (DMF) (1 mL) were put into <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (5.0 g, 20 mmol), and agitated and refluxed for 4 hours. After an excessive amount of thionyl chloride (SOC12) was removed by the vacuum distillation, N-methylpyrolidyne (NMP) (20 mL), and N-phenyl-1,2-diaminobenzene (3.7 g, 20 mmol) were put into the reaction mixture and agitated at 160C for 12 hours. The temperature was lowered to normal temperature, and an excessive amount of water was added thereto to form a solid. It was filtered, washed with water and ethanol, and dried to prepare the compound B-18 (6.2 g, 78%). MS:[M]+=399
78%
PREPARATION EXAMPLE II-9 Preparation of Compound B-9 (0096) (0097) To <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (5.0 g, 20 mmol), 20 mL of thionyl chloride (SOCl2), and dimethylformaldehyde (DMF, 1 mL) were added, and the mixture was stirred under heating for 4 hours. An excessive amount of thionyl chloride (SOCl2) was distilled off in vacuum, and then to the reaction mixture, 20 mL of N-methylpyrrolidine (NMP), and N-phenyl-1,2-diaminobenzene (3.7 g, 20 mmol) were added, and the mixture was stirred at 160 C. for 12 hours. The mixture was cooled to ambient temperature, and then an excessive amount of water was added thereto to form a solid. The solid was filtered, washed with water and then ethanol, and dried to prepare a compound B-9 (6.2 g, yield 78%). (0098) MS [M+H]+=399
To a methylene chloride (80 ml) solution of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (20 mmol) was added oxalyl chloride (80 mmol), N,N-dimethylformamide (8 dot) under anhydrous condition, the reaction mixture was stirred at room temperature for 1h. Solvent distilled under reduced pressure and dried under high vacuum to remove oxalyl chloride. Reaction mass was dissolved in tetrahydrofuran (70 mL). The solution was added dropwise to the precooling ammonia solution (60 mL). Meanwhile, the liquor was allowed to stir vigorously at -10 C for 1 h and then stirred at room temperature overnight. The solvent was removed in vacuo and the crude residue was washed with water. The product was collected by filtration to give compound 1: 6-Bromo-naphthalene-2-carboxylic acid amide, white solid, yield, 96%, 1H NMR (600 MHz, DMSO-d6) delta 8.47 (s, 1H), 8.25 (s, 1H), 7.96 (qd, J=8.5, 3.7 Hz, 3H), 7.68 (dd, J=8.8, 1.7 Hz, 1H), 8.13+7.49 (Brs, 2H, NH2).
With diphenylphosphoranyl azide; triethylamine; In toluene; at 100℃; for 4h;
Diphenylphosphoryl azide (17.09 mL, 79 mmol) was added to a solution of 6- bromo-2-naphthoic acid (16.5 g, 65.7 mmol), triethylamine (18.32 mL, 131 mmol), and tert-butylalcohol (7.54 mL, 79 mmol) in toluene (225 mL) and stirred for 4 h at 100 C. The volatiles were removed by rotary evaporation and the residue taken up in EtOAc (500 mL) and washed with water and brine. A precipitate formed upon concentration which was isolated by filtration and washed with 1 : 1 Et20/Hex to give Example J.9gl (10.5 g). A second crop of less pure product was isolated upon concentration of the mother liquor (9.8 g); combined yield (93%). LC/MS (Cond. J2): RT = 3.44 min. LC/MS Anal. Calcd. for [M+Na Ci5H16BrN02: 345.02; found 345.03.
85%
With diphenyl phosphoryl azide; triethylamine; at 100℃;
Section VIIExample VII-IPreparation of Compound 201 and 202General Procedure VII-A6-Bromonaphthalene-2-carboxylic acid (VII-IA) (11 g, 44 mmol) in t-BuOH (50 mL) containing Et3N (4.86 g, 48 mmol) was treated with DPPA (13.2 g, 48 mmol) and stirred at 100 C. overnight. After cooling to r.t., the mixture was poured into water and extracted with EtOAc (100 mL×3), the organic layer was combined and washed with brine, dried over anhydrous sodium sulfate, concentrated in vacuo. The residue was purified on silica gel column chromatography, eluting by petroleum ether and ethyl acetate (7:1), to afford compound VII-IB (12 g, yield 85%).
With diphenyl phosphoryl azide; triethylamine; at 80℃; for 16h;
Step 1 : tert-butyl-N-(6-bromo-2-naphthyl)carbamate To a cooled solution of <strong>[5773-80-8]6-bromonaphthalene-2-carboxylic acid</strong> (50 g, 0.2 mol) in t- butanol (350 ml.) triethylamine (30 g, 0.3 mol) was added slowly. Diphenylphosphoryl azide (60 g, 0.22 mol) was then added and the reaction mixture was stirred at 80C for 16 h. The mixture was allowed to cool to room temperature, then poured into saturated NaHC03 solution. The precipitate that had formed was filtered to give tert-butyl-N-(6- bromo-2-naphthyl)carbamate (65 g) which was used without further purification.
A mixture of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (80.3 g, 319 mmol), diphenylphosphoryL azide (71 mL, 352 mmol) and triethylamine (50 mL, 358 mmol) in fert-butanol (400 mL) was heated to reflux and allowed to stir at this temperature for 15 hours. The reaction mixture was then cooled to room temperature and poured over saturated aqueous NaHC03 solution (6Theta0 mL) and stirred vigorouslyfor 30 minutes. The resulting suspension was filtered, washed with water (200 mL) and dried in vacuo at 65 C. The resulting white solid was suspended in MeOH (500 mL) and cooled to -78 C, then HCl gas was bubbled into the mixture until saturated. The reaction mixture was then allowed to stir at room -temperature for 15 hours, after which time the resulting solids were collected by filtration, then washed with- ice-cold MeOH (100 mL) to provide Compound Int-6a as an off-white solid (74.8 g, 91%), which was used without further purification. l NM (DMSO-_¾) 6 10.5-10.0 (br s, 3H), 8.23 (s, 1H), 7.99 (d, J= 9.0 Hz, 1H), 7.92 (d, J= 9.0 Hz, 1H), 7.84 (s, 1H), 7.68-7.65 (m, 1H), 7.56-7.51 (m, 1H . "LRMS: (M+2H)+ = 223.
91%
Preparation of Compound 1A1AA mixture of 6-bromo-2--naphthoic acid-(80.3 g, 319 mmol),diphenylphosphorylazide (71 mL, 352 mmol) and triethylamine (50 mL, 358 mmol) in fer/-butanol (400 mL) was heated to reflux for 15 hours then cooled to room temperature. The reaction mixture was poured over saturated NaHC03 (600 mL) and stirred vigorously for 30 minutes. The solids were filtered, washed with water (200 mL) and dried under vacuum at 65 C. The resulting. white solid was suspended in MeOH (500 mL), then HCl(g) was bubbled into the mixture at -78 C until saturated. The reaction mixture was stirred at room temperature for 15 hours, then the resulting solids were collected by filtration, and washed with ice-cold MeOH (100 mL) to provide Compound 1A as an off- white solid (74.8 g, 91%). 1H NMR (DMSO-<4) delta 10.5-10.0 (br s, 3H), 8.23 (s, 1H), 7.99 (d, J= 9.0 Hz, 1H), 7.92 (d, J= 9.0 Hz, 1H), 7.84 (s, 1H), 7.68-7.65 (m, 1H), 7.56-7.51 (m, 1H). LRMS: (M+2H)+ = 223.
Step A - Synthesis of Intermediate Compound Int-17a (0328) (0329) A mixture of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (80.3 g, 319 mmol), diphenylphosphoryl azide (71 mL, 352 mmol) and triethylamine (50 mL, 358 mmol) in tert-butanol (400 mL) was heated to reflux and allowed to stir at this temperature for 15 hours. The reaction mixture was then cooled to room temperature and poured over saturated aqueous NaHCO3 solution (600 mL) and stirred vigorously for 30 minutes. The resulting suspension was filtered, washed with water (200 mL) and dried in vacuo at 65 C. The resulting white solid was suspended in MeOH (500 mL) and cooled to -78 C, then HCl gas was bubbled into the mixture until saturated. The reaction mixture was then allowed to stir at room temperature for 15 hours, after which time the resulting solids were collected by filtration, then washed with ice-cold MeOH (100 mL) to provide Compound Int-17a as an off-white solid (74.8 g, 91 %), which was used without further purification. 1H NMR (DMSO-d6) delta 10.5-10.0 (br s, 3H), 8.23 (s, 1H), 7.99 (d, J= 9.0 Hz, 1H), 7.92 (d, J= 9.0 Hz, 1H), 7.84 (s, 1H), 7.68-7.65 (m, 1H), 7.56-7.51 (m, 1H). LRMS: (M+2H)+ = 223.
With palladium 10% on activated carbon; sodium carbonate; In methanol; water; at 78℃;Inert atmosphere;
A suspension of <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (3a) (12.55 g, 0.05 mol), 4-methoxyphenylboronic acid (8) (8.5 g, 0.056 mol), anhydrous sodium carbonate (12.0 g, 0.113 mol) and 10% palladium on activated carbon (0.5 g) in water (70 mL) and methanol (70 mL) was vigorously stirred under nitrogen and gradually warmed to reach reflux temperature (78 C). After refluxing for 3 h, the reaction was stopped by slow addition of 37% HCl (18.8 mL), followed by 99% formic acid (5 mL) to a final pH value of 1. The mixture was refluxed for 0.5 h then cooled to room temperature and the gray solid was filtered. The filtrate was repeatedly washed with water and then suspended in water (200 mL); triethylamine (14.5 g, 0.14 mol) was added and the suspension warmed to form the water-soluble triethylammonium salt of the acid 2a. The catalyst was recovered from the warm mixture by filtration on a paper filter and washed with an aqueous solution of triethylamine. The aqueous solution of the triethylammonium salt of 3a was poured into a round-bottom flask, warmed at 60 C and 99% formic acid (12.2 g, 10 mol) was dropped into the solution in 30 min (final pH 2). After refluxing (15 min), the solution was cooled at room temperature and the precipitate filtered, then washed with water and finally with methanol. Pure acid 2a (13.1 g, 0.047 mol) was obtained in 94% yield. Mp 266-278 C (decomp.); HRMS (ESI+): m/z [M+1]+ Calcd for C18H15O3: 279.31351. Found: 279.31348 1H NMR (DMSO-d6) delta = 3.82 (3 H, s, ArOCH3), 7.08 (2 H, d, J = 8.7 Hz, 3- and 5-phenyl H), 7.79 (2 H, d, J = 8.7 Hz, 2- and 6-phenyl H), 7.90 (1 H, dd, J = 8.6 and 2.0 Hz, 7-naphthyl H), 7.99 (1 H, dd, J = 8.5 and 1.3 Hz, 4-naphthyl H), 8.03 (1 H, d, J = 8.6 Hz, 8-naphthyl H), 8.16 (1 H, d, J = 8.6 Hz, 3-naphthyl H), 8.23 (1 H, s, 5-naphthyl H), 8.61 (1 H, s, 1-naphthyl H), 13.13 (1H, br. s, COOH); 13C NMR (125.76 MHz, DMSO-d6) delta = 55.70, 114.99, 124.54, 126.08, 126.16, 128.41, 128.75, 130.35, 130.70, 131.50, 132.23, 135.86, 139.84, 159.81, 167.97.
With 4-methyl-morpholine; benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃;Cooling with ice;
Intermediate (iv)6-Bromo-naphthalene-2-carboxylic acid (tetrahydro-pyran-4-yl)-amide Bromo-naphthalene-2-carboxylic acid (0.63g, 2.5 mmol) was dissolved in DCM (20 ml_) and DMF (5ml_) and the solution was cooled to an ice-water bath. To this solution was added a solution of tetrahydropyran-4-yl amine (0.26 g, 2.5 mmol, 1 equiv.) in 1 ml_ of DCM, followed by N-methylmorpholine (0.575 g, 0.8 ml_, 7.5mmol, 3 equiv.), 1 -Hydroxylbenzotriazole (HOBT) (0.44 g, 3.25 mmol, 1 .3 equiv.), sequentially, and finally EDC.HCI (0.625g, 3.25 mmol, 1 .3 equiv. ). The resultant clear light brown solution was stirred at r.t. overnight. TLC (10% MeOH in DCM) and LC/MS detected the product peak at retention time of 3.608 min with MS 334/336. The reaction was quenched with saturated sodium bicarbonate aqueous solution (10 ml_) and 10 mL of DCM. The two layers were separated, and the aqueous layer was extracted with DCM (15 ml_x2). The combined DCM extracts were washed with sodium bicarbonate (10 mL), and brine (10 mL), dried (anhydrous potassium carbonate), filtered, and concentrated in vacuo to get 0.85g ( 100% yield) of the title compounds.TLC (5% MeOH in DCM) Rf = 0.7.LCMS: RT = 2.85 minutes, MS: 335 (M+H+).1 H NMR (CDCIs, 300MHz), delta (ppm): 8.24 (s, 1 H), 8.04 (s, 1 H), 7.82 (m, 3H), 7.62 (m, 1 H), 6.13 (m, 1 H), 4.03 (m, 2H), 3.56 (dt, 2.2Hz, 1 1 .7Hz, 2H), 2.06 (m, 2H), 1 .62 (m, 1 H).
100%
With 4-methyl-morpholine; benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 16h;Cooling with ice;
General procedure: 4-Bromo-2-methylbenzoic acid (5.38g, 25mmol) was dissolved in DCM (100mL) and DMF (25mL) and the solution was cooled to an ice-water bath. To this solution was added a solution of tetrahydropyran-4-yl amine (2.60g, 25mmol, 1.0equiv) in 10mL of DCM, followed by N-methylmorpholine (5.75g, 8mL, 75mmol, 3.0equiv), 1-hydroxylbenzotriazole (HOBt) (4.40g, 32.5mmol, 1.3equiv), sequentially, and finally EDC·HCl (6.25g, 32.5mmol, 1.3equiv). The resultant clear light brown solution was stirred at rt overnight. TLC (10% MeOH in DCM) and LCMS detected a product peak with m/z of 299 (M+H+). The reaction was quenched with saturated sodium bicarbonate aqueous solution (10mL) and DCM (10mL). The two layers were separated, and the aqueous layer was extracted with DCM (15mL×2). The combined DCM extracts were washed with sodium bicarbonate aqueous solution (10mL) and brine (10mL), and dried (anhydrous potassium carbonate), filtered, and concentrated in vacuo to get 6.87g (yield 92%) of the title compound as a white solid
Example 12 Preparation of Compound 12 6-Bromo-naphthalene-2-carbonyl chloride: 6-Bromonaphthalene-2-carboxylic acid (25.1 g) was suspended in thionyl chloride (200 mL), stirred at 60 C. for 16 hours and evaporated under vacuum. Solid was dissolved in dichloromethane (50 mL) and evaporated under vacuum, giving 6-bromonaphthalene-2-carbonyl chloride (27.0 g, crude) as a white solid. 1-(6-Bromo-naphthalen-2-yl)-2-diazo-ethanone:
Step 1 6-bromo-2-naphthoic acid (72.3 g, 282 mmol, 1.0 equiv.) was suspended in dichloro-methane (600 mL) and DMF (catalytic, 5 drops) was added. Oxalylchloride (71.6 g, 564 mmol, 2.0 equiv.) was added portion wise during 1 hour. The reaction mixture was stirred overnight with a CaCl2 drying tube mouthed on the flask. Complete dissolution occurred. The reaction mixture was concentrated, dichloromethane (100 mL) was added and the solvent was evaporated again, yielding 6-bromo-2-naphthoyl chloride (76.1 g, 100%) as an oil which was used as such in next step.
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium acetate; In N,N-dimethyl-formamide; at 90℃;
General procedure: Aryl halide (1 equiv), bis-pinacolato-diboron (1.5-3 equiv), Pd(dppf)Cl2 (0.05-0.1 equiv), and KOAc (3-6 eq.) were suspended in DMF (2-6 mL). The mixture was then heated at 90 C for 2-6 h, cooled to rt, diluted with H2O (10 mL) and extracted with EtOAc (3 × 5 mL). The organic layer was dried (MgSO4) and the solvent removed in vacuo. The residue was purified by chromatography using a stepped gradient of 0-10% EtOAc in heptane to yield the desired boronic ester.
Sodium; 6-bromo-naphthalene-2-carboxylate[ No CAS ]
[ 5773-80-8 ]
Yield
Reaction Conditions
Operation in experiment
95.5%
With hydrogenchloride; In water; at 50℃; for 1h;Inert atmosphere;
Subsequently, the residue was put into a glass container of 200 ml provided with a temperature measurement tube and an electromagnetic stirrer and nitrogen atmosphere therein. In addition, 97 g of water was put into the glass container and the temperature therein was raised to 50 degrees C. to dissolve the residue. A weight of 5.4 g of 36% hydrochloric acid was dropped into the aqueous solution in the glass container, and after the completion of the drop, the aqueous solution was held at 50 degrees C. during one hour. The temperature in the glass container was then decreased to 25 degrees C. to obtain a precipitate. The obtained precipitate was subjected to filtration to be recovered as a residue, and the residue was washed by 110 g of water. The residue after the washing was dried under reduced pressure of 5 mmHg and 40 degrees C. until being of constant mass. [0046] A yellowish-white refined BNA product was thus obtained with a purity of 99.9% and a weight of 7.78 g. The yield of the refined BNA product was 95.5% relative to the crude BNA product.
With N-ethyl-N,N-diisopropylamine; N-[(dimethylamino)-3-oxo-1H-1,2,3-triazolo[4,5-b]pyridin-1-yl-methylene]-N-methylmethanaminium hexafluorophosphate; In N,N-dimethyl-formamide; at 20℃; for 4h;
At room temperature, <strong>[5773-80-8]6-bromo-2-naphthoic acid</strong> (AA_072-1, 10 g, 39.83 mmol), N,O-dimethyl hydroxylamine hydrochloride (5.05 g, 51.76 mmol) and DIPEA (15.44 g, 119.49 mmol) were dissolved in DMF (100 mL), HATU (23 g, 59.7 mmol) was added. The reaction mixture was stirred at room temperature for 4 h. After the reaction was complete as detected by TLC, methyl tert-butyl ether (300 mL) was added and the reaction mixture was washed with H2O and saturated brines. The organic phase was dried over anhydrous sodium sulfate. After filtration, the filtrate was concentrated by a rotary evaporator to remove the solvent thereby giving Weinreb amide (5.8 g, yield 90%). The product was directly used for the next step without purification. The Weinreb amide (5.8 g, 35.9 mmol) was dissolved in THF (25 mL), cooled to 0 C., a solution of methyl magnesium bromide in ethyl ether (3 mol/L, 7.9 mL, 23.7 mmol) was dripped slowly. After dripping, the reaction mixture was stirred at room temperature for 2 h. After the reaction was complete as detected by TLC, the reaction was quenched with saturated ammonium chloride solution (50 mL) and extracted with ethyl acetate (50 mL×3). The organic phases were combined and dried over anhydrous sodium sulfate. After filtration, the filtrate was concentrated by a rotary evaporator to remove the solvent thereby delivering the target compound AA_072-2 (white solid, 4 g, yield: 40.4%). The product was directly used for the next step without purification. 1H NMR (CDCl3, 400 MHz): delta 8.43 (s, 1H), 8.06 7.85-7.80 (m, 2H), 7.85-7.80 (m, 2H), 7.65 (d, J=2.0 Hz, 1H).
With palladium 10% on activated carbon; potassium acetate; bis(pinacol)diborane; In ethanol; at 60℃; for 6h;Inert atmosphere;
General procedure: To a solution of brominated aromatic compounds (1 equiv),bis(pinacolate)diboron (1.5 equiv) and anhydrous ethanol (15 mL)were added10%palladium-carbon (0.01 equiv), followed by potassium acetate (3 equiv) under argon. The mixture was heated in a 60 C with stirring for the indicated time. Thereactor was cooled to room temperature, and the reaction mixture was filtered, and thefiltrate was concentrated under reduced pressure. The residue was extracted with dichloromethane (20 mL×3), and organic layer was washed with water (20 mL×2)and once with brine (25 mL), dried over magnesium sulfate and concentrated in vacuo.The product was purified by silica gel flash chromatography on silica gel usingpetroleum as eluent.
With (4,4'-di-tert-butyl-2,2'-dipyridyl)-bis-(2-phenylpyridine(-1H))-iridium(III) hexafluorophosphate; palladium diacetate; caesium carbonate; N-ethyl-N,N-diisopropylamine; DavePhos In dimethyl sulfoxide at 20℃; for 36h; Irradiation; Green chemistry; chemoselective reaction;
N-(2-phenylquinazolin-4-yl)-benzene-1,4-diamine[ No CAS ]
[ 5773-80-8 ]
6-bromo-N-(3-((2-phenylquinazolin-4-yl)amino)phenyl)-2-naphthamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
81%
General procedure: To the mixture of substituted benzoic acids (8a-i and 12a-f,1 mmol) and HATU (1 mmol), DMF (3 mL) was added slowly undernitrogen atmosphere. The reaction mixture was then stirred for 20 min at 0 C, followed by the addition of substituted 2-aryl quinazoline (7a-c, 11 and 15, 1 mmol). The reaction mixturewas stirred for 20 min at room temperature, followed by the addition of DIPEA. Upon completion of the reaction as monitored byTLC, crushed ice was added to the reaction mixture. The resulting solid was then subjected to vacuum filtration; excess of water was used to wash off the insoluble solids to obtain crude powder which was purified using column chromatography (elution with hexane/EtOAc = 7:3). The pure products were collected as white colours olids in good yields.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






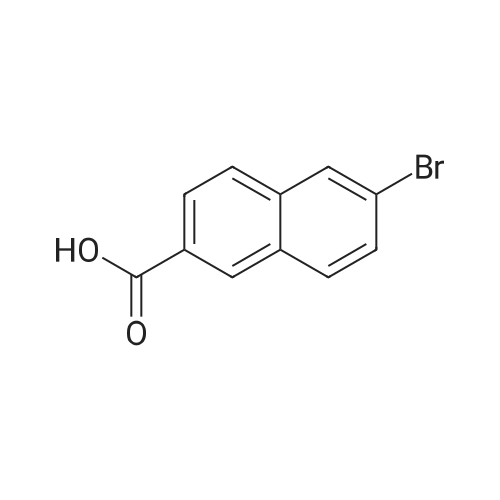

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping