* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Synlett, 2005, # 13, p. 2043 - 2046
[2] Australian Journal of Chemistry, 1998, vol. 51, # 5, p. 389 - 396
[3] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2891 - 2898
[4] Journal of the Chemical Society, 1949, p. 1855,1862[5] Journal of the Chemical Society, 1946, p. 676,679
[6] Journal of Medicinal Chemistry, 2012, vol. 55, # 12, p. 5720 - 5733
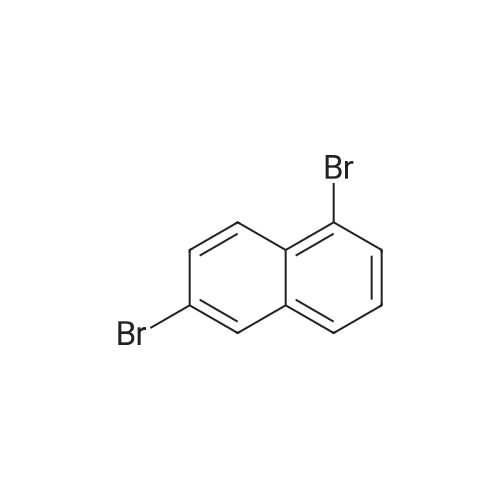
With phosphorus tribromide In acetonitrile for 48 h;
Example 20 : Preparation of compound 50; [235] <n="79"/>[236] 20-A. Preparation of compound 20a; [237] To 1,6-dihydroxynaphthalene (1.2g, 7.68 mmol), acetonitrile (50 mL), PBr (2.9 Ig,10.8 mmol) was added, and heated under stirring for 48 hours. The mixture was cooled to normal temperature and then was added methanol (100 mL) to precipitate a solid. After the solid was filtered, washed with methanol sufficiently and dried to prepare a compound 20a 1,6-dibromonaphthalene (1.6 g, 74percent). [M] = 286
Reference:
[1] Patent: WO2007/86695, 2007, A1, . Location in patent: Page/Page column 76-77
[2] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 1, p. 267 - 273
14
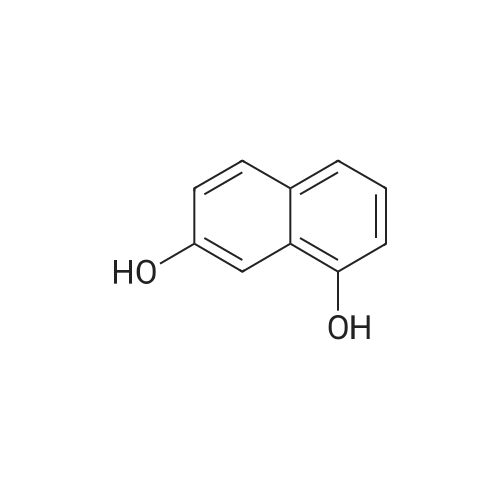
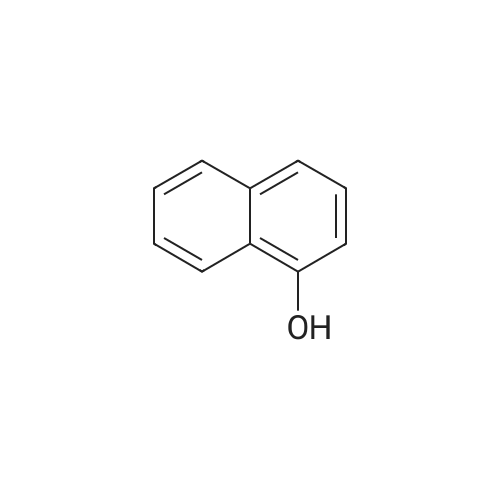
[ 575-44-0 ]
[ 74-88-4 ]
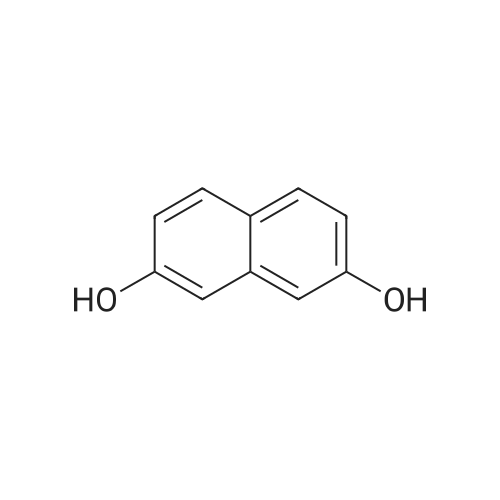
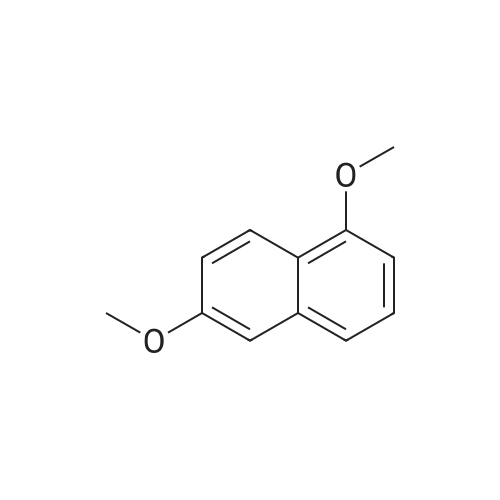
[ 3900-49-0 ]
Yield
Reaction Conditions
Operation in experiment
90%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 2 h;
Into a round-bottom flask equipped with a stirring apparatus, 1.6 g (10.0 mmol) of 1,6-dihydroxynaphthalene (Compound 39: produced by Tokyo Chemical Industry Co., Ltd.), and 15 mL of N,N-dimethylformamide (DMF) were added for dissolution, and thrther 14.2 g (100.0 mmol) of methyl iodide (produced by Wako Pure Chemical Industries, Ltd.), and 13.8 g (100.0 mmol) of potassium carbonate (produced by Wako Pure Chemical Industries, Ltd.) were added, and the reaction was carried out at room temperature for 2 hours. Afier completion of the reaction, dichloromethane and water were added, and then an organic layer, obtained by solution separation, was washed with water, and the solvent was removed from the reaction solution by concentration under reduced pressure to obtain 1 .7 g (yield:90percent) of a colorless liquid methyl derivative (Compound40).
Reference:
[1] Organic Process Research and Development, 2009, vol. 13, # 3, p. 647 - 651
[2] Synlett, 2005, # 13, p. 2043 - 2046
[3] Journal of Medicinal Chemistry, 2009, vol. 52, # 18, p. 5590 - 5602
[4] Journal of the American Chemical Society, 2017, vol. 139, # 51, p. 18522 - 18535
[5] Journal of the Chemical Society, 1949, p. 1855,1862[6] Journal of the Chemical Society, 1946, p. 676,679
[7] Journal fuer Praktische Chemie (Leipzig), 1916, vol. <2> 94, p. 3
[8] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2891 - 2898
[9] Journal of Organic Chemistry, 1986, vol. 51, # 26, p. 5252 - 5258
[10] Australian Journal of Chemistry, 1998, vol. 51, # 5, p. 389 - 396
[11] Journal of Medicinal Chemistry, 2007, vol. 50, # 22, p. 5293 - 5300
[12] Journal of Medicinal Chemistry, 2012, vol. 55, # 12, p. 5720 - 5733
16
[ 67-56-1 ]
[ 575-44-0 ]
[ 3900-49-0 ]
[ 22604-07-5 ]
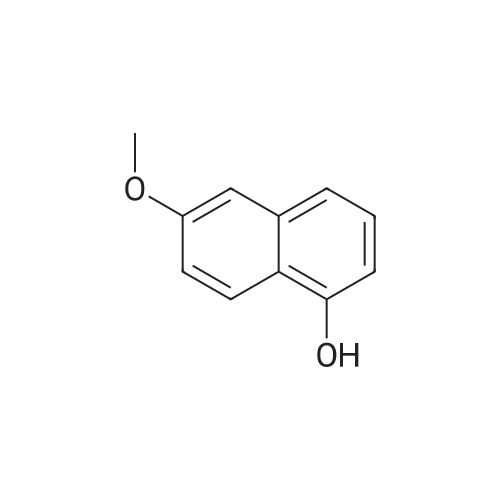
[ 150712-57-5 ]
Reference:
[1] Australian Journal of Chemistry, 1993, vol. 46, # 5, p. 731 - 737
17
[ 575-44-0 ]
[ 4018-91-1 ]
Reference:
[1] Journal of Medicinal Chemistry, 1993, vol. 36, # 20, p. 2891 - 2898
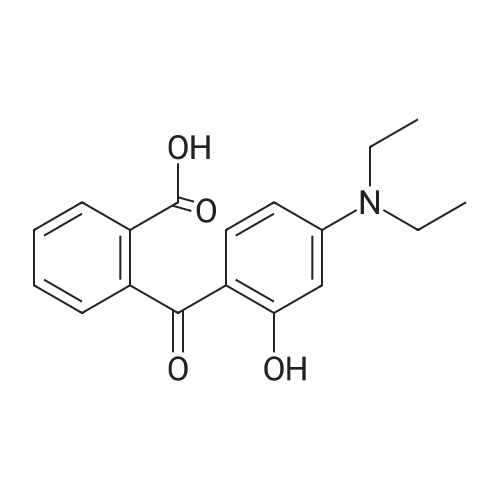
According to the literature,46 phthalic anhydride (0.890 g, 6.0mmol) and <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (2.40g,15mmol) were added in a round-bottom flask. After adding methanesulfonic acid (15mL), the mixture was refluxed for 12h at 135C. Next, the mixture was poured into ice-water and filtered. The crude product was purified by silica gel column chromatography (CH2Cl2/CH3OH=125:1) to give compound M as a aubergine solid. Yield: 0.983g (37.8%). 1H NMR (500MHz, DMSO-d6) delta 10.17 (s, 2H), 8.70 (d, J=9.1Hz, 2H), 8.09 (m, 1H), 7.76 (m, 2H), 7.46 (d, J=8.8Hz, 2H), 7.37 (dd, J=9.1, 2.4Hz, 2H), 7.30 (d, J=6.7Hz, 1H), 7.21 (d, J=2.4Hz, 2H), 6.71 (d, J=8.8Hz, 2H).
In N,N-dimethyl-formamide; at 100℃; for 4h;Inert atmosphere;
129.6 mg (0.81 mmol) of 1,6 dihydroxynapthalene and 142.9 mg (0.62 mmol) of 2 were dissolved in 3 mL of dimethylformamide (DMF) and heated to 1009 C under a blanket of nitrogen. The reaction was allowed to proceed for 4 hours monitored via thin layer chromatography (TLC) (dichloromethane (DCM):methanol (MeOH): 95:5) (Scheme 1). The solvent was removed under reduced pressure and the product was purified by column chromatography, eluting with chloroform (CHCI3):Methanol (MeOH):Triethylamine (TEA), 100:3:2). ESI+ calculated for C20H19N2O3: 335.14 m/z; measured: 335.14 m/z.
In N,N-dimethyl-formamide; at 140℃; for 5h;Inert atmosphere;
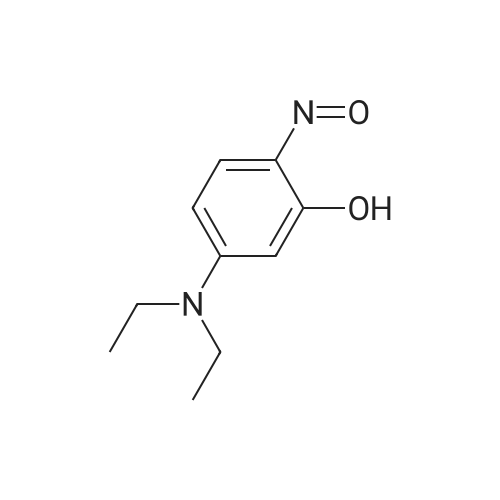
Under the ice bath, Take 5-diethylamino-2-nitrosophenol hydrochloride (0.5g, 2.6mmol) and<strong>[575-44-0]1,6-Dihydroxynaphthalene</strong> (0.41g, 2.6mmol) was dissolved in 8mL of N, N-dimethylformamide, and the reaction was refluxed under argon at 140 C for 5 hours, and the reaction was completed.After the reaction solution was cooled to room temperature, 100 mL of H2O was added and extracted with dichloromethane.Dry over anhydrous sodium sulfate, spin dry,Separation and purification by chromatography column,The Nile Red derivative with hydroxyl group is finally obtained.
With potassium carbonate; In methanol; N,N-dimethyl-formamide;
Step 1 A mixture of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (5.02 g, 31.3 mmole), anhydrous potassium carbonate (52.0 g, 376 mmole), N,N-dimethylformamide (50 ml) and iodoethane (15 ml, 188 mmole) was stirred in a 35 C. oil bath for 24 hours. The reaction mixture was filtered and the solid residue was rinsed thoroughly with ethyl ether. The filtrate and the washings were combined and concentrated in vacuo to remove most of the solvents. The brown residue was partitioned between water and ether and the layers were separated. The ether layer was washed with water. The combined aqueous layers were back extracted with ether. The ether extracts were combined, washed with brine and dried over magnesium sulfate. Filtration and concentration gave a crude brown solid (6.74 g, 99% yield). Recrystallization of the crude product from hot methanol gave 1,6-diethoxynaphthalene (4.36 g, 64% yield, first crop) as a light brown solid. 1H NMR (CDCl3) delta 8.20 (1H, d, phenyl), 7.06-7.36 (4H, m, phenyl), 6.66 (1H, dd, phenyl), 4.10-4.23 (4H, 2 sets of q, 2 CH2), 1.45-1.56 (6H, 2 sets of t, 2 CH3).
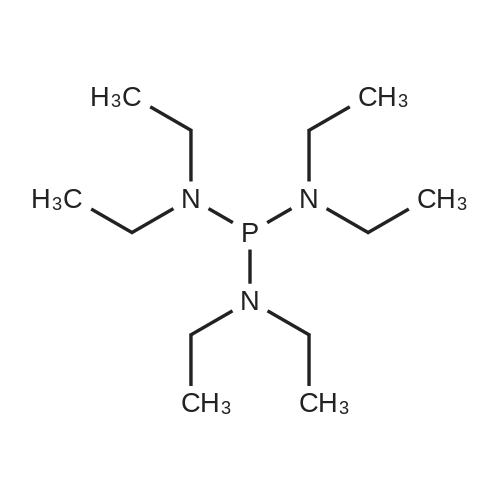
Stage #1: 1,7-Dihydroxynaphthalene; hexaethylphosphoric triamide In acetonitrile at 20℃; for 0.333333h;
Stage #2: 1,6-dihydroxynaphthalene In acetonitrile at 20℃; for 48h;
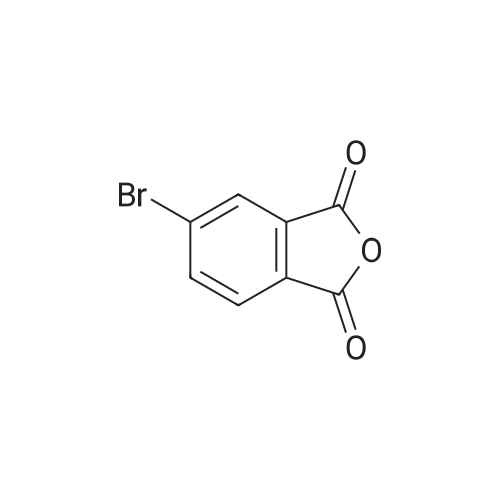
1,6-Dihydroxy naphthalene (1.3g, 8.0mmol) and 1,2,4-benzoic anhydride (0.78g, 4.0mmol) were dissolved in methanesulfonic acid and the mixture was stirred at 100C for 20h. After that, the mixture was cooled to room temperature and poured into ice water slowly. Then, the obtained mixture was filtrated under reduced pressure and the residue was dissolved in 20% sodium hydroxide solution after washing with water for three times. The atrovirens solution was stirred for 30min under room temperature and the pH value was further adjusted to faintly acid with hydrochloric acid. The crude product was obtained by filtration which was purified by column chromatography with methanol/dichloromethane (1/9, v/v, 0.4% glacial acetic acid) as eluent to compound 1 as dark red solid (0.83g, 1.7mmol 40% yield). 1H NMR (600MHz, DMSO-d6): delta (ppm): 10.18 (s, 2H), 8.70 (d, J=9.0Hz, 2H), 8.50 (s, 0.5H), 8.29-8.25 (m, 1H), 8.21 (d, J=7.9Hz, 0.5H), 7.71 (s, 0.5H), 7.45 (t, J=9.3Hz, 2.5H), 7.37 (d, J=9.0Hz, 2H), 7.21 (s, 2H), 6.76 (t, J=9.2Hz, 2H), 1.91 (s, 3H) (Fig. S1). 13C NMR (151MHz, DMSO-d6): delta (ppm): 168.4, 168.3, 166.5, 160.2, 152.4, 152.3, 137.8, 136.6, 133.3, 131.4, 130.0, 129.7, 129.6, 127.3, 126.0, 125.8, 125.1, 124.9, 113.2, 109.4, 109.3, 102.8 (Fig. S2).
With hydrogenchloride; In methanol; for 7h;Reflux;
General procedure: Compound 2 (2.12 g, 6.8 mmol), 1-naphthylamine (0.98 g,6.8 mmol), and HCl (2.0 mL, 12.0 M) in 50 mL methanol were refluxed for 7 h. The solvent was evaporated and the residue was isolated by column chromatography on silica eluting with the gradient elution of dichloromethane:methanol =60:1~40:1~30:1 to give the title compound 3 (2.29 g, 80.8%)as metallic green powder.
The synthesis and characterization of Dye A wasperformed based on our previous reports [29]. <strong>[575-44-0]1,6-Dihydroxynaphthalene</strong>(0.176 g, 1.1 mmol) and 3-hydroxy-4-nitroso-N,N-diethylaniline(0.194 g, 1.0 mmol) were added to a 100 mL round bottom flask with magnetic beads and DMF (10 mL). The mixture was degassed and heatedto 140 C in a magnetic stirrer to form a deep red solution. The reactionwas monitored once per hour with a silica gel TLC plate (eluent:dichloromethane/methanol 50:1, v/v) until the starting material wasconsumed completely. The solution was cooled to room temperature.The organic layer was washed with deionized water, extracted withmethylene chloride, dried with anhydrous magnesium sulfate and theDMF removed. The crude material was further purified with silica gelcolumn and eluted with methylene chloride: methanol 50:1 (v/v).This led to Dye A, which was a dark red solid (0.22 g, 0.66 mmol). Theyield was 60%. m.p.: 270-272 C. IR (KBr) : 2976, 2932, 1567, 1409,1117 cm 1. 1H NMR (400 MHz, DMSO-d6) 10.44 (s, 1H), 7.98 (d, J 8.5 Hz, 1H), 7.89 (d, J 2.2 Hz, 1H), 7.59 (d, J 9.1 Hz, 1H), 7.10 (dd,J 8.5, 2.3 Hz, 1H), 6.81 (d, J 6.6 Hz, 1H), 6.66 (s, 1H), 6.16 (s, 1H),3.51 (q, J 7.0 Hz,4H),1.17 (t, J 6.9 Hz, 6H). 13C NMR (100 MHz,DMSO-d6) 181.99, 161.04, 155.95, 153.83, 151.96, 151.09, 146.77,139.23, 134.17, 131.17, 127.85, 127.19, 124.29, 124.13, 118.76,117.37, 117.25, 110.28, 108.95, 108.62, 105.72, 104.57, 96.48, 44.84,12.88. HRMS(ESI) calcd. for C20H18N2O3 [M]: 335.1396, found:335.1385.
54%
In N,N-dimethyl-formamide; at 140℃; for 10h;
(1) 0.194 g of compound IV and 0.176 g of 1,6 dihydroxynaphthalene are added to 10 ml of DMF,After reacting at 140 C for 10 h, the resulting solution was washed with deionized water to remove DMF.After that, the organic layer was rotary distilled to remove the solvent, and the solid was dissolved in dichloromethane.Separation by column chromatography with 50:1 by volume of dichloromethane and methanol.Compound III was obtained in a yield of 54%.
4-methyl-3,10-dihydroxy-spiro[7H-benzo[c]xanthen-7,1'(3'H)-isobenzofuran]-3'-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
79%
In methanesulfonic acid; water;
EXAMPLE 2 4-Methyl-3,10-dihydroxy-spiro[7H-benzo[c]xanthen-7,1'(3'H)-isobenzofuran]-3'-one (2) 2'-Carboxy-3-methyl-2,4-dihydroxybenzophenone (1, 8.16 g, 30.0 mmol) and <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (4.81 g, 30.0 mmol) were combined in methanesulfonic acid (120 mL) and sealed in a thick-walled glass tube. The resulting viscous mixture was stirred at 90 C. for 24 h. The reaction was poured into ice-cold water (1 L) and the precipitate was filtered and washed with water (3*200 mL). Purification by flash column chromatography (silica gel, 9:1 dichloromethane/methanol) furnished semi-naphthofluorescein 2 as a brick red powder (9.40 g, 79% yield). 1H NMR (CD3OD, 500 MHz): delta 8.41 (1 H, d, J=9.0 Hz), 8.00 (1 H, d, J=7.0 Hz), 7.69 (2 H, m), 7.27 (1 H, dd, J1=8.5 Hz, J2=3.5 Hz), 7.23 (1 H, d, J=10.0 Hz), 7.16 (1 H, d, J=7.5 Hz), 7.09 (1 H, br s), 6.59 (2 H, m), 6.46 (1 H, d, J=8.5 Hz), 2.48 (3 H, s). HRMS (ESI) calcd for [M+H]+ 397.1071, found 397.1057.
Fmoc-(D,L)-5-ethoxy-2-aminotetraline-2-carboxylic acid (Fmoc-(D,L) 5-EtOAtc-OH)[ No CAS ]
[ 3955-85-9 ]
Yield
Reaction Conditions
Operation in experiment
99%; 64%
With potassium carbonate; In N,N-dimethyl-formamide;
EXAMPLE 12 Preparation of Fmoc-(D,L)-5-ethoxy-2-aminotetraline-2-carboxylic acid (Fmoc-(D,L) 5-EtOAtc-OH) A mixture of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (5.02 g, 31.3 mmole), anhydrous potassium carbonate (52.0 g, 376 mmole), N,N-dimethylformamide (50 ml) and iodoethane (15 ml, 188 mmole) was stirred in a 35 C. oil bath for 24 hours. The reaction mixture was filtered and the solid residue was rinsed thoroughly with ethyl ether. The filtrate and the washings were combined and concentrated in vacuo to remove most of the solvents. The brown residue was partitioned between water and ether and the layers were separated. The ether layer was washed with water. The combined aqueous layers were back extracted with ether. The ether extracts were combined, washed with brine and dried over magnesium sulfate. Filtration and concentration gave a crude brown solid (6.74 g, 99% yield). Recrystallization of the crude product from hot methanol gave 1,6-diethoxynaphthalene (4.36 g, 64% yield, first crop) as a light brown solid. 1H NMR (CDCl3) delta8.20 (1H, d, phenyl), 7.06-7.36 (4H, m, phenyl), 6.66 (1H, dd, phenyl), 4.10-4.23 (4H, 2 sets of q, 2 CH2), 1.45-1.56 (6H, 2 sets if t, 2 CH3).
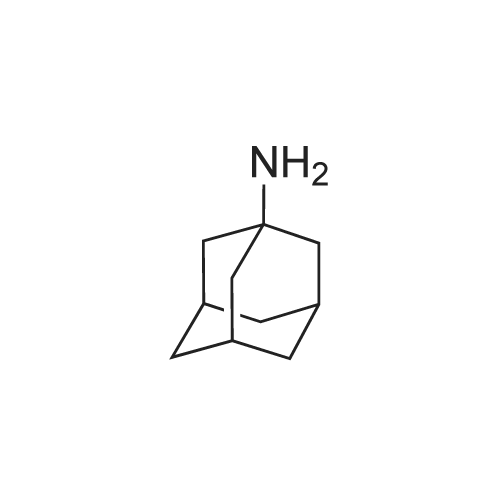
1,6-Dihydroxy-2,5-bis-(1-adamantanaminomethyl)-naphthalene dihydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; formaldehyd; water;
Step A: 2:9-di-(1-Adamantyl)-1:2:3:4:7:8:9:10-octahydro-2:9-diaza-4:7-dioxachrysen To a mixture of 1-adamantanamine (7.6 g, 0.050 mol) and methanol (40 ml) cooled in an ice-bath were added with stirring 37% formaldehyde (wt. % solution in water, 8.1 ml, 0.10 mol) followed by a solution of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (4.0 g, 0.025 mol) in methanol (25 ml). The reaction mixture was allowed to attain room temperature and was stirred overnight. The resulting solid was filtered and washed with methanol. Purification was achieved by vacuum filtration through a pad of silica gel (Merck beta 7734) and elution initially with dichloromethane and subsequently 100:1 and 25:1 dichloromethane-diethyl ether; yield 6.8 g (53%); m/z (e.i.) 347 (M-163).
In methanol; formaldehyd; water;
Step A: 2:9-di-(1-Adamantyl)-1:2:3:4:7:8:9:10-octahydro-2:9-diaza-4:7-dioxachrysen To a mixture of 1-adamantanamine (7.6 g, 0.050 mol) and methanol (40 ml) cooled in an ice-bath were added with stirring 37% formaldehyde (wt. % solution in water, 8.1 ml, 0.10 mol) followed by a solution of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (4.0 g, 0.025 mol) in methanol (25 ml). The reaction mixture was allowed to attain room temperature and was stirred overnight. The resulting solid was filtered and washed with methanol. Purification was achieved by vacuum filtration through a pad of silica gel (Merck #7734) and elution initially with dichloromethane and subsequently 100:1 and 25:1 dichloromethanediethyl ether; yield 6.8 g (53%); m/z (e.i.) 347 (M-163).
In methanol; formaldehyd; water;
Step A: 2:9-di(1-Adamantyl)-1:2:3:4:7:8:9:10-octahydro-2:9-diaza-4:7-dioxachrysene To a mixture of 1-adamantanamine (7.6 g, 0.050 mol) and methanol (40 ml) cooled in an ice-bath were added with stirring 37% formaldehyde (wt. % solution in water, 8.1 ml, 0.10 mol) followed by a solution of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (4.0 g, 0.025 mol) in methanol (25 ml). The reaction mixture was allowed to attain room temperature and was stirred overnight. The resulting solid was filtered and washed with methanol. Purification was achieved by vacuum filtration through a pad of silica gel (Merck #7734) and elution initially with dichloromethane and subsequently 100:1 and 25:1 dichloromethanediethyl ether; yield 6.8 g (53%); m/z (e.i.) 347 (M-163).
In methanol; formaldehyd; water;
Step A: 2:9-di-(1-Adamantyl)-1:2:3:4:7:8:9:10-octahydro-2:9-diaza-4:7-dioxachrysen To a mixture of 1-adamantanamine (7.6 g, 0.050 mol) and methanol (40 ml) cooled in an ice-bath were added with stirring 37% formaldehyde (wt. % solution in water, 8.1 ml, 0.10 mol) followed by a solution of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (4.0 g, 0.025 mol) in methanol (25 ml). The reaction mixture was allowed to attain room temperature and was stirred overnight. The resulting solid was filtered and washed with methanol. Purification was achieved by vacuum filtration through a pad of silica gel (Merck #7734) and elution initially with dichloromethane and subsequently 100:1 and 25:1 dichloromethanediethyl ether; yield 6.8 g (53%); m/z (e.i.) 347 (M-163).
In methanol; formaldehyd; water;
Step A: 2:9-di-(1-Adamantyl)-1:2:3:4:7:8:9:10-octahydro-2:9-diaza-4:7-dioxachrysene To a mixture of 1-adamantanamine (7.6 g, 0.050 mol) and methanol (40 ml) cooled in an ice-bath were added with stirring 37% formaldehyde (wt. % solution in water, 8.1 ml, 0.10 mol) followed by a solution of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (4.0 g, 0.025 mol) in methanol (25 ml). The reaction mixture was allowed to attain room temperature and was stirred overnight. The resulting solid was filtered and washed with methanol. Purification was achieved by vacuum filtration through a pad of silica gel (Merck # 7734) and elution initially with dichloromethane and subsequently 100:1 and 25:1 dichloromethanediethyl ether; yield 6.8 g (53%); m/z (e.i.) 347 (M-163).
3,10-dihydroxy-spiro[7H-benzo[c]xanthen-7,1'(3'H)-isobenzofuran]-3'-one[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
With methanesulfonic acid; trifluoroacetic acid; at 80℃; for 24h;
General procedure: Hydroxybenzophenone (918 mumol) and 1,8-naphthalene derivative (1380 mumol) are dissolved in 1.5 mL of methanesulfonic acid, then 1.5 mL of trifluoroacetic acid (TFA) are added. The mixture is heated and stirred at 80 C. for 16-24 hours. The reaction mixture is allowed to warm to room temperature, and then poured into 50 mL of deionized (DI) water. The mixture is neutralized to pH 6-7 by portion-wise addition of solid NaHCO3. The resulting precipitate is filtered, washed with DI water and air dried. The target compound is isolated by flash column chromatography on silica gel.
Zinc chloride; In methanol; potassium hydroxide; chloroform; water; acetic anhydride;
EXAMPLE 1 Preparation of 3,10-dihydroxy-spiro[7H-benzo[c]xanthen-7,1'(3'H)-isobenzofuran]-3'-one, 1 and its diacetate 16. A mixture of 6.0 g 2-(2',4'-dihydroxybenzoyl)benzoic acid, 4.2 g <strong>[575-44-0]1,6-dihydroxynaphthalene</strong>, and 6.1 g anhydrous zinc chloride was heated at 160 to 165 C. for 1 hour. The cooled melt was pulverized and washed several times with boiling water and air dried. The crude dye was refluxed in 50 ml acetic anhydride for 11/2 hours and concentrated to about 25 ml by boiling. The residue was poured into water and stirred at about 50 for 30 minutes. The warm water was decanted and the residue was stirred in water at room temperature for 1 hour. The solid was collected by filtration and air dried. The crude diacetate was recrystallized once from toluene/isopropyl alcohol then once from chloroform/isopropyl alcohol to give 6.1 g slightly orange tinged crystals of 16 (MP=124-126 C.). A portion of the diacetate, 16, (2.5 g) was dissolved in 20 ml of 2M potassium hydroxide in methanol and 10 ml water was added. The mixture was stirred at about 50 C. for 2 hours. After addition of 40 ml water the methanol was removed by distillation. Acidification with glacial acetic acid, filtration and drying gave 1.86 g of compound 1 as an orange powder. Rf =0.36 with methanol:chloroform (1:9).
With hydrogenchloride;aluminium trichloride; In nitromethane;
EXAMPLE 22 Preparation of 1-(1,6-dihydroxy-2-naphthalenyl)ethanone To a solution of 8.3 g of aluminum chloride and 3.6 ml of acetyl chloride in 60 ml of anhydrous nitromethane at room temperature was added 8.0 g of 1,6dihydroxynaphthalene. The reaction mixture was stirred under argon at 23 for 20 hours and then concentrated in vacuo. The residue was treated with 6N hydrochloric acid and extracted with ethyl acetate. The extract was washed with sodium bicarbonate solution, dried (magnesium sulfate) and concentrated in vacuo. The crude product was chromatographed on 250 g of silica gel. Elution with 2% ethyl acetatetoluene gave 3.6 g (36% yield), mp 195-200, of 1-(1,6-dihydroxy-2-naphthalenyl)ethanone.
With phosphorus tribromide; In acetonitrile; for 48h;
Example 20 : Preparation of compound 50; [235] <n="79"/>[236] 20-A. Preparation of compound 20a; [237] To <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (1.2g, 7.68 mmol), acetonitrile (50 mL), PBr (2.9 Ig,10.8 mmol) was added, and heated under stirring for 48 hours. The mixture was cooled to normal temperature and then was added methanol (100 mL) to precipitate a solid. After the solid was filtered, washed with methanol sufficiently and dried to prepare a compound 20a 1,6-dibromonaphthalene (1.6 g, 74%). [M] = 286
A solution of 2-carboxy-3,4,5,6-chloro-3'-dimethylamino-2'-hydroxybenzophenone (178 mg, 0.407 mmol) and <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (95.0 mg, 0.593 mmol) in 0.5 mL of methanesulfonic acid was refluxed for 10 min. The reaction mixture was neutralized with sat. NaHCO3 aq., diluted with AcOEt and washed with sat. NaHCO3 aq. The solvent was removed in Vacuo, and the crude residue was purified by column chromatography on silica gel (CH2Cl2/AcOEt: gradient from1:0 to 0:1(v/v)) to give SNARF-Cl in 48 % yield (107 mg, 0.196 mmol). 1H NMR (DMSO-d6, 400MHz) : delta 8.35 (d, 1H, J = 9.28 Hz), 7.32 (d, 1H, J = 9.04 Hz), 7.19 (dd, 1H, J = 9.28 Hz, 2.20 Hz), 7.11 (d, 1H, J =2.20 Hz), 6.87-6.91 (m, 2H ), 6.71 (d, 1H, J = 2.44 Hz), 6.49 (dd, 1H, J = 8.88 Hz, 2.44 Hz), 2.94 (s, 6H). Anal. Calcd for C26H15Cl4NO4 : C, 57.07; H, 2.76; N, 2.56. Found: C, 56.91; H, 3.01; N, 2.47. FAB-HRMS (m-NBA): calcd for C26H16Cl4NO4+ ([M+H]+) 545.9827, found 545.9842.
With dmap; triethylamine; In dichloromethane; at 0 - 20℃; for 13h;Inert atmosphere;
To a solution of diol A61 (16.1 mmol), triethylamine (24.1 mmol), and DMAP (0.16 mmol) in CH2CI2 (120 ml) was added TBDMSCI15 (19.3 mmol) in 5 portions over 1 h at 0 C. The resulting heterogeneous reaction mixture was gradually warmed tort. The mixture wasstirred for 12 h before dilution with water and CH2CI2. The organic layer was washed successively with solutions of saturated aqNaHC03, saturated aq NH4CI, water and brine, dried over MgS04, filtered, and concentrated in vacuo. The pale yellow oil was purifiedby vacuum chromatography to give the intermediate silyl ether.A mixture of the intermediate silyl ether (1.0 mmol) and catalyst (Rh or Ru on activated carbon, N.E. Chemcat; 10 wt% of substrate) in20 iPrOH (1 ml) in a sealed tube was stirred at 60 oc at 5 atm H2. After cooling to room temperature, the reaction mixture was diluted with MeOH (20 ml) and the catalyst was removed by filtration through a membrane filter (Millipore, Millex-LH, 0.45 mm). The filtratewas concentrated in vacuo to give the corresponding decalin.To a solution of the intermediate decal in alcohol (8 mmol) in pyridine (6 ml) was added portionwise TsCI (8 mmol) with efficient stirringduring 30 min at 20 C. After being stirred for 30 min, the reaction mixture was maintained for 20 h at room temperature, diluted withCH2CI2 (1 00 ml), and washed with 2 N aq. HCI until the aqueous washings became acidic. The H20 layer was extracted with CH2CI2(3 X 50 ml). The combined organic extract was dried (Na2S04) and concentrated to give tosylate AGO.A solution of tosylate AGO (0.67 mmol) in 5 ml of dry HMPA was cooled in an ice bath under N2. This mixture was added to a coldsolution of NaSBn (1 0 mmol) in 20 ml of dry HMPA (prepared from 400 mg of sodium and excess BnSH in dry ether, which wassubsequently removed and replaced with HMPA). After the addition, the solution was stored in the freezer (-15 "C) for 14 h. It was then1 0 treated with 1 00 ml of water and extracted three times with ether. The ether extracts were washed four times with water and driedover MgS04. The solvent was then removed under vacuum and the residue (140 mg) chromatographed over silica gel to give theintermediate silyl ether.The intermediate silyl ether (0.235 mmol) was dissolved in THF (1 ml) at room temperature, followed by addition of 70% HF·pyridine(0.2 ml). After stirring for two days, the reaction mixture was carefully quenched with sat. aq. NaHC03 and the resulting solution was15 diluted with EtOAc. The organic layer was washed with brine, dried over Na2S04, filtered and concentrated in vacuo. The residue waspurified by silica gel column chromatography to give alcohol A59.
In 1,2-dichloro-benzene; at 200℃; for 3h;Sealed tube; Irradiation; Inert atmosphere;
General procedure: A vessel charged with 6a (72 mg, 0.50 mmol), 10e (400 mg, 1.52 mmol), and 1,2-dichlorobenzene (390 L) was sealed and irradiated with microwaves at normal absorption level at 200 C for 3 h. The reaction mixture was cooled to room temperature and was diluted with acetone. After the mixture was concentrated, remaining 1,2-dichlorobenzene was azeotropically removed with toluene (x3). The residue was purified by flash column chromatography on silica gel (5 g, CH2Cl2 only to CH2Cl2/EtOAc 30:1) to afford 12e (130 mg,0.319 mmol) and 13e (13 mg, 0.032 mmol) in 64% and 6% yields, respectively
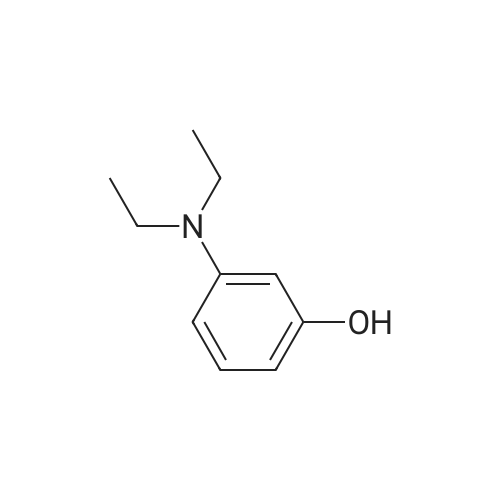
EXAMPLE 13A A solution containing <strong>[91-68-9]3-diethylaminophenol</strong> (15.0 g, 90.7 mmol) in concentrated HCl (100 mL) was prepared and cooled to 0 C. in an ice-bath. NaNO2 (6.9 g, 100.0 mmol) in water (50 mL) was added slowly over 40 minutes such that no brown NOx vapours were observed. The reaction was left to stir for 2 hours, after which the thick precipitate was filtered using a Buechner funnel and washed with small portions of water (3×50 mL). After drying the solid for 1 hour on the Buechner funnel, the solid material was dissolved into EtOH (70 mL), Et2O (35 mL) was added and the solution stored at -20 C. overnight to allow for crystallization. The next day, the solid material was collected by vacuum filtration, using a Buechner funnel and air dried. The product was an orange/red solid (8.8 g, 50%). EXAMPLE 14A The compound of Example 13A (7.0 g, 36.0 mmol) and 1,6-dihydroxynaphthelene (5.9 g, 36.1 mmol) were refluxed in DMF (100 mL) for 3 hours. After the solvents were removed under vacuum, the residue was redissolved in methanol and adsorbed onto silica and purified using silica gel column chromatography (MeOH:NEt3=50:1) to afford the title compound as a dark purple solid (11.1 g, 92%).
General procedure: To a solution of 2-chloro-1,3-dimethylimidazolinium chloride 7(228 mg, 1.35mmol) in acetonitrile (2 mL), sodium azide (99.4mg, 1.5mmol) and 15-crown-5 ether (0.06ml, 0.3mmol) was added at 20C and the mixture was stirred for 30 min. 1,5-Naphthalene diol 15b (144 mg, 0.9mmol) and triethylamine(0.25 mL, 1.8mmol) in THF (4 mL) was added to the mixture,which was stirred for 1 h. To the mixture, acetic anhydride (0.17 mL, 1.8mmol) and triethylamine (4.0 mL, 29mmol) was added, and the mixture was stirred until diazonaphthoquinone was consumed by monitoring with TLC. The reaction was quenched with water, and organic materials were extracted three times with CH2Cl2. The combined extracts were washed with water and brine, and then, dried over anhydrous sodium sulfate. The solvent was removed in vacuo to afford crude compounds. The crude materials were purified by flash column chromatography (silica gel: hexane/ethyl acetate = 4/1) to give 17 in 56% yield.
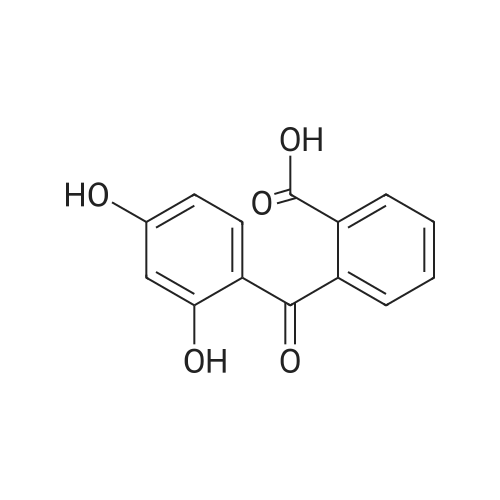
4-(2,4-dihydroxybenzoyl)isophthalic acid[ No CAS ]
C25H14O7[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
79%
With methanesulfonic acid; trifluoroacetic acid; at 20℃; for 24h;
General procedure: 4-(2,4-Dihydroxybenzoyl)isophthalic acid (1) (500 mg, 1.65 mmol) and the appropriate dihydroxynaphthalene (275 mg, 1.75 mmol) were dissolved in methanesulfonic acid (10 mL) or a mixture of TFA (5 mL) and methanesulfonic acid (5 mL), and stirred at room temperature overnight. The reaction was quenched with H2O (25 mL) and the resulting dark purple precipitate collected by centrifugation. After decantation, the precipitate was dissolved in NaOH(aq) (2 M, 15 mL) and precipitated with HCl (aq) (2 M, 20 mL). After decantation, the precipitate was washed with H2O (2 x 35 mL), dissolved in EtOH (10 mL) and precipitated with H2O. After decantation and washing with H2O (2 x 35 mL), the compound was dried in vacuo yielding a dark purple powder. If required, further purification by silica gel dry column vacuum chromatography (2% AcOH in CH2Cl2/MeOH with 5% increments) was performed by dissolution of the compound in MeOH and NaOH (aq) (12 M, 2 drops) and evaporation onto Celite in vacuo.
With methanesulfonic acid; trifluoroacetic acid; at 20℃;
General procedure: 4-(2,4- Dihydroxybenzoyl)isophthalic acid (500 mg, 1.65 mmol) and the appropriate dihydroxynaphthalene (275 mg, 1.75 mmol) were dissolved in TFA (5 mL) and methanesulfonic acid (5 mL). The reaction mixture was stirred at room temperature overnight. The reaction was quenched by adding H20 (25 mL) and the resulting dark purple precipitate was collected by centrifugation. After decantation the sediment was dissolved in NaOH(aq) (2 M, 15 mL) and precipitated with HCI(aq) (2 M, 20 mL). After decantation the sediment was washed with H20 (2 x 35 mL) and re-precipitated by dissolving in EtOH (10 mL) and precipitated with H20 (ad H20 until precipitation). After decantation and washing with H20 (2 x 35 mL) the crude compound was dried in vacuo yielding a dark purple powder. Further purification by silica gel dry column vacuum chromatography was performed by dissolving the crude compound in MeOH and 2 drops of 12 M NaOH(aq), evaporation on celite in vacuo, using 2 % AcOH in CH2CI2/MeOH with 5 % increments was done if required.
With toluene-4-sulfonic acid; In 1,2-dichloro-benzene; at 130℃; for 3h;
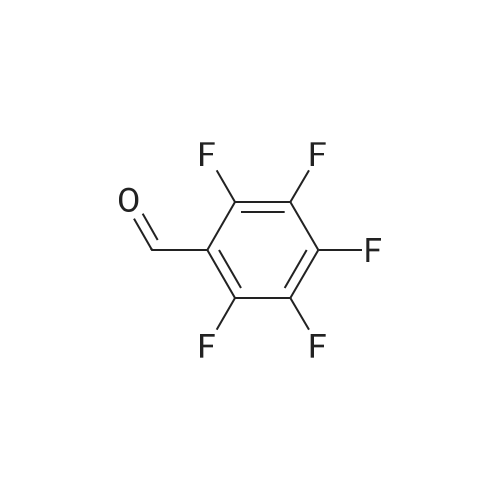
General procedure: Extended fluorescein dyes of structure 440 were synthesized in reactions of 1,6-dihydroxy napthalene derivatives 320 and benzaldehyde derivatives as outlined in FIG. 4. Following the alternative general dye synthesis conditions described for 340, 1,6-dihydroxynathalene 420 and benzaldehyde derivatives 130 were suspended in dichlorobenezene with 10 equivalents of p-toluene sulfonic acid and the reaction mixture was heated with stirring at 130 C. for 3 hours. Following this general procedure, dye 19 below was produced from reactions of 1,6-dihydroxynapthalene 420 with compounds 130 where R11-R15=fluorine.
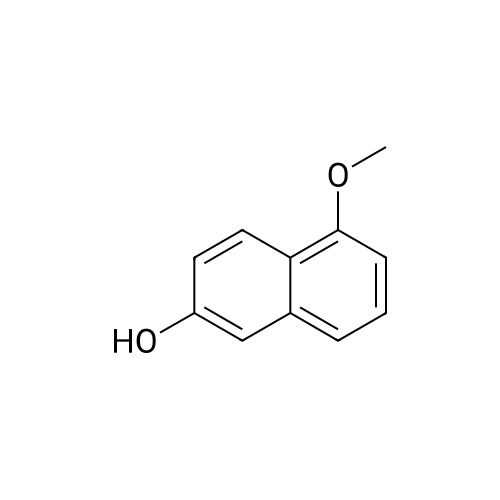
<strong>[575-44-0]1,6-dihydroxynaphthalene</strong> 5.00g (31.2mmol, 1.0eq.) Was dissolved in pyridine 80 mL, under cooling with ice trifluoromethylsulfonate anhydride 5.8 mL (34. 5mmol, 1.1eq.) was dropped slowly.After stirring for 1 hour under ice-cooling, and the mixture was stirred at room temperature for 1 hour.Water was added, then extracted 3 times with toluene, summarized toluene layer was dried over anhydrous sodium sulfate.Aftersodium sulfate, concentrated, toluene - through silica gel column chromatography with ethyl acetate as eluent.The fractions containing the desired product was collected, and concentrated to give, to obtain the desired product "1Np6OTf" and "6Np1OTf" as both a white solid.1Np6OTf Yield: 2.67g, yield: 29.3 percent, purity: 98.4Pasento (HPLC) 6Np1OTf yield: 2.58g, yield: 28.3%, purity: 99.0 Pasento (HPLC)
2-(10-(diethylamino)-3-oxo-3H-benzo[c]xanthen-7-yl)benzoic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
In methanesulfonic acid; at 90℃; for 7h;
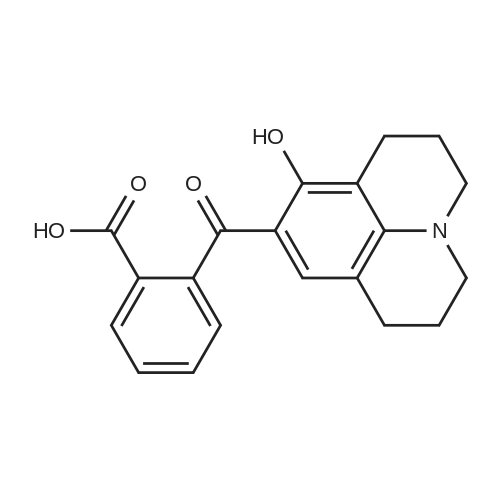
In a 100 mL round bottomed flask, Compound 1 (949 mg, 3 mmol)and <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (460 g, 3 mmol) were dissolved in40 mL methanesulfonic acid. The resulting mixture was stirred at 90 Cfor 7 h. After cooling to the room temperature, the reaction mixturewas poured into NaCl solution (100 mL, 10%). The resulting purplishred precipitate was collected by filtration and dried under vacuum. Theresidue was further purified by silica gel chromatography using petroleumether/dichloromethane/absolute alcohol (v/v/v, 3:3:1) as an eluentto give pure compound RN with yield of 60%. 1H NMR (400 MHz,DMSO d6) delta (ppm) 10.10 (s, 1H), 8.45 (d, J = 9.0 Hz, 1H), 8.04 (d, J =7.4 Hz, 1H), 7.80-7.71 (m, 2H), 7.37 (d, J = 8.8 Hz, 1H), 7.28-7.24 (m,2H), 7.14 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 1.7 Hz, 1H), 6.6 (d, J=8.8Hz,1H), 6.56-6.50 (m, 2H),3.43 (q, J = 7.0 Hz, 4H), 1.13 (t, J = 7.0 Hz, 6H).HRMS (ESI): Calc. Mass for [M + H]+ 438.1705, found 438.1702.
45.7%
With methanesulfonic acid; at 90℃; for 12h;
Firstly, 2-(4-diethylamino-2-hydroxybenzoyl) benzoic acid was prepared accordingto the previous literature [32]. Then, 2-(4-diethylamino-2-hydroxybenzoyl) benzoicacid (469 mg, 1.5 mmol) and <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (240 mg, 1.5 mmol) wereadded into methanesulfonic acid (10 mL) and stirred at 90 C for 12 h. After thereaction mixture was cooled to room temperature, it was further poured into ice-coldwater (45 mL). The obtained precipitate was filtered, washed with brine(3 9 15 mL), and dried under vacuum. Finally, the target compound RH wasisolated by SiO2 column chromatography using a mixed solution of dichloromethaneand methanol (20:3, v/v) as the eluent. Compound RH: 2-(10-(diethylamino)-3-oxo-3H-benzo[c]xanthen-7-yl) benzoic acid, dark red crystals, yield: 300 mg, 45.7%. 1HNMR (400 MHz, d6-DMSO) d 10.13 (1 H, s), 8.45 (1 H, d, J = 8 Hz), 8.03 (1 H, d,J = 8 Hz), 7.75 (2 H, m), 7.36 (1 H, d, J = 8 Hz), 7.26 (2 H, m), 7.15 (1 H, d,J = 4 Hz), 6.73 (1 H, s), 6.55 (3 H, m), 1.13 (6 H, t, J = 8 Hz) (Fig. S1). 13C NMR(100 MHz, d6-DMSO) d 168.83, 157.28, 151.97, 149.35, 146.94, 135.88, 135.47,130.02, 128.65, 126.35, 124.61, 124.19, 124.01, 123.67, 121.83, 118.90, 117.12,109.71, 109.29, 97.13, 55.98, 54.91, 43.81, 28.98, 18.51, 12.34. (Fig. S2). HRMS(ESI): calcd. For [C28H24N1O4]? 438.1705; found 438.1705 (Fig. S5).
With methanesulfonic acid; 3-mercaptopropionic acid; In 1,2-dichloro-ethane; at 50 - 60℃; for 6h;Inert atmosphere;
10.0 of indeno[1,2-b]fluorene-6,12-dione, 34.3 g of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong>, and 120 mL of 1,2-dichloroethane were mixed to form a homogeneous solution at 50 C. in a nitrogen atmosphere. Then, a mixed liquid of 17.8 g of methanesulfonic acid and 1.5 g of 3-mercaptopropionic acid was added dropwise thereto gently, and the mixture was stirred under heating at 60 C. for 6 hours. After cooling to room temperature, 300 g of methyl isobutyl ketone and 100 g of pure water were added thereto. An insoluble matter was then removed by filtration, the water layer was removed, and the organic layer was washed with 100 g of pure water four times. After washing, the organic layer was evaporated under reduced pressure to dryness and dissolved in 80 g of ethyl acetate. Then, it was added to 320 g of toluene to precipitate a crystal. The crystal was collected by filtration with Kiriyama funnel, washed with 100 g of toluene twice, and then dried at 60 C. under vacuum to obtain 15.7 g of Compound X4. The obtained compound was identified as Compound X4 shown by the above structural formula by analysis with IR, 1H-NMR, and 13C-NMR. IR (ATR method): nu=3317, 3057, 1631, 1608, 1574, 1520, 1479, 1443, 1379, 1352, 1277, 1260, 1197, 1154, 1104, 953, 856, 818 cm-1 1H-NMR (600 MHz in DMSO-d6): delta=8.67 (4H, d), 8.60 (4H, -OH), 7.71 to 7.74 (4H, m), 7.27 (4H, d-d), 7.08 (2H, m), 7.00 to 7.10 (12H, m), 6.35 (4H, d) ppm 13C-NMR (150 MHz in DMSO-d6): delta=157.43, 157.35, 157.29, 146.94, 141.52, 140.67, 136.37, 128.65, 128.22, 126.79, 126.68, 123.85, 123.34, 120.88, 119.89, 118.87, 118.57, 116.26, 109.96, 54.68 ppm
spiro[fluorene-9,9'-(2',7'-dihydroxybenzoxanthene)][ No CAS ]
Yield
Reaction Conditions
Operation in experiment
62.9%
With sulfuric acid; 3-mercaptopropionic acid; In 1,4-dioxane; at 60℃; for 3h;
9 parts by weight of 9-fluorenone (0.05 mole, manufactured by Osaka Gas Chemicals Co., Ltd.), 24 parts by weight (1,55 moles, <strong>[575-44-0]1,6-dihydroxynaphthalene</strong>, Manufactured by Kanto Chemical Co.,Ltd.),0.3 parts by weight (0.003 mol)of 3-mercaptopropionic acid and 40 parts by weight of 1,4-dioxane, 13.2 parts by weight (0.013 mol) of sulfuric acid was added dropwise, and at 60 C. 3h. And stirred for a while. The residual amount of 9-fluorenone as a raw material was measured by HPLC (Hitachi HPLC D-7000, solvent: acetonitrile: 0.1 wt% phosphoric acid aqueous solution = 55/45 to 95/5) and the conversion was 99.5 % Or more was confirmed. After confirmation, 100 parts by weight of methyl isobutyl ketone (MIBK) was added to and dissolved in the reaction solution and washed three times with water. After concentrating the organic layer, washing the obtained black solid with toluene, 14.6 parts by weight (yield 62.9%spiro [fluorene 9,9 '- (2', 9'-dihydroxydibenzoxanthene)] was obtained. ) as a gray solid.
62.9%
With sulfuric acid; 3-mercaptopropionic acid; In 1,4-dioxane; at 60℃; for 3h;
In a 300 mL separable flask,9 parts by weight of 9-fluorenone (0.05 mol, manufactured by Osaka Gas Chemicals Co., Ltd.), 24 parts by weight of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (0.15 mol, manufactured by Kanto Kagaku Co., Ltd.), 3-mercaptopropionic acid 0.3 Parts by weight and 1,4-dioxane 40 parts by weight, 13.2 parts by weight of sulfuric acid was added dropwise, and the mixture was stirred at 60 C. for 3 hours. The residual amount of 9-fluorenone as a raw material was measured by HPLC (Hitachi HPLC D-7000, solvent: acetonitrile: 0.1 wt% phosphoric acid aqueous solution = 55/45 to 95/5) and the conversion was 99.5 % Or more was confirmed. After confirmation, 100 parts by weight of methyl isobutyl ketone (MIBK) was added to and dissolved in the resultant reaction solution, and washed with water. After concentrating the organic layer, washing the obtained black solid with toluene, 14.6 parts by weight (yield 62. spiro [fluorene 9,9 '- (2', 9 '- dihydroxydibenzoxanthene)]. 9%) as a gray solid
With sodium hydroxide; In water; toluene; at 20℃; for 0.5h;
1.60 g (10 mmol) of <strong>[575-44-0]2,5-dihydroxynaphthalene</strong> was dispersed in 20 g of deaerated water in a 100 ml three-necked flask equipped with a thermometer and a stirrer, then an aqueous 3 g of water of 0.48 g (12 mmol) of sodium hydroxide In addition, a black dark solution was obtained.Next, a solution of 1.56 g (15 mmol) of methacryloyl chloride in toluene was added, and a thin purple solid precipitated.After stirring at room temperature for 30 minutes, the reaction product was suction filtered, washed with water and washed with toluene.After drying, a gray powder of 1.00 g (4.4 mmol) of 5-hydroxy-2-naphthyl methacrylate was obtained.The isolated yield based on <strong>[575-44-0]2,5-dihydroxynaphthalene</strong> as a raw material was 44 mol%.
12-(2-carboxyphenyl)-N,N-diethyl-8-hydroxy-3H-5-oxatetraphen-3-iminium[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
With trifluoroacetic acid; for 24h;Reflux;
(Scheme 3) 1.6017 g (10 mmol) of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong>,3.7602 g (12 mmol) of 4-diethylaminonic acid was dissolved(10 mL) trifluoroacetic acid (TFA) and then heated to reflux for 24 h, Followed by the addition of 15mL ethyl acetate heated reflux 15min,Cooled to room temperature,After recrystallization, the pure product was 4.0734 g,The yield was 93%.
With caesium carbonate; In N,N-dimethyl-formamide; at 100℃; for 12h;
Step 1 (0342) To a solution of 4a (28 g, 113 mmol) in DMF (500 mL) was added naphthalene- 1,6-diol (22 g, 136 mmol) and CS2CO3 (74 g, 226 mmol). The resulting solution was stirred at 100C for 12 h, before it was poured into water and extracted with EA. The combined organic phases were dried over Na2S04. After filtration and concentration, the residue was purified by Si02 chromatography, eluting with petroleum ether: ethyl acetate (100/1-10/1) to give compound 4b (25 g, 59.5%).
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 2h;
Into a round-bottom flask equipped with a stirring apparatus, 1.6 g (10.0 mmol) of <strong>[575-44-0]1,6-dihydroxynaphthalene</strong> (Compound 39: produced by Tokyo Chemical Industry Co., Ltd.), and 15 mL of N,N-dimethylformamide (DMF) were added for dissolution, and thrther 14.2 g (100.0 mmol) of methyl iodide (produced by Wako Pure Chemical Industries, Ltd.), and 13.8 g (100.0 mmol) of potassium carbonate (produced by Wako Pure Chemical Industries, Ltd.) were added, and the reaction was carried out at room temperature for 2 hours. Afier completion of the reaction, dichloromethane and water were added, and then an organic layer, obtained by solution separation, was washed with water, and the solvent was removed from the reaction solution by concentration under reduced pressure to obtain 1 .7 g (yield:90%) of a colorless liquid methyl derivative (Compound40).
Take 50 ml round-bottom flask will be 313 mg, 1 mmol of 4 - diethylamino-ketoacid (compound 1, CAS: 5809 - 23 - 4,) and 160 mg, 1 mmol of 1 - 6 naphthol (compound 2, CAS: 59335 - 81 - 8,) is added to the bottle, add 4 ml concentrated sulfuric acid as the solvent, for 90 C reflux 6 h. Waiting for the reaction cooling to room temperature, the flask is put into the low-temperature environment (- 5 - 0 C) the height of the perchloric acid is slowly dropped to precipitate, filtering, drying and over-column purification, the polarity of the eluant (V(DCM): V(MeOH)=10:1) to obtain a red solid product is the type (II) compound, yield 58%.
3,3′-((2-hydroxy-5-oxo-5H-benzo[a]phenoxazin-9-yl)azanediyl)dipropionic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
58.8%
With hydrogenchloride; In methanol; for 7h;Reflux;
General procedure: The solution of compound 8 (1.21 g, 4.3 mmol), 1-naphthylamine (0.61 g, 4.3 mmol) and HCl (1.25 mL,12.0 M) in methanol (35 mL) was refluxed for 7 h with stirring. The mixture was concentrated, and subjected to column chromatography on silica with dichloromethane: methanol =40:1 to yield metallic green powder (1.57 g, 91.3%)
With potassium carbonate In butanone for 20h; Reflux;
1
J. Org. Chem. , 34 (12), 4060 (1969). Synthesized according to 4 -Bromo-1,2-butylene oxide 7.30 g (48.3 mmol), 3.22 g (20.1 mmol) of 1,6-dihydroxynaphthalene, K2CO 36.68 g (48.3 mmol) The mixture was refluxed in 84 ml of 2-butanone solution for 20 hours. After cooling the reaction solution, 200 ml each of pure water and toluene was added. After separating the organic layer, the organic layer was washed twice with the same amount of pure water and dried with 00544. After filtration and distillation of the solvent under reduced pressure, the obtained product was purified by column chromatography (silica gel, toluene / ethyl acetate = 10/1) to obtain a polymerizable compound (1-2a). Yield 4.47 g (yield 74%). The polymerizable compound was liquid at room temperature.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping