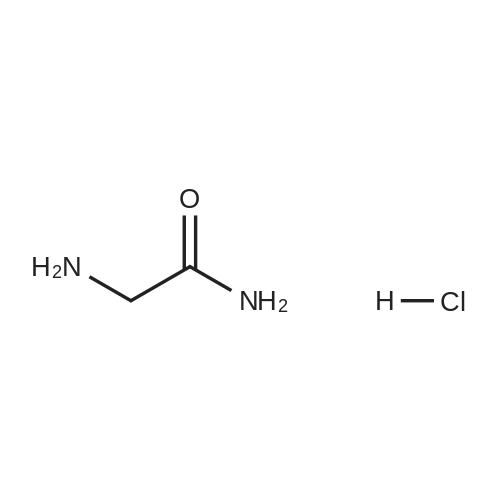
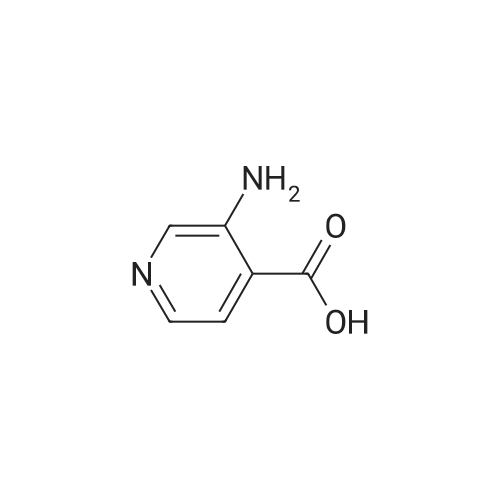
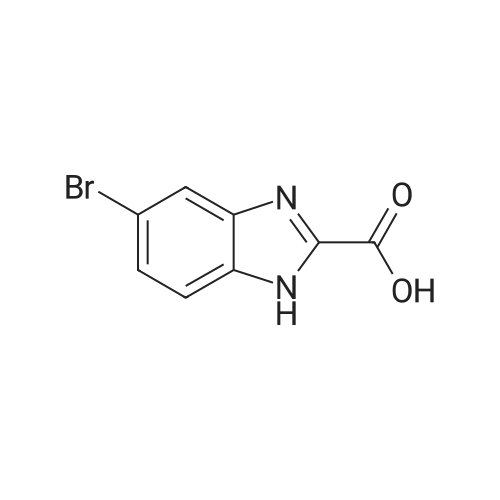
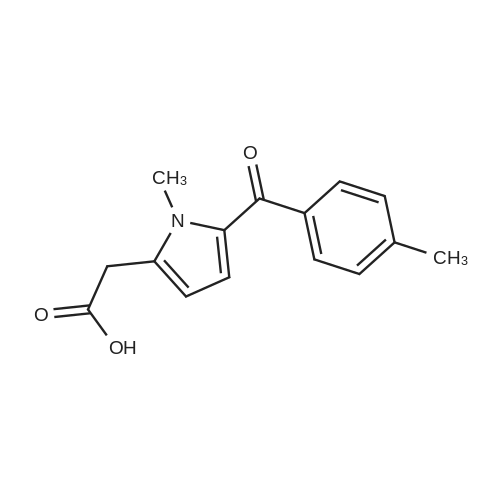
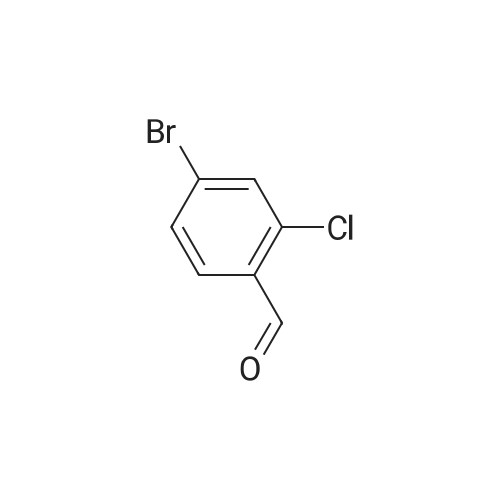
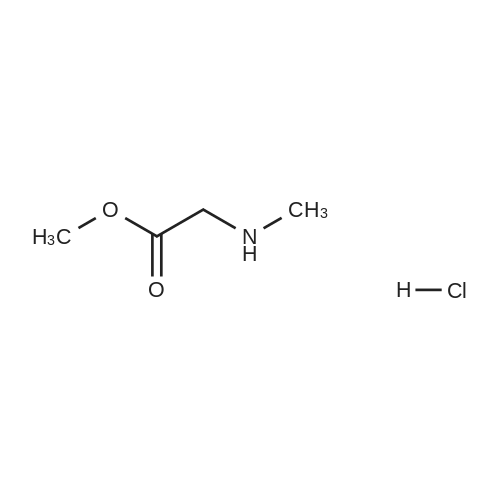
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
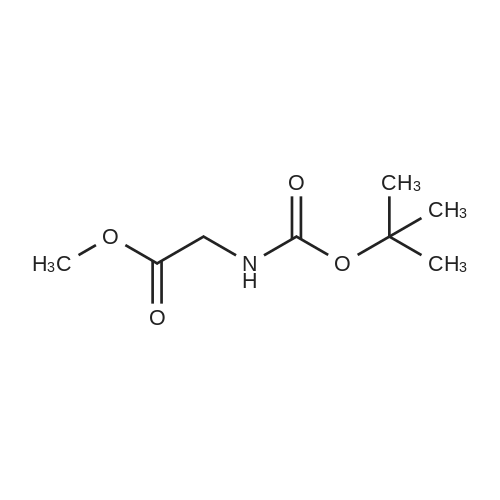
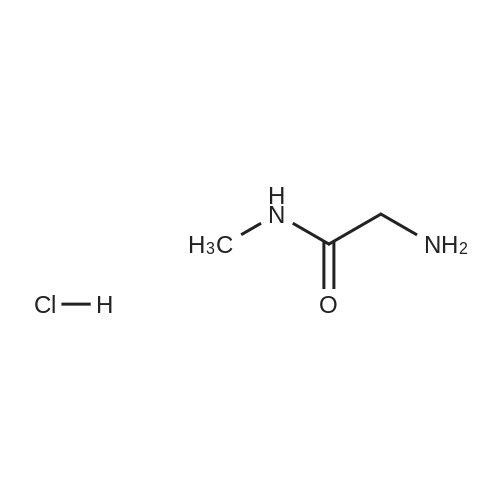
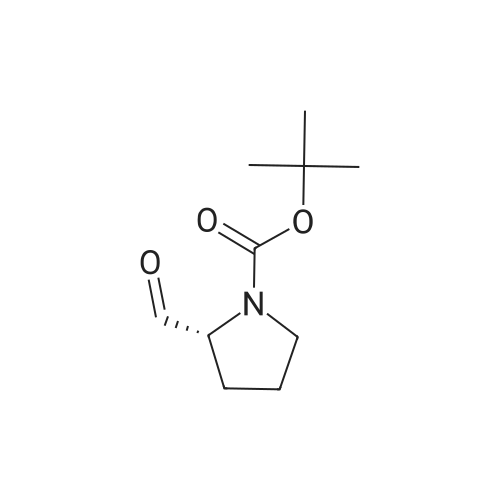
Stage #1: With sodium hydrogencarbonate In acetonitrile at 0 - 20℃; for 21 h; Inert atmosphere Stage #2: With hydrazine hydrate In ethanol at 20℃; for 24 h;
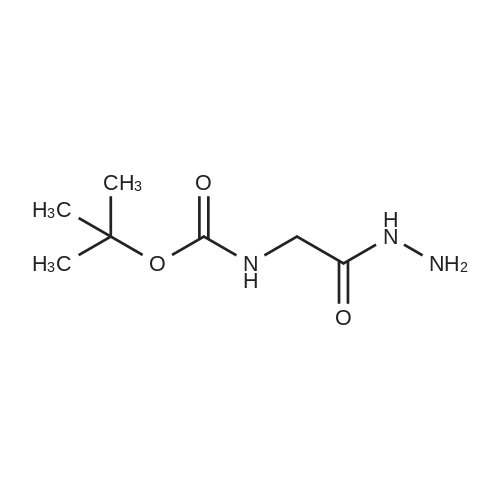
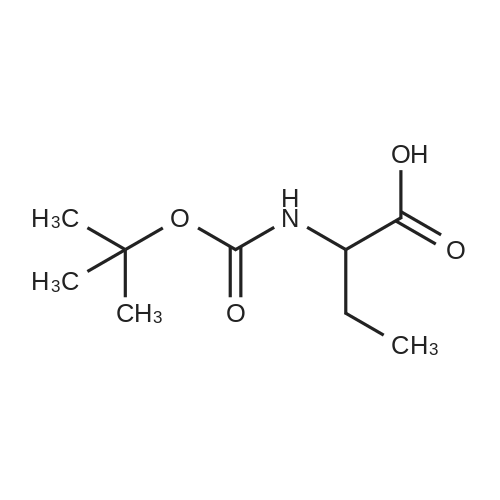
The compound 2-aminoacetic acid methyl ester hydrochloride was added(28.1 g, 224 mmol) and sodium bicarbonate (36.4 g, 433 mmol)Suspended in acetonitrile (400 mL)And then di-tert-butyl dicarbonate was added to the stirred reaction solution under the reaction of 0 ° C under nitrogen(45 mL, 200 mmol). The mixture was stirred at room temperature for 21 hours and then concentrated under reduced pressure. The residue was diluted with H2O (40 mL) to give The mixture was extracted with DCM (50 mL x 3). The combined organic phases were washed with saturated brine (50 mL), dried over anhydrous sodium sulfate, Concentrated under reduced pressure to give a white solid which was used directly in the next step.The white solid was dissolved in ethanol (100 mL) and then at room temperature, Hydrazine hydrate (27 mL) was added to the solution, and the resulting mixture was stirred at room temperature for 24 hours and then concentrated under reduced pressure. The residue was diluted with H? O (50 mL) and the resulting mixtureDCM (100 mL x 3). The combined organic phases were washed with saturated brine (60 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure A white solid was obtained which was used directly in the next step (31 g, 73percent) without further purification.
Reference:
[1] Patent: CN105924434, 2016, A, . Location in patent: Paragraph 0883; 0884; 0885; 0886
11
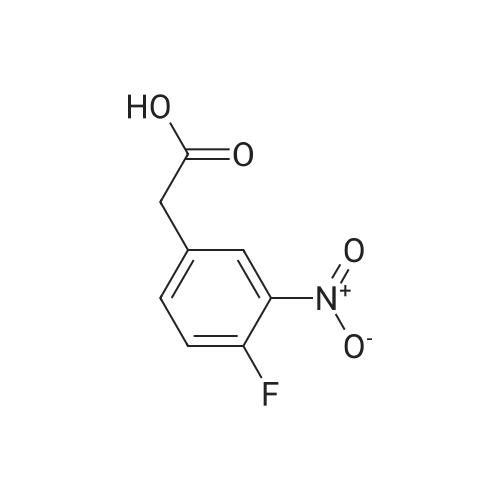
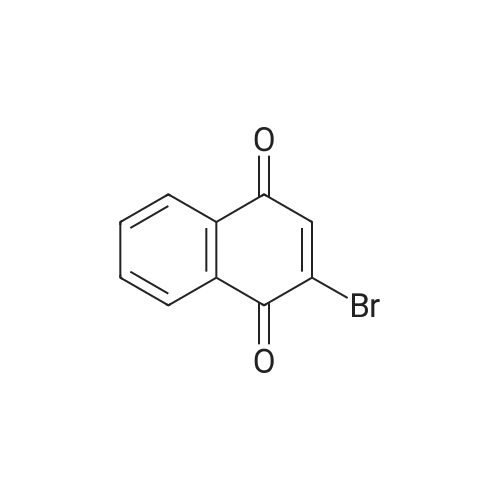
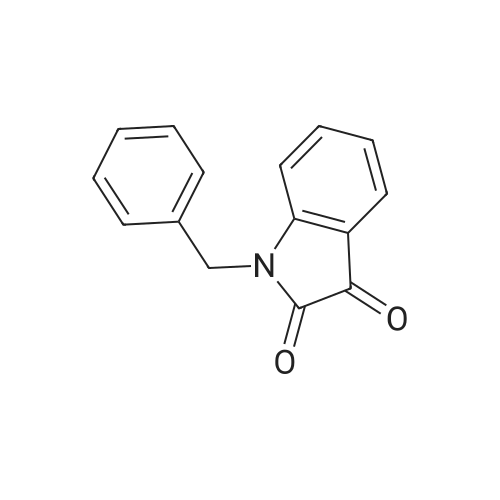
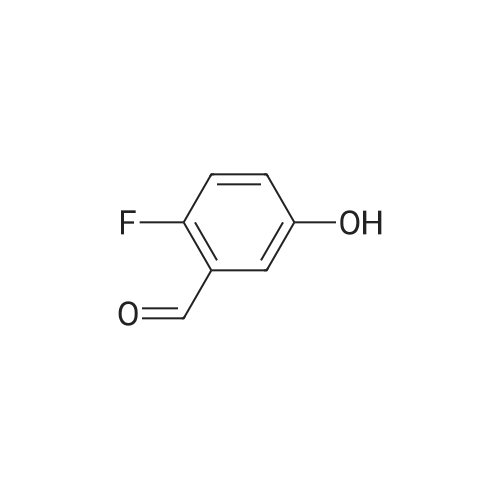
[ 5680-79-5 ]
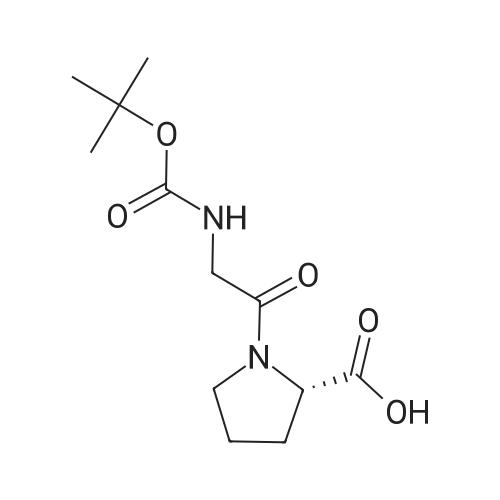
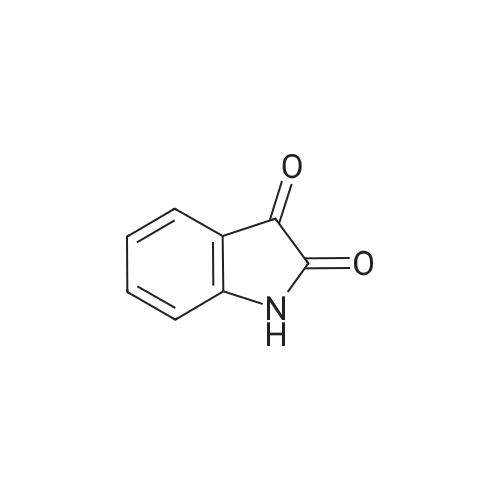
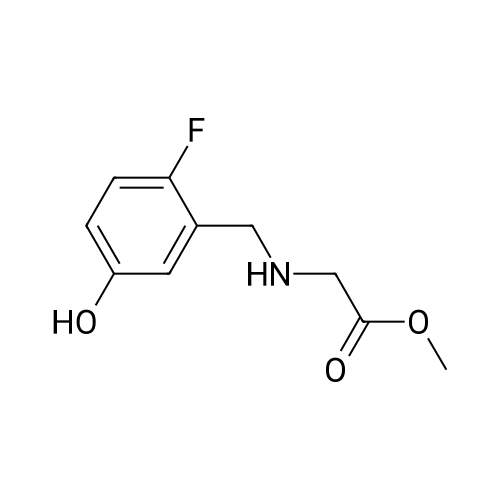
[ 3321-03-7 ]
Reference:
[1] Chemistry - A European Journal, 2015, vol. 21, # 34, p. 11980 - 11983
12
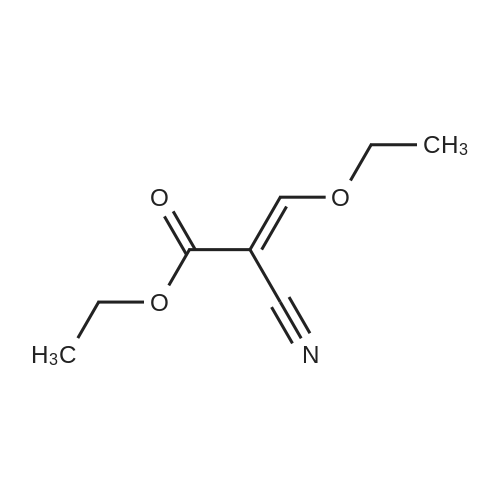
[ 5680-79-5 ]
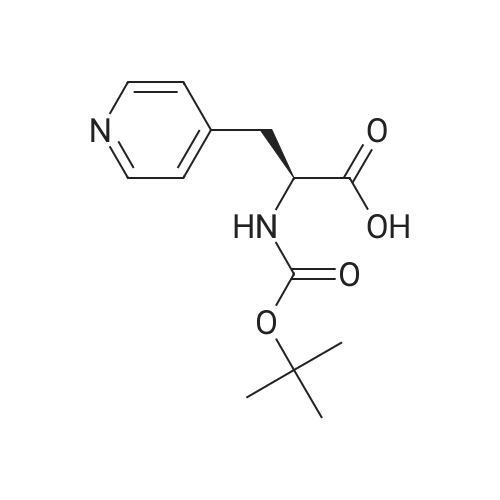
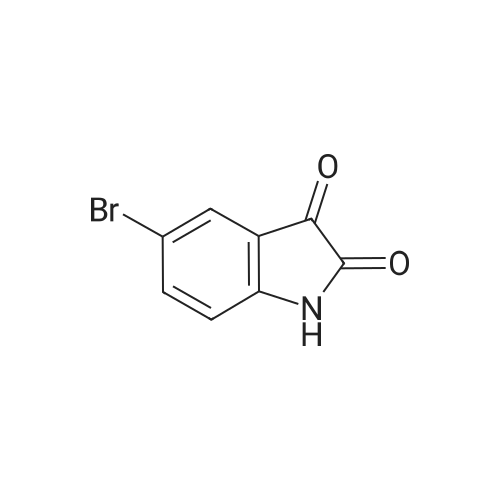
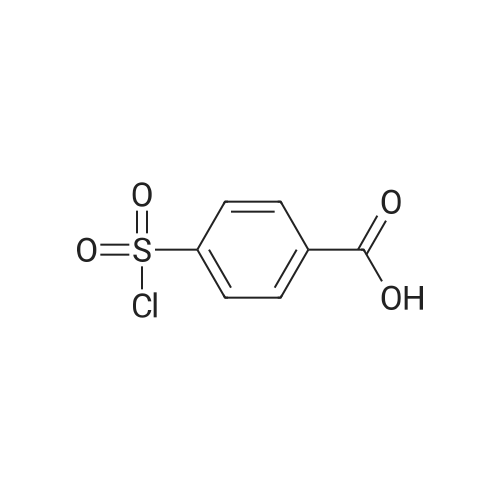
[ 98-59-9 ]
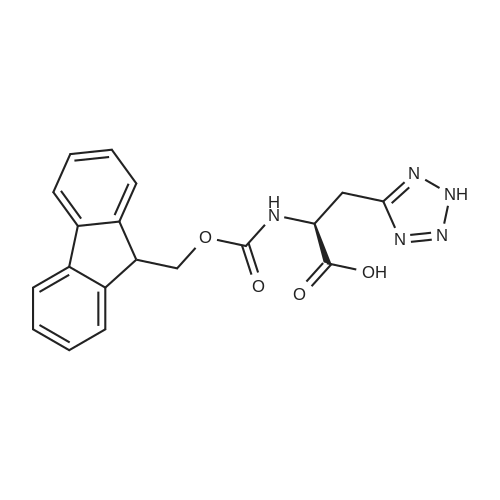
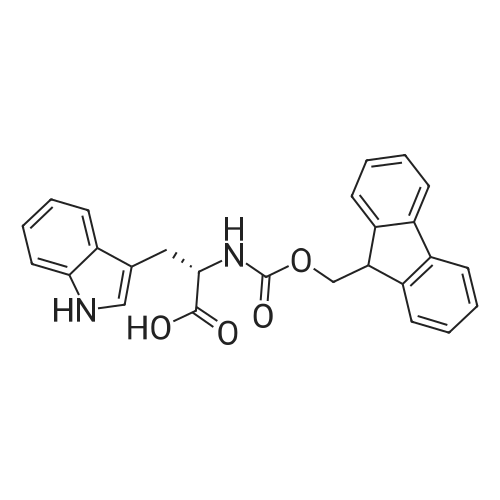
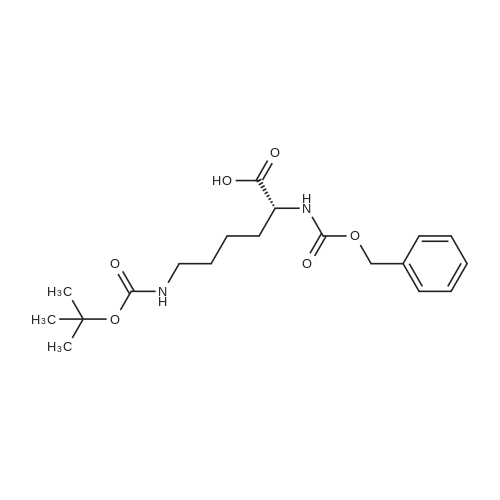
[ 2645-02-5 ]
Yield
Reaction Conditions
Operation in experiment
78%
With sodium carbonate In dichloromethane at 20℃; Cooling with ice
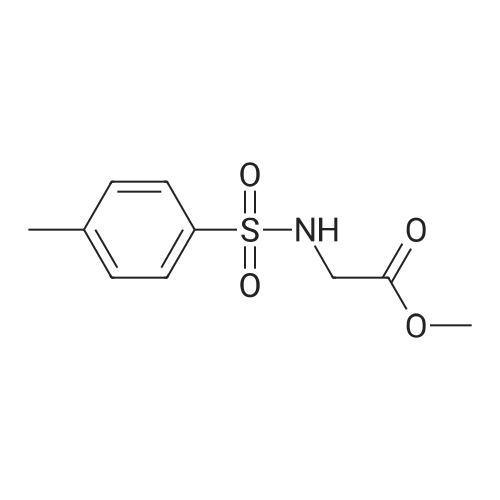
General procedure: Syntheses of G(2–4) and P(2–4): General experimental procedure is as follows: Glycine or L-phenyl alanine derivative (as HCl salt) (10 mmol) and Na2CO3 (2.1 g, 20 mmol) were dissolved in anhydrous dichloromethane (50 mL) and cooled in an ice bath before tosyl chloride (1.9 g, 10 mmol) was added. The resultant reaction mixture was stirred overnight at room temperature. After the reaction, more solvent was added and the residue was washed sequentially with 0.1 M HCl, saturated NaHCO3 and brine. Organic portion was dried with Na2SO4 and concentrated in vacuo to give crude product. The product was purified by column chromatography on silica with hexane and EtOAc (1:1). Compound G2: Yield 78percent as white solid. 1H NMR (400 MHz,CDCl3): δ 7.74 (d, J= 8.3 Hz, 2H, ArH), 7.31 (d, J= 8.3 Hz, 2H, ArH), 5.04 (t, J= 5.1 Hz, 1H, –NH–), 3.78 (d, J= 5.1 Hz, 2H, –CH2–), 3.64 (s, 3H, –OCH3), 2.42 (s, 3H, –CH3) ppm. 13C NMR (100 MHz, CDCl3): δ 169.4, 144.0, 136.3, 129.9, 127.4, 52.7, 44.2, 21.7 ppm. Anal.Calcd. [C10H13NO4S]: C, 49.37; H, 5.39; N, 5.76; S, 13.18. Found:C, 50.63; H, 5.09; N, 5.36; S, 12.87.
Reference:
[1] Organic Letters, 2005, vol. 7, # 25, p. 5717 - 5719
[2] ChemMedChem, 2011, vol. 6, # 8, p. 1459 - 1470
[3] Journal of Medicinal Chemistry, 2013, vol. 56, # 18, p. 7190 - 7200
[4] Tetrahedron, 2008, vol. 64, # 52, p. 11884 - 11895
[5] ACS Medicinal Chemistry Letters, 2015, vol. 6, # 9, p. 982 - 986
[6] Chemical Biology and Drug Design, 2011, vol. 77, # 3, p. 189 - 198
[7] Journal of Organic Chemistry USSR (English Translation), 1989, vol. 25, # 6.2, p. 1162 - 1167[8] Zhurnal Organicheskoi Khimii, 1989, vol. 25, # 6, p. 1292 - 1298
[9] Bioorganic and Medicinal Chemistry, 2015, vol. 23, # 23, p. 7353 - 7358
[10] Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999), 1972, p. 627 - 633
[11] Tetrahedron, 1989, vol. 45, # 6, p. 1691 - 1702
[12] Journal of the American Chemical Society, [13] Journal of the American Chemical Society, 2009, vol. 131, p. 3832 - 3833
[14] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 8, p. 2162 - 2167
13
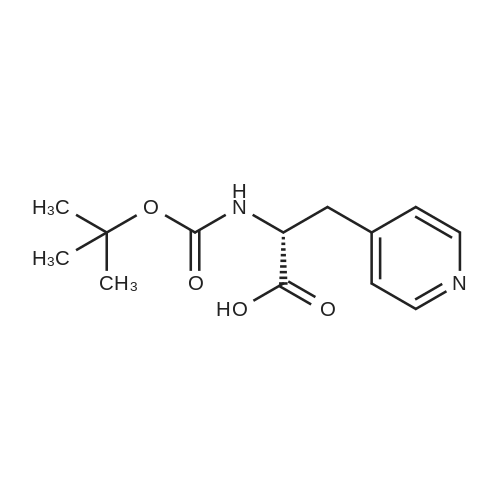
[ 5680-79-5 ]
[ 100-52-7 ]
[ 53386-64-4 ]
Reference:
[1] Journal of Organic Chemistry, 2017, vol. 82, # 12, p. 6210 - 6222
[2] Patent: WO2017/59191, 2017, A1, . Location in patent: Paragraph 001095
Stage #1: for 1 h; Cooling with ice Stage #2: at 20 - 66℃; for 6.5 h;
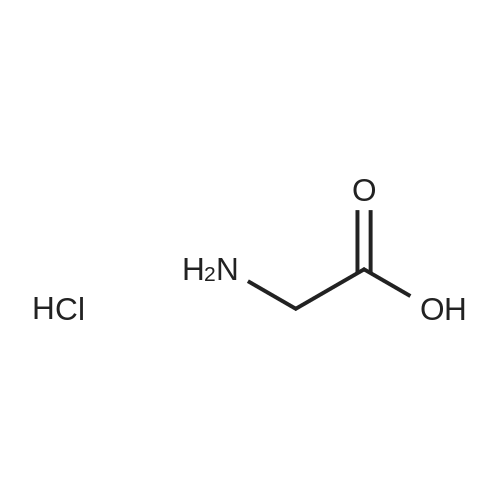
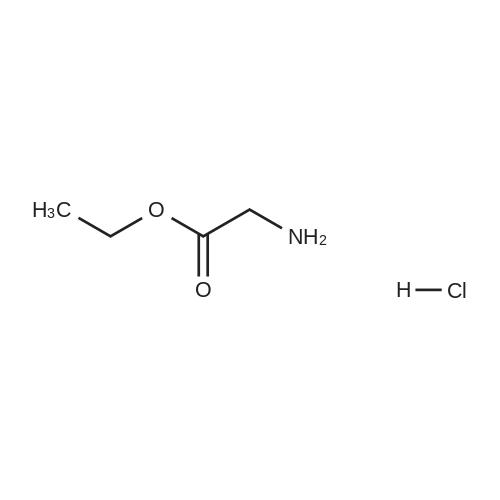
(2) Preparation of glycine methyl ester hydrochloride Under ice bath, 100 ml round-bottom flask was added with 60 ml of methanol, and then slowly added with 4 ml of SOCl2 through constant pressure dropping funnel (with a drying tube on the top), and NaOH solution was used to absorb exhaust. After stirring for 1 h, 8 mmol of glycine was added and stirred at room temperature for 30 min, and then refluxed at 66° C. for 6 h. The reaction was tracked by TLC until the raw materials disappeared, with a solution of 2percent ninhydrin in ethanol as chromogenic reagent. The solvent was evaporated out to obtain glycine methyl ester hydrochloride. Yield: 100percent.
99%
at -5 - 20℃; for 3 h; Inert atmosphere
Under N2 dryCH3OH (100 mL) was cooled down to -5 °C and SOCl2 (21 mL,0.3mol) was added dropwise. Glycine (7.5 g, 0.1mol) was added to this solutionand stirring was continued at rt for 3 h. The solvent and gaseous reactionproducts were removed under reduced pressure to obtain the pure product ascolorless crystals, mp 180 °C (ref. 175-178 oC)9, yield12.4 g (99 percent). C3H8ClNO2 (125.5 g / mol).FT-IR (neat): v (cm-1) =2879 (s, vC-H aliphatic), 1744 (s, vC=O ester), 1582/1496(m, vN-H). 1H NMR(400MHz, CD3OD): δ (ppm) = 3.84 (s, 3H, CO2CH3),3.89 (s, 2H, CH2). The signal for NH3+-protonsis not seen in the 1H NMR-spectrum. 13C NMR (101MHz, CD3OD):δ (ppm) = 40.2 (1C, CH2), 53.5 (1C, CO2CH3), 168.8 (1C, C=O).
98%
at -5 - 20℃;
General procedure: A suspension of L-amino acid (50mmol) in methanol (50mL) was stirred under ice cold conditions. Thionyl chloride (5mL) was slowly dropped to the solution at−5°C. Then, the mixture was allowed to slowly warm to room temperature while being stirred. The reaction was monitored by TLC (N-butanol: water: acetic acid=4:1:1). When the reaction was completed, the solvent was evaporated under reduced pressure to afford the crude product, which was recrystallized from methanol/ether to give a white solid.
96%
at 0 - 20℃;
To a solution of SOCl2(2.33mL; 3.2 10-2 mol) in MeOH at 0°C, the glycine (2g, 2.6610-2mol) was added. The mixture was stirred at rt overnight. Reaction mixture was evaporated under vacuum. The residue was dissolved in MeOH and evaporated again under vacuum. This procedure was repeated three times. The residue was recrystallized in a mixture of MeOH and Et2O to provide a fluffy powder (2.27g, 96percent). IR (ATR): 3109,3012, 2882, 2687, 2635, 1743, 1584, 1557 cm-1. 1H NMR(300 MHz, DMSO D6) δ ppm 8.64(s, 3H), 3.76 (s, 2H), 3.72 (s, 3H). 13C NMR (75MHz, DMSO D6) δ 167.9,52.4, 39.3.
90%
at 20℃; for 24 h; Sealed tube
Amino acid is placed into a 50 ml one-port flask and added with 2.0 eq. TMSC1 dropwise before adding 5 mL of dry methanol as the solvent. The reaction mixture is sealed and allowed to react at room temperature with stirring for 24 h. Upon completion of the reaction, the solvent is evaporated and methanol (3X10 mL) is further added to co-evaporate the residual hydrochloride. Ethyl ether is then added to the oily product obtained after the evaporation and a white solid is precipitated and further recrystallized with methanol-ethyl ether.
90%
at 20℃; for 24 h; Sealed tube
Example 4 [0068] Synthesis of amino acid methyl ester hydrochloride is described as follows. Amino acid is placed into a 50 ml one-port flask and added with 2.0 eq. TMSCl dropwise before adding 5 mL of dry methanol as the solvent. The reaction mixture is sealed and allowed to react at room temperature with stirring for 24 h. Upon completion of the reaction, the solvent is evaporated and methanol (3×10 mL) is further added to co-evaporate the residual hydrochloride. Ethyl ether is then added to the oily product obtained after the evaporation and a white solid is precipitated and further recrystallized with methanol-ethyl ether. Experimental data for the products are shown in the table below.
90%
for 2 h; Reflux
General procedure: 26.2 g (200 mmol) trans-4-hydroxy-L-proline (1) in methanol(300 ml) was treated with dry hydrogen chloride until homogeneous.The solution was heated to the reflux temperature for 2 h and concentrated in vacuo. Upon cooling, the product was crystallizedfrom the solvent, collected by filtration, washed with acetoneand ether, and dried to yield trans-4-hydroxy-L-proline methylesterhydrochloride (2) as white crystal (32.7 g, 90percent), mp 159–162 C.
90.68%
Stage #1: at -5 - 5℃; for 1 h; Stage #2: at 70℃; for 5 h;
Control the temperature of -5 ~ 5 ° C, acetyl chloride 23.56g drop by drop to no waterMethanol 60ml, the drip after the incubation reaction lh, divided into two batches of glycine 7.51g, add the system temperature to 70 ° C reaction 5h, minusPressure to remove the excess solvent,The residue was further added with acetone 30 ml for 5 h,filter,The filter cake was dried in vacuo to give 11.39 g of glycine methyl ester hydrochloride in a yield of 90.68percent and a melting point of 174-176 ° C.
81%
at 0 - 20℃; for 16 h;
To a stirred solution of 2-aminoacetic acid 8-1 (2 g, 26.64 mmol) in MeOH (20 mL) was added thionyl chloride (5.8 mL, 79.92 mmol) at 0 °C, and stirred at RT for 16 hr. The solvents were evaporated under reduced pressure and the crude residue was purified by washings with pentane/ether to afford 9-10A (2.7 g, 21.6 mmol, 81percent yield) as a brown solid.
4.93 g
at 65 - 70℃; for 1.25 h;
In the first step, glycine methyl ester hydrochloride is prepared by weighing 3 g of glycine, adding 150mL of anhydrous methanol, heating in an oil bath at 65 ° C to 70 ° C, continuously feeding dry Hcl gas,completely dissolving glycine in about 60 min, and continuing to pass Hcl gas. After 15 min, the reactionwas stopped, concentrated under reduced pressure, and the residue was stirred with acetone to givewhite crystals 4.93g
Reference:
[1] Synthetic Communications, 1998, vol. 28, # 3, p. 471 - 474
[2] European Journal of Organic Chemistry, 2012, # 29, p. 5774 - 5788,15
[3] European Journal of Organic Chemistry, 2012, # 29, p. 5774 - 5788
[4] Patent: US2014/206741, 2014, A1, . Location in patent: Paragraph 0131
[5] European Journal of Organic Chemistry, 2017, vol. 2017, # 3, p. 695 - 703
[6] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2006, vol. 45, # 8, p. 1942 - 1944
[7] Bioorganic and Medicinal Chemistry Letters, 2016, vol. 26, # 3, p. 889 - 893
[8] Tetrahedron Asymmetry, 1992, vol. 3, # 11, p. 1431 - 1440
[9] Journal of Medicinal Chemistry, 2006, vol. 49, # 20, p. 6094 - 6103
[10] European Journal of Medicinal Chemistry, 2016, vol. 112, p. 196 - 208
[11] Organic Preparations and Procedures International, 2001, vol. 33, # 4, p. 341 - 349
[12] Organic Preparations and Procedures International, 2002, vol. 34, # 1, p. 87 - 94
[13] Archiv der Pharmazie, 2011, vol. 344, # 5, p. 320 - 332
[14] Molecules, 2008, vol. 13, # 5, p. 1111 - 1119
[15] European Journal of Organic Chemistry, 2010, # 22, p. 4276 - 4287
[16] European Journal of Medicinal Chemistry, 2015, vol. 103, p. 163 - 174
[17] ACS Medicinal Chemistry Letters, 2017, vol. 8, # 4, p. 428 - 432
[18] Journal of Labelled Compounds and Radiopharmaceuticals, 2006, vol. 49, # 2, p. 109 - 123
[19] Chemical Biology and Drug Design, 2016, p. 542 - 555
[20] J. Gen. Chem. USSR (Engl. Transl.), 1991, vol. 61, # 1.1, p. 104 - 108
[21] Patent: EP2615101, 2013, A1, . Location in patent: Paragraph 0062
[22] Patent: US2013/303747, 2013, A1, . Location in patent: Paragraph 0068
[23] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 11, p. 3055 - 3064
[24] Patent: CN105820102, 2016, A, . Location in patent: Paragraph 0032
[25] Biomedicine and Pharmacotherapy, 2017, vol. 88, p. 1163 - 1172
[26] Journal of Medicinal Chemistry, 2005, vol. 48, # 19, p. 5932 - 5941
[27] Synthetic Communications, 2010, vol. 40, # 8, p. 1161 - 1179
[28] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1994, # 21, p. 3079 - 3088
[29] Organic and Biomolecular Chemistry, 2008, vol. 6, # 9, p. 1594 - 1600
[30] Research on Chemical Intermediates, 2013, vol. 39, # 2, p. 621 - 629
[31] RSC Advances, 2016, vol. 6, # 49, p. 42836 - 42844
[32] Patent: WO2016/154241, 2016, A1, . Location in patent: Paragraph 401; 409
[33] Journal of the Indian Chemical Society, 1985, vol. 62, # 6, p. 457 - 459
[34] Bioorganic and Medicinal Chemistry, 2009, vol. 17, # 8, p. 3061 - 3071
[35] Tetrahedron, 2010, vol. 66, # 9, p. 1661 - 1666
[36] Archiv der Pharmazie, 2011, vol. 344, # 8, p. 494 - 504
[37] Journal of Chemical Research, Miniprint, 1981, # 9, p. 3261 - 3278
[38] Tetrahedron, 1990, vol. 46, # 15, p. 5325 - 5332
[39] Heterocycles, 1991, vol. 32, # 10, p. 1879 - 1895
[40] Chemistry of Natural Compounds, 1990, vol. 26, # 2, p. 199 - 201[41] Khimiya Prirodnykh Soedinenii, 1990, # 2, p. 245 - 248
[42] J. Gen. Chem. USSR (Engl. Transl.), 1984, vol. 54, p. 933 - 936[43] Zhurnal Obshchei Khimii, 1984, vol. 54, # 5, p. 1045 - 1048
[44] Tetrahedron, 1989, vol. 45, # 6, p. 1691 - 1702
[45] J. Gen. Chem. USSR (Engl. Transl.), 1983, vol. 53, # 6, p. 1109 - 1114[46] Zhurnal Obshchei Khimii, 1983, vol. 53, # 6, p. 1243 - 1249
[47] J. Gen. Chem. USSR (Engl. Transl.), 1985, vol. 55, # 7, p. 1417 - 1423[48] Zhurnal Obshchei Khimii, 1985, vol. 55, # 7, p. 1594 - 1600
[49] Synthetic Communications, 1992, vol. 22, # 7, p. 979 - 985
[50] Archiv der Pharmazie, 1992, vol. 325, # 11, p. 709 - 715
[51] Collection of Czechoslovak Chemical Communications, 1997, vol. 62, # 10, p. 1562 - 1576
[52] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 4, p. 387 - 398
[53] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 1999, vol. 38, # 4, p. 505 - 507
[54] Russian Journal of General Chemistry, 1999, vol. 69, # 11, p. 1846 - 1847[55] Zhurnal Obshchei Khimii, 1999, vol. 69, # 11, p. 1929 - 1930
[56] Bioorganic and Medicinal Chemistry Letters, 2005, vol. 15, # 6, p. 1629 - 1632
[57] Journal of Medicinal Chemistry, 2006, vol. 49, # 24, p. 7215 - 7226
[58] Tetrahedron, 2007, vol. 63, # 31, p. 7334 - 7348
[59] Patent: WO2007/74390, 2007, A2, . Location in patent: Page/Page column 6-7
[60] Tetrahedron Letters, 2008, vol. 49, # 24, p. 3943 - 3945
[61] Journal of Pharmacy and Pharmacology, 2008, vol. 60, # 5, p. 607 - 613
[62] Acta Poloniae Pharmaceutica - Drug Research, 2007, vol. 64, # 6, p. 509 - 516
[63] European Journal of Medicinal Chemistry, 2009, vol. 44, # 7, p. 2796 - 2806
[64] Zeitschrift fur Anorganische und Allgemeine Chemie, 2010, vol. 636, # 1, p. 236 - 241
[65] Bioorganic and Medicinal Chemistry Letters, 2009, vol. 19, # 2, p. 406 - 409
[66] Journal of Polymer Science, Part A: Polymer Chemistry, 2010, vol. 48, # 23, p. 5364 - 5374
[67] Bioorganic and Medicinal Chemistry, 2011, vol. 19, # 10, p. 3105 - 3119
[68] Chemistry - An Asian Journal, 2011, vol. 6, # 1, p. 84 - 97
[69] Chinese Journal of Chemistry, 2011, vol. 29, # 5, p. 1011 - 1016
[70] Journal of Enzyme Inhibition and Medicinal Chemistry, 2011, vol. 26, # 5, p. 688 - 695
[71] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 12, p. 3807 - 3815
[72] Asian Journal of Chemistry, 2012, vol. 24, # 3, p. 1237 - 1241
[73] Asian Journal of Chemistry, 2012, vol. 24, # 3, p. 1227 - 1236
[74] Biomacromolecules, 2012, vol. 13, # 8, p. 2446 - 2455
[75] Medicinal Chemistry Research, 2012, vol. 21, # 11, p. 3361 - 3368
[76] Chinese Chemical Letters, 2013, vol. 24, # 8, p. 767 - 769
[77] , 2018, vol. 67, # 2, p. 307 - 319
[78] Journal of Heterocyclic Chemistry, 2013, vol. 50, # 5, p. 1067 - 1070
[79] ACS Medicinal Chemistry Letters, 2015, vol. 6, # 7, p. 764 - 769
[80] European Journal of Medicinal Chemistry, 2016, vol. 108, p. 166 - 176
[81] Journal of Polymer Science, Part A: Polymer Chemistry, 2016, vol. 54, # 8, p. 1065 - 1077
[82] Tetrahedron, 2016, vol. 72, # 51, p. 8486 - 8492
[83] ACS Medicinal Chemistry Letters, 2016, vol. 7, # 12, p. 1185 - 1190
[84] CrystEngComm, 2017, vol. 19, # 9, p. 1286 - 1293
[85] Patent: CN106632193, 2017, A, . Location in patent: Paragraph 0055
[86] Patent: CN106478645, 2017, A, . Location in patent: Paragraph 0040; 0043
[87] Chirality, 2017, vol. 29, # 11, p. 737 - 746
[88] Russian Chemical Bulletin, 2017, vol. 66, # 1, p. 136 - 142[89] Izv. Akad. Nauk, Ser. Khim., 2017, # 1, p. 136 - 142,6
[90] New Journal of Chemistry, 2017, vol. 41, # 11, p. 4390 - 4399
[91] Journal of Chemical Sciences, 2018, vol. 130, # 5,
[92] FEBS Journal, 2018, vol. 285, # 12, p. 2292 - 2305
[93] Patent: WO2018/104962, 2018, A1, . Location in patent: Page/Page column 22
[94] Patent: CN107827815, 2018, A, . Location in patent: Paragraph 0084; 0085; 0123; 0124; 0143; 0144
[95] Patent: CN107936020, 2018, A, . Location in patent: Paragraph 0007; 0008
[96] Journal of Organometallic Chemistry, 2018, vol. 876, p. 1 - 9
[97] Amino Acids, 2018, vol. 50, # 10, p. 1461 - 1470
16
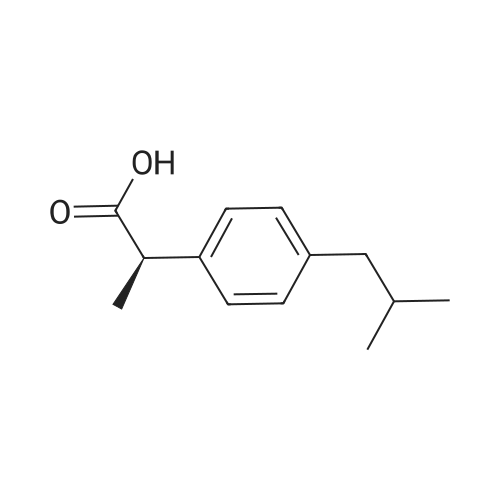
[ 616-34-2 ]
[ 5680-79-5 ]
Yield
Reaction Conditions
Operation in experiment
99.5%
With thionyl chloride In methanol at -10℃; for 0.166667 h;
IIIa the compound (3g, 40mmol) dissolved in MeOH (30 ml) in, salt bath cooling to -10 °C, stirring slowly dripping SOCl2(29 ml, 400mmol), reaction after the end of the dropping 10 min, to remove salt bath, the reaction at room temperature overnight, TLC detection reaction, concentrating under reduced pressure, then adding 20mLCH2Cl2, repeatedly two times concentrated under reduced pressure, the solvent turns on lathe does, drying, the obtained product 5g, the yield is 99.5percent, product without purification, used directly in the next reaction.
Reference:
[1] Patent: CN105732683, 2016, A, . Location in patent: Paragraph 0122; 0123; 0124; 0125; 0126
Reference:
[1] Journal of the American Chemical Society, 1992, vol. 114, # 12, p. 4834 - 4843
[2] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 9, p. 2456 - 2458
[3] Journal of the American Chemical Society, 2015, vol. 137, # 38, p. 12369 - 12377
19
[ 82208-51-3 ]
[ 5680-79-5 ]
Reference:
[1] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1987, vol. 36, p. 1076 - 1077[2] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1987, vol. 36, # 5, p. 1162 - 1164
20
[ 70036-25-8 ]
[ 5680-79-5 ]
Reference:
[1] Bulletin de la Societe Chimique de France, 1992, # 1, p. 71 - 75
Reference:
[1] Bulletin of the Chemical Society of Japan, 2004, vol. 77, # 6, p. 1187 - 1193
27
[ 67-56-1 ]
[ 6000-43-7 ]
[ 5680-79-5 ]
Reference:
[1] Journal fuer Praktische Chemie (Leipzig), 1888, vol. <2>37, p. 163
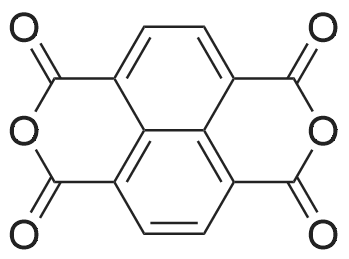
[2] Journal of Materials Chemistry C, 2018, vol. 6, # 5, p. 1071 - 1082
28
[ 67-56-1 ]
[ 4746-64-9 ]
[ 5680-79-5 ]
Reference:
[1] Journal of Physical Organic Chemistry, 2004, vol. 17, # 5, p. 448 - 457
29
[ 64-17-5 ]
[ 120879-96-1 ]
[ 5680-79-5 ]
Reference:
[1] Chemische Berichte, 1921, vol. 54, p. 1541
[2] Bulletin de la Societe Chimique de France, 1900, vol. <3> 23, p. 661
30
[ 7647-01-0 ]
[ 128666-05-7 ]
[ 5680-79-5 ]
[ 65-85-0 ]
Reference:
[1] Helvetica Chimica Acta, 1925, vol. 8, p. 881
[2] Acta Chemica Scandinavica (1947-1973), 1949, vol. 3, p. 647[3] Chem. Zentralbl., 1950 II, 1574,
31
[ 1013-88-3 ]
[ 5680-79-5 ]
[ 81167-39-7 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 20℃; for 24 h;
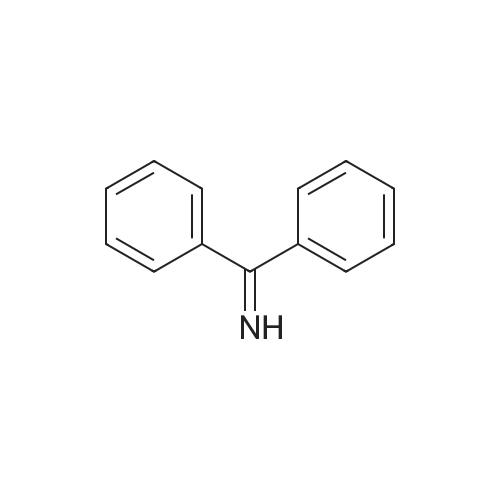
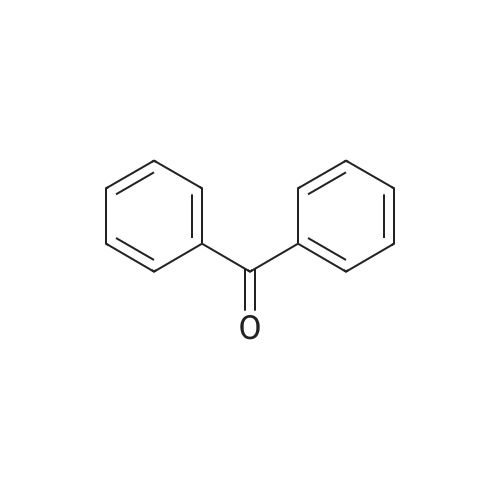
To a stirring solution of glycine methyl ester hydrochloride (17.49 g, 139 mmol) in dry dichloromethane (150 mL) under N2 at room temperature was added diphenylimine (25.00 g, 137 mmol) in one portion. The reaction mixture was stirred for 24 h, during which time ammonium chloride precipitated. Water (20 mL) was added and the layers were separated. The organic layer was washed with saturated Na2CO3 solution (2 x 20 mL) and brine (20 mL). The organic layer was dried (Na2SO4), filtered and concentrated by rotary evaporation to give ~ 35 g of a thick light brown syrup (99 percent pure) in ~ 100 percent yield. This was taken on to the next reaction without further purification.
100%
at 20℃; for 24 h;
To a stirring solution of methyl glycine ester hydrochloride (17.49 g, 139 mmol) in dry dichloromethane (150 mL) under N2 at room temperature was added diphenylimine (25.00 g, 137 mmol) in one portion. The reaction mixture was stirred for 24 h, during which time ammonium chloride precipitated. Water (20 mL) was added and the layers were separated. The organic layer was washed with saturated Na2C03 solution (2 x 20 mL) and brine (20 mL). The organic layer was dried (Na2SC>4), filtered and concentrated by rotary evaporation to give ~35 g of a thick light brown syrup (99percent pure) in ~ 100percent yield. This was taken on to the next reaction without further purification.
92%
at 20℃; for 24 h;
Benzophenone imine (50.0 g, 0.276 mol) was added in one batch to a solution of glycine methyl ester hydrochloride (39.4 g, 0.314 mol) in 300 mL dichloromethane under stirring, and the resulting reaction solution was stirred at room temperature for 1 day. The resulting solid was removed by filtration, and the filtrate was washed sequentially with water, sodium carbonate solution and saturated brine, and was concentrated by evaporation to give methyl 2-((diphenylmethylene)amino)acetate (64.2 g) as an oily product, which was solidified after being cooled, and used directly for next step. Yield: 92percent. 1H-NMR (400 MHz, CDCl3): δ= 7.66 (2H, m), 7.45 (4H, m), 7.35 (2H, m), 7.17 (2H, m), 4.22 (2H, s), 3.74 (3H, s).
81%
at 25℃; for 24 h; Inert atmosphere
To a stirred suspension containing 5.00 g (39.9 mmol) of glycine methyl ester hydrochloride in 20 mL of anhydrous CH2Cl2 was added dropwise 6.70 mL (7.20 g, 39.9 mmol) of benzophenone imine.The milky white mixture was stirred at 25 °C for 24 h under an argon atmosphere. The reaction mixture was filtered and concentrated under diminished pressure. The crude product was crystallized from ether–hexanes to afford 3 as colorless crystals: yield 8.20 g (81percent); silica gel TLC Rf 0.22 (1:9 ethyl acetate−hexanes); 1H NMR (CDCl3) δ 3.73 (s, 3H), 4.21 (s, 2H), 7.15-7.18(m, 2H), 7.30-7.45 (m, 6H) and 7.63-7.66 (m, 2H); 13C NMR (CDCl3)δ 52.0, 55.6, 127.6, 128.1, 128.69,128.75, 128.83, 130.5, 135.9, 139.2, 171.1 and 171.9; mass spectrum (APCI), m/z 254.1182 (M + H)+ (C16H16NO2 requires m/z 254.1181).
81%
at 25℃; for 24 h; Inert atmosphere
[0067] Methyl 2-(diphenylmethyleneamino)acetate (41). To a stirred suspension containing 5.00 g (39.9 mmol) of glycine methyl ester hydrochloride (40) in 20 mL of anhydrous CH2CI2 was added 6.70 mL (7.20 g, 39.9 mmol) of benzophenone imine drop wise. The white mixture was stirred at 25 °C for 24 h under argon atmosphere. The reaction mixture was filtered and concentrated under diminished pressure. [0068] The crude product was crystallized from ether-hexanes to afford 41 as white crystals; yield 8.20 g (81percent); silica gel TLC R{ 0.22 (1 :9 ethyl acetate-hexanes); XH NMR (CDCI3) δ 3.73 (s, 3H), 4.21 (s, 2H), 7.15-7.18 (m, 2H), 7.30-7.45 (m, 6H) and 7.63-7.66 (m, 2H); 1 C NMR (CDCI3) δ 52.0, 55.6, 127.6, 128.05, 128.69, 128.75, 128.83, 130.5, 135.9, 139.2, 171.1 and 171.9; mass spectrum (APCI), m/z 254.1182 (M + H)+ (Ci6Hi6N02 requires m/z 254.1181).
Reference:
[1] Bioorganic and Medicinal Chemistry Letters, 2001, vol. 11, # 15, p. 1975 - 1979
[2] Patent: WO2004/5293, 2004, A2, . Location in patent: Page 69-70
[3] Patent: WO2006/23630, 2006, A2, . Location in patent: Page/Page column 39-40; 65
[4] Chemistry - A European Journal, 2012, vol. 18, # 12, p. 3773 - 3779
[5] Organic Process Research and Development, 2018, vol. 22, # 10, p. 1458 - 1460
[6] Patent: EP3257857, 2017, A1, . Location in patent: Paragraph 0084; 0085
[7] Journal of Organic Chemistry, 1982, vol. 47, # 13, p. 2663 - 2666
[8] Chemistry - A European Journal, 2010, vol. 16, # 4, p. 1153 - 1157
[9] European Journal of Organic Chemistry, 2008, # 14, p. 2375 - 2387
[10] Bioorganic and Medicinal Chemistry Letters, 2015, vol. 25, # 21, p. 4715 - 4718
[11] Journal of Organic Chemistry, 2015, vol. 80, # 23, p. 11755 - 11767
[12] Patent: WO2016/118877, 2016, A1, . Location in patent: Paragraph 0067; 0068
[13] Organic and Biomolecular Chemistry, 2011, vol. 9, # 6, p. 1823 - 1830
[14] Angewandte Chemie - International Edition, 2018, vol. 57, # 19, p. 5350 - 5354[15] Angew. Chem., 2018, vol. 130, # 19, p. 5448 - 5452,5
[16] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 5, p. 1688 - 1691
[17] Journal of Medicinal Chemistry, 2005, vol. 48, # 19, p. 5932 - 5941
[18] Phosphorus and Sulfur and the Related Elements, 1987, vol. 34, p. 93 - 104
[19] Journal of Organic Chemistry, 2009, vol. 74, # 4, p. 1627 - 1631
[20] Patent: US2005/239857, 2005, A1, . Location in patent: Page/Page column 62
[21] Organic Letters, 2010, vol. 12, # 4, p. 708 - 711
[22] Journal of the American Chemical Society, 2016, vol. 138, # 1, p. 265 - 271
32
[ 119-61-9 ]
[ 5680-79-5 ]
[ 81167-39-7 ]
Reference:
[1] European Journal of Organic Chemistry, 2017, vol. 2017, # 3, p. 695 - 703
[2] Patent: WO2010/123792, 2010, A1, . Location in patent: Page/Page column 11-13
[3] Revue Roumaine de Chimie, 2010, vol. 55, # 10, p. 689 - 695
[4] Angewandte Chemie - International Edition, 2013, vol. 52, # 49, p. 12942 - 12945[5] Angew. Chem., 2013, vol. 125, # 49, p. 13180 - 13183,4
[6] Journal of Materials Chemistry C, 2018, vol. 6, # 5, p. 1071 - 1082
With sodium carbonate; In dichloromethane; at 20℃;Cooling with ice;
General procedure: Syntheses of G(2-4) and P(2-4): General experimental procedure is as follows: Glycine or L-phenyl alanine derivative (as HCl salt) (10 mmol) and Na2CO3 (2.1 g, 20 mmol) were dissolved in anhydrous dichloromethane (50 mL) and cooled in an ice bath before tosyl chloride (1.9 g, 10 mmol) was added. The resultant reaction mixture was stirred overnight at room temperature. After the reaction, more solvent was added and the residue was washed sequentially with 0.1 M HCl, saturated NaHCO3 and brine. Organic portion was dried with Na2SO4 and concentrated in vacuo to give crude product. The product was purified by column chromatography on silica with hexane and EtOAc (1:1). Compound G2: Yield 78% as white solid. 1H NMR (400 MHz,CDCl3): delta 7.74 (d, J= 8.3 Hz, 2H, ArH), 7.31 (d, J= 8.3 Hz, 2H, ArH), 5.04 (t, J= 5.1 Hz, 1H, -NH-), 3.78 (d, J= 5.1 Hz, 2H, -CH2-), 3.64 (s, 3H, -OCH3), 2.42 (s, 3H, -CH3) ppm. 13C NMR (100 MHz, CDCl3): delta 169.4, 144.0, 136.3, 129.9, 127.4, 52.7, 44.2, 21.7 ppm. Anal.Calcd. [C10H13NO4S]: C, 49.37; H, 5.39; N, 5.76; S, 13.18. Found:C, 50.63; H, 5.09; N, 5.36; S, 12.87.
The four-mouth flask equipped with a stirrer was added methyl 3-chlorosulfonyl-2-thiophenecarboxylate 8 g and water 20 ml. At room temperature, add dropwise glycine methyl ester hydrochloride 4.5 g aqueous solution (10 ml). At the same time, add dropwise 10% acid binding agent aqueous solution 30 ml, control pH to 7-8. After the completion of the addition, reaction at room temperature for 6 hours, filtering, the filter cake after the water washing, drying after recrystallization with methanol, getting white solid TNXK-1 to 9 g, yield 92.3%.
With triethylamine; In dimethyl sulfoxide; at 140℃; for 0.5h;Microwave irradiation;
Example-88 (6-Methyl-5-nitro-2-pyridin-2-ylamino)-butionic methyl ester 6-chloro-3-nitro-2-picoline (600 mg, 3.5 mmol) was coupled with glycine methyl ester hydrochloride (880 mg, 7 mmol), triethylamine (1.5 ml, 10.5 mmol) in DMSO 3 mL at 140 °C for 30 min in microwave (Parameters : high absorbance, fixed holding time, pre- stirring 25 seconds). The crude mixture was treated with a saturated aqueous solution of NH4Cl. The aqueous solution of was extracted with EtOAc, washed with water and brine. The crude product was purified on a silica column with CH2Cl2-MeOH as mobile phase. This gave 39 mg (51percent) of 580 mg (74percent) of (6-methyl-5-nitro-2-pyridin-2-ylarcino)- budonic methyl ester as a yellow solid.
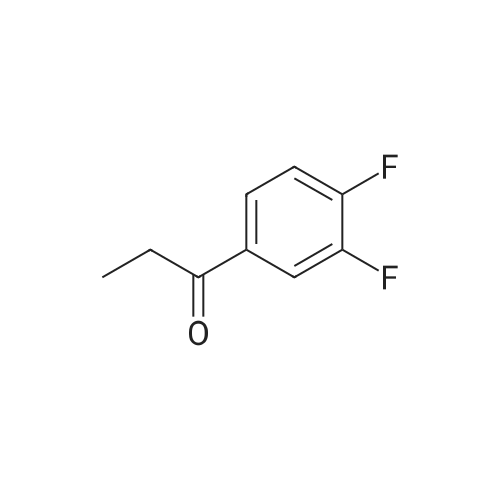
a. Preparation of intermediate 35 Following procedure was done 3 x. [A mixture of <strong>[23384-72-7]1-(3,4-difluorophenyl)-1-propanone</strong> (0.43 mol), glycine methyl ester, hydrochloride (0.5 mol) and KF (0.43 mol) in CH30H (700 ml) was hydrogenated at 50 C (in Parr apparatus) with Pd/C 10% (5 g) as a catalyst in the presence of thiophene solution (2 ml). After uptake of H2 (1 equivalent) was complete, the catalyst was filtered off and the filtrate was evaporated. The residue was stirred in water, then treated with NaHC03 (q.s.) and the product was extracted with CH2Cl2. The organic layer was separated, dried, filtered and the solvent evaporated The residue was dissolved in 2-propanol (1000 ml) and converted into the hydrochloric acid salt with 6 N HCl/2-propanol. The precipitate was filtered off, washed with DIPE (to remove the 2-propanol), then stirred in water. CH2Cl2 was added and the mixture was treated with K2C03 (q.s.). The layers were separated The organic layer was dried, filtered and the solvent evaporated. ] Yield: 221 g of intermediate 35.
N-[ (2-oxo-1-pyrrolidinyl)acetyl]glycine methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With triethylamine; In N-methyl-acetamide;
Reference Example 2 Preparation of N-[(2-oxo-1-pyrrolidinyl)acetyl]glycine (the starting carboxylic acid used in Example 5) 55 g of (2-oxo-1-pyrrolidinyl)acetic acid was dissolved in 1 l of dimethylformamide and 39.3 g of triethylamine was added thereto. After the solution was cooled to -8° C., 55.1 g of isobutyl chloroformate was added at -8° to -1° C. Then the reaction liquid was stirred at 0° to -7° C. for 10 minutes, 50.7 g of glycine methyl ester hydrochloride was added at -7° to -4° C. and stirred at -4° to -8° C. for 6 minutes. Next, 40.8 g of triethylamine was added at -8° to -3° C. The reaction liquid was allowed to stand so as to rise its temperature to room temperature over 2 hours and triethylamine hydrochloride was filtered off. Dimethylformamide was distilled off under reduced pressure and the residue was purified through the chromatography on silica gel (silica gel 1200 g, chloroform-1 to 3percent of methanol as the eluent) to obtain 70.5 g of N-[(2-oxo-1-pyrrolidinyl)acetyl]glycine methyl ester as the oily substance.
,1 -Dimethylethyl (2f?)-2-formyl-1-pyrrolidinecarboxylate (1. g, 5.02 mmol) was dissolved in 20 ml of anhydrous dichloroethane. Methyl glycinate hydrochloride (0.95 g, 7.57 mmol) was added, followed after 30 min by sodium triacetoxyborohydride (2.2 g, 10.38 mmol). The reaction was stirred for 8hrs at room temperature. The reaction mixture was diluted with methanol to obtain a clear solution, which was passed through a SCX cartridge. The product was eluted with 1 M methanolic ammonia. The product was isolated by EPO <DP n="43"/>chromatography (silica, dichloromethane/methanol 95/5) as colourless oil: 800 mg, 2.94 mmol.MS (ES/+): m/z = 273 [M+H]+.NMR (CDCI3): delta (ppm): 4.01 (bs, 1 H); 3.75 (s, 3H); 3.53-3.30 (m, 4H); 2.94-2.53 (m, 3H);1.92-1.76 (m, 3H); 1.48 (s, 9H).Rf: 0.40 (cyclohexane/EtOAc 1/1 )
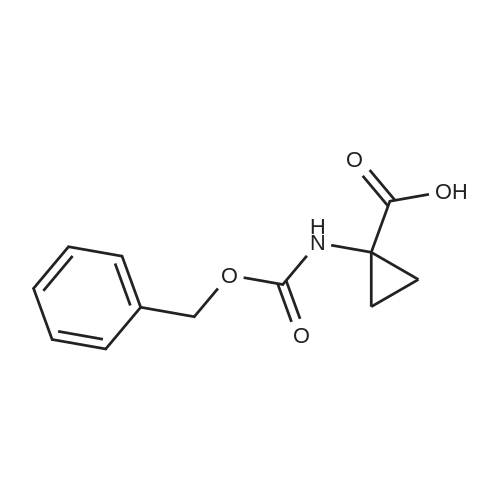
Example 2 Benzyl-5-(5-[(2-aminophenyl)amino]carbonyl}pyridin-2-yl)-2,5-diazabicyclo[4.2.0]octane-2-carboxylate bis-trifluoroacetateA solution of the Cbz-protected cyclopropylglycine (5.00 g, 21.3 mmol) in 25 mL of DMF was treated with EDC (5.50 g, 29.0 mmol), HOBt (3.50 g, 25.9 mmol), i-Pr2NEt (6 mL, 34 mmol) and glycine methyl ester hydrochloride (3.50 g, 24.0 mmol) and finally stirred for 15 h. The reaction mixture was poured into EtOAc and washed with 2 N HCl, 2 N NaOH, brine, dried Na2SO4 and concentrated giving a white foam. The material was dissolved in 25 mL of EtOH and treated with 5% Pd/C (2.00 g, 0.94 mmol), vacuum/hydrogen gas exchange, then stirred with H2 balloon for 5 h, filtered and concentrated. The foamy residue was heated to 200 C. for 5 min giving a solid diketopiperazine.1H NMR (600 MHz, DMSO-d6) delta 8.23 (br s, 1H), 7.99 (br s, 1H), 3.82 (d, J=2.1 Hz, 2H), 1.12 (dd, J=7.9 Hz, 4.7 Hz, 2H), 0.88 (dd, J=7.3 Hz, 4.1 Hz, 2H).
A solution of the Cbz-protected cyclopropylglycine (5.00 g, 21.3 mmol) in 25 mL of DMF was treated with EDC (5.50 g, 29.0 mmol), HOBt (3.50 g, 25.9 mmol), /-Pr2NEt (6 mL, 34 mmol) and glycine methyl ester hydrochloride (3.50 g, 24.0 mmol) and finally stirred for 15 h. The reaction mixture was poured into EtOAc and washed with 2 N HCl, 2 N NaOH, brine, dried Na2SO4 and concentrated giving a white foam. The material was dissolved in 25 mL of EtOH and treated with 5% Pd/C (2.00 g, 0.94 mmol), vacuum/hydrogen gas exchange, then stirred with H2 balloon for 5 h, filtered and concentrated. The foamy residue was heated to 200 0C for 5 min giving a solid diketopiperazine.1H NMR (600 MHz, DMSO-J6) delta 8.23 (br s, IH), 7.99 (br s, IH), 3.82 (d, J= 2.1 Hz, 2H), 1.12 (dd, J= 7.9 Hz, 4.7 Hz, 2 H), 0.88 (dd, J= 7.3 Hz, 4.1 Hz, 2H).The diketopiperazine (100 mg, 0.71 mmol) was dissolved in DMF (10 mL) and treated with BoC2O (450 mg, 2.06 mmol), NEt3 (0.300 mL, 2.16 mmol) and DMAP (25 mg, 0.20 mmol). The mixture was stirred for 25 h, poured into EtOAc, washed with 2 N HCl, 2 N NaOH, brine, dried (Na2SO4), filtered and concentrated. A portion of this bis Boc-protected diketopiperazine (100 mg, 0.294 mmol) in 3 mL of THF was treated at -78 0C with 1 M DIBAL-H in toluene (1.50 mL, 1.50 mmol) and stirred for 1 h before quenching with methanol and allowing to warm to RT. The mixture was diluted with EtOAc and then washed with Rochelle's salt solution, brine, dried (Na2SO4), filtered and concentrated. The oily residue was dissolved in 3 mL Of CH2Cl2, cooled to -78 0C and treated with Et3SiH (0.25 mL, 1.60 mmol) and BF3»OEt2 (0.20 mL, 1.65 mmol). The reaction mixture was stirred for 2 h, then quenched with sat'd NaHCO3, dried (Na2SO4) and concentrated. The Boc groups were removed by stirring in 2 mL of 1:1 TFA/CH2C12 giving the bis-TFA salt of 2,5-diazabicyclo[4.2.0]octane: 1H NMR (600 MHz, CD3OD) delta 4.17 (m, 2H), 3.58 (m, 2H), 3.41 (m, 2H), 2.45-2.35 (m, 4H). The residue was treated with the nicotinyl chloride methyl ester (50 mg, 0.29 mmol) andNEt3 (0.20 mL, 1.44 mmol) in 2 mL of DMSO and stirred at 100 0C for 1 h. When cool, CbzCI (0.10 mL) and NEt3 (0.20 mL) were added and the mixture stirred overnight, poured into EtOAc, washed with sat'd NaHCO3, dried (Na2SO4), filtered and concentrated. The residue was treated with LiOH (40 mg, 0.95 mmol) in 2 mL of 1:1 THF/water and stirred for 15 h, then concentrated and azeotropically dried <n="55"/>with methanol and benzene. A solution in 3 mL of DMF was stirred with EDC (225 mg, 1.18 mmol), HOBT (100 mg, 0.74 mmol) and 1,2-phenylenediamine (130 mg, 1.20 mmol) for 12 h and concentrated. Purification by reverse phase chromatography (10-100% MeCN/water with 0.05% TFA) gave the bis- TFA salt.1HNMR (600 MHz, CD3OD) delta 8.80 (d, J= 2.4 Hz, IH), 8.29 (dd, J= 9.4, 2.6 Hz, IH), 7.50-7.42 (m, 4H), 7.40-7.30 (m, 5H), 6.87 (d, J= 9.4 Hz, IH), 5.16 (AB, 2H), 4.70 (q, J= 7.0 Hz, IH), 4.52 (m, IH), 4.12 (m, IH), 3.60 (m, IH), 2.38 (m, IH), 2.28 (m, 2H), 2.02 (m, IH); MS (ESI+): cal'd [M+H]+ 458, exp. 458.
With potassium carbonate; In acetonitrile;Heating / reflux;
Reference Example 6 (6-ChIoro-2-morpholin-4-yl-Pyrimidin-4-ylamino)- acetic acid methyl ester (Intermediate Fl)Intermediate Al (400mg, 1.71mmol) and glycine methyl ester hydrochloride(1.3 equiv., 279mg) were refluxed in acetonitrile (6ml) with potassium carbonate (2.5 equiv., 591mg) overnight. The reaction mixture was then cooled to room temperature and diluted with water. The precipitate was filtered, washed with water, air-dried to give (6-chloro-2-morpholin-4-yl-pyrimidin-4-ylamino)-acetic acid methyl ester as a white solid (417mg).
Example 1: Synthesis of [l-(l,4-dimethyl-lH-indol-3-ylmethyl)-2,4-dioxo-l,4-dihydro-2H- pyrido[3,4-rf]pyrimidin-3-yl]-acetic acid; To a solution of lOOmg of 3-amino-isonicotinic acid in pyridine (2.5 mL) is added 117 mg of l,l'-carbonyldiimidazole and the mixture is stirred for 20 min at 55 0C. Then the mixture is cooled to room temperature and 91 mg of glycine methylester hydrochloride is added and the resulting mixture is stirred for 30 min at 55 0C and 1 h at room temperature. Then the solvent is removed under vaccum and the residue is purified by flash chromatography on silica gel to give 100 mg (66 percent) of [(3-amino-pyridine-4-carbonyl)-amino]-acetic acid methyl ester.
With triethylamine; In ethanol; at 50℃; for 1h;Inert atmosphere;
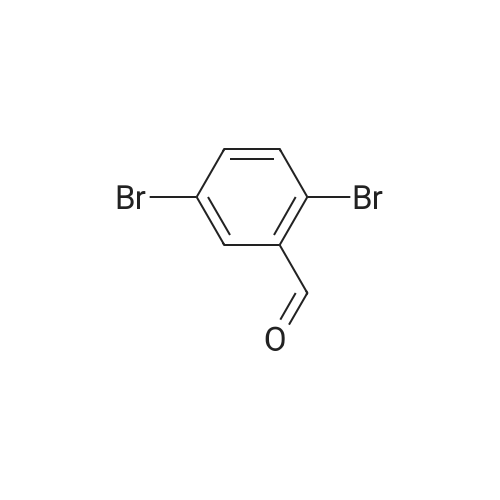
<strong>[158435-41-7]4-bromo-2-chlorobenzaldehyde</strong> (2B1 ) (4.56mmol, 1.0g) and glycine methyl ester hydrochloride (2C2) (4.56mmol, 572mg) were dissolved in ethanol (15 ml) under nitrogen with stirring and triethylamine (135mmol, 19.0ml, 13.7g) was added. The reaction mixture was heated to 50C for 1 hour, then evaporated to dryness and the residue taken up in methanol (15ml) and cooled in an ice bath. Sodium borohydride (5.01 mmol, 0.190g) was added portionwise and stirring continued until no starting material remained as determined by TLC. The reaction mixture was concentrated to dryness and partitioned between DCM and water and the phases separated. The organic layer was dried over sodium sulphate, filtered and concentrated before purification on silica eluting with 15% EtOAc/heptane to yield methyl 2-(4-bromo-2-chlorobenzylamino)acetate (2C3) 420mg (31 %); 1H NMR (400MHz, CDCIs) delta 7.53 (s, 1 H) 7.38 (d, 1 H) 7.31 (d, 1 H) 3.86 (s, 2H) 3.73 (s, 3H) 3.43 (s, 2H).
2-(Methoxycarbonylmethyl-carbamoyl)-morpholine-4-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
64%
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In tetrahydrofuran; dichloromethane; at 20℃; for 48h;
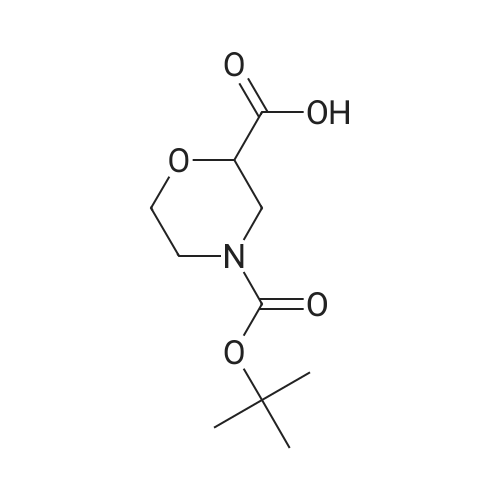
To a solution of Morpholine-2,4-dicarboxylic acid 4-tert-butyl ester (0.3 g, 1.29 mmol), glycine methyl ester hydrochloride (0.162 g, 1.29 mmol) and diisopropylethylamine (0.167 g, 1.29 mmol) in dichloromethane (8 mL) and THF (4 mL) was added EDCI (0.274 g, 1.43 mmol) followed by DMAP (0.048 g, 0.39 mmol).The reaction mixture was stirred at room temperature for 48 hours and the solvent evaporated in vacuo to give a solid. The solid was partitioned between dichloromethane and an aqueous 5% citric acid solution. The dichloromethane layer was washed with aqueous saturated sodium carbonate, brine, dried over Na2SO4, filtered, and the solvent evaporated in vacuo to yield the title compound as a colorless gum (0.25 g, 64% yield).1H NMR (400 MHz, CDCl3) delta 7.06 (br s, 1H), 4.33 (br s, 1H), 4.07 (d, 2H), 3.99-3.93 (m, 3H), 3.77 (s, 3H), 2.93-2.91 (m, 1H), 2.81-2.75 (m, 1H), 1.47 (s, 9H).LC/MS (m/z) [MNa]+ 325.2 (calculated for C13H22N2O6, 302.15).
With triethylamine; In methanol; at 60℃; for 3.0h;
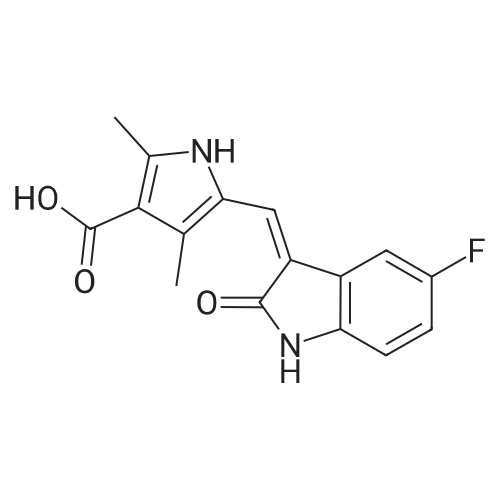
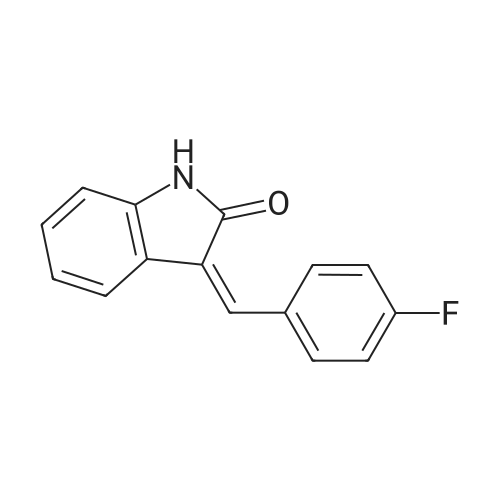
General procedure: A mixture of isatin (0.1 mmol), methyl glycinate 2a (1.0 eq.), methyleneindolinone 3a (1.0 eq.), Et3N (1.0 eq.), in methanol (1.0 mL) was stirred for 3 h at 60 C. After completion of the reaction (TLC), the solvent was removed under vacuum. The crude product was subjected to column chromatography on silica gel using pet/ethyl acetate (2:1) as the eluent to give 4a (43.4 mg, 99% yield).
With triethylamine; In methanol; at 60℃; for 3.0h;
General procedure: A mixture of isatin (0.1 mmol), methyl glycinate 2a (1.0 eq.), methyleneindolinone 3a (1.0 eq.), Et3N (1.0 eq.), in methanol (1.0 mL) was stirred for 3 h at 60 C. After completion of the reaction (TLC), the solvent was removed under vacuum. The crude product was subjected to column chromatography on silica gel using pet/ethyl acetate (2:1) as the eluent to give 4a (43.4 mg, 99% yield).
With triethylamine; In methanol; at 60℃; for 3.0h;
General procedure: A mixture of isatin (0.1 mmol), methyl glycinate 2a (1.0 eq.), methyleneindolinone 3a (1.0 eq.), Et3N (1.0 eq.), in methanol (1.0 mL) was stirred for 3 h at 60 C. After completion of the reaction (TLC), the solvent was removed under vacuum. The crude product was subjected to column chromatography on silica gel using pet/ethyl acetate (2:1) as the eluent to give 4a (43.4 mg, 99% yield).
In toluene; at 200℃; for 5h;Microwave irradiation;
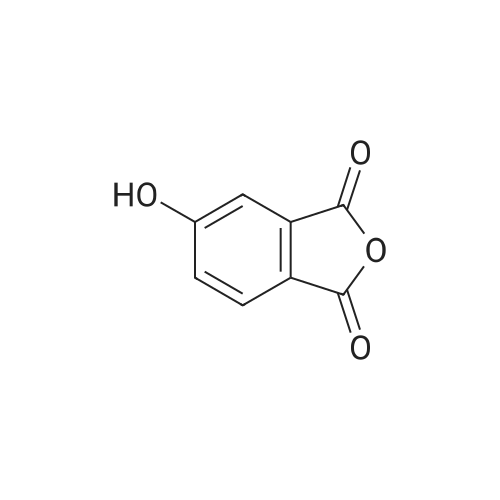
In a MW vial <strong>[27550-59-0]5-hydroxyisobenzofuran-1,3-dione</strong> (561 mg, 3.42 mmol) and methyl 2-aminoacetate hydrochloride (386 mg, 3.08 mmol) were suspended in toluene (15 ml). The obtained mixture was heated under microwave irradiation at 200 C. for 5 hours. A mixture of the desired compound with an undesired byproduct (about 1:2 ratio) was obtained. After filtration, the obtained beige solid was purified by silica gel flash chromatography (petroleum ether/EtOAc 1/1 to EtOAc) to give 7 methyl 2-(5-hydroxy-1,3-dioxoisoindolin-2-yl)acetate (78 mg, 0.332 mmol, 9.70% yield, MS/ESI+ 235.9 [MH]+).
In toluene; at 200℃; for 5h;Microwave irradiation;
Step 2: Preparation of methyl 2-(5-hydroxy-1 ,3-dioxoisoindolin-2- yl)acetate (245): In a MW vial 5-hydroxyisobenzofuran-1 ,3-dione (561 mg, 3.42 mmol) and methyl 2-aminoacetate hydrochloride (386 mg, 3.08 mmol) were suspended in toluene (15 ml). The obtained mixture was heated under microwave irradiation at 200C for 5 hours. A mixture of the desired compound with an undesired byproduct (about 1 :2 ratio) was obtained. After filtration, the obtained beige solid was purified by silica gel flash chromatography (petroleum ether/EtOAc 1 /1 to EtOAc) to give 7 methyl 2-(5-hydroxy-1 ,3-dioxoisoindolin-2-yl)acetate (78 mg, 0.332 mmol, 9.70% yield, MS/ESI+ 235.9 [MH] +).
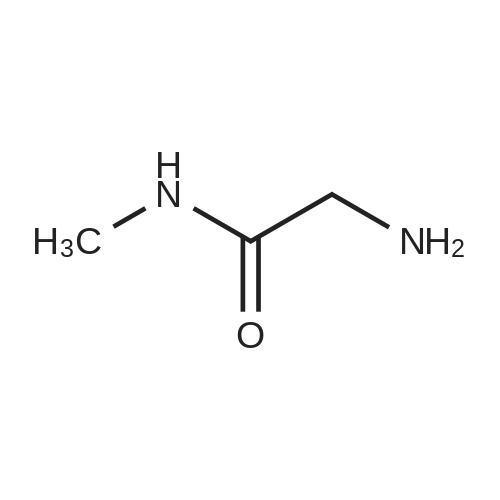
Into a 250-mL round-bottom flask, a solution of methyl 2-aminoacetate hydrochloride (2.0 g, 15.93 mmol, 1.00 equiv) in 100 mL of anhydrous methanol was added CH3NH2 (2 M in THF, 35.7 mL) and the resulting solution was stirred for 48 h at room temperature under nitrogen. After completion of the reaction, the resulting mixture was concentrated under reduced pressure to afford 2-amino-N-methylacetamide hydrochloride (2.5 g, crude) as a colorless oil containing large amounts of methylamine.
methyl N-(3-nitro-5-(trifluoromethyl)pyridin-2-yl)glycinate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
5.09 g
With triethylamine; In ethanol; for 5h;Reflux;
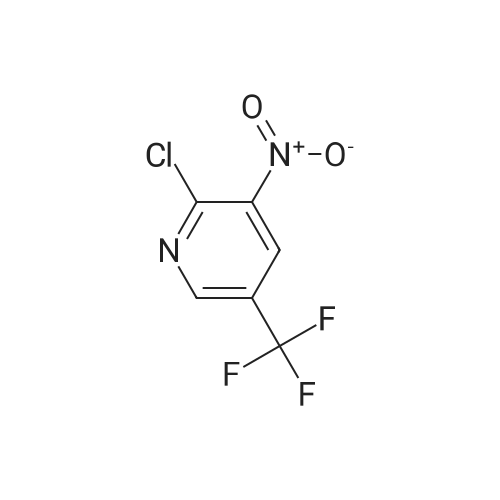
A) methyl N-(3-nitro-5-(trifluoromethyl)pyridin-2-yl)glycinate To a solution of methyl glycinate hydrochloride (13.9 g) and <strong>[72587-15-6]2-chloro-3-nitro-5-(trifluoromethyl)pyridine</strong> (5.0 g) in ethanol (100 mL) was added triethylamine (15.4 mL). The reaction mixture was heated at reflex for 5 hr, and the solvent was evaporated under reduced pressure. The residue was triturated with water, and the solid was collected by filtration, and washed with ethanol to give the title compound (5.09 g). 1H NMR (400 MHz, CDCl3) delta 3.82 (3H, s), 4.43 (2H, d, J = 5.2 Hz), 8.63 (1H, d, J = 2.0 Hz), 8.68 (1H, d, J = 2.4 Hz), 8.74 (1H, s).
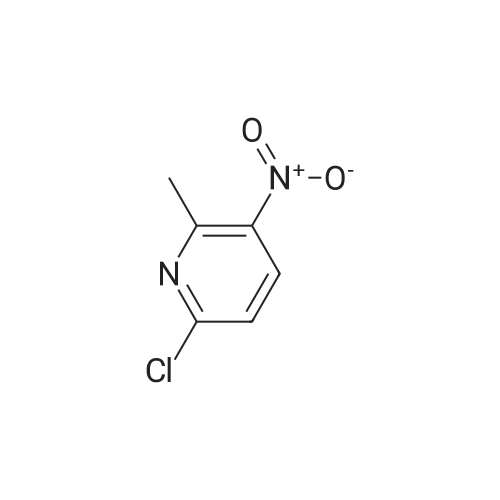
methyl N-(6-methyl-3-nitropyridin-2-yl)glycinate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
2.65 g
With triethylamine; In N,N-dimethyl-formamide; at 80℃;
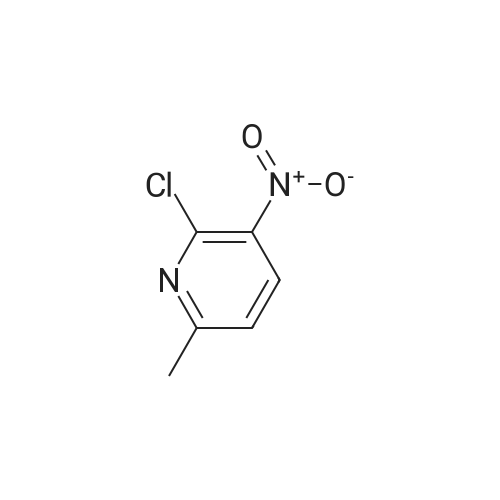
To a solution of methyl glycinate hydrochloride (1.91 g) and <strong>[56057-19-3]<strong>[56057-19-3]2-chloro-6-methyl-3-nitropyridin</strong>e</strong> (2.5 g) in N,N-dimethylformamide (16 mL) was added triethylamine (5.05 mL). The reaction mixture was stirred overnight at 80C, and the solvent was evaporated under reduced pressure. The residue was diluted with ethyl acetate, and the mixture was washed successively with water and saturated brine. The organic layer was dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give the title compound (2.65 g). 1H NMR (300 MHz, DMSO-d6) delta 2.38 (3H, s), 3.66 (3H, s), 4.29 (2H, d, J = 6.1 Hz), 6.70 (1H, d, J = 8.3 Hz), 8.34 (1H, d, J= 8.7 Hz), 8.76 (1H, t, J = 5.7 Hz).
4-amino-3-(N-(methoxycarbonylmethyl)-glyoxylamido)-pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
54%
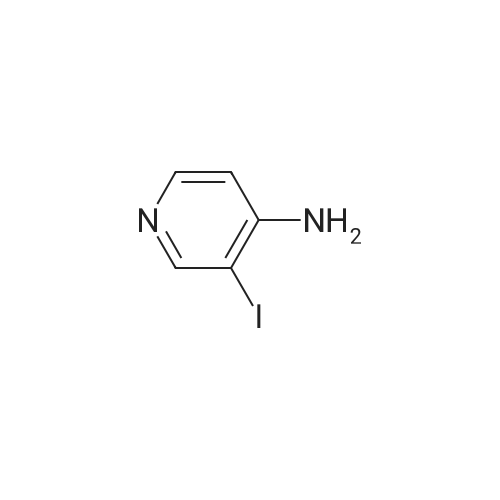
With palladium diacetate; triethylamine; triphenylphosphine; In N,N-dimethyl-formamide; at 50℃; under 30003.0 Torr; for 96h;Inert atmosphere; Autoclave;
General procedure: In a typical experiment Pd(OAc)2 (5.6 mg, 0.025 mmol), triphenylphosphine (13.2 mg, 0.05 mmol), <strong>[88511-27-7]4-amino-3-iodopyridine</strong>(1) (220 mg, 1.00 mmol), tert-butylamine (0.315 mL, 3.00 mmol)(or the given amount of primary or secondary amine (Table 1)) and triethylamine (0.5 mL) were dissolved in DMF (10 mL) under argon in a 100 mL autoclave. The atmosphere was changed to carbonmonoxide and pressurized to 40 bar. The reaction was conducted for the given reaction time upon stirring at 50C and analysed by TLC. The autoclave was vented, the reaction mixture was concen-trated and evaporated to dryness. The residue was dissolved in chloroform (20 mL) and washed with water (320 mL). The organic phase was dried over Na2SO4, filtered and evaporated to a solid material. All compounds were subjected to column chromatography ((Silicagel 60 (Merck), 0.063 e 0.200 mm); MeOH/CHCl3 orMeOH/EtOAc/CHCl3 (the exact ratios are specied in Characterization (4.4.) for each compound)).
With potassium carbonate; In 1,4-dioxane; water; at 20℃;
General procedure: Glycine methyl ester hydrochloride (1 eq) was dissolved in dioxane/water mixture (8:2 v/v; 10 ml/mmol). Potassium carbonate (3 eq) and the respective sulfonyl chloride (1.2 eq) were added at room temperature. The mixture was stirred vigorously overnight. The reaction was quenched by addition of water, acidified by means of diluted hydrochloric acid and extracted with EtOAc (3x25 ml). The combined organic layers were dried over Na2SO4 and evaporated. The residue could usually be used without purification or could be purified by flash chromatography (silica, heptane/EtOAc gradient) if necessary.
With benzotriazol-1-ol; O-(1H-benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate; triethylamine; In acetonitrile; at 20℃;Inert atmosphere;
rac-(2R,3S,4R,5S)-methyl 3-(tert-butyl)-5-(3-chloropyridin-2-yl)-4-nitropyrrolidine-2-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
37%
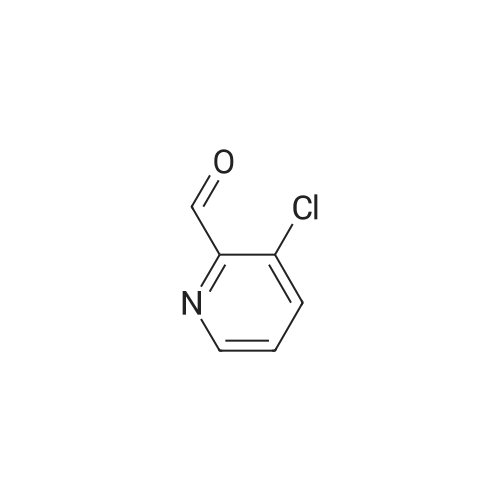
Methyl 2-aminoacetate hydrochloride (1.06 g, 8.48 mmol, 1.2 eq) was suspended in dry toluene (25 mL) and triethylamine (2.17 mL, 15.5 mmol, 2.2 eq) was added and the reaction mixture was stirred for 10 minutes. Then <strong>[206181-90-0]3-chloropyridine-2-carbaldehyde</strong> (1 g, 7.06 mmol, 1.00 eq) was added and the mixture was stirred at room temperature for 2 hours. The resulting mixture was filtered and filtrate was cooled to 0 C. in an ice bath. Triethylamine (986 muL, 7.1 mmol, 1 eq) was added. (E)-3,3-Dimethyl-1-nitrobut-1-ene (684.3 mg, 5.30 mmol, 0.75 eq) was added followed by silver acetate (AgCO2CH3, 1.18 g, 7.06 mmol, 1.00 eq). After 2 hours, the reaction was quenched with aqueous ammonium chloride solution. The reaction mixture was extracted with dichloromethane. The organic layer was passed through phase separator and concentrated in vacuo. The crude material was purified by flash chromatography (cyclohexane/ethyl acetate 100/0 to 40/60) to provide the title compound (900 mg, 37% over 2 steps). LC/MS (ESI+) m/z 342.21 (M+H)+.
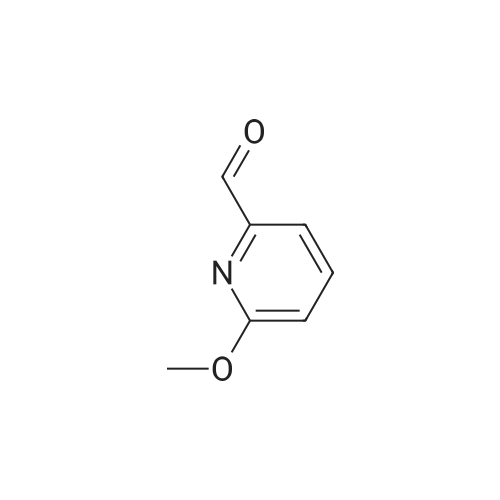
Glycine methyl ester hydrochloride (925 mg, 7.37 mmol, 1.1 eq) was added to a solution of triethylamine (1.07 mL, 8.04 mmol, 1.2 eq) in toluene (20 mL). The reaction mixture was stirred for 30 minutes on 3 A molecular sieves. Then <strong>[54221-96-4]6-methoxy-2-pyridinecarboxaldehyde</strong> (0.92 g, 6.70 mmol, 1.0 eq) was added and the reaction mixture was stirred at room temperature overnight. The reaction mixture was filtered, diluted with toluene and cooled to 0° C., and used in the next step without further purification.
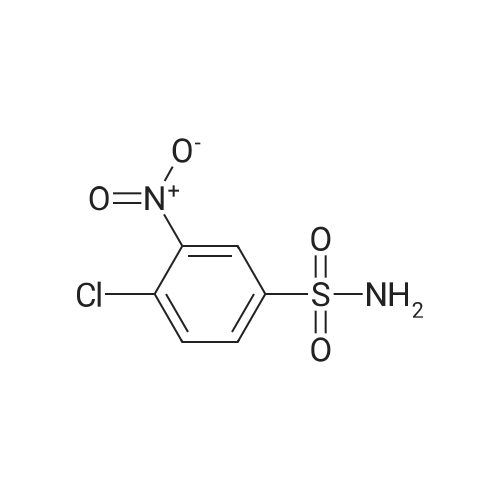
(2-nitro-4-sulfamoylphenyl)glycine methyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
76.7%
With N-ethyl-N,N-diisopropylamine; In acetonitrile; at 80℃; for 10h;
700mg 4- chloro-3-nitrobenzenesulfonyl chloride was dissolved in 20ml of acetonitrile were added 800mg glycine methyl ester hydrochloride and 1.64g DIEA, reacted at 80 10 hours; after cooling to room temperature the reaction with ethyl acetate. diluted with 1M citric acid and the organic phase was washed with saturated brine were washed twice, dried over anhydrous magnesium sulfate, and spin dry; purified by silica gel column chromatography to give a yellow solid 920 mg, 76.7% yield;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping