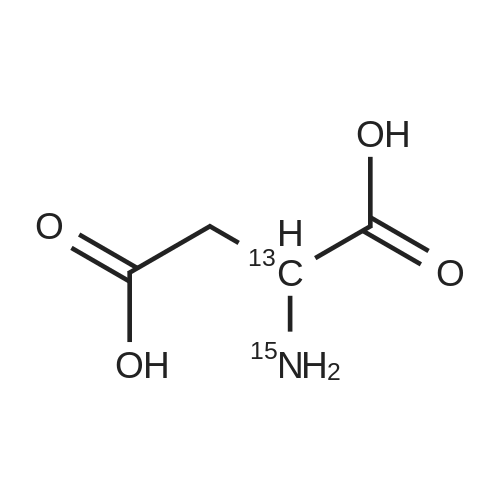
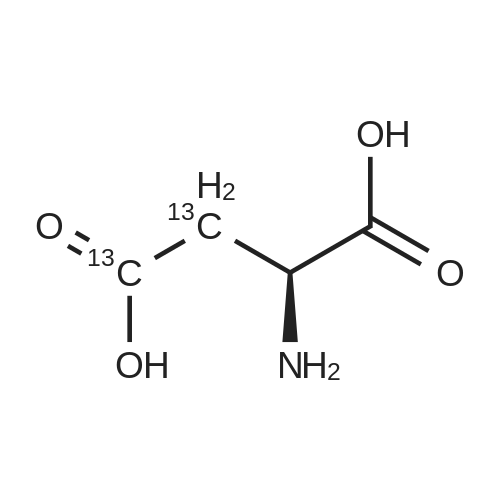
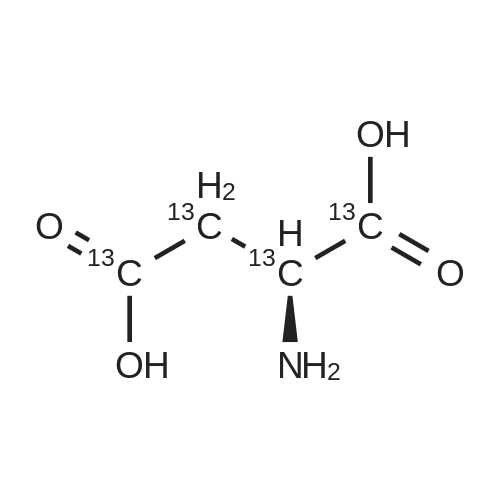
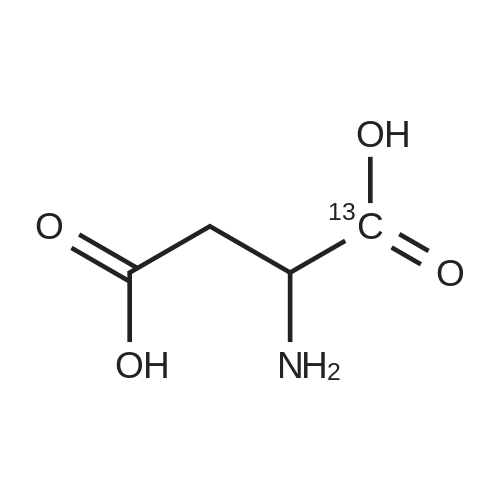
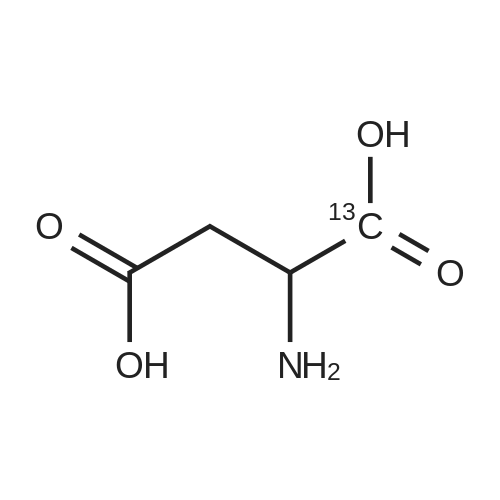
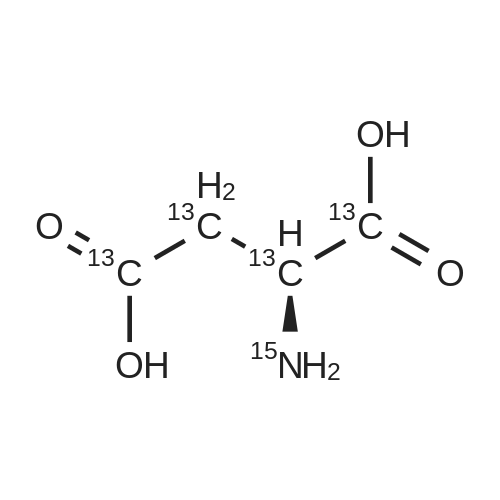
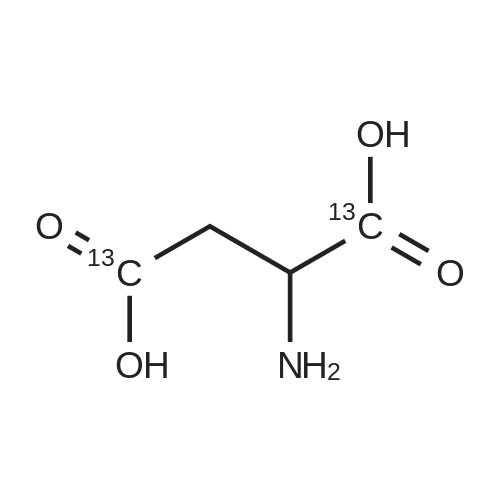
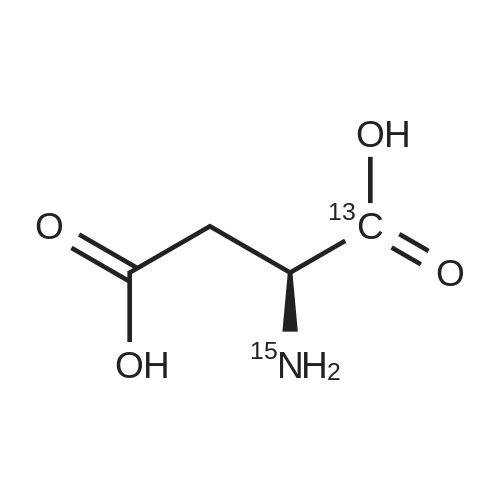
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of Physical Organic Chemistry, 2012, vol. 25, # 11, p. 939 - 945
17
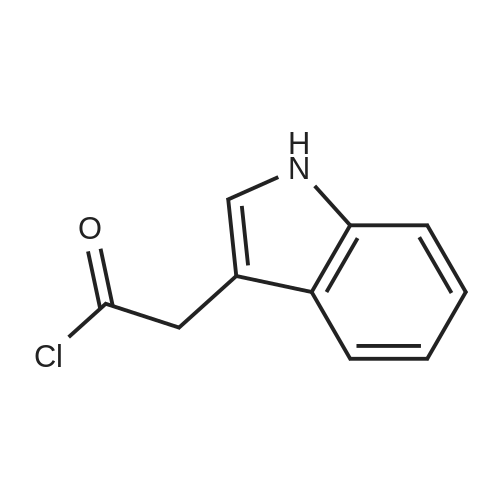
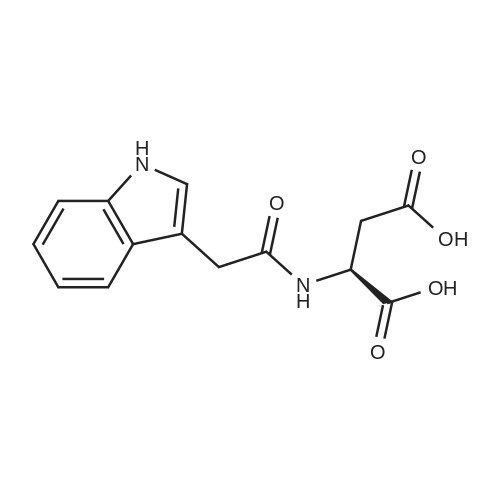
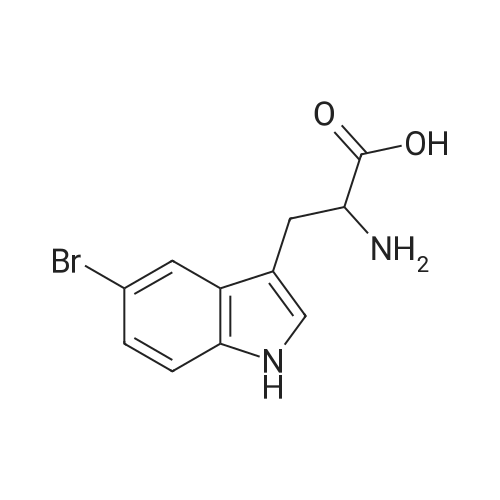
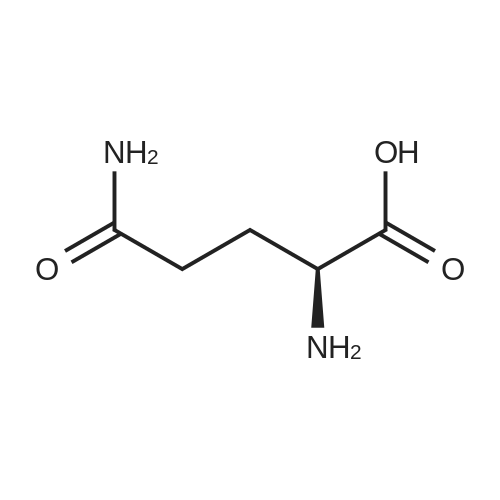
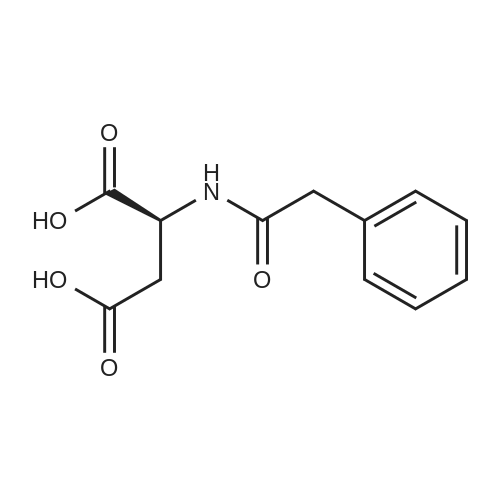
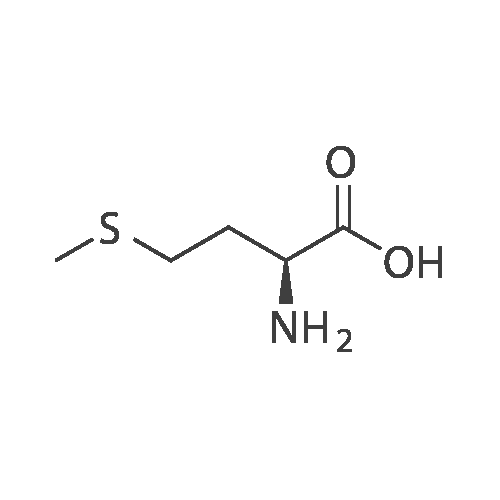
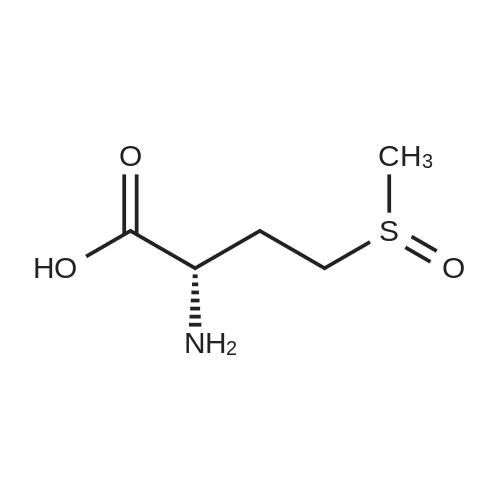
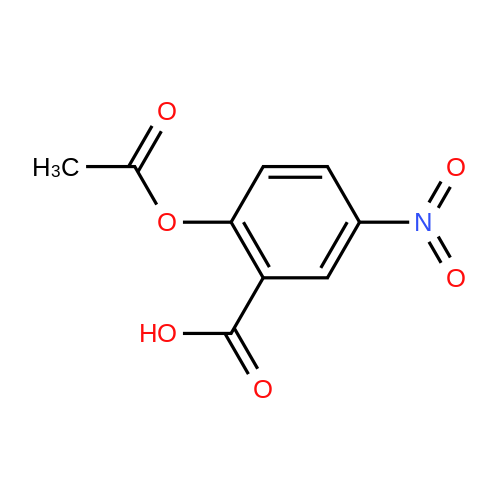
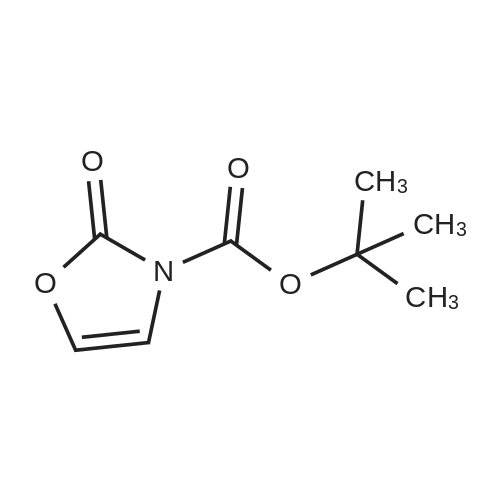
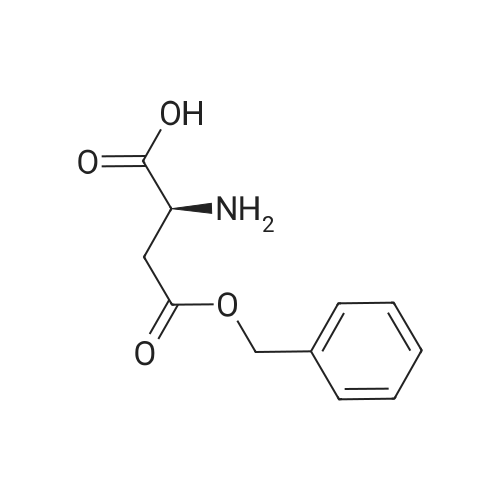
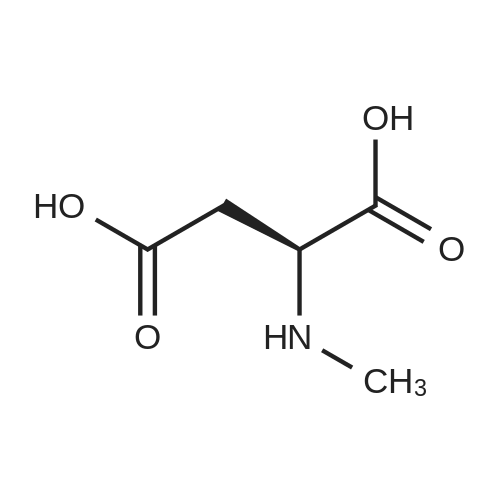
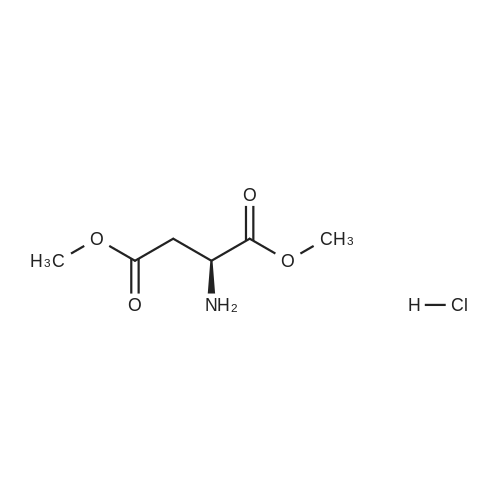
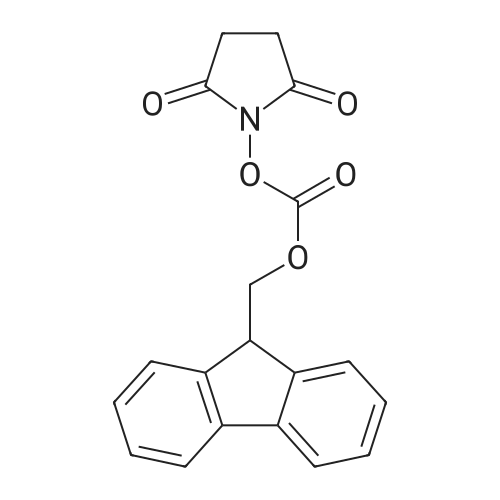
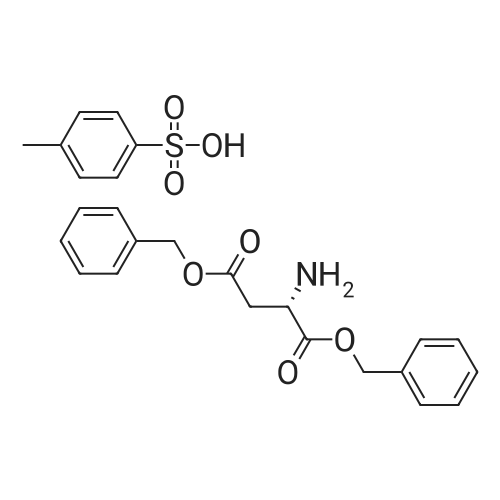
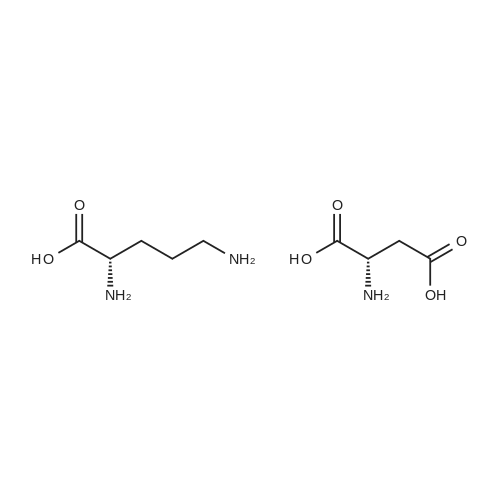
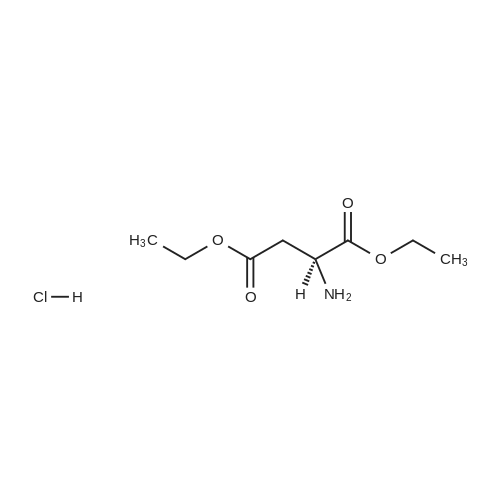
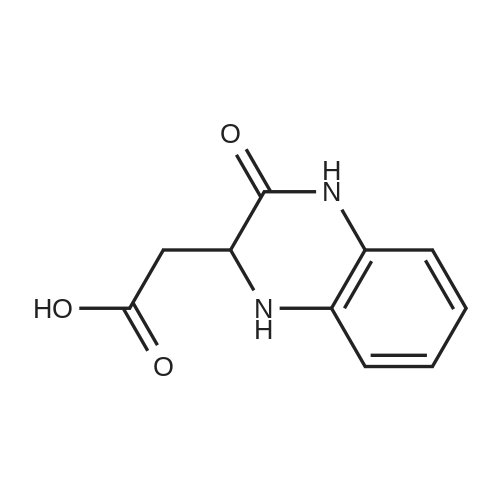
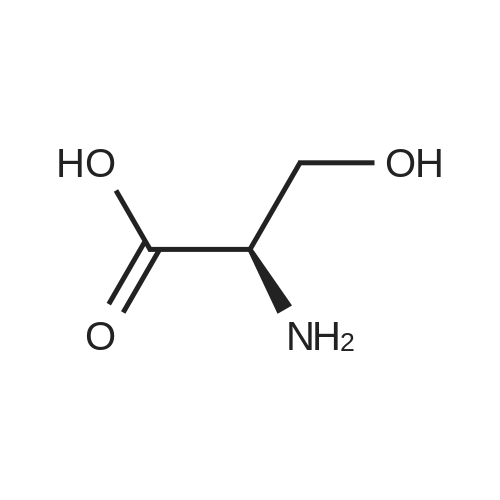
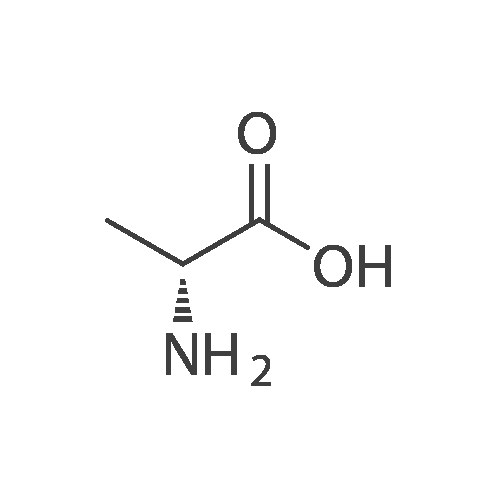
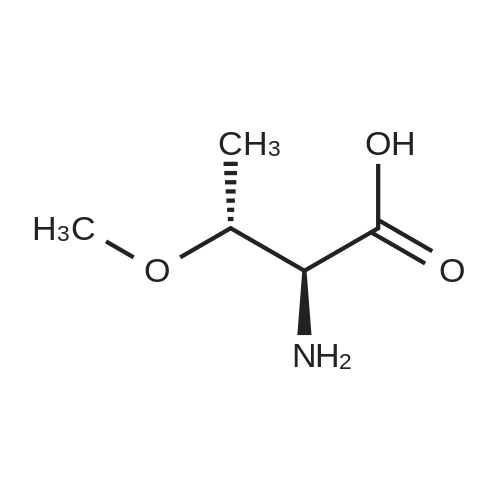
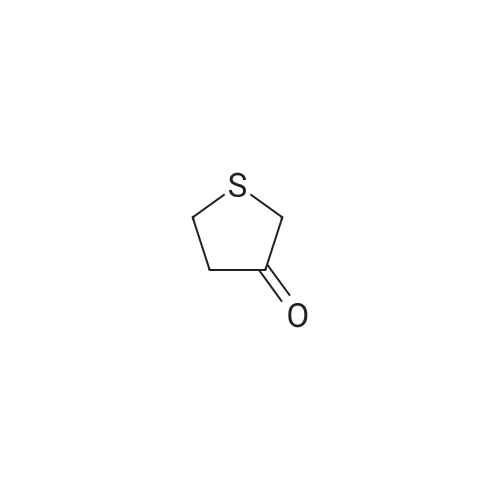
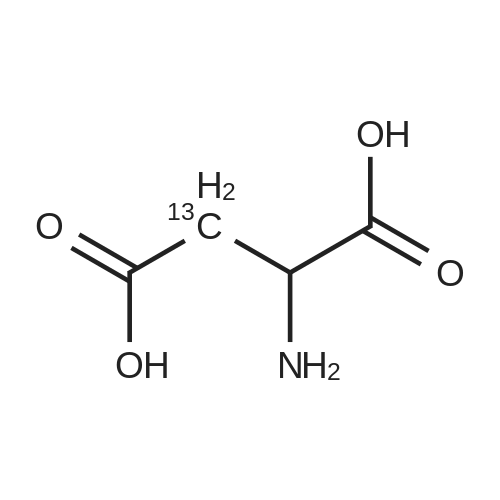
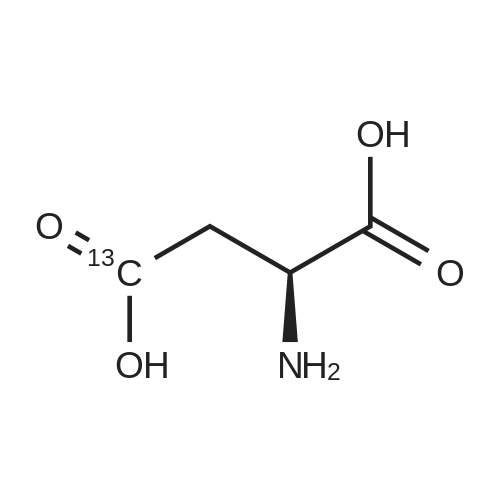
[ 56-84-8 ]
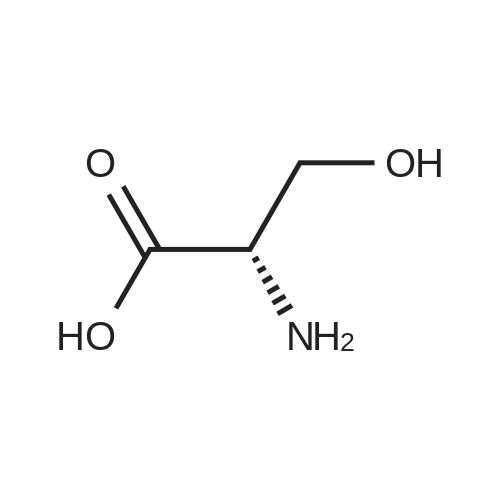
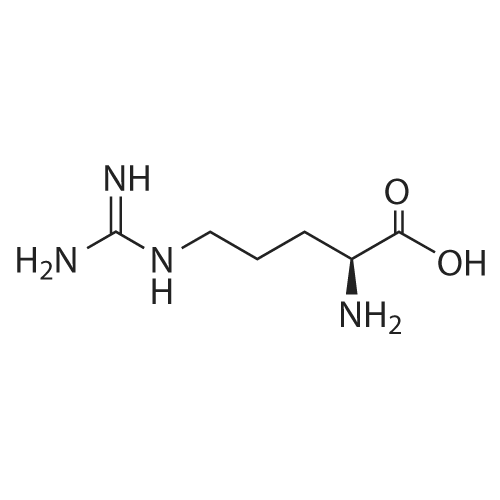
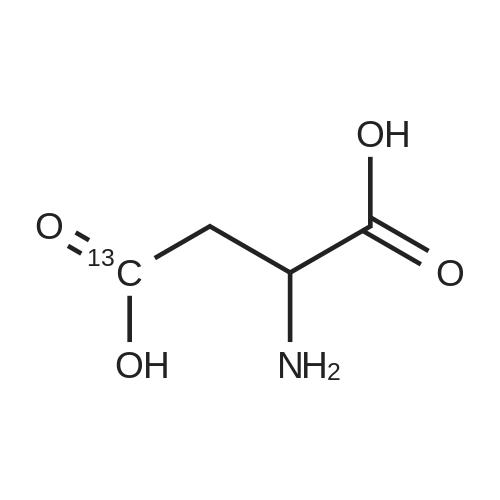
[ 1734-62-9 ]
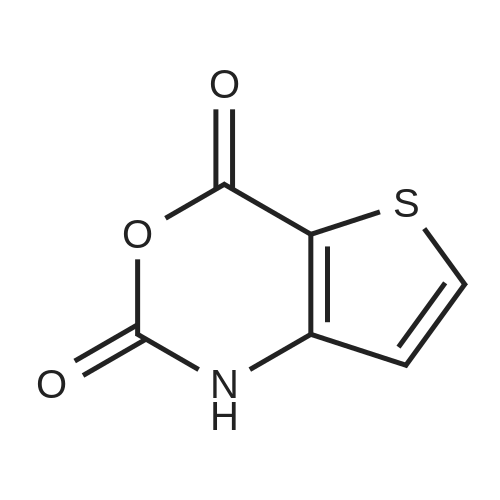
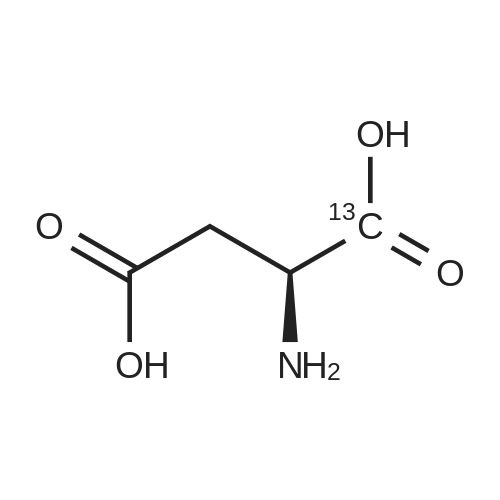
[ 997-55-7 ]
Reference:
[1] Journal of Physical Organic Chemistry, 2012, vol. 25, # 11, p. 939 - 945
18
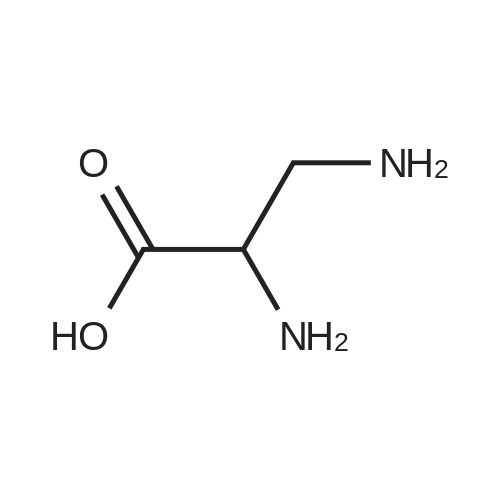
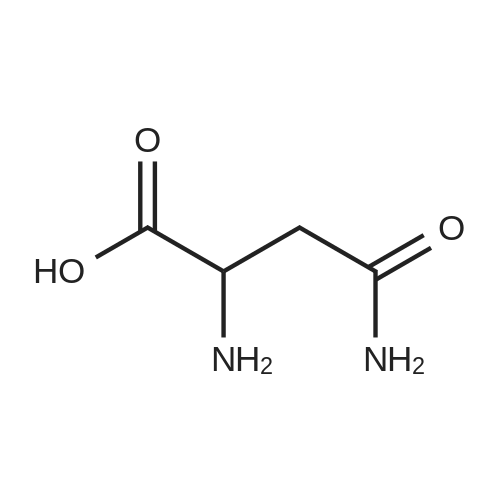
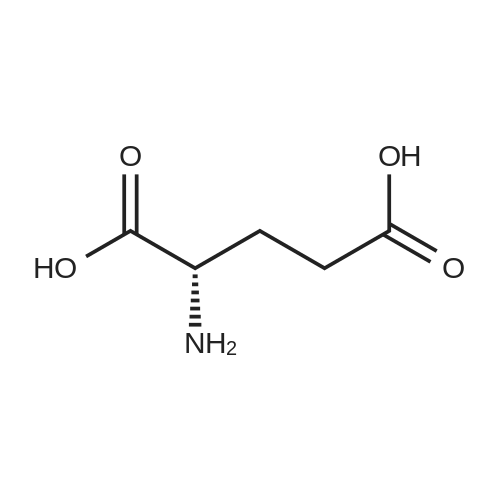
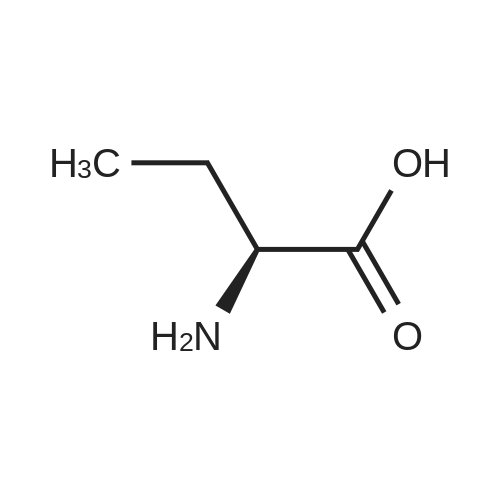
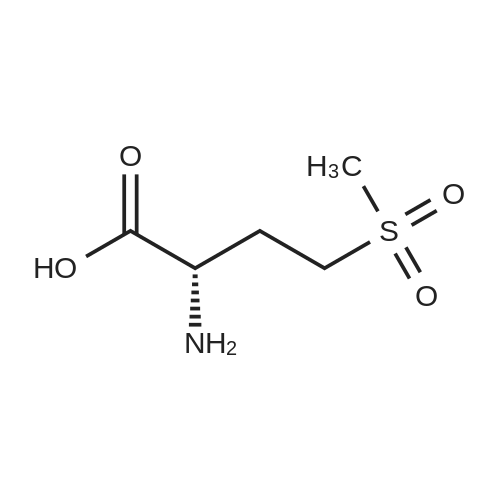
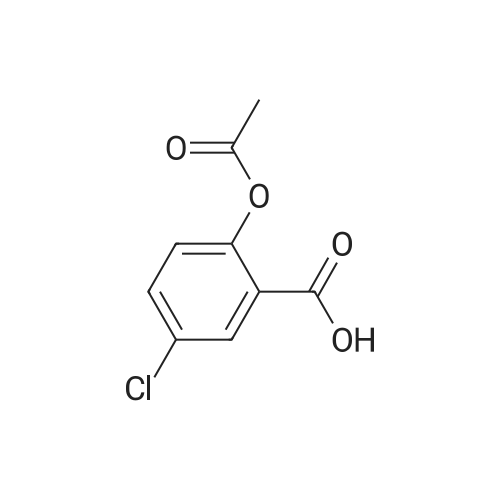
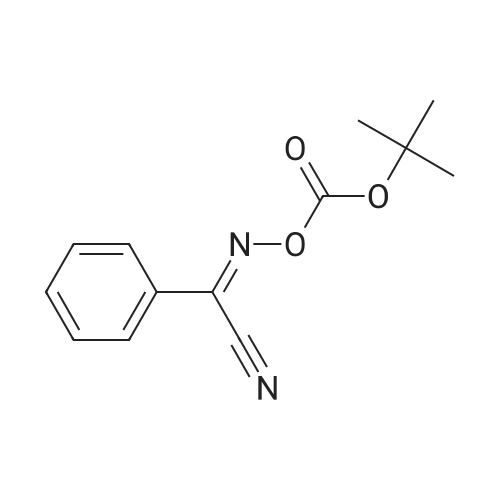
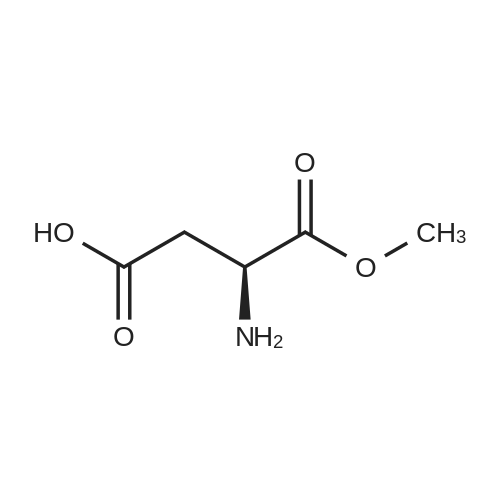
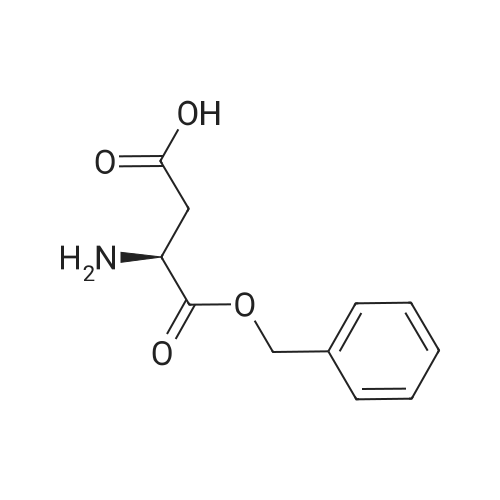
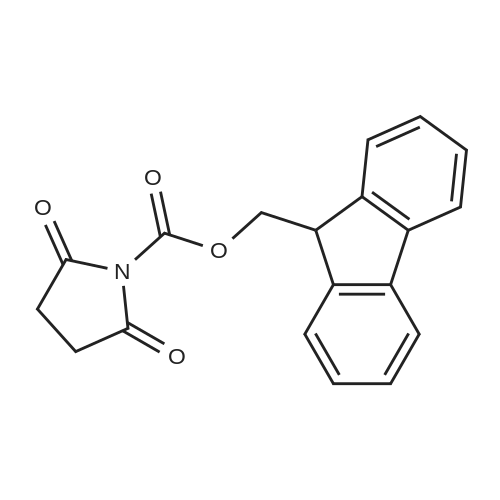
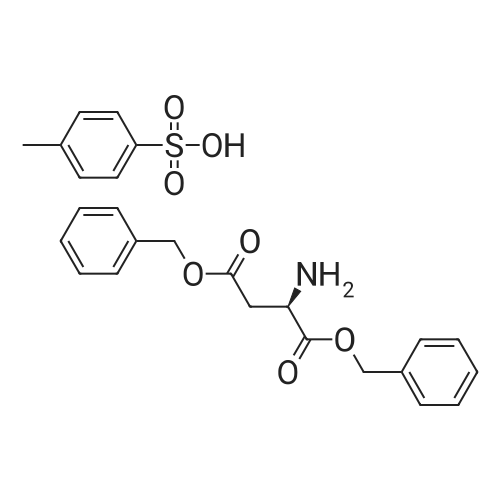
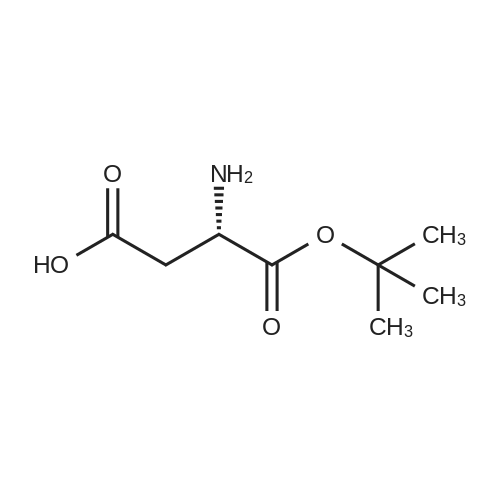
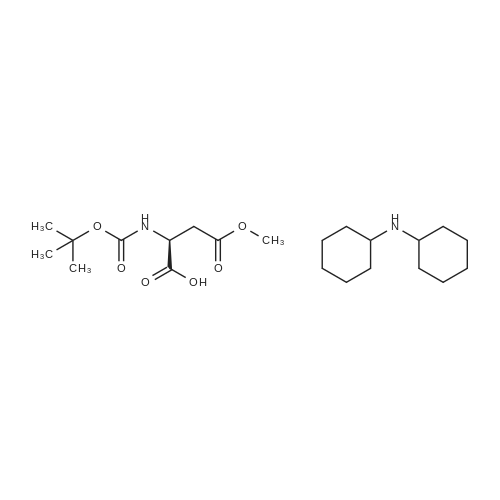
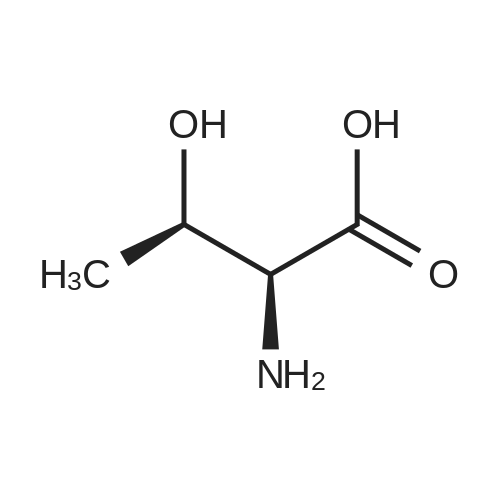
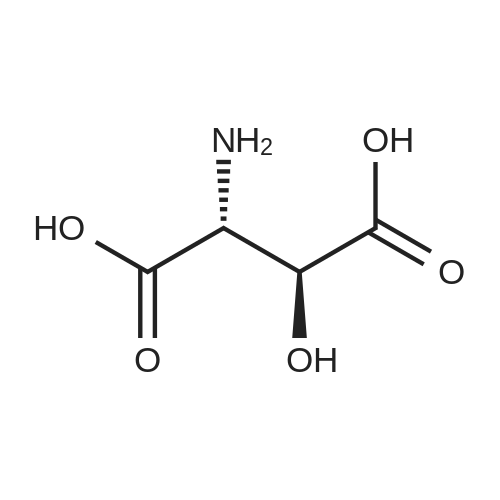
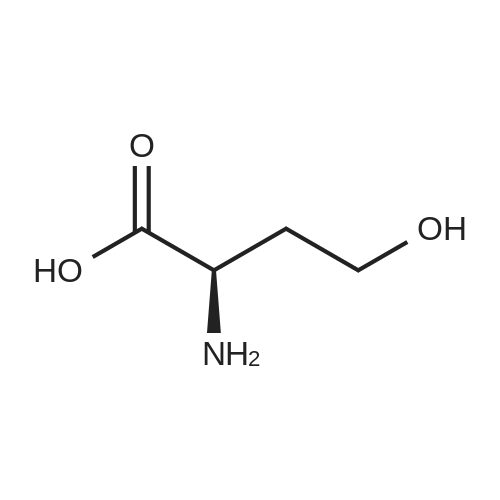
[ 56-84-8 ]
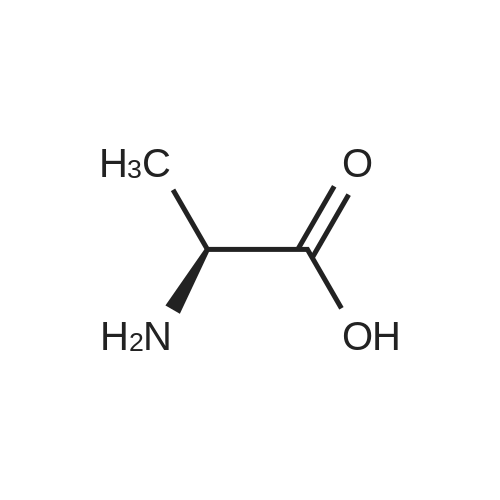
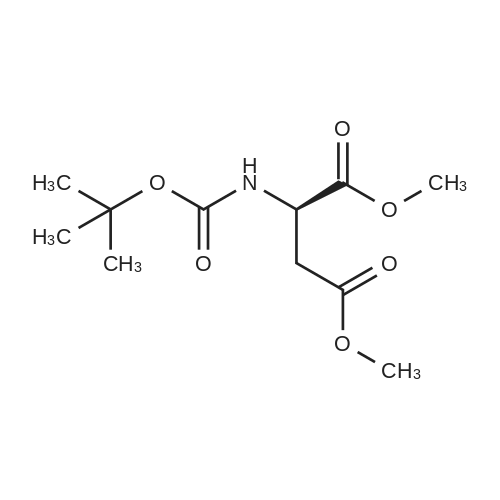
[ 50-78-2 ]
[ 997-55-7 ]
Reference:
[1] Journal of Physical Organic Chemistry, 2012, vol. 25, # 11, p. 939 - 945
19
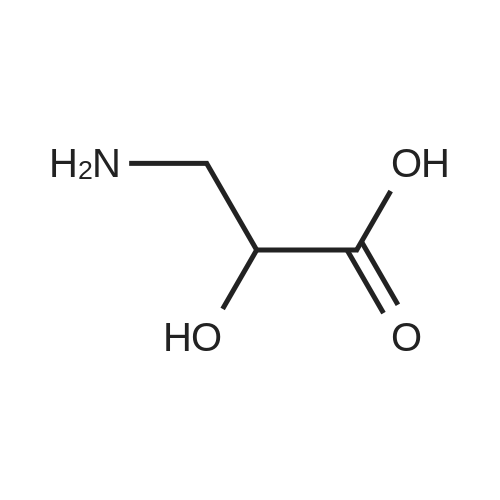
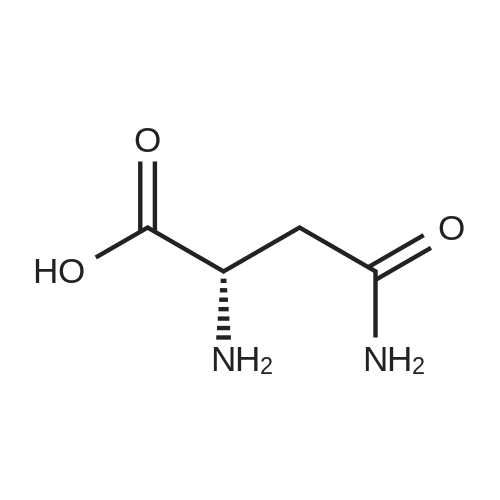
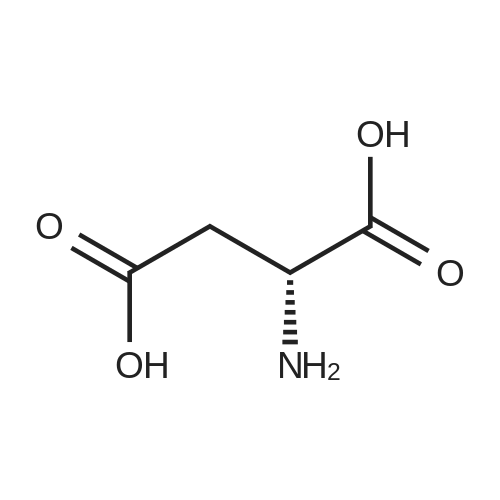
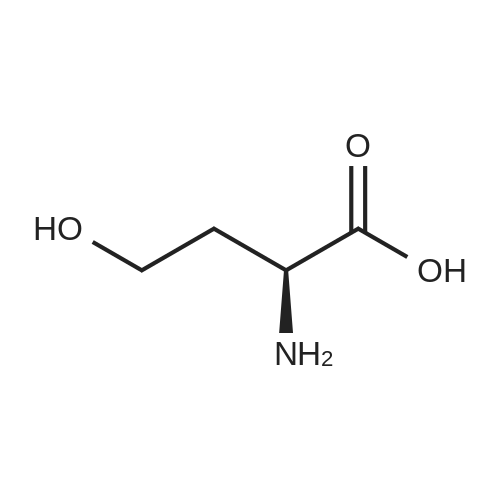
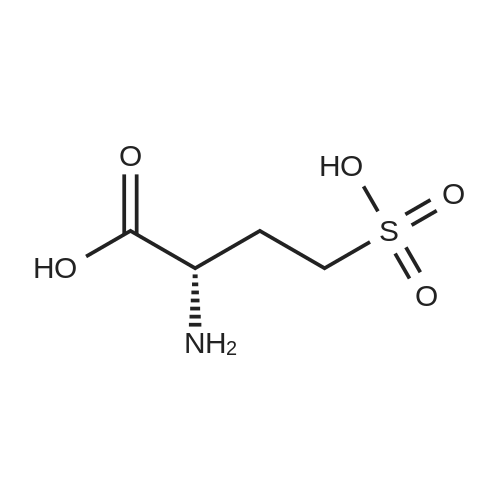
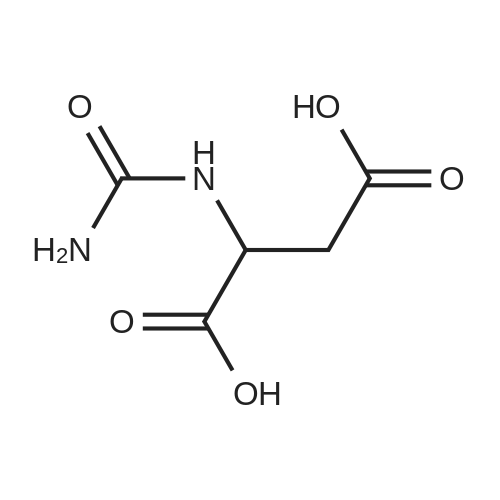
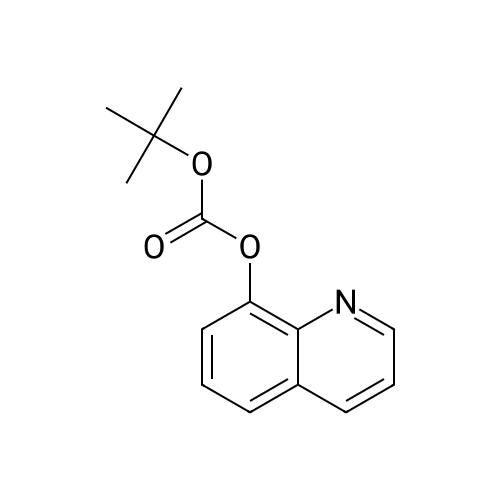
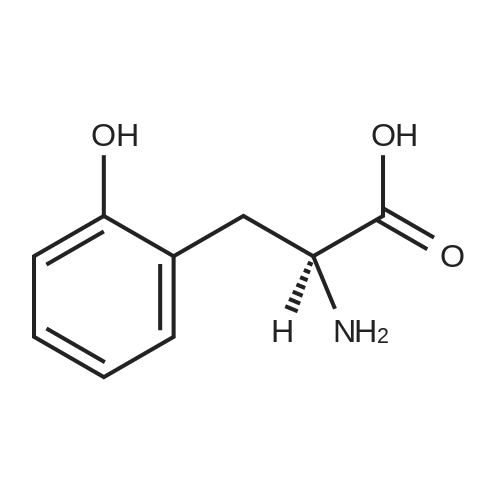
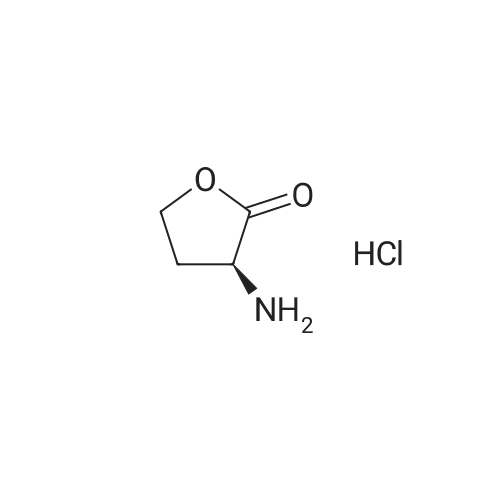
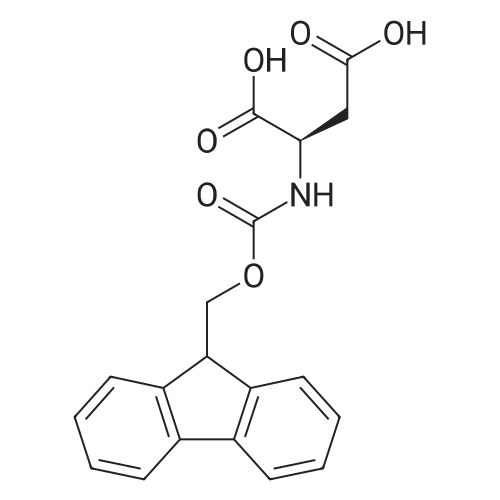
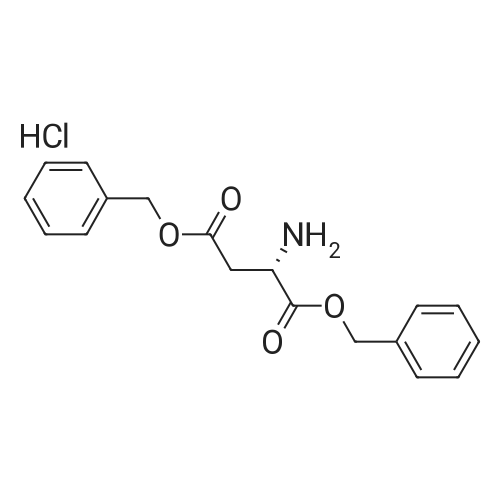
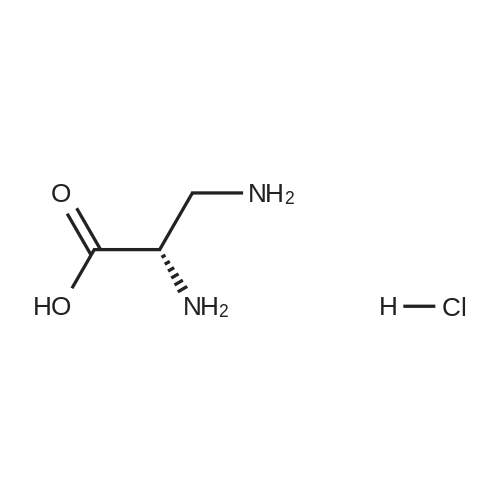
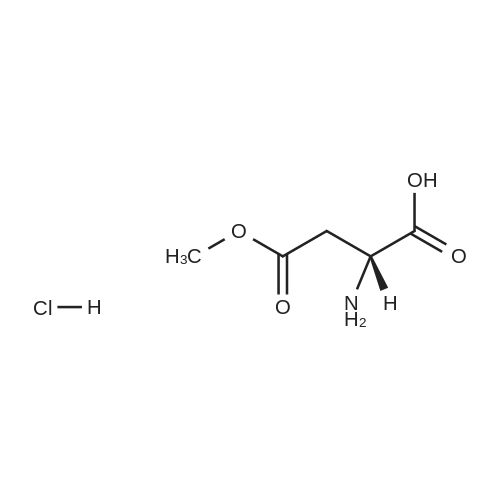
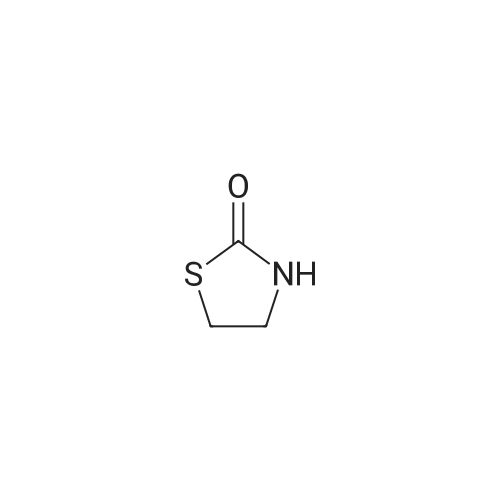
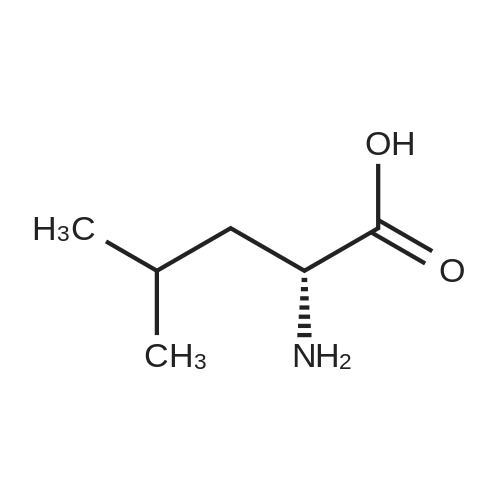
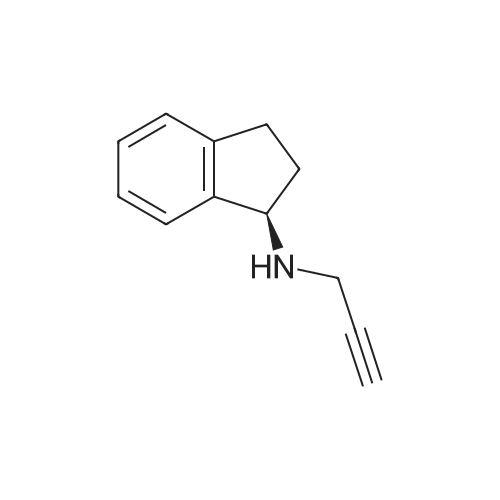
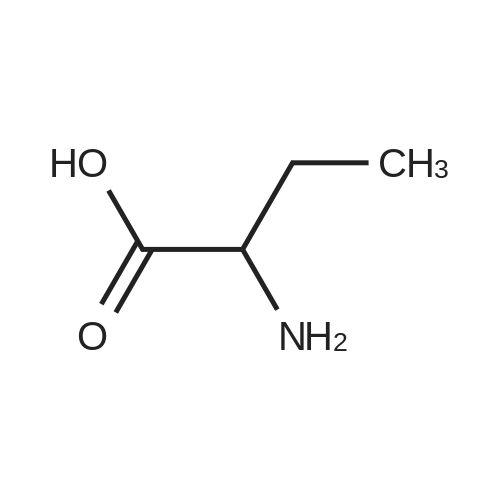
[ 56-84-8 ]
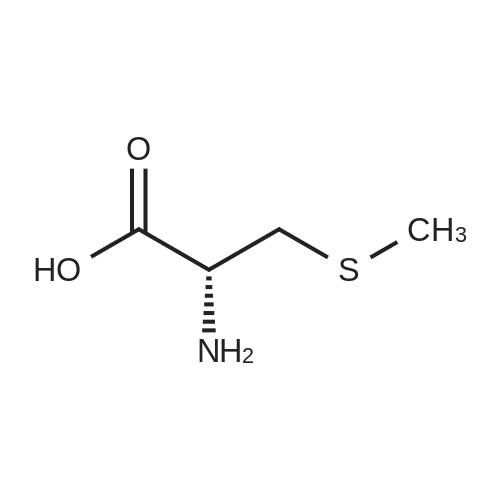
[ 57-13-6 ]
[ 923-37-5 ]
Reference:
[1] Gazzetta Chimica Italiana, 1886, vol. 16, p. 278[2] Gazzetta Chimica Italiana, 1887, vol. 17, p. 185
[3] Gazzetta Chimica Italiana, 1887, vol. 17, p. 185
[4] Chemische Berichte, 1908, vol. 41, p. 2979[5] Hoppe-Seyler's Zeitschrift fuer Physiologische Chemie, 1914, vol. 90, p. 124
20
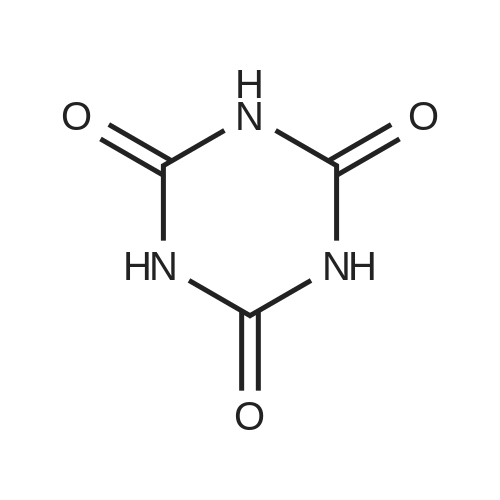
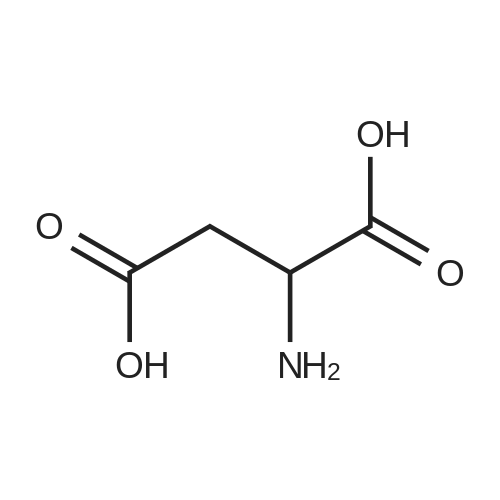
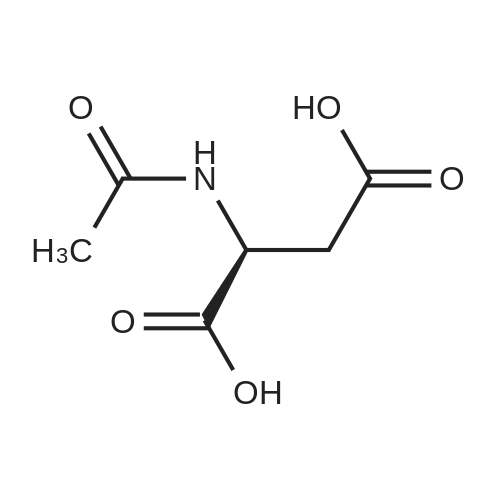
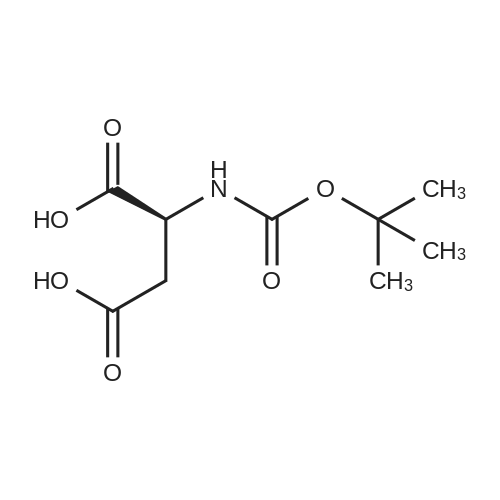
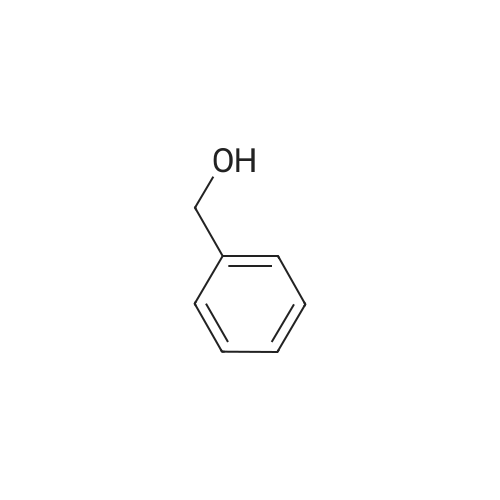
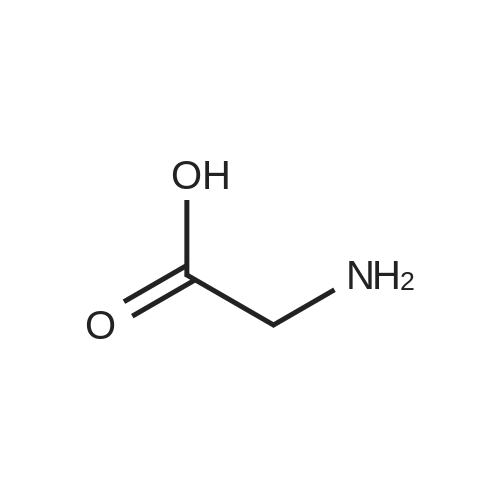
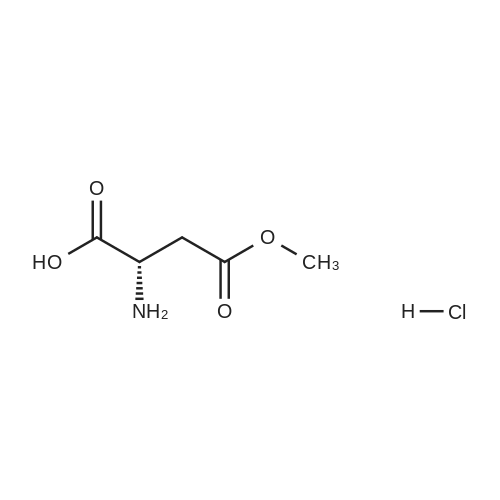
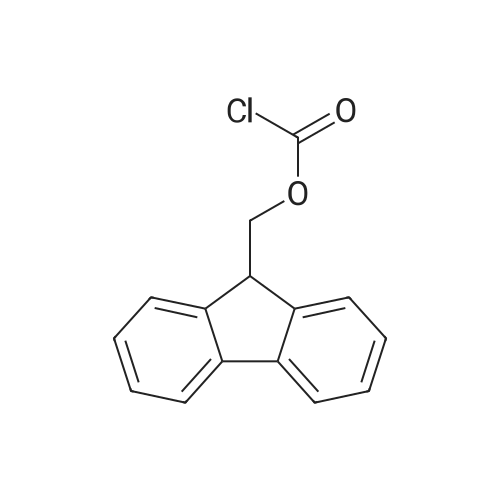
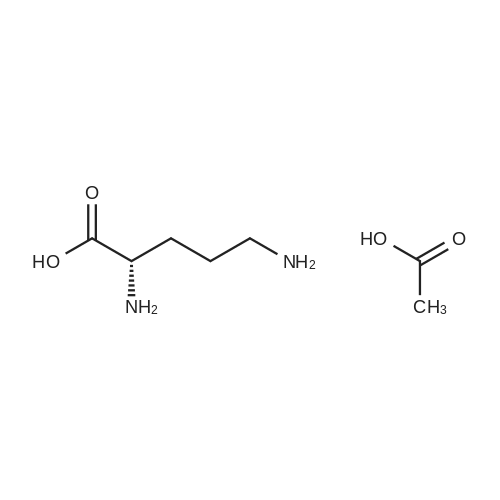
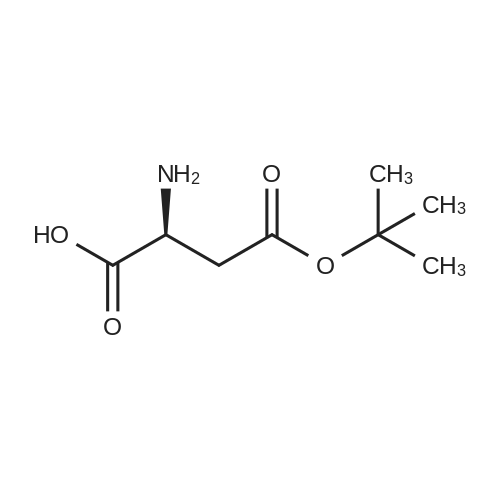
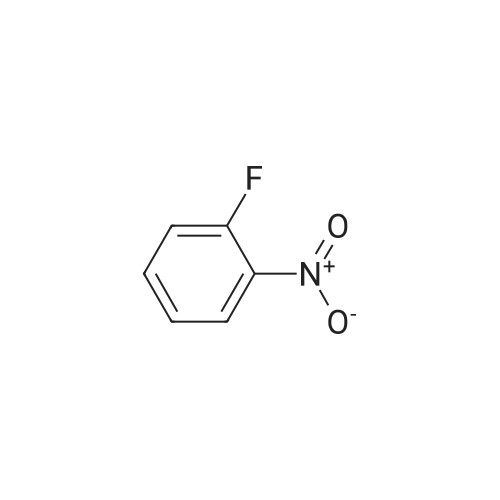
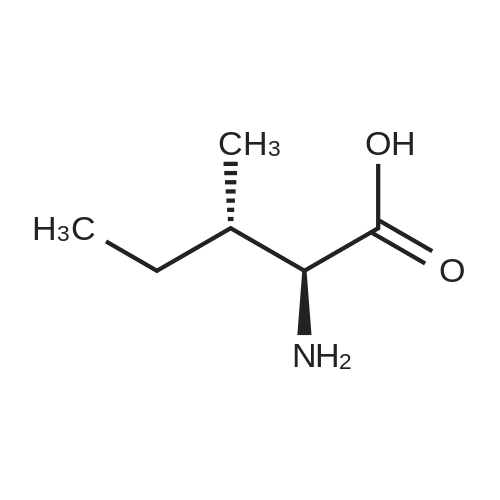
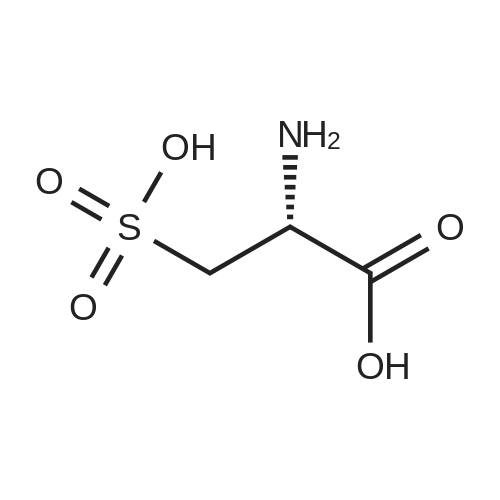
[ 56-84-8 ]
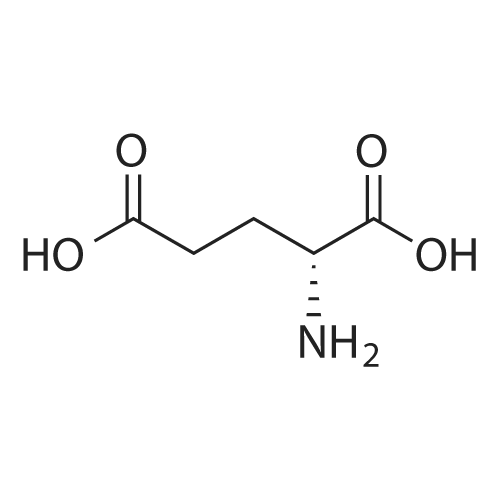
[ 24424-99-5 ]
[ 13726-67-5 ]
Yield
Reaction Conditions
Operation in experiment
85%
Stage #1: With sodium hydroxide In 1,4-dioxane; water Stage #2: at 20℃;
Example 2. Compound 1 (Boc-Asp) (0203) In a 500 mL one-neck round bottom flask, aspartic acid (Asp, 4 g, 30 mmol), 1,4-Dioxane (120 mL) and H2O (60 mL) were mixed yielding a heterogeneous solution. An aqueous solution of NaOH (1 M) was added to the mixture with constant stirring until the solution became homogeneous and clear indicating that the aspartic acid was completely dissolved. The solution was cooled using an ice-bath, then di-tert-butyl dicarbonate (Boc2O, 7.2 g, 33 mmol) dissolved in 1,4-Dioxane (20 mL) was added dropwise. The reaction mixture was then stirred at room temperature overnight. Once the reaction was complete, the solvent was partially evaporated, using a rotary evaporator, to a final volume of 30 mL. Then, EtOAc (20 mL) was added and the water layer was acidified under ice-cold conditions using an aqueous solution of KHSO4 to pH2. The solution mixture was transferred to a separatory funnel and the product was extracted using EtOAc (volume, 3 times). The organic layers were combined and dried over Na2SO4. The solvent was evaporated using rotary evaporator, and further vacuum drying was applied overnight to yield the compound as white solid (6 g; reaction yield85percent). (0204) 1H NMR (600 MHz, DMSO-d6): δ 1.38 (s, 9H), 2.51-2.55 (m, 1H), 2.65-2.69 (m, 1H), 4.24-4.28 (m, 1H), 7.05-7.07 (d, 1H, J=12 Hz), 12.5 (s, 2H).
76.5%
With sodium hydroxide In 1,4-dioxane; water at 0 - 20℃; for 5 h;
L-aspartic acid (10 g, 0.075 mol) and sodium hydroxide (6.0 g, 0.15 mol) are dissolved in distilled water (100 mL). The solution is cooled in ice bath to 0°C and dioxane (100 mL) was added. (Boc)2O (18.0 g, 0.083 mol) is added dropwise during 1 h. Reaction mixture is stirred in ice bath for 2 hours and then another 2 h at room temperature. Reaction mixture is concentrated on vacuum evaporator. The unreacted (Boc)2O is removed by extraction into the diethyl ether. The aqueous layer is separated, acidified to pH=2 with saturated solution of sodium hydrogen sulfate and the product is extracted into the ethyl acetate. Organic phase is separated, dried with sodium sulfate and concentrated on vacuum evaporator. Boc-L-Asp-OH was crystallized from mixture ethyl acetate/hexane. Yield: 13.4 g (76.5percent), m.p. 116-118°C.
Reference:
[1] Journal of the American Chemical Society, 2015, vol. 137, # 51, p. 16084 - 16097
[2] Patent: US2017/168042, 2017, A1, . Location in patent: Paragraph 0203-0204
[3] Asian Journal of Chemistry, 2014, vol. 26, # 15, p. 4716 - 4722
[4] Patent: EP1647283, 2006, A1, . Location in patent: Page/Page column 5
[5] Chemistry Letters, 1988, # 10, p. 1643 - 1646
[6] Tetrahedron Letters, 2001, vol. 42, # 43, p. 7599 - 7603
[7] Tetrahedron, 2007, vol. 63, # 29, p. 6932 - 6937
[8] Synthesis, 2010, # 15, p. 2512 - 2514
[9] Bulletin of the Korean Chemical Society, 2013, vol. 34, # 7, p. 2011 - 2015
21
[ 56-84-8 ]
[ 13726-67-5 ]
Yield
Reaction Conditions
Operation in experiment
80%
With sodium hydroxide In water; acetonitrile at 25℃; for 15 h;
General procedure: L-Phenylalanine (1.0 g, 6.05 mmol) and sodium carbonate (0.71 g, 6.70 mmol) in water (10 mL) was reacted with a solution of 2 (1.89 g, 6.36 mmol) in acetone (10 mL). The reaction mixture was stirred at room temperature till the reaction completes, as monitored by TLC. The resulting solution was concentrated, to the residue was added water (10 mL) and ethyl acetate (10 mL), pH adjusted to 6.0 with 10percent KHSO4 and stirred for 5 minutes. The organic layer was removed and the aqueous layer was acidified to pH 2.0 with 10percent KHSO4 at 0-5°C and extracted with ethyl acetate (2 x 10 mL). The combined organic layers were washed with 5percent NaHCO3 solution, water, brine and dried over anhydrous Na2SO4. The ethyl acetate layer was concentrated and the residue was crystallized from EtOAc/ n-hexane (2:8) mixture to give the product as a white solid (1.53g, 95percent yield).
EXAMPLE 28 Synthesis of tert-butyloxycarbonyl-L-aspartic acid To a solution of 1.33 g (10 mmol) of L-aspartic acid in a dioxane/water (1:1) mixture (30 ml), 4.2 ml (30 mmol) of triethylamine are added, and the mixture is stirred until solution is complete (approximately 10 min). 2.85 g (10 mmol) of tert-butyl 1,2,2,2-tetrachloroethyl carbonate are then added and the mixture is stirred for 6 hours at 20° C. 50 ml of water are then added and the mixture is extracted with 2*20 ml of ethyl acetate. The aqueous phase is acidified (pH 2-3) with NHCl, and then extracted with 3*30 ml of ethyl acetate. The extract is washed with saturated NaCl solution, dried over MgSO4 and evaporated. The product obtained is crystallized in ethyl acetate and petroleum ether. 1.4 g (60percent yield) of the expected acid is obtained. STR60
Reference:
[1] Synthesis, 1986, # 8, p. 627 - 632
[2] Journal of Organic Chemistry, 1985, vol. 50, # 20, p. 3951 - 3953
[3] Patent: US4725680, 1988, A,
23
[ 56-84-8 ]
[ 13726-67-5 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1989, vol. 62, # 10, p. 3103 - 3108
24
[ 56-84-8 ]
[ 75844-68-7 ]
[ 13726-67-5 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1984, vol. 32, # 6, p. 2174 - 2181
25
[ 1070-19-5 ]
[ 56-84-8 ]
[ 13726-67-5 ]
Reference:
[1] Chemische Berichte, 1972, vol. 105, # 11, p. 3650 - 3657
[2] Justus Liebigs Annalen der Chemie, 1964, vol. 673, p. 208 - 220
[3] Journal of medicinal chemistry, 1971, vol. 14, # 1, p. 24 - 30
[4] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 1977, vol. 15, p. 80 - 81
26
[ 56-84-8 ]
[ 58632-95-4 ]
[ 13726-67-5 ]
Reference:
[1] Bulletin of the Chemical Society of Japan, 1977, vol. 50, p. 718 - 721
27
[ 18595-55-6 ]
[ 56-84-8 ]
[ 13726-67-5 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1968, vol. 716, p. 216 - 218
28
[ 56-84-8 ]
[ 18595-34-1 ]
[ 13726-67-5 ]
Reference:
[1] Justus Liebigs Annalen der Chemie, 1968, vol. 716, p. 175 - 185
29
[ 56-84-8 ]
[ 100-51-6 ]
[ 2177-63-1 ]
Yield
Reaction Conditions
Operation in experiment
90%
Stage #1: With sulfuric acid In diethyl ether Stage #2: at 25℃; for 24 h;
In a 500mL beaker by adding 100mL of anhydrous ether, and then slowly add 20mL concentrated sulfuric acid, while the side of the edge of the stirring, such as coldBut to room temperature,Add 150mL benzyl alcohol, stir well,The ether was removed under reduced pressure and then 26 g of aspartic acid was added in 5 batchesTo the reaction flask. Room temperature stir the reaction 24h,Then add 300 mL of 95percent ethanol and drop 80 mL with a dropping funnelPyridine, edge dripping vigorously stirring, and then frozen through the refrigerator overnight, filter to get solid, solid 80 ethanol stirring dissolved, heatFiltered, the filtrate was frozen for 8 h, filtered and lyophilized to obtain pure β-aspartic acid benzyl ester (90percent yield).
64%
Stage #1: at 60℃; for 12 h; Stage #2: With ammonia In ethanol; water at 40 - 60℃; for 2 h;
EXAMPLE 3 Preparation of β-benzyl L-aspartate 100 g (0.75 mol, 1 eq.) of L-aspartic acid and 162 g (1.5 mol, 2 eq.) of benzyl alcohol are introduced into a fitted-out 1 litre reactor and the mixture is stirred. 86.4 g (0.9 mol, 1.2 eq.) of methanesulphonic acid are subsequently introduced gradually while allowing the temperature to rise.The temperature is brought by heating to 60° C., stirring of the mixture is continued for 12 hours and then the mixture is cooled to 40° C. 200 ml of water are subsequently added, then 300 ml of ethanol are added and subsequently 95 ml of 22° B aqueous ammonia are added, so as to precipitate the ester at a PH of 6.5-7. The mixture is subsequently heated to 60° C. and is stirred at this temperature for 2 hours to improve the crystalline form of the product. It is cooled to a temperature of 5° C.-10° C. The crystalline precipitate is filtered off and washed twice with 100 ml of ethanol and 3 times with 100 ml of water. A wet product is collected and is dried under vacuum. 108 g (64percent yield) of β-benzyl L-aspartate are thus obtained, the characteristics of which are as follows: Appearance: white powder. Melting point: 212° C. [α]D20: +27.6° (read at 1percent in HCl 1N).
41%
With sulfuric acid In water at 70℃; for 2.5 h;
L-Aspartic acid (1b) (50 g, 0.38 mol), benzyl alcohol (125 g, 1.16 mol), a mixture of concentrated sulfuric acid (40.5 g) and water (10 g) were mixed in a water bath at 70 °C for 0.5 h to give a clear solution, which was stirred for s further 2 h. The resulting solution was evaporated in vacuo to an oily product, which was added to a cold solution containing sodium hydrogen carbonate (70 g) and water (250 mL) at 0 °C. The obtained cooled solution was mixed with ether (150 mL) to give a precipitate, which was filtered and washed with cold water to give another precipitate (88 g). This was recrystallized twice with water to give 34 g (41 percent). m.p. 218-219 °C (lit.23) 222 °C).
Reference:
[1] Journal of Natural Products, 2017, vol. 80, # 7, p. 2136 - 2140
[2] Patent: CN107129501, 2017, A, . Location in patent: Paragraph 0009; 0010; 0011
[3] Synthesis and Reactivity in Inorganic, Metal-Organic and Nano-Metal Chemistry, 2011, vol. 41, # 5, p. 459 - 464
[4] Patent: US2004/133033, 2004, A1, . Location in patent: Page 4-5
[5] Chemistry - A European Journal, 2018, vol. 24, # 54, p. 14373 - 14377
[6] Synthesis, 1987, # 7, p. 635 - 637
[7] Archiv der Pharmazie, 2001, vol. 334, # 6, p. 189 - 193
[8] Archiv der Pharmazie, 2006, vol. 339, # 6, p. 283 - 290
[9] Asian Journal of Chemistry, 2014, vol. 26, # 19, p. 6541 - 6548
[10] Nippon Kagaku Zasshi, 1958, vol. 79, p. 420,423[11] Chem.Abstr., 1960, p. 4408
[12] Canadian Journal of Chemistry, 1962, vol. 40, p. 570 - 572
[13] Bulletin of the Chemical Society of Japan, 1967, vol. 40, # 7, p. 1709 - 1715
[14] Indian Journal of Chemistry, 1965, vol. 3, p. 554 - 558
[15] Journal of pharmaceutical sciences, 1963, vol. 52, p. 847 - 851
[16] Phosphorus, Sulfur and Silicon and the Related Elements, 1995, vol. 105, # 1-4, p. 129 - 144
[17] Doklady Chemistry, 2006, vol. 408, # 1, p. 57 - 60
[18] Patent: CN104725645, 2017, B, . Location in patent: Paragraph 0040-0043
[19] Russian Chemical Bulletin, 2017, vol. 66, # 11, p. 2057 - 2065[20] Izv. Akad. Nauk, Ser. Khim., 2017, vol. 66, # 11, p. 2057 - 2065,9
30
[ 56-84-8 ]
[ 100-39-0 ]
[ 2177-63-1 ]
Reference:
[1] Synthesis, 1982, # 9, p. 744 - 747
31
[ 67-56-1 ]
[ 56-84-8 ]
[ 17812-32-7 ]
Reference:
[1] Patent: CN103864642, 2016, B, . Location in patent: Paragraph 0026-0028
[2] Chemical Biology and Drug Design, 2018, vol. 91, # 4, p. 893 - 901
32
[ 63-91-2 ]
[ 56-84-8 ]
[ 7423-92-9 ]
[ 587-33-7 ]
[ 60-18-4 ]
[ 56-40-6 ]
Reference:
[1] Chemistry Letters, 1984, p. 1629 - 1632
33
[ 56-84-8 ]
[ 77-78-1 ]
[ 4226-18-0 ]
Reference:
[1] Journal of Medicinal and Pharmaceutical Chemistry, 1962, vol. 5, p. 1187 - 1199
34
[ 56-84-8 ]
[ 100-51-6 ]
[ 7362-93-8 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2006, vol. 14, # 20, p. 6998 - 7010
35
[ 56-84-8 ]
[ 2185-03-7 ]
Reference:
[1] Journal of Organic Chemistry, 1990, vol. 55, # 15, p. 4763 - 4765
36
[ 67-56-1 ]
[ 56-84-8 ]
[ 16856-13-6 ]
Yield
Reaction Conditions
Operation in experiment
85%
at -10 - 20℃; for 2 h;
To 3.8 mL (41.2 mmol) of SOCl2 in 26 mL of dry methanol, were added at -10°C, 5 g of L-aspartic acid (37.6 mmol). The mixture was stirred two hours at room temperature and 75 mL of diethyl ether were added. The white solid was filtered and washed with 2 x 50 mL of diethyl ether to afford (S)-aspartate methyl monoester chlorhydrate in 85percent yield. White solid. 1H NMR (300 MHz, DMSO): δ (ppm) = 3.05 (dd, J = 3.4, 4.7 Hz, 2H, CH2), 3.78 (s, 3H, OCH3), 4.31-4.35 (m, 1H, CHN)
45%
at -20℃; for 1.5 h;
Example 50 (L)-5-(1,3-Dioxo-1,3-dihydro-isoindol-2-ylmethyl)-2-(oxalyl-amino)-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3-carboxylic acid; To a solution of L-aspartic acid (120 g, 0.90 mol) in methanol (600 ml) cooled to -20° C. was added thionylchoride (93 ml, 1.29 mol) dropwise over 0.5 h. The cooling bath was removed and the mixture was stirred for 1 h, before diethyl ether (1.8 L, containing 50 ml 1 N hydrochloric acid in diethyl ether) was added upon cooling. The resulting precipitate was filtered off and washed with diethyl ether. The product was recrystallized twice:First recrystallization: The product was dissolved in warm methanol (600 ml) and reprecipitated with 1.8 ml diethyl ether (containing 50 ml 1 N hydrochloric acid in diethyl ether).Second recrystallization: The product was dissolved in warm methanol (250 ml) and reprecipitated with 1.0 m diethyl ether (containing 50 ml 1 N hydrochloric acid in diethyl ether).This afforded 75 g (45percent) of L-aspartic acid β-methyl ester hydrochloride as a solid.To a solution of the above β-methyl ester (50 g, 0.27 mol) in water (120 ml) cooled to 0° C. was added triethylamine (95 ml, 0.68 mol) and methyl acrylate (74 ml, 0.82 mol). The reaction mixture was stirred for 3 hours before the cooling bath was removed. After stirring for an additional 1 h the mixture was washed with petrol ether (2.x.400 ml), before tert-butanol (40 ml) and di-tert-butyl dicarbonate (74 g, 0.34 mol) was added and the reaction mixture was stirred for 16 h. The mixture was washed with petrol ether (2.x.400 ml), cooled to 0° C. and the pH was adjusted to 3 with concentrated hydrochloric acid. After extraction with ethyl acetate (3.x.200 ml) the organic phase was washed with brine (200 ml), dried (MgSO4), filtered and the volatiles evaporated in vacuo. The residue was subjected to column chromatography on silicagel using a mixture of ethyl acetate/hexane/methanol/acetic acid (25:25:2.5:1) as eluant. Pure fractions were collected and the solvent evaporated in vacuo which afforded 60 g (66percent) of 2-(tert-butoxycarbonyl-(2-methoxycarbonyl-ethyl)-amino)-succinic acid 4-methyl ester as a solid.To a solution of the above di-ethyl ester (96.9 g, 0.29 mol) in dry degassed tetrahydrofuran (1.01) was added sodium methoxide (161 ml, 30percent solution in methanol) and the reaction mixture was refluxed under nitrogen for 16 h with mechanical stirring. The reaction mixture was cooled to room temperature, the volatiles remove in vacuo until a wet cage was observed. Water (500 ml) was added and the reaction mixture was refluxed for 16 h. The remaining organic solvents were evaporated in vacuo before the pH was adjusted to 2.5 with concentrated hydrochloric acid. The aqueous phase was extracted with ethylacetate (3.x.3 ml) and the combined organic phases were washed with brine (100 ml), dried (MgSO4) and filtered. tert-Butyl amine (25.36 g, 0.350 mol) was added dropwise under stirring whereupon a off white precipitate was formed. The precipitate was filtered off and washed with ethyl acetate, dried in vacuo affording 74.4 g (81percent) of 4-oxo-piperidine-1,2-dicarboxylic acid 1-tert-butyl ester, tert-butyl amine salt as a solid.Analytically pure compound can be obtained from recrystallisation of the crude product from ethanol-diisopropyl ether by heating the compound in ethanol (ca 100 ml per 10 g compound) and while still hot diisopropyl ether is added (ca 250 ml per 10 g compound). Yield in recrystallisation is approximately 50percent.A solution of the above 4-oxo-piperidine-1,2-dicarboxylic acid 1-tert-butyl ester, tert-butyl amine salt (3.0 g, 9.48 mmol), tert-butyl cyanoacetate (2.01 g, 14.22 mmol), sulfur (0.456 g, 14.22 mmol) and diisopropyl-ethylamine (1.64 ml, 9.48 mmol) was heated to 50° C. under nitrogen for 12 h. The orange-yellow solution was allowed to cool to room temperature before a small precipitate was filtered off. The filtrate was evaporated in vacuo and the residue was divided between ethyl acetate (50 ml) and saturated ammonium chloride (100 ml). The aqueous phase was extracted with ethyl acetate (3.x.50 m) and the combined organic phases were washed with brine (50 ml), dried (MgSO4), filtered and the solvent evaporated in vacuo. The residue was subjected to column chromatography using a mixture of petrol ether/ethyl acetate/methanol (8:4:1) as eluant. Pure fractions were collected and the solvent evaporated in vacuo affording 2.22 g (58percent) of 2-amino-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3,5,6-tricarboxylic acid 3,6-di-tert-butyl ester as a solid.To a solution of the above 3,5,6-tricarboxylic acid 3,6-di-tert-butyl ester (0.63 g, 1.58 mmol) in dimethoxyethane (10 ml) cooled to -20° C. was added N-methylmorpholine (174 ml, 1.58 mmol) followed by isobutylchloroformate (205 ml, 1.58 mmol) and the reaction mixture was stirred for two min. before a precipitate was filtered off. The precipitate was rapidly washed with dimethoxyethane (2.x.2.5 ml), recooled to -20° C. and a solution of sodium borohydride (90 mg, 2.37 mmol) in water (1 ml) was added in one lot. (Caution-gas evolution).The reaction mixture was stirred until gas evolution ceases (app. 3 min.) and the mixture was poured into water (25 ml) and extracted with ethyl acetate (10 ml), washed with brine (5 ml), dried (MgSO4), filtered and the solvent evaporated in vacuo affording 0.40 g (66percent) of 2-amino-5-hydroxymethyl-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3,6-dicarboxylic acid di-tert-butyl ester as a solid.To a mixture of the above 2-amino-5-hydroxymethyl-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3,6-dicarboxylic acid di-tert-butyl ester (2.00 g, 5.20 mmol), phthalimide (0.92 g, 6.24 mmol) and triphenylphosphine (1.64 g, 6.24 mmol) in dry tetrahydrofuran (30 ml) cooled to 0° C. under a nitrogen atmosphere was added diethyl azodicarboxylate (DEAD) (0.98 ml, 6.24 mmol). The reaction mixture was allowed to stir overnight, slowly warming to room temperature. Next day the reaction mixture was again cooled to 0° C. and phthalimide (0.46 g, 3.12 mmol), triphenylphosphine (0.82 g, 3.12 mmol) and diethyl azodicarboxylate (DEAD) (0.49 ml, 3.12 mmol) was added in sequence and the reaction mixture was allowed to stir overnight, slowly warming to room temperature. The volatiles were evaporated in vacuo and the resultant solid dissolved in dichlorormethane (20 ml). The residue was subjected to flash column chromatography using a mixture of ethyl acetate/hexane (1:2) as eluant. Fractions were collected affording after evaporation in vacuo 1.0 g of the desired compound contaminated with phthalimide. Recrystallization from ethanol gave 0.23 g (9percent) of pure 2-amino-5-(1,3-dioxo-1,3-dihydro-isoindol-2-ylmethyl)-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3,6-dicarboxylic acid di-tert-butyl ester as a solid.To the above di-tert-butyl ester (0.20 g, 0.39 mmol) dissolved in dichloromethane (4 ml) was added a mixture of imidazol-1-yl-oxo-acetic acid tert butyl ester (0.23 g, 1.17 mmol) in dichloromethane (1 ml) under nitrogen. The reaction mixture was allowed to stir at room temperature overnight. The reaction mixture was added dichlorormethane (5 ml) and washed with 1percent hydrochloric acid (10 ml), dried (Na2SO4), filtered and the organic phase evaporated in vacuo affording 0.25 g (100percent) of 2-(tert-butoxyoxalyl-amino)-5-(1,3-dioxo-1,3-dihydro-isoindol-2-ylmethyl)-4,5,6,7-tetrahydro-thieno[2,3-c]pyridine-3,6-dicarboxylic acid di-tert-butyl ester.The above tri-tert-butyl ester (0.25 g, 0.39 mmol) was dissolved in 20percent trifluoroacetic acid in dichloromethane (5 ml). The reaction was stirred at room temperature for 24 h. before diethyl ether (5 ml) was added. The precipitate was filtered off, washed with diethyl ether, dried in vacuo to give 0.150 g of a solid. NMR revealed the presence of a trace amount of material arising from incomplete deprotection. 0.100 g of the crude product was redissolved in 20percent trifluoroacetic acid in dichloromethane (5 ml), and stirred at room temperature for 24 h. before diethyl ether (5 ml) was added. The product was filtered off and washed with diethyl ether and dried in vacuo to give 0.05 g (40percent) of the title compound as a solid.M.p.: dec.>240° C.Calculated for C19H15N3O7S..C2HF3O2.1/2H2O; C, 49.58percent; H, 3.46percent; N, 8.82percent. Found: C, 49.84percent; H, 3.83percent; N, 8.99percent.
Reference:
[1] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1989, p. 833 - 834
[2] Journal of Organic Chemistry, 1988, vol. 53, # 9, p. 1900 - 1903
[3] Patent: WO2013/30193, 2013, A1, . Location in patent: Paragraph 30
[4] Journal of Organic Chemistry, 1985, vol. 50, # 24, p. 4982 - 4984
[5] Chemical Communications, 2001, # 18, p. 1710 - 1711
[6] Chemical and Pharmaceutical Bulletin, 1989, vol. 37, # 4, p. 883 - 886
[7] Journal of the Chemical Society. Perkin Transactions 1, 2001, # 17, p. 2022 - 2034
[8] Journal of the Chemical Society, Perkin Transactions 1: Organic and Bio-Organic Chemistry (1972-1999), 1983, # 11, p. 2811 - 2814
[9] Patent: US7115624, 2006, B1, . Location in patent: Page/Page column 108-110
[10] Journal of the American Chemical Society, 1957, vol. 79, p. 5697,5701
[11] RSC Advances, 2015, vol. 5, # 88, p. 71868 - 71872
[12] Tetrahedron Letters, 2017, vol. 58, # 12, p. 1194 - 1197
[13] Tetrahedron, 2017, vol. 73, # 27-28, p. 3838 - 3847
Reference:
[1] Journal of Organic Chemistry, 1990, vol. 55, # 10, p. 3068 - 3074
39
[ 56-84-8 ]
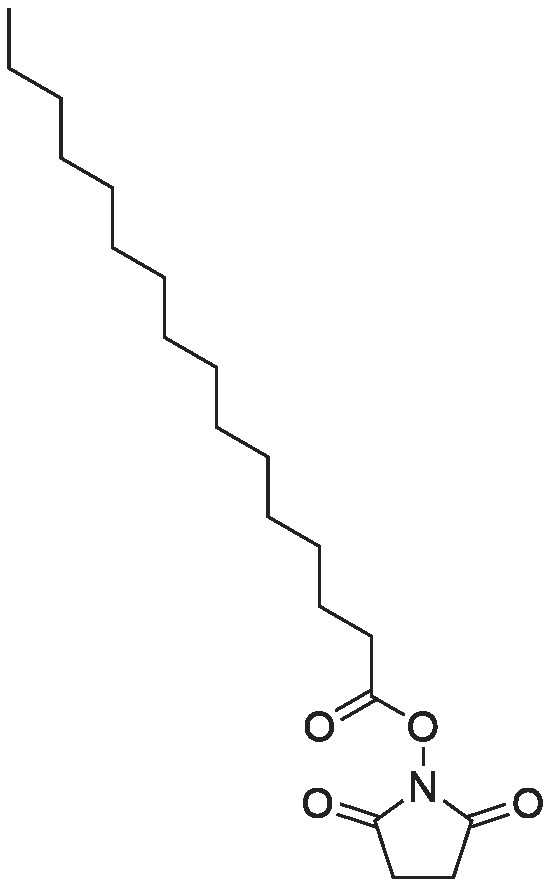
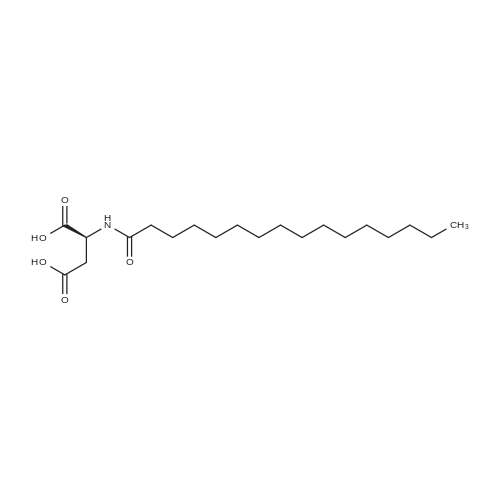
[ 102774-86-7 ]
[ 136083-57-3 ]
Yield
Reaction Conditions
Operation in experiment
92%
With sodium carbonate In water; N,N-dimethyl-formamide at 0 - 20℃; for 1 h;
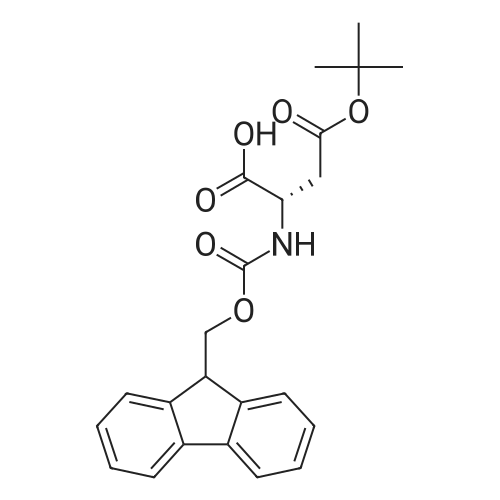
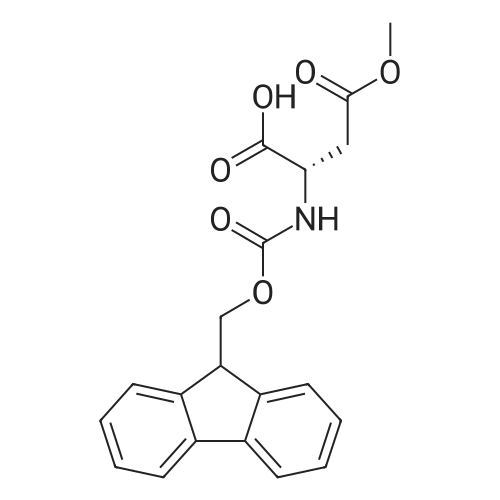
3.03 g (22.8 mmol; 1.2 eq.) of L-aspartic acid (Fluka) are dissolved in 54 ml (68.8 mmol; 3.6 eq.) of a 13.5percent (m/v) aqueous sodium carbonate solution, in a dry 250 ml round-bottomed flask. The medium is cooled in an ice bath at 0° C., then a solution of 6.41 g (19.0 mmol; 1 eq.) of N-(9-fluorenylmethoxycarbonyloxy)succinimide (N-Fmoc) dissolved in 44 ml of DMF is added with vigorous stirring (a precipitate forms in the reaction medium). The stirring is maintained for 1 hour at ambient temperature. The mixture is then diluted in 665 ml of water, and extracted with ether (1*80 ml) then with ethyl acetate (2*60 ml). The resulting aqueous phase is cooled in an ice bath and acidified to pH 2 with concentrated (6 N) hydrochloric acid. The aqueous phase containing the precipitated product (in the form of an oil) is extracted with ethyl acetate (6*60 ml). The organic phase derived from the extraction is washed with a saturated aqueous sodium chloride solution (3*35 ml), and then with water (2*35 ml), dried over sodium sulphate, and concentrated in a rotary evaporator (35° C.) until a small residual volume is obtained. Compound 17 is recrystallized by adding petroleum ether (approximately 10 times the residual volume) with vigorous stirring. After having allowed the mixture to separate by settling out for 2 hours at 4° C., the precipitate is filtered off and then dried for 24 hours in a vacuum oven. 6.22 g (17.5 mmol) of compound 17 are isolated in the form of a fine white powder. Empirical formula: C19H17NO6, M=355.35 g.mol-1 Yield: 92percent M.p.: 181° C. TLC: Rf=0.8 eluent: 60percent AcOH/BuOH 4/6 (v/v) ESI-MS +: m/z measured at 378.1 [M+Na]+, calculated at 378.1 for C19H17NO6Na 1H NMR (dmso-d6, 500.13 MHz) δ (ppm): 12.60 (broad s, 2H, COOH); 7.89 (d, 2H, H-4/H-4', 3J4-3=3J4'-3'=7.5 Hz); 7.72 (d, 1H, NαH); 7.70 (d, 2H, H-1/H-1', 3J1-2=3J1'-2'=7.5 Hz); 7.42 (t, 2H, H-3/H-3', 3J3-2=3J3-4=3J3'-2'=3J3'-4'=7.5 Hz); 7.33 (t, 2H, H-2/H-2', 3J2-1=3J2-3=3J2'-1'=3J2'-3'=7.5 Hz); 4.34 (m, 1H, H-α); 4.29 (d, 2H, H-8); 4.22 (t, 1H, H-7); 2.73 (dd, 1H, H-β,3Jβ-α=5.5 Hz, 3Jβ-β'=16.4 Hz); 2.58 (dd, 1H, H-β', 3Jβ'-α=8.3 Hz, 3J=16.4 Hz) 13C NMR (dmso-d6, 125.77 MHz) δ (ppm): 172.8, 171.8 (CαH-COOH, CβH2-COOH); 155.9 (C-9); 143.9 (C-5/C-5'); 140.8 (C-6/C-6'); 127.7 (C-3/C-3'); 127.2 (C-2/C-2'); 125.4 (C-1/C-1'); 120.2 (C-4/C-4'); 65.8 (C-8); 50.6 (C-α); 46.7 (C-7); 36.1 (C-β)
Reference:
[1] Journal of Organic Chemistry, 2011, vol. 76, # 16, p. 6825 - 6831
[2] Journal of the Chinese Chemical Society, 2011, vol. 58, # 4, p. 509 - 515
41
[ 56-84-8 ]
[ 82911-69-1 ]
[ 136083-57-3 ]
Reference:
[1] Chemical and Pharmaceutical Bulletin, 1988, vol. 36, # 10, p. 3915 - 3919
[2] Tetrahedron Letters, 2009, vol. 50, # 46, p. 6351 - 6354
42
[ 56-84-8 ]
[ 102774-86-7 ]
[ 115-11-7 ]
[ 71989-14-5 ]
[ 129460-17-9 ]
[ 129460-09-9 ]
Reference:
[1] Synthesis, 1990, # 7, p. 571 - 572
43
[ 56-84-8 ]
[ 104-15-4 ]
[ 100-51-6 ]
[ 2886-33-1 ]
Reference:
[1] Journal of Organic Chemistry, 1993, vol. 58, # 9, p. 2369 - 2376
[2] Organic Process Research and Development, 2015, vol. 19, # 7, p. 878 - 883
[3] European Journal of Organic Chemistry, 2014, vol. 2014, # 29, p. 6467 - 6480
[4] European Journal of Medicinal Chemistry, 2008, vol. 43, # 12, p. 2699 - 2716
[5] Chemistry - An Asian Journal, 2011, vol. 6, # 5, p. 1163 - 1170
[6] Journal of the Chemical Society - Perkin Transactions 1, 1999, # 8, p. 855 - 866
[7] Tetrahedron Asymmetry, 1999, vol. 10, # 2, p. 391 - 401
[8] Tetrahedron Letters, 1987, vol. 28, # 27, p. 3167 - 3168
44
[ 56-84-8 ]
[ 104-15-4 ]
[ 100-51-6 ]
[ 2886-33-1 ]
[ 4079-64-5 ]
Reference:
[1] Organic Process Research and Development, 2015, vol. 19, # 7, p. 878 - 883
45
[ 67-56-1 ]
[ 56-84-8 ]
[ 32213-95-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 20℃; for 48 h;
EXAMPLE 1; This example demonstrates the synthesis of 3-benzyl L-aspartic acid (Compound II).; L-Aspartate dimethyl ester hydrochloride:; To L-aspartic acid (13.5 g, 100 mmol) in 100 ml methanol was added dropwise neat thionyl chloride and stirred at room temp for 48 hrs. The reaction was then concentrated in vacuo and chased with methanol (3.x.30 ml) and methylene chloride (3.x.30 ml) using the rotovapor to yield L-aspartate dimethyl ester hydrochloride (19.5 g, quant. yield) as a white powder.
82.5%
at -10℃; for 5.5 h; Heating / reflux
Methanol (100 mL), dried with calcium hydride, is put into 250 mL two neck round bottom flask equipped with stirrer and condenser. Methanol is cooled to -10°C and the thionyl chloride (16.4 mL, 0.225 mol) is added dropwise during 30 min. L-Aspartic acid (15.0 g, 0.112 mol) is added and a reaction mixture was stirred and refluxed for 5 h. The reaction mixture is cooled to room temperature and concentrated on vacuum evaporator. The final product is crystallized from a mixture methanol/diethyl ether. Yield: 18.3 g (82.5percent), m.p. 115-117°C.
Reference:
[1] Organic and Biomolecular Chemistry, 2009, vol. 7, # 20, p. 4309 - 4316
[2] Patent: US2007/142467, 2007, A1, . Location in patent: Page/Page column 5
[3] Chemical Communications, 2011, vol. 47, # 23, p. 6569 - 6571
[4] Journal of Medicinal Chemistry, 2012, vol. 55, # 23, p. 10460 - 10474
[5] Organic and Biomolecular Chemistry, 2015, vol. 13, # 15, p. 4514 - 4523
[6] Australian Journal of Chemistry, 2008, vol. 61, # 11, p. 914 - 919
[7] Journal of Organic Chemistry, 1990, vol. 55, # 10, p. 3068 - 3074
[8] Bioorganic and Medicinal Chemistry, 2007, vol. 15, # 18, p. 6273 - 6290
[9] Langmuir, 2012, vol. 28, # 40, p. 14172 - 14179,8
[10] Synthetic Communications, 2010, vol. 40, # 8, p. 1161 - 1179
[11] Research on Chemical Intermediates, 2013, vol. 39, # 2, p. 621 - 629
[12] Patent: EP1647283, 2006, A1, . Location in patent: Page/Page column 5
[13] Phytochemistry, 1980, vol. 19, # 5, p. 959 - 961
[14] Journal of Medicinal Chemistry, 2006, vol. 49, # 24, p. 7215 - 7226
[15] Journal of Organic Chemistry, 2001, vol. 66, # 1, p. 206 - 215
[16] Heterocycles, 2006, vol. 70, p. 321 - 334
[17] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 4, p. 1764 - 1774
[18] Bioorganic and Medicinal Chemistry, 2008, vol. 16, # 16, p. 7740 - 7748
[19] Patent: WO2007/56143, 2007, A2, . Location in patent: Page/Page column 55-56
[20] Organic Letters, 2012, vol. 14, # 17, p. 4518 - 4521
[21] Tetrahedron Asymmetry, 2013, vol. 24, # 11, p. 663 - 668
[22] Journal of Medicinal Chemistry, 2016, vol. 59, # 11, p. 5505 - 5519
[23] Chemical Communications, 2017, vol. 53, # 2, p. 447 - 450
[24] CrystEngComm, 2017, vol. 19, # 9, p. 1286 - 1293
[25] Bioorganic and Medicinal Chemistry, 2017, vol. 25, # 3, p. 870 - 885
[26] Asian Journal of Chemistry, 2014, vol. 26, # 20, p. 7009 - 7015
[27] Patent: WO2017/172476, 2017, A1, . Location in patent: Paragraph 00150; 00151
46
[ 56-84-8 ]
[ 32213-95-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
With thionyl chloride In methanol
Example 1 L-Aspartic acid dimethyl ester hydrochloride To a stirred suspension of L-aspartic acid (100.9 g, 758.0 mmol) in methanol (560.0 mL) was added thionyl chloride (125.0 g, 1050.7 mmol, 76.6 mL) dropwise over a 1 h period with ice cooling bath. After the addition was complete the ice bath was removed and the resulting clear solution was stirred for 40 h. The solvent was removed under reduced pressure and the residue was solidified by trituration with diethyl ether (100 mL). The solid was filtered, washed with cold ether and dried under vacuum to provide the dimethyl L-aspartate hydrochloride (150.5 g, 100percent) which is pure enough for further reaction. The aspartate hydrochloride can be further crystallized from acetone (128.1 g, 85.1percent) when a high quality sample is needed. Yield: 100percent. Mp: 115-116° C. [α]22 D: +6.0° (c=1.0 in H2 O).
Reference:
[1] Patent: US5322942, 1994, A,
[2] Patent: US4215070, 1980, A,
47
[ 75-77-4 ]
[ 56-84-8 ]
[ 32213-95-9 ]
Yield
Reaction Conditions
Operation in experiment
100%
at 20℃; for 16 h; Cooling with ice
General procedure: Under ice cooling, chlorotrimethylsilane (33.2ml, 263.0mmol) was added dropwise to a suspension of (S)-aspartate (10g, 75.1mmol, 98percent ee) in methanol abs. (150ml) and the reaction mixture was stirred for 16h at room temperature. The solvent and volatile byproducts were evaporated in vacuo. The residue was suspended in methanol (1×30ml) and the solvent was removed in vacuo. Then diethyl ether was added (30ml) and the solvent was removed under reduced pressure. This procedure was repeated three times. The product was dried in high-vacuum. Colorless solid, mp 112–115°C, yield 15g (100percent). C6H12ClNO4, Mr=197.6. Specific rotation: [α]20D[α]D20 +10.3 (c 1.00; H2O). 1H NMR (CD3OD): δ [ppm]=3.07 (d, J=5.3Hz, 2H, CHCH2CO2CH3), 3.75 (s, 3H, OCH3), 3.84 (s, 3H, OCH3), 4.40 (t, J=5.4Hz, 1H, CHCH2CO2CH3). The signal for the protons of the NH3+-group is not seen. 13C NMR (CD3OD): δ [ppm]=35.0 (1C, CHCH2CO2CH3), 50.4 (1C, CHCH2CO2CH3), 53.0 (1C, CO2CH3), 54.0 (1C, CO2CH3), 169.8 (1C, CO2CH3), 171.2 (1C, CO2CH3). MS (EI): m/z [percent]=162 (M–Cl, 22). IR (neat): ν [cm−1]=2864 (C–Haliph.), 1736 (C=Oester).
Reference:
[1] Bioorganic and Medicinal Chemistry, 2014, vol. 22, # 1, p. 221 - 233
[2] Journal of Medicinal Chemistry, 2014, vol. 57, # 7, p. 2884 - 2894
48
[ 56-84-8 ]
[ 75-36-5 ]
[ 32213-95-9 ]
Yield
Reaction Conditions
Operation in experiment
160 g
at -78 - 20℃; for 72 h;
Preparation 71 Dimethyl L-aspartate hydrochloride Acetyl chloride (160 mL) was added dropwise to MeOH (600 mL) at -78°C followed by L-aspartic acid (100 g, 0.75 mol) and the reaction stirred warming to room temperature for 3 days. The reaction was concentrated in vacuo to furnish the title compound as a white solid as the hydrochloride salt (160 g, quant). 1H NMR (400MHz, D2O): δ ppm 3.04 (m, 2H), 3.64 (s, 3H), 3.73 (s, 3H), 4.39 (m, 1 H).
Reference:
[1] Bioorganic and Medicinal Chemistry, 2010, vol. 18, # 17, p. 6220 - 6229
52
[ 56-84-8 ]
[ 60259-81-6 ]
[ 3230-94-2 ]
Yield
Reaction Conditions
Operation in experiment
18.3 g
With ammonium carbonate In methanol; water at 70 - 80℃; for 4.16667 h;
22.5 g of L-ornithine acetate (monohydrate),13.3 g of L-aspartic acid,Add 20mL of purified water,Stir.A solution of ammonium carbonate (9.9 g of ammonium carbonate dissolved in 40 mL of purified water) was added dropwise to the above suspension and CO2 gas evolution accompanied by a slight exotherm (control of dropping rate).The suspension becomes clear.Plus,Continue stirring for 10 minutes,The pH of the reaction solution is about 7.0-8.0.Heated to 70-80 ,Slowly add methanol 150mL,A solid precipitation.Insulation crystallization 2h,Slowly down to room temperature,Continue stirring 2h.filter,Solid first with methanol: water = 4: 1 mixture of about 50mL rinse once,Then rinsed with methanol (50 mL x 2).Drained, had wet solid 21.7g. No drying, directly for refining.Aspartic acid ornithine wet crude 21.7g,Add purified water 42mL,Stir to dissolve.Warmed to 60-70 ° C,Add about 0.6g activated carbon,Thermal decolorization 30 minutes.filter,The filtrate was warmed to 70-80 ° C,Slowly add methanol dropwise about 84mL.A solid precipitation.After all drops,Continue stirring 2h.Slowly down to room temperature,Continue stirring 2h.filter.Methanol: water = 4: 1 mixture (50 ml × 2),Methanol wash (50 mL × 2).Drained and dried under reduced pressure to give 18.3 g.Specific rotation + 28.1 °, content of 99.56percent.
Reference:
[1] Patent: CN106699586, 2017, A, . Location in patent: Paragraph 0030; 0031; 0032; 0033
53
[ 56-84-8 ]
[ 1482-97-9 ]
Reference:
[1] Journal of Organic Chemistry, 1997, vol. 62, # 12, p. 3880 - 3889
54
[ 540-88-5 ]
[ 56-84-8 ]
[ 4125-93-3 ]
Reference:
[1] Patent: CN106045883, 2016, A, . Location in patent: Paragraph 0060-0062
Reference:
[1] Organic Letters, 2005, vol. 7, # 7, p. 1423 - 1426
[2] Journal of Organic Chemistry, 2001, vol. 66, # 1, p. 206 - 215
[3] Patent: WO2006/94770, 2006, A2, . Location in patent: Page/Page column 12-13
[4] Organic Process Research and Development, 2011, vol. 15, # 6, p. 1328 - 1335
[5] Organic Letters, 1999, vol. 1, # 11, p. 1745 - 1747
[6] European Journal of Medicinal Chemistry, 2011, vol. 46, # 1, p. 11 - 20
[7] Chemical Communications, 2014, vol. 50, # 38, p. 4908 - 4910
57
[ 56-84-8 ]
[ 16115-68-7 ]
Reference:
[1] Patent: US4215070, 1980, A,
58
[ 67-56-1 ]
[ 56-84-8 ]
[ 24424-99-5 ]
[ 101-83-7 ]
[ 135941-84-3 ]
Reference:
[1] Synthesis, 1991, # 7, p. 526 - 528
59
[ 67-56-1 ]
[ 56-84-8 ]
[ 22728-89-8 ]
Yield
Reaction Conditions
Operation in experiment
4.73 g
at 0 - 20℃; for 2 h;
In 50ml three-necked flask, at RT, 5g(39.59mmol, 1.0eq) L- aspartic acid was dissolved in 30ml of methanol. Cool to0 deg.C, slowly add dropwise 4.43g (39.6mmol, 1.0eq) SOCl2. Naturally warm toroom temperature the reaction stirred for 2h. To the reaction system was added 60ml of diethyl ether, vacuum filtration, the filter cake washed with ether,dried to give the product cake was 4.73g.
Reference:
[1] Patent: CN105294669, 2016, A, . Location in patent: Paragraph 0233; 0234; 0235
In a 500mL beaker by adding 100mL of anhydrous ether, and then slowly add 20mL concentrated sulfuric acid, while the side of the edge of the stirring, such as coldBut to room temperature,Add 150mL benzyl alcohol, stir well,The ether was removed under reduced pressure and then 26 g of aspartic acid was added in 5 batchesTo the reaction flask. Room temperature stir the reaction 24h,Then add 300 mL of 95% ethanol and drop 80 mL with a dropping funnelPyridine, edge dripping vigorously stirring, and then frozen through the refrigerator overnight, filter to get solid, solid 80 ethanol stirring dissolved, heatFiltered, the filtrate was frozen for 8 h, filtered and lyophilized to obtain pure beta-aspartic acid benzyl ester (90% yield).
64%
EXAMPLE 3 Preparation of beta-benzyl L-aspartate 100 g (0.75 mol, 1 eq.) of L-aspartic acid and 162 g (1.5 mol, 2 eq.) of benzyl alcohol are introduced into a fitted-out 1 litre reactor and the mixture is stirred. 86.4 g (0.9 mol, 1.2 eq.) of methanesulphonic acid are subsequently introduced gradually while allowing the temperature to rise.The temperature is brought by heating to 60 C., stirring of the mixture is continued for 12 hours and then the mixture is cooled to 40 C. 200 ml of water are subsequently added, then 300 ml of ethanol are added and subsequently 95 ml of 22 B aqueous ammonia are added, so as to precipitate the ester at a PH of 6.5-7. The mixture is subsequently heated to 60 C. and is stirred at this temperature for 2 hours to improve the crystalline form of the product. It is cooled to a temperature of 5 C.-10 C. The crystalline precipitate is filtered off and washed twice with 100 ml of ethanol and 3 times with 100 ml of water. A wet product is collected and is dried under vacuum. 108 g (64% yield) of beta-benzyl L-aspartate are thus obtained, the characteristics of which are as follows: Appearance: white powder. Melting point: 212 C. [alpha]D20: +27.6 (read at 1% in HCl 1N).
41%
With sulfuric acid; In water; at 70℃; for 2.5h;
L-Aspartic acid (1b) (50 g, 0.38 mol), benzyl alcohol (125 g, 1.16 mol), a mixture of concentrated sulfuric acid (40.5 g) and water (10 g) were mixed in a water bath at 70 C for 0.5 h to give a clear solution, which was stirred for s further 2 h. The resulting solution was evaporated in vacuo to an oily product, which was added to a cold solution containing sodium hydrogen carbonate (70 g) and water (250 mL) at 0 C. The obtained cooled solution was mixed with ether (150 mL) to give a precipitate, which was filtered and washed with cold water to give another precipitate (88 g). This was recrystallized twice with water to give 34 g (41 %). m.p. 218-219 C (lit.23) 222 C).
28%
A solution of 167 ml benzyl alcohol, 167 ml diethyl ether and 16.7 ml conc. H2SO4 prepared at room temperaturewas evaporated without heating, by removing diethyl ether. To this solution at room temperature under vigorous stirring22.2 g (166.8 mmol) H-Asp-OH was added in small portions. The resulting suspension was stirred vigorously at 20Cuntil a completely homogeneous clear solution was obtained which was allowed to stand at room temperature for 2 days.Then 335 ml ethyl alcohol and 85 ml pyridine were added under vigorous stirring. The reaction mixture was stirred at20C for 1 h. A gradual formation of a white finely dispersed precipitate took place. The solution containing the precipitatewas allowed to stand at 0C for 1 day. Then the precipitate was filtered off and washed with water. The resulting crystallinemass was recrystallized from 335 ml water supplemented with 335 ml pyridine. The recrystallization was performed froma hot solution (80-85C) by allowing it to cool to room temperature and to stand for 1 day. The resulting crystals werefiltered off, washed with acetone and dried to yield 10.5 g (28%) of the product as white crystals. Rf 0.28 (E), Rf 0.15 (I);m.p. 214-215C. 1H NMR (DMSO-d6 - CF3COOD): 2.95 and 2.99 (2H, 2d, CH2 Asp), 4.24 (1H, m, CH Asp), 5.14 (2H, s,-CH2C6H5), 7.35 (5H, m, -CH2C6H5), 8.42 (av. s, NH2 amide, OH).
Take 500 m L single mouth bottle,Add 100 mL of anhydrous ether,Then add 10 mL of H2SO4 (98%),While adding edge mixing,After cooling to room temperature,Add 100 mL of benzyl alcohol (1 mol, 1.04 g / mL, 108.06 g / mol)Stir well and spin off the ether.A total of 13.3 g (0.1 mol, 133.04 g / mol) of L-aspartic acid was added in multiple portions.After stirring for 24 h at room temperature, 200 mL of 95% ethanol was added, and then 50 mL of pyridine was added dropwise with vigorous stirring.Then placed in the refrigerator frozen overnight, suction filtration, the filter cake was washed with ultrapure water several times, and then washed twice with anhydrous ether.Drained, placed in 500 mL flask, add 300 ~ 400 mL of water, 70 heating and stirring to dissolve, water can be added to 400 ~ 500 mL, all dissolved within 1.5 h, the solution becomes clear.Heat filtration, the filtrate refrigerated overnight. Filtered and recrystallized to obtain pure aspartic acid benzyl ester.
In sulfuric acid; at 70℃; for 0.75h;
L-Aspartic acid (1.33 g,10 mmol) and benzyl alcohol (1.08 g, 10 mmol) were disolved in 60% sulfuric acid (1.65 g) and heated at 70 C for 45 min. Clear gel gained by vacuum concentration at 70 C was neutralized using sodium bicarbonate and the white sample thus obtained was dried over a period of 16 h. The sample (0.60 g)was mixed with salicylaldehyde (0.24 g, 2 mmol) in methanol and the mixture was heated at 40 for 3 h. Copper(II) acetate monohydrate (0.20 g, 1 mmol) was added, the mixture was stirred for an additional 3 h, and a green sample was then obtained by concentration. Complex 3 was recrystallized from MeOH.
With sulfuric acid; In diethyl ether; at 20℃; for 12h;
Synthesis of benzyl beta-aspartate is first synthesized. A 500 mL single-mouth eggplant-shaped flask was taken, and 100 mL of anhydrous diethyl ether, 12 mL of H2SO4 (98%) and 120 mL of benzyl alcohol (1.2 mol, 1.04 g/mL, 108.06 g/mol) were successively added, and the mixture was stirred and evaporated to remove diethyl ether. A total of 16 g (0.12 mol, 133.04 g/mol) of L-aspartic acid was added in three portions with stirring. After stirring for 12 h at room temperature, it was diluted with 240 mL of 95% ethanol, and then 60 mL of pyridine was added dropwise to precipitate a white precipitate. After freezing in a refrigerator overnight, the solid was withdrawn and washed with pure water. The obtained crude product was recrystallized twice with pure water at 80 C to give purified benzyl aspartate.
(1) Prepare a single-mouth bottle (500m L), add 100mL of anhydrous ether,Add 10 mL of H2SO4 (98%) slowly and stir vigorously while adding dropwise.After cooling to room temperature, 100 mL of benzyl alcohol was added, and after stirring well, the ether was distilled off by a rotary evaporator;(2) Then add 13.3 g of L-aspartic acid to the reaction flask in three portions;Stir the reaction evenly at room temperature for 24 h.Then add 200 mL of 95% ethanol, and add 50 mL of pyridine dropwise with a dropping funnel, and vigorously stir while dropping;Then, it was frozen overnight in a refrigerator, and the solid after the reaction was stirred and dissolved at 80 C.Heat filtration, the filtrate was refrigerated overnight, and the solution was recrystallized twice as described above.After lyophilization, pure beta-benzyl-L-aspartate is obtained.The reaction process is as follows:
Example 2. Compound 1 (Boc-Asp) (0203) In a 500 mL one-neck round bottom flask, aspartic acid (Asp, 4 g, 30 mmol), 1,4-Dioxane (120 mL) and H2O (60 mL) were mixed yielding a heterogeneous solution. An aqueous solution of NaOH (1 M) was added to the mixture with constant stirring until the solution became homogeneous and clear indicating that the aspartic acid was completely dissolved. The solution was cooled using an ice-bath, then di-tert-butyl dicarbonate (Boc2O, 7.2 g, 33 mmol) dissolved in 1,4-Dioxane (20 mL) was added dropwise. The reaction mixture was then stirred at room temperature overnight. Once the reaction was complete, the solvent was partially evaporated, using a rotary evaporator, to a final volume of 30 mL. Then, EtOAc (20 mL) was added and the water layer was acidified under ice-cold conditions using an aqueous solution of KHSO4 to pH2. The solution mixture was transferred to a separatory funnel and the product was extracted using EtOAc (volume, 3 times). The organic layers were combined and dried over Na2SO4. The solvent was evaporated using rotary evaporator, and further vacuum drying was applied overnight to yield the compound as white solid (6 g; reaction yield85%). (0204) 1H NMR (600 MHz, DMSO-d6): delta 1.38 (s, 9H), 2.51-2.55 (m, 1H), 2.65-2.69 (m, 1H), 4.24-4.28 (m, 1H), 7.05-7.07 (d, 1H, J=12 Hz), 12.5 (s, 2H).
76.5%
With sodium hydroxide; In 1,4-dioxane; water; at 0 - 20℃; for 5h;
L-aspartic acid (10 g, 0.075 mol) and sodium hydroxide (6.0 g, 0.15 mol) are dissolved in distilled water (100 mL). The solution is cooled in ice bath to 0C and dioxane (100 mL) was added. (Boc)2O (18.0 g, 0.083 mol) is added dropwise during 1 h. Reaction mixture is stirred in ice bath for 2 hours and then another 2 h at room temperature. Reaction mixture is concentrated on vacuum evaporator. The unreacted (Boc)2O is removed by extraction into the diethyl ether. The aqueous layer is separated, acidified to pH=2 with saturated solution of sodium hydrogen sulfate and the product is extracted into the ethyl acetate. Organic phase is separated, dried with sodium sulfate and concentrated on vacuum evaporator. Boc-L-Asp-OH was crystallized from mixture ethyl acetate/hexane. Yield: 13.4 g (76.5%), m.p. 116-118C.
To a solution of L-aspartic acid (1.0 g, 7.51 mmol) in dioxane/H2O(1:1, 40 mL) NaOH was added (1.2 g, 4 eq). The reaction mixture was stirred at room temperature for 0.5 h, and then cooled to 0 C (ice bath). To the cooled reaction mixture (Boc)2O (2.5 g, 1.5 eq) was added and allowed to warm up to room temperature and further stirred for 18 h. After completion of the reaction, the mixture was diluted with water (200 mL) and titrated to pH 3-4 with 1 N HCl. The aqueous solution was extracted with EtOAc (100 mL× 3) and the EtOAc layers were all combined. The organic layer was washed with brine (100 mL × 2), water (100 mL × 2),and then evaporated in vacuo. The given BocNH-Asp-OH as colorless syrup was directly used in the next step.
EXAMPLE 28 Synthesis of tert-butyloxycarbonyl-L-aspartic acid To a solution of 1.33 g (10 mmol) of L-aspartic acid in a dioxane/water (1:1) mixture (30 ml), 4.2 ml (30 mmol) of triethylamine are added, and the mixture is stirred until solution is complete (approximately 10 min). 2.85 g (10 mmol) of tert-butyl 1,2,2,2-tetrachloroethyl carbonate are then added and the mixture is stirred for 6 hours at 20 C. 50 ml of water are then added and the mixture is extracted with 2*20 ml of ethyl acetate. The aqueous phase is acidified (pH 2-3) with NHCl, and then extracted with 3*30 ml of ethyl acetate. The extract is washed with saturated NaCl solution, dried over MgSO4 and evaporated. The product obtained is crystallized in ethyl acetate and petroleum ether. 1.4 g (60% yield) of the expected acid is obtained. STR60
The N-Boc-protected (5)-2-amino-l,4-butandiol 7 was prepared from aspartic acid in 3 steps in 95% overall yield using previously reported methods, shown in Scheme 7 below.
With thionyl chloride; at -5 - 78℃; for 1.5h;Reflux;
General procedure: A mixture of alanine (3.56 g, 0.04 mol) in ethanol (30 mL), with thionyl chloride (3.6 mL) dropwise added at -5 C, was refluxed in ethanol at 78 C under stirring within 1.5 h. Subsequently, the solution was distilled in vacuum to afford Alanine ethyl ester hydrochloride 2a in >80% yield. The physical constants of 2a-2l are identical with the corresponding data in reference [24].
Chlorotrimethylsilane (21.2 mL, 165.4 mmol) was added slowly to a stirred suspension of l-aspartic acid (5) (5.00 g, 37.6 mmol) in methanol (100 mL) at 0 C. The reaction mixture was allowed to stir for 1 h at 0 C and then at room temperature for 24 h. Triethylamine (34.0 mL, 244.4 mmol) and di-tert-butyl dicarbonate (9.02 g, 41.4 mmol) were added slowly and the mixture stirred for 2 h. The reaction mixture was concentrated in vacuo and the resulting residue dissolved in diethyl ether (200 mL) and filtered to remove the white precipitate. The filtrate was concentrated in vacuo and purified using flash column chromatography (35% ethyl acetate in petroleum ether) to yield dimethyl (2S)-2-(tert-butoxycarbonylamino)butane-1,4-dioate (6) as a white solid (9.81 g, 100%). Mp 58-60 C; numax (NaCl) 3406 (NH), 2927 (CH), 2360, 1704 (C=O), 1458, 1159, 1045 cm-1; [alpha]D25 +35.8 (c 1.0, CHCl3), lit. 9a [alpha]D25 +30.8 (c 2.1, CHCl3); deltaH (400 MHz, CDCl3) 1.47 (9H, s, tBu), 2.85 (1H, dd, J 16.8, 4.2 Hz, 3-HH), 3.03 (1H, dd, J 16.8, 4.2 Hz, 3-HH), 3.72 (3H, s, OMe), 3.78 (3H, s, OMe), 4.58-4.61 (1H, m, 2-H), 5.50 (1H, br d, J 8.4 Hz, NH); deltaC (101 MHz, CDCl3) 28.3 (3*CH3), 36.7 (CH2), 49.9 (CH), 52.1 (CH3), 52.8 (CH3), 80.2 (C), 155.4 (C), 171.5 (C), 171.6 (C); m/z (CI) 262 (MH+, 15%), 206 (100), 162 (36), 85 (13); HRMS (CI): MH+, found 262.1293. C11H20NO6 requires 262.1291.
92%
L-Aspartic acid (1 ; 5.002 g, 37.58 mmol) was added to an oven-dried round-bottom flask and placed under an atmosphere of dry N2(g). The starting material was then dissolved partially with 60 mL of anhydrous methanol, and the mixture was cooled to 0 C. Once the desired temperature was reached, thionyl chloride (8.2 mL, 1 10 mmol) was added drop-wise. After the addition was complete, the reaction mixture became homogenous, and was warmed slowly to room temperature and left to stir for 14 h. The reaction mixture was then concentrated under reduced pressure, and the resulting diester was dissolved in 150 mL of DCM and 100 mL of water. To this biphasic solution was added sodium bicarbonate (4.212 g, 50.14 mmol) and di-f-butyl dicarbonate (9.841 g, 45.09 mmol), and the reaction mixture was heated at reflux for 4 h. After the reaction was confirmed to be complete by TLC, the reaction mixture was allowed to cool to room temperature. The organic layer was separated, and the aqueous layer was extracted three times with 150 mL of DCM. The organic extracts were combined, washed with 250 mL of saturated NaCI(aq), dried over MgS0 (s), and concentrated under reduced pressure. Flash chromatography (35% v/v ethyl acetate in hexanes) was used to isolate 2 [(S)-dimethyl 2-(teri-butoxycarbonylamino)succinate] as a white solid (9.080 g, 92%, 2 steps). [00159] 1H NMR (400 MHz, CDC13) delta = 5.49 (d, J = 8.3 Hz, 1H), 4.60-4.57 (m, 1H), 3.76 (s, 3H), 3.70 (s, 3H), 3.01 (dd, J = 17, 4.4 Hz, 1H), 2.83 (dd, J = 17.0, 4.7), 1.45 (s, 9H); 13C NMR (75 MHz, CDC13) delta = 171.6, 171.5, 155.5, 80.3, 52.8, 52.1, 50.0, 36.8, 28.4; HRMS (ESI) calculated for [CnHi9N06Na]+ (M+Na+) requires mlz = 284.1105, found 284.1113
A 0 C suspension of L-aspartic acid (10.1 g, 76.0 mmol) in anhydrous methanol (100 mL) was treated with trimethylsilyl chloride (24.7 g, 228 mmol) via rapid dropwise addition. The cooling bath was removed and the resulting solution stirred at ambient temperature overnight (16 h) and then concentrated under reduced pressure. The resulting oil was taken up in dichloromethane (100 mL), treated with di-tert-butyl dicarbonate (17.4 g, 80.1 mmol) and diisopropylethylamine (26.5 mL, 152 mmol) and stirred at ambient temperature overnight (16 h). The resulting solution was washed with 0.25 N aqueous hydrochloric acid (3x50 mL), 0.25 N aqueous sodium hydroxide (3x50 mL), and brine (1x50 mL); dried over MgSO4 and concentrated under reduced pressure to afford (S)-dimethyl 2-((tert-butoxycarbonyl)amino)succinate (15.9 g, 60.9 mmol, 80% crude yield) as a near colorless oil. The crude product was used in the next step without further purification.
EXAMPLE 10 L-Aspartic acid, diethyl ester, hydrochloride Hydrogen chloride was bubbled into a stirred suspension of L-aspartic acid (133 g.; 1.00 mole) in absolute ethanol (1.95 l.) at room temperature for 2 hours. The resulting solution was heated at reflux for 5 hours, cooled to room temperature, then evaporated in vacuo. The residue was dissolved in benzene (500 ml.). The solution was heated to reflux, and the water present was removed by means of a Dean-Stark trap. The solution was then concentrated at reduced pressure to an oil which slowly crystallized on standing. The solid was suspended in ether (1.3 l.), collected on a filter, washed with ether (800 ml.), then dried; yield of L-aspartic acid, diethyl ester, hydrochloride, 216 g. (95.7%); m.p., 99-103. This material was recrystallized from 1.25 l. of acetone-ether (4:1) to give 164.6 g.(76.2% recovery) of product suitable for further transformation; m.p., 106.5-107.5.
With triethylamine; In 1,4-dioxane; ethyl acetate;
L-Aspartic acid, N--(phosphonoacetyl)--, tetraethyl ester To a cool (10), stirred suspension of L-aspartic acid, diethyl ester, hydrochloride (19.2 g.; 0.0850 mole) in dioxane (350 ml.) was added, dropwise, triethylamine (18.2 g.; 0.180 mole) during 20 minutes. The mixture was stirred at 10 for 15 minutes, then a solution of phosphonoacetyl chloride, P,P-diethyl ester, prepared from 0.090 mole of acid, in dioxane (90 ml.) was added, dropwise, during 1 hour. The temperature was maintained at 8-10 during the addition. The cooling bath was removed, and the reaction mixture was stirred for 3.5 hours. The insoluble material was collected on a filter then washed with dioxane (100 ml.) and ether (200 ml.). The filtrate was spin-evaporated in vacuo, and the residue was dissolved in ethyl acetate (300 ml.). The organic solution was washed with water (3*100 ml.), dried over magnesium sulfate, then concentrated in vacuo to an oil; yield of L-aspartic acid, N--(phosphonoacetyl)--, tetraethyl ester, 31.1 g. (99.6%). A 25.9 g. portion of the product was dissolved in ether (500 ml.), and the solution was extracted with water (5*150 ml.). The aqueous extracts were combined and concentrated at reduced pressure to an oil. This pale yellow material was dried in vacuo over phosphorus pentoxide to give 20.1 g. (77.6% recovery) of analytically pure product. The compound moves as a single spot on silica gel (Eastman Chromagram Sheet 13181) developed with acetone, chloroform, or ethyl acetate; detection by iodine vapors.
Referential Example 1 Synthesis of 3(S)-[(t-butoxycarbonyl)amino]-gamma-butyrolactone Into 800 ml of water were dissolved 66.6 g of L-aspartic acid and 90.9 g of potassium carbonate, and 46.4 ml of carbobenzoxy chloride were added dropwise. After stirring the mixture for 2 hours at room temperature, 40.0 g of potassium carbonate were added and 46.4 ml of carbobenzoxy chloride were added dropwise, which was further stirred for 4 hours at room temperature. After washing the reaction liquor with ether, the pH was brought to 2 with concentrated hydrochloric acid, which was extracted with ethyl acetate. The ethyl acetate layer was separated, dried over anhydrous Glauber's salt, and then concentrated. The residue was crystallized with ether-n-hexane and the crystals deposited were collected by filtration, and then washed with n-hexane to obtain 108.6 g of 2(S)-[(benzyloxycarbonyl)amino]butanoic diacid.
2.00 g of desvenlafaxine base is dissolved in 50 ml of absolute ethanol at reflux conditions and 1.12 g of L-aspartic acid is added. The solution is further stirred at reflux conditions for 10 minutes and cooled down to -10 C. The mixture is further stirred for one hour at -10 C, the precipitated white crystals are filtered, washed with mother liquor and dried under vacuum at ambient temperature to yield 2.68 g (89.0%) of L-aspartate salt.
82 mg L-aspartic acid was dissolved in 7 ml water at 700C. A solution of 100 mg (0.58 mmol) <strong>[136236-51-6]rasagiline</strong> in 2 ml isopropanol was added at this temperature. The solution remained clear, was cooled to O0C and stirred at this temperature for 1 h. Since no turbidity was observed the solvents were removed in vacuo. The residual clear oil partially crystallised on standing. After a slurry wash with acetone, a white crystalline material was obtained (40 mg). Analysis revealed that this material was aspartic acid.
General procedure: [00416] Formula I free base (ca. 30 mg) was suspended in selected solvents (THF, nitromethane, acetonitrile and MEK; 50 volumes, 1.5 mL) at 50C with stirring. The corresponding acid (1 equivalent) was added and the samples were stirred for 30 minutes, prior to ramping at l C -min_1 to 25C. The samples were placed in a shaker and subjected to heating/cooling cycles, between room temperature and 50C, with four hours at each condition. Details about the acids used for these experiments, including input materials and amounts, are summarized in Table 3 below.Table 3. Input materials used for the salt screensolution in THF)[00417] After 16 hours, an aliquot was taken, isolated by vacuum filtration, dried by suction and analyzed by XRPD. Further solvent (25 volumes) was added, and the suspensions were allowed to cycle for four more days. An aliquot was taken, isolated by vacuum filtration, dried by suction and analyzed by XRPD. Further acid (0.2 equivalents, 17 ??) and solvent (25 volumes) were added to those samples showing the presence of free base in the XRPD pattern, suggesting incomplete salt formation. The samples were stored in a shaker and subjected to heating/cooling cycles, between room temperature and 50C, with four hours at each condition for one week. An aliquot was taken, isolated by vacuum filtration, dried by suction and analyzed by XRPD. Water (10% for MeCN, and 5% for THF, MEK and nitromethane, based on ca. 1.5 mL solvent) was added and the samples were cycled for a further 5 days. An aliquot was taken, isolated by vacuum filtration, dried by suction and analyzed by XRPD.[00418] For samples exhibiting new XRPD patterns, the remaining solid was isolated by vacuum filtration, dried by suction and further characterization was undertaken as follows.
With thionyl chloride; sodium hydrogencarbonate; In methanol; water; ethyl acetate;
A. L-Aspartic acid (1; 5.002 g, 37.58 mmol) was added to an oven-dried round-bottom flask and placed under an atmosphere of dry N2(g). The starting material was then dissolved partially with 60 mL of anhydrous methanol, and the mixture was cooled to 0 C. Once the desired temperature was reached, thionyl chloride (8.2 mL, 110 mmol) was added drop-wise. After the addition was complete, the reaction mixture became homogenous, and was warmed slowly to room temperature and left to stir for 14 h. The reaction mixture was then concentrated under reduced pressure, and the resulting diester was dissolved in 150 mL of DCM and 100 mL of water. To this biphasic solution was added sodium bicarbonate (4.212 g, 50.14 mmol) and di-t-butyl dicarbonate (9.841 g, 45.09 mmol), and the reaction mixture was heated at reflux for 4 h. After the reaction was confirmed to be complete by TLC, the reaction mixture was allowed to cool to room temperature. The organic layer was separated, and the aqueous layer was extracted three times with 150 mL of DCM. The organic extracts were combined, washed with 250 mL of saturated NaCl(aq), dried over MgSO4(s), and concentrated under reduced pressure. Flash chromatography (35% v/v ethyl acetate in hexanes) was used to isolate 2 [(S)-dimethyl 2-(tert-butoxycarbonylamino)succinate] as a white solid (9.080 g, 92%, 2 steps). 1H NMR (400 MHz, CDCl3) delta=5.49 (d, J=8.3 Hz, 1H), 4.60-4.57 (m, 1H), 3.76 (s, 3H), 3.70 (s, 3H), 3.01 (dd, J=17, 4.4 Hz, 1H), 2.83 (dd, J=17.0, 4.7), 1.45 (s, 9H); 13C NMR (75 MHz, CDCl3) delta=171.6, 171.5, 155.5, 80.3, 52.8, 52.1, 50.0, 36.8, 28.4; HRMS (ESI) calculated for [C11H19NO6Na]+ (M+Na+) requires ink=284.1105, found 284.1113.
With perchloric acid; In methanol; water; at 0℃;pH 1.5;Resolution of racemate;
General procedure: The sample was dissolved in each of the moving phases used in the measurements to provide a solution with a sample concentration of approximately 1 mg/1 mL and was analyzed by HPLC under the conditions indicated below and the retention factor, separation factor, and separation degree were determined. The results are given in Tables 1 to 6. B-5: aqueous perchloric acid solution (pH 1.5)/methanol=80/20 (v/v), 0 C.
General procedure: Separation Between the D-Body and L-Body of Arginine (Arg)20 muL of a 200 mM (pH 8.0) buffer solution of sodium borate with a pH of 8.0 and 5 muL of a 40 mM solution of 4-fluoro-7-nitro-2,1,3-benzoxadiazole in anhydrous methyl cyanide were added to 50 pmol of a mixture of the D-body and the L-body of Arginine with a mass ratio of 1:4, and subsequently, heating was conducted at 60 C. for 2 minutes to cause fluorescent derivatization thereof. Then, 95 muL of a 0.5% aqueous solution of trifluoroacetic acid was added thereto, so that a measurement sample was obtained.The column was mounted on a high performance liquid chromatograph SI-2 (produced by Shiseido Co., Ltd.), and subsequently, 2 muL of the measurement sample was injected thereto on the following conditions, so that the D-body and the L-body of arginine were separated.Temperature: 25 C.Mobile phase: methanol Flow rate of the mobile phase: 150 L/min
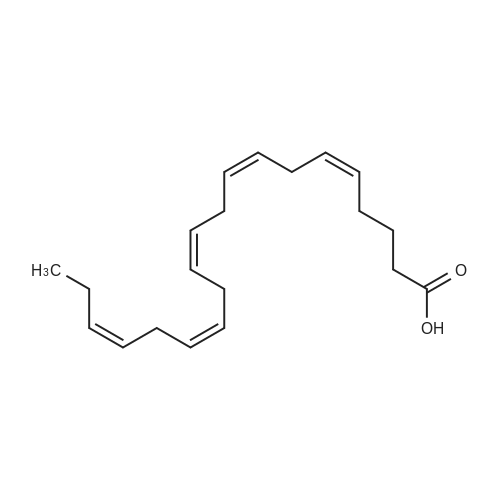
bis[[(dimethylamino)(imino)methyl]amino}(imino)methanaminium]-(5Z,8Z,11Z,14Z,17Z)-eicosa-5,8,11,14,17-pentaenoate-(3S)-3-ammonio-3-carboxypropanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
In methanol; at 60℃; for 1.5h;
A mixture of L-aspartic acid (2.90 g, 21.8 mmol), (5Z,8Z,l lZ,14Z,17Z)-eicosa- 5,8,11,14,17-pentaenoic acid (6.60 g, 21.8 mmol) and N,N-dimethylimidodicarbonimidic diamide (5.64 g, 43.7 mmol) in methanol (300 mL) was warmed in a 60 C oil bath for 30 minutes. Solids all dissolved. After stirring for another hour at 60 C, the mixture was allowed to cool to RT, evaporated in vacuo and left to dry on high vac overnight to yield a tan solid, 14.93g (99%). 1H NMR (300 MHz, MeOD) delta ppm 0.97 (t, 7=7.54 Hz, 3 H) 1.53 - 1.78 (m, 2 H) 1.96 - 2.28 (m, 6 H) 2.57 (dd, 7=17.23, 10.15 Hz, 1 H) 2.73 - 2.92 (m, 9 H) 3.04 (s, 12 H) 3.73 (dd, 7=10.15, 3.54 Hz, 1 H) 4.89 (br. s., 15 H) 5.18 - 5.51 (m, 10 H). MS (ESI-) for C4H7NO4 m/z 132 (M-H)". MS (ESI-) for C20H30O2 m/z 301 (M-H)~. MS (ESI+) for C4H11N5 m/z 130 (M+H)+. Anal Calcd for C32H59Ni i06: C, 55.39; H, 8.57; N, 22.20. Found: C, 55.03 ; H, 8.81 ; N, 21.76.
A 0 C suspension of L-aspartic acid (10.1 g, 76.0 mmol) in anhydrous methanol (100 mL) was treated with trimethylsilyl chloride (24.7 g, 228 mmol) via rapid dropwise addition. The cooling bath was removed and the resulting solution stirred at ambient temperature overnight (16 h) and then concentrated under reduced pressure. The resulting oil was taken up in dichloromethane (100 mL), treated with di-t-butyl dicarbonate (17.4 g, 80.1 mmol) and diisopropylethylamine (26.5 mL, 152 mmol) and stirred at ambient temperature overnight (16 h). The resulting solution was washed with 0.25 N aqueous hydrochloric acid (3x50 mL), 0.25 N aqueous sodium hydroxide (3x50 mL), and brine (1x50 mL); dried over MgSC^ and concentrated under reduced pressure to afford (^-dimethyl 2-{{tert- butoxycarbonyl)amino)succinate (15.9 g, 60.9 mmol, 80% crude yield) as a near colorless oil. The crude product was used in the next step without further purification. 1H NMR (400MHz, CDCh) delta ppm 5.50 (br. s., 1H), 4.60 (br. s., 1H), 3.78 (s, 3H), 3.71 (s, 3H), 3.02 (dd, J=16.9, 4.4 Hz, 1H), 2.84 (dd, J=16.8, 4.8 Hz, 1H), 1.47 (s, 9H).
In 50ml three-necked flask, at RT, 5g(39.59mmol, 1.0eq) L- aspartic acid was dissolved in 30ml of methanol. Cool to0 deg.C, slowly add dropwise 4.43g (39.6mmol, 1.0eq) SOCl2. Naturally warm toroom temperature the reaction stirred for 2h. To the reaction system was added 60ml of diethyl ether, vacuum filtration, the filter cake washed with ether,dried to give the product cake was 4.73g.
With thionyl chloride; In dichloromethane; at -10 - 20℃;
50ml of methanol was added to a 100ml round bottom flask, placed in a cold trap 15min, a thermometer measuring the flask of methanol keeping the temperature below -10 . Weigh 1.5gL- aspartic acid, into a round bottom flask. Monitoring at any time along the sidewall, times slowly added thionyl chloride 1.5eq liquid, the bottle during the temperature kept below 0 . After stirring in the cold trap and then to aspartate dissolved, stirring was continued IH, then transferred to room temperature and stirred overnight. Drained under reduced pressure to give a yellow viscous liquid. Quantitative dichloromethane was added to give a quantitative solution. 50ml round bottomed flask was added 40mg rhein, 38.4mgEDC · HCl, 20mgHOBt, an appropriate amount of aspartic acid methyl ester, and then adding an appropriate amount of dichloromethane, then added 4-5 eq of triethylamine, stirred and dissolved, to purple, clear liquid. At room temperature was stirred 5h, TCL (chloroform as eluant) to monitor the reaction. Through the column, eluted with chloroform, collecting the second strip, rhubarb spin done aspartate ester. Rhubarb aspartic acid methyl ester to the acid, the solution was added 1MNaOH, IH After stirring for 45 , 10% hydrochloric acid was added dropwise, shallow purple solution, to precipitate a pale yellow solid, add two drops, to the solution was stirred for longer purple. Filtered, washed twice with water, ethanol and rinsed again, and dried to give a yellow solid rhubarb aspartate. TCL (toluene: ethyl acetate: formic acid = 7.5:2:0.5) have only one large polar than the starting material rhein yellow dot. Yield 28.9%,
tert-butyl (2,4-dioxo-3-azaspiro[5,5]undecan-3-yl) carbonate[ No CAS ]
[ 13726-67-5 ]
Yield
Reaction Conditions
Operation in experiment
80%
With sodium hydroxide; In water; acetonitrile; at 25℃; for 15h;
General procedure: L-Phenylalanine (1.0 g, 6.05 mmol) and sodium carbonate (0.71 g, 6.70 mmol) in water (10 mL) was reacted with a solution of 2 (1.89 g, 6.36 mmol) in acetone (10 mL). The reaction mixture was stirred at room temperature till the reaction completes, as monitored by TLC. The resulting solution was concentrated, to the residue was added water (10 mL) and ethyl acetate (10 mL), pH adjusted to 6.0 with 10% KHSO4 and stirred for 5 minutes. The organic layer was removed and the aqueous layer was acidified to pH 2.0 with 10% KHSO4 at 0-5C and extracted with ethyl acetate (2 x 10 mL). The combined organic layers were washed with 5% NaHCO3 solution, water, brine and dried over anhydrous Na2SO4. The ethyl acetate layer was concentrated and the residue was crystallized from EtOAc/ n-hexane (2:8) mixture to give the product as a white solid (1.53g, 95% yield).
Take 46.5 mg of the <strong>[1118567-05-7]THR1442</strong> solid sample obtained in Preparation Example 1, and add 2.0 mL of methanol to dissolve.Take 13.3 mg of L-aspartic acid and add 3 mL of water to form a clear solution.The L-aspartic acid aqueous solution was added to a methanol solution of <strong>[1118567-05-7]THR1442</strong>, and the resulting solution was allowed to volatilize at 10 C.57.0 mg of <strong>[1118567-05-7]THR1442</strong> L-aspartic acid cocrystal was obtained in a yield of 95%.
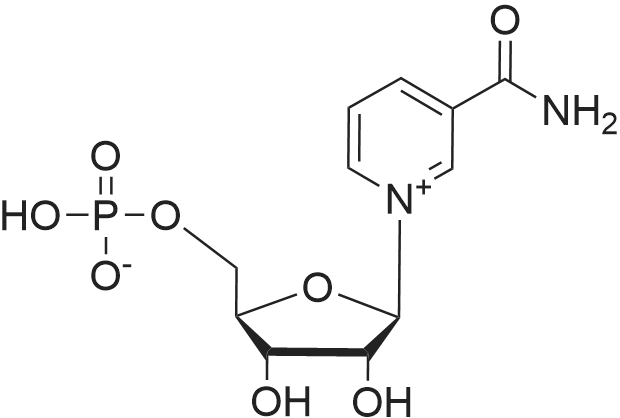
(S)-1,2-dicarboxyethan-1-aminium-((2R,3S,4R,5R)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl phosphate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
In water;pH 2.99 - 3.39;Inert atmosphere; Cooling with ice;
A 100 mL 3N RBF was charged with NMN (0.5 g, 1.492 mmol, 1.0 eq) and 25 ml of distilled deionised water and mixed to form a solution under nitrogen. This solution was cooled using an ice/water bath (pH of solution = 3.39). To this solution was then added dropwise 14.92 ml of 0.1 M Aspartic Acid solution slowly via syringe so as to prevent the temperature from increasing. After this addition the pH was 2.99. The mixture was transferred to a 250 mL RBF and the flask was then removed and the colourless solution frozen using liquid nitrogen. While the flask was frozen it was connected to the freeze dryer. This will slowly remove water. Once dried the product is rendered as a colourless to faint yellow solid (hygroscopic). Yield: Quantitative Melting point: 72 C (degradation), outgassing at 128 C Analytical data. 1 H-NMR (400 MHz, D20) d = 9 40 (s, 1H), 9.30 (d, 1 H), 8.97 (d, 1H), 8.25 (app t, 1H), 6.22 (d, 1H), 4.62 (p, 1H), 4.56 (t, 1H), 4.44 (m, 1H), 4.27 (dq, 1H), 4.15 (dq, 1H), 4.05 (q, 1H), 3.0 (dq, 2H) ppm.
General procedure: p-TsCl catalysed benzyl esterification of mono- and di- carboxylic acid is described in Scheme 1. For L-phenylalanine, since it contains monocarboxylic group, half equivalents of benzyl alcoholand p-TsCl were taken than those used for dicarboxylicamino acids l-aspartic acid and l-glutamic acid. Reaction mixture contained 1 gm (6.8 mmol) l-glutamic acid, 1.56gm (8.2 mmol) p-TsCl, 4 ml (38.6 mmol) benzyl alcoholand 20 ml (excess) cyclohexane. The reaction mixture wasrefluxed overnight under continuous stirring and progressof reaction was monitored through TLC. On completion,the reaction mixture was cooled to room temperature andethyl acetate was added till complete precipitation of products1a-1c. The precipitate obtained was filtered, washedwith cold ethyl acetate and dried to yield benzyl-protectedp-toluenesulfonate salts of respective amino acids.
With sodium hydroxide; In ethanol; at 139.84℃; for 72h;pH Ca. 10;High pressure; Autoclave;
A mixture of Cu(NO3)2·3H2O (241.6mg, 1.0mmol), D- AspH334.6 mg, 0.5 mmol), and L-AspH3 (34.6 mg, 0.5 mmol)was added to a mixed solvent of ethanol/H2O (volumeratio 1:3.8mL), and then 5 muL of a 6 NaOH solution wasadded to adjust the pH value of the solution to ~10. The resulting aqueous solution was transferred into a 25-mL Teflon-lined stainless steel reactor and kept at T=413 K for 3 days, and then slowly cooled to room temperatureat a rate of 10K h-1. Blue block crystals of 1 were obtainedin 60% yield (about 217mg) based on the H3Asp ligand.Analysis calculated (%) for C12H20Cu4N8O12 (Cu4(Asp)2(Ta)2(H2O)2·2H2O): C 19.95, H 2.79, N 15.51; Found C 20.03, H2.86, N 15.47. FT-IR (KBr, 4000-400cm-1): =3421(br, w),1574(s), 1547(s), 1520(s), 1435(w), 1393(s), 1309(w), 1292(w),1171(m), 1119(w), 1092(s), 1003(w), 978(w), 906(m), 824(w),721(m), 663(m), 611(w), 575(w) cm-1.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping