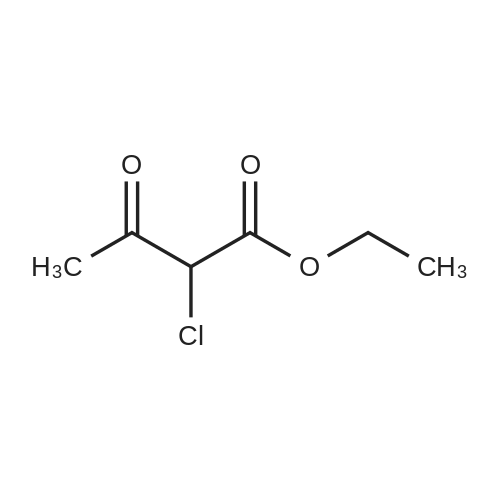
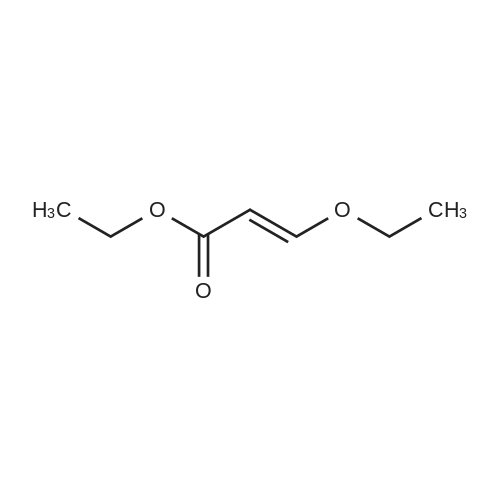
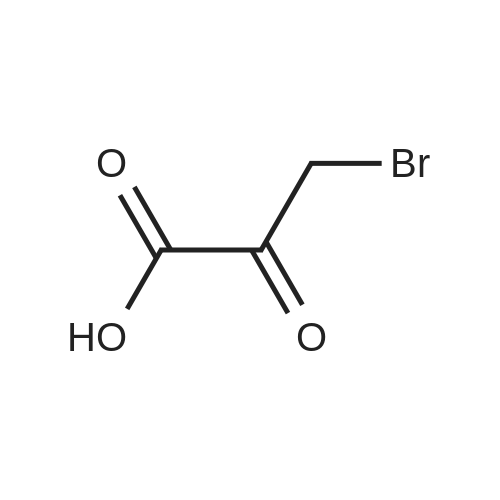
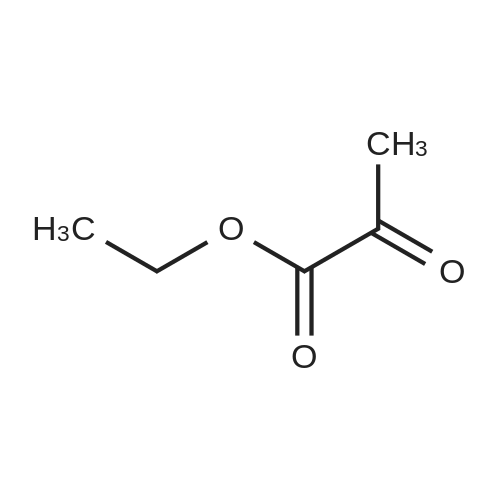
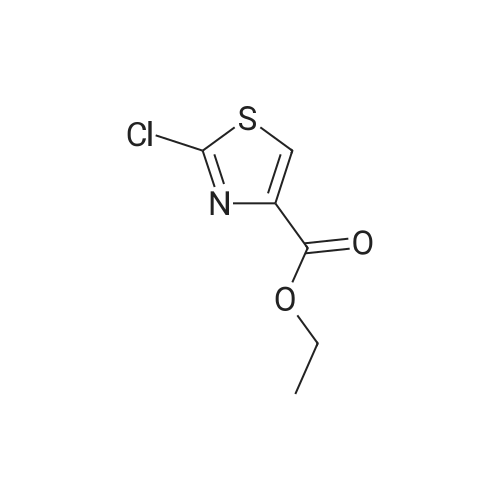
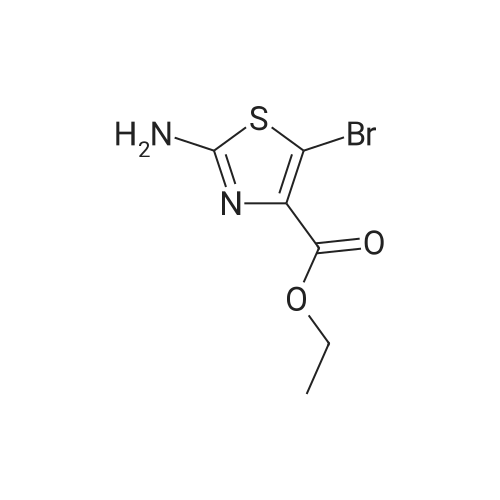
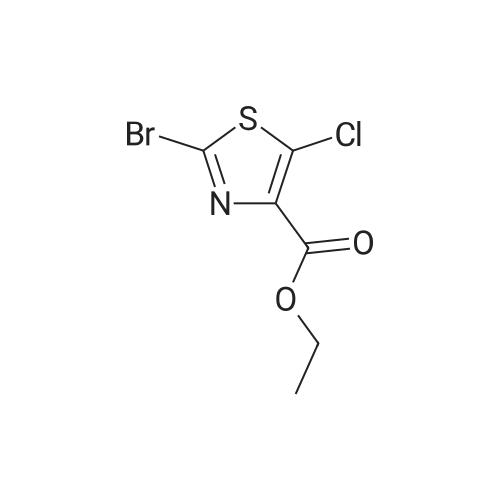
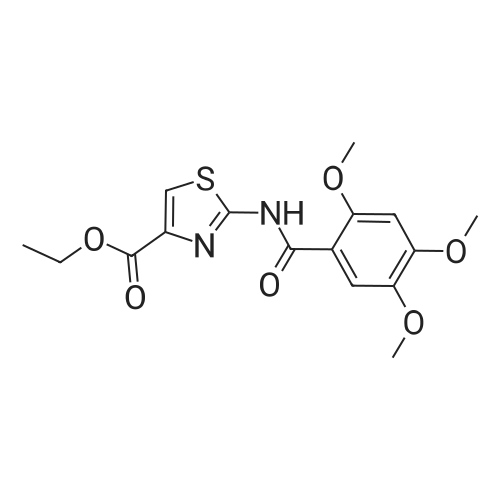
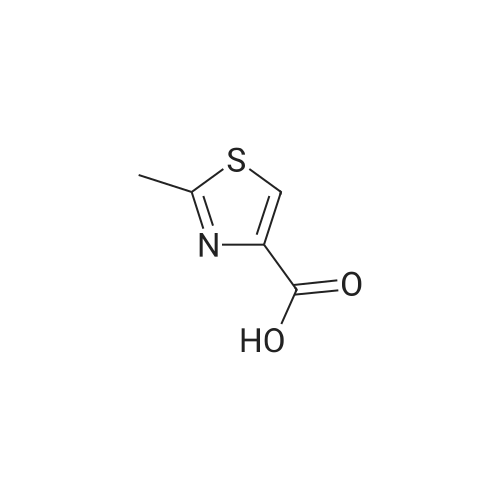
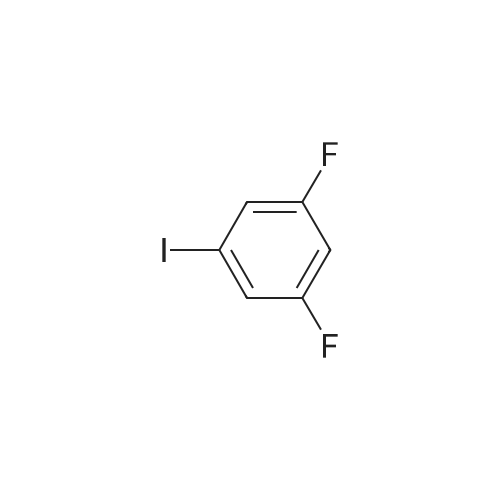
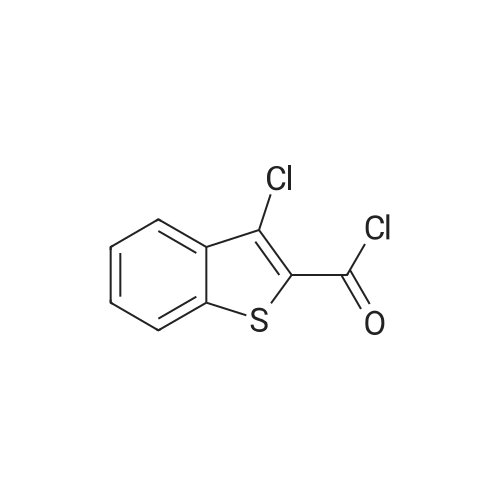
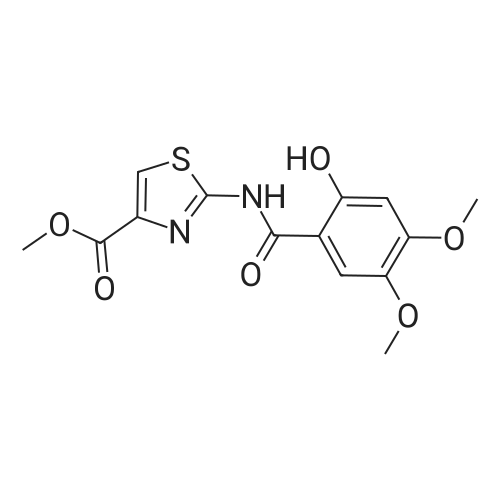
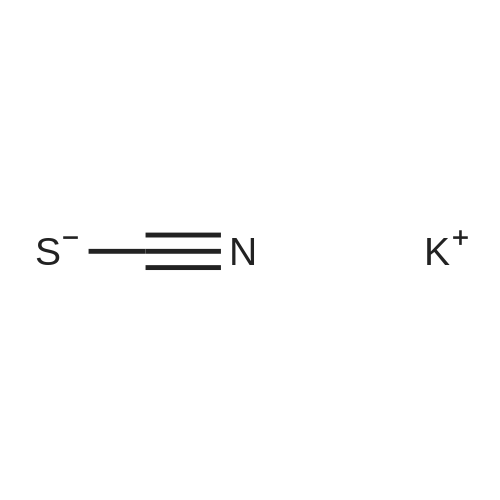| 92% |
In dichloromethane Inert atmosphere; |
|
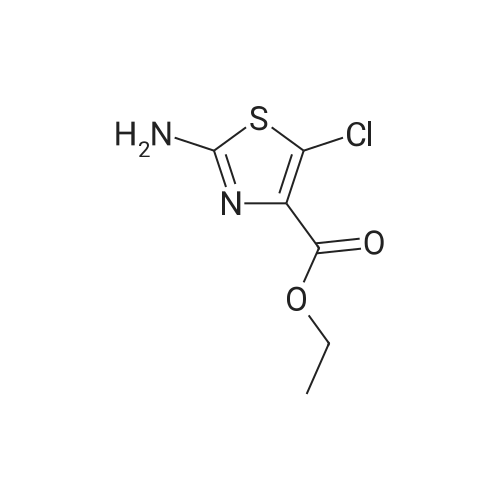
| 91.6% |
With triethylamine In tetrahydrofuran at 0 - 20℃; for 5h; |
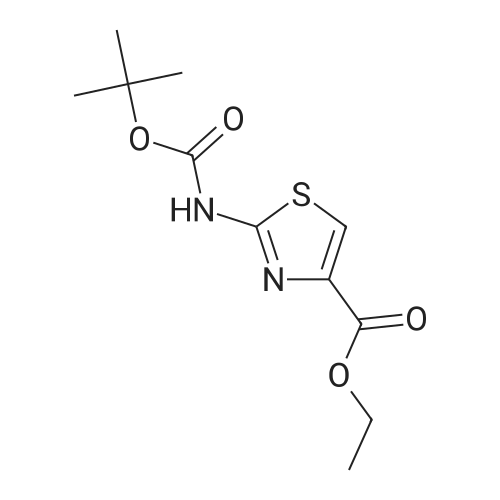
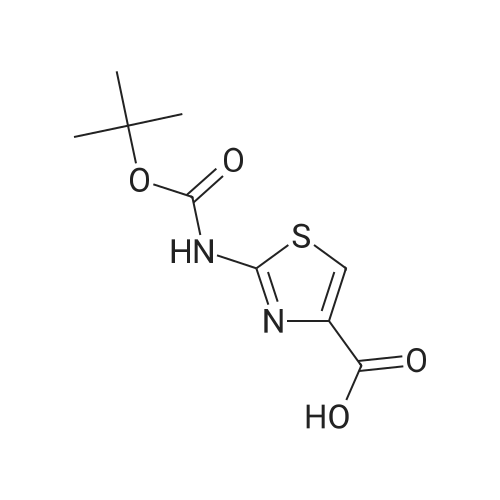
4.3. Procedure for the preparation of tert-butyl-4-(ethoxycarbonyl)thiazol-2-carbamate (2)
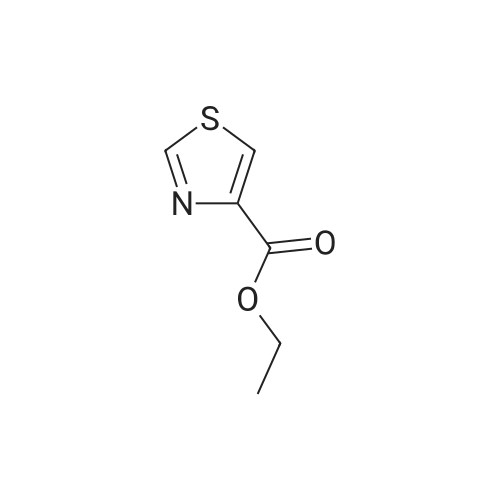
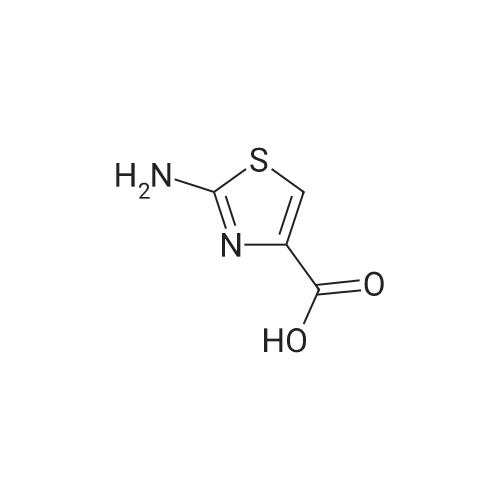
The intermediate ethyl-2-aminothiazole-4-carboxylate (20 g, 0.116 mol) in tetrahydrofuran (300 mL) was stirred for 5 min, cooled the reaction mass to 0 °C and charged triethylamine (48.54 mL, 0.348 mol), the added BOC anhydride (28.26 mL, 0.123 mol) slowly to the reaction mixture, during the addition temperature was maintained at 0-10 °C, after complete addition reaction temperature was raised to ambient temperature and stirred for 5 h. Reaction completion was monitored by TLC. Reaction was complete. The reaction mass was filtered and the THF was concentrated under reduced pressure. The crude obtained was dissolved in ethylacetate (250 mL) and the organic layer was washed with water (3×100 mL). The organic layer was dried over sodium sulfate and concentrated under reduced pressure to afford (2) (29 g, 91.6%) as an off white solid. M.p. = 151-152 °C; 1H NMR (DMSO) δ ppm = 1.26 (t, 3H, CH3), 1.48 (s, 9H, BOC), 4.26 (m, 2H, CH2), 8.0 (s, 1H, ArH), 11.80 (s, 1H, NH); LC/MS(ESI-MS)m/z = 273.1 (M + 1); Anal. Calcd for C11H16N2O4S; C, 48.52; H, 5.92; N, 10.29; Found C, 48.64; H, 5.97 N, 10.38. |
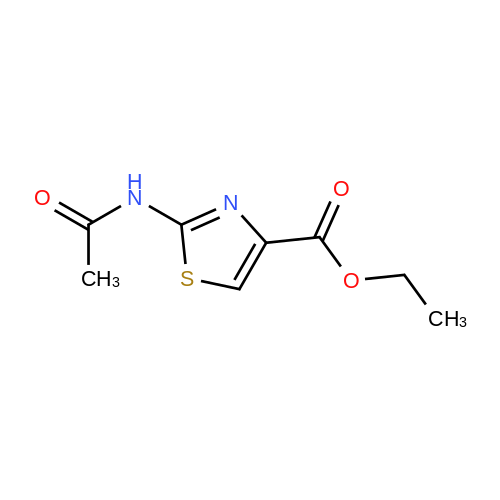
| 74% |
With triethylamine In tetrahydrofuran; dichloromethane for 72h; Reflux; |
4.1.4. BocHN-Tz-COOEt (4c)
Thiazole 4b (0.439 g 2.5 mmol) was dissolved in a mixture ofCH2Cl2:THF (1:1) (10 mL). (Boc)2O (0.577 g, 2.65 mmol) and TEA (0.744 g, 7.36 mmol) were added. The mixture was refluxed for 72 h and then concentrated under reduced pressure. The residue was disolved in AcOEt, washed with HCl 5% v/v (3 x 15 mL), dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by silica flash cromatography AcOEt:EP (3:7). White solid. Yield 74%. 1H NMR ((CD3)2CO, 400 MHz): δ 1.33 (t, 3H,J =7.2 Hz), 1.54 (s, 9H), 4.30 (q, 2H, J = 7.2 Hz), 7.22 (s, 1H), 10.33 (s,1H). 13C NMR ((CD3)2CO, 100 MHz): δ 14.6, 28.2, 61.1, 82.2, 122.4,143.0, 160.4, 161.8. |
| 74% |
With dmap; triethylamine In tetrahydrofuran; dichloromethane for 72h; Inert atmosphere; Reflux; |
4.5.7. Ethyl 2-((tert-butoxycarbonyl)amino)thiazole-4-carboxylate(8)
Compound 5 (0.439 g, 2.5 mmol) is dissolved in 10 mL inDCM:THF (1:1) under a N2 atmosphere. (Boc)2O (0.577 g,2.65 mmol), TEA (0.744 g, 7.36 mmol) and 4-DMAP (cat) wereadded and the reaction was refluxed for 72 h, then the solvent wasremoved in vacuo and redissolved in EtOAc, washed with HCl [5%(v/v); 3 15 mL], dried over Na2SO4, filtered and concentrated invacuo. The residue was purified using flash chromatography usingEtOAc:Hexanes (3:7) as eluent. White solid. Rf 0.38. R 74%. M.p.:105-106 °C. 1H NMR (400 MHz, (Acetone-d6): δ 1.33 (t, J 7.2 Hz,3H),1.54 (s, 9H), 4.30 (q, J 7.2 Hz, 2H), 7.92 (s, 1H),10.37 (s, 1H). 13CNMR (100 MHz, (Acetone-d6): δ 14.6, 28.2, 61.1, 82.2, 122.4, 142.8,153.1, 160.4, 161. |
| 74% |
With dmap; triethylamine In tetrahydrofuran at 60℃; Inert atmosphere; |
|
| 72% |
With dmap; triethylamine In tetrahydrofuran at 25 - 60℃; for 1h; |
3 Thiazole carbamate 61
Thiazole carbamate 61 : To a stirred solution of aminothiazole ester 60 (500 mg, 2.90 mmol, 1.0 equiv.) in tetrahydrofuran (9.7 mL) at 25 °C was added triethylamine (0.53 mL, 3.77 mmol, 1.3 equiv.), 4- (dimethylamino)pyridine (35 mg, 0.29 mmol, 0.1 equiv.), and di-/er/-butyl-dicarbonate (696 mg, 3.19 mmol, 1.1 equiv.) sequentially. The reaction mixture was heated to 60 °C for 1 h, then allowed to cool to 25 °C and quenched with a saturated aqueous solution of ammonium chloride (5 mL). The two phases were separated, and the aqueous layer was extracted with ethyl acetate (3 x 5 mL). The combined organic layers were dried with anhydrous sodium sulfate and concentrated in vacuo. Purification by flash column chromatography (25% ethyl acetate in hexanes) afforded pure thiazole carbamate 61 (569 mg, 2.10 mmol, 72%) as a white solid. 61 : Rf = 0.24 (silica gel, 25% ethyl acetate in hexanes); FT-IR (neat) v 3168, 3068, 2980, 2935, 1713, 1553, 1478, 1455, 1393, 1368, 1331, 1294, 1235, 1207, 1154, 1098, 1071, 1021, 957, 915, 875, 802, 734, 682 cm"1; NMR (600 MHz, CDC13) δ = 8.67 (br s, 1 H), 7.77 (s, 1 H), 4.35 (q, J = 7.2 Hz, 2 H), 1.52 (s, 9 H), 1.36 (t, J = 7.2 Hz, 3 H) ppm; 13C NMR (151 MHz, CDC13) δ = 161.5, 159.8, 152.3, 142.1, 121.7, 83.3, 61.4, 28.3, 14.5 ppm; HRMS (ESI) calcd for CnHigNzC S [M+Na]+ 295.0723, found 295.0712. |
| 72% |
With dmap; triethylamine In tetrahydrofuran at 60℃; for 1h; Inert atmosphere; |
|
| 72% |
With dmap; triethylamine In tetrahydrofuran at 60℃; for 1h; |
3 Ethyl 2-[(fert-butoxycarbonyl)amino]-l,3-thiazole-4-carboxylate (S13):
To a stirred solution of aminothiazole ester S12 (0.500 g, 2.90 mmol, 1.0 equiv) in tetrahydrofuran (9.7 mL) at 25 °C was added triethylamine (0.53 mL, 3.8 mmol, 1.3 equiv), 4-(dimethylamino)pyridine (35 mg, 0.29 mmol, 0.1 equiv), and di-fert-butyl-dicarbonate (696 mg, 3.19 mmol, 1.1 equiv) sequentially, and the reaction mixture was heated to 60 °C. After 1 h, the reaction mixture was allowed to cool to 25 °C, and quenched with saturated aqueous ammonium chloride solution (15 mL). The two phases were separated, and the aqueous layer was extracted with ethyl acetate (3 x5 mL). The combined organic layers were dried with anhydrous sodium sulfate and concentrated under reduced pressure. The obtained residue was purified by flash column chromatography (25% ethyl acetate in hexanes) to afford pure thiazolyl carbamate S13 (569 mg, 2.10 mmol, 72% yield) as a white solid. S13: i= 0.24 (silica gel, 25% ethyl acetate in hexanes); FT-IR (film) vmax 3168, 3068, 2980, 2935, 1713, 1553, 1478, 1455, 1393, 1368, 1331, 1294, 1235, 1207, 1154, 1098, 1071, 1021, 957, 915, 875, 802, 734, 682 cm"1; NMR (600 MHz, CDC13) 5 = 8.67 (br s, 1 H), 7.77 (s, 1 H), 4.35 (q, /= 7.2 Hz, 2 H), 1.52 (s, 9 H), 1.36 (t, /=7.2 Hz, 3 H) ppm; 13C NMR (151 MHz, CDCI3) 5 = 161.5, 159.8, 152.3, 142.1, 121.7, 83.3, 61.4, 28.3, 14.5 ppm; HRMS (ESI) calcd for CiiHi6N204S+ [M+Na]+ 295.0723, found 295.0712. |
| 66% |
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
122.1 Synthesis of ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate:
Ethyl 2-aminothiazole-4-carboxylate (10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (12.67 g, 58.1 mmol) and 4- (dimethyl)aminopyridine (DMAP) (10.0 mg, 0.082 mmol). The reaction mixture was stirred at 50 °C for 4 h and then at room temp for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate (10.5 g, 66 % yield) |
| 66% |
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
122.1 Synthesis of ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate
Ethyl 2-aminothiazole-4-carboxylate (10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (12.67 g, 58.1 mmol) and 4-(dimethyl)aminopyridine (DMAP) (10.0 mg, 0.082 mmol). The reaction mixture was stirred at 50 °C for 4 h and then at room temp for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate (10.5 g, 66 % yield). |
| 62% |
With dmap In dichloromethane at 0 - 20℃; |
11
2-tert-butoxycarbonylamino-thiazole-4-carboxylic acid ethyl ester; Di-tert-butyl dicarbonate (57 g, 0.26 mol) in dichloromethane (100 mL) was slowly added to a solution of 2- amino-thiazole-4-carboxylic acid ethyl ester (45 g, 0.26 mole) and dimethyl-pyridin-4-yl-amine (1 g, 0.008 mol) in dichloromethane (500 mL) at 0 °C. The mixture was stirred at room temperature overnight and then diluted with water. The organic layer was washed with a saturated aqueous solution of sodium chloride, dried over sodium sulfate, filtered, and evaporated to dryness to give a crude product. The product was purified by column chromatography to afford the pure product (43 g, 0.16 mol, 62 %) as a white solid |
| 61% |
With dmap; triethylamine In tetrahydrofuran at 20℃; |
247 ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate (1):
To a stirring solution of ethyl 2-aminothiazole-4-carboxylate SM 1 (0.5 g, 3 mmol) and(Boc)20 (0.65 g, 3 mmol) in THF (10 mL) was TEA (0.6 g, 6 mmol) and DMAP (36 mg, 0.3mmol). The resulting reaction mixture was stirred at RT for overnight. The mixture was quenched with water and extracted with EtOAc, dried and evaporated, purified by combiflash (methanol:DCM = 1:20) to give compound 1(0.5 g, 61%). LC-MS: m/z = 172.0 [M+H-Boc] |
| 57% |
In tetrahydrofuran for 36h; Heating; |
|
| 54% |
In pyridine for 6h; Reflux; |
Tetradecyl 2-aminothiazole-4-carboxylate ( A2 )
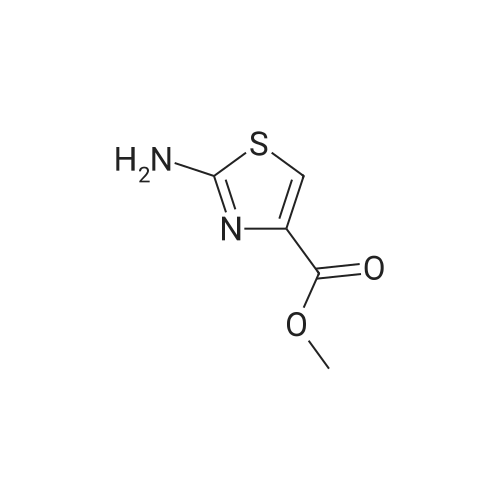
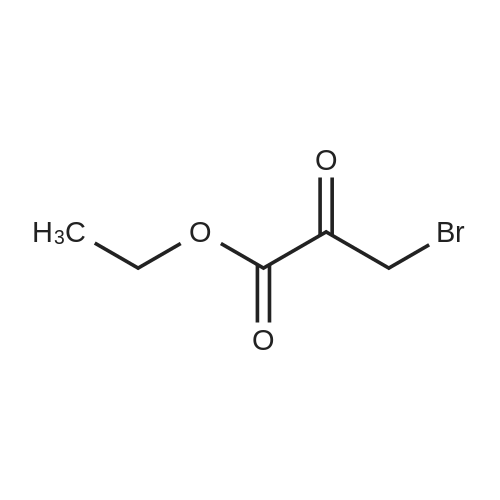
Thiourea (0.76 g, 10.0 mmol) was added to ethanol (10 mL), stirred at room temperature for 10 min, and ethyl bromopyruvate (1.25 mL, 10.0 mmol) was slowly added, at which time heat release was intense. The reaction mixture was stirred at 78°C for 4 h, and the reaction completion was monitored by TLC. The reaction mixture was filtered over Celite and the ethanol layer was concentrated under reduced pressure to give pale-yellow solids. The crude product was recrystallized by ethyl acetate/cyclohexane to obtain intermediate (2) as yellow crystal (2.3g, 91%). 1H NMR (400 MHz, DMSO): δ 7.45 (s, 1H), 7.20 (s, 2H), 4.18 (q, J = 7.2 Hz, 2H), 1.24 (t, J = 7.2 Hz, 3H). ESI-MS (m/z): [M+H]+= 173.0. The intermediate (2) (1.42 g, 5.1 mmol) was dissolved in anhydrous pyridine (30 mL), and (Boc)2O (1.84 g, 8.4 mmol) was added, the mixture was refluxed for 6 h. Pyridine was removed by vacuum evaporation, the residua was diluted with ethyl acetate (80 mL), followed by washed with saturated NaCl solution three times. The organic phase was dried over Na2SO4, filtered, and then concentrated. The crude mixture was purified by column chromatography on silica gel to afford intermediate (3) as a white solid (0.83 g, 54%). 1H NMR (400 MHz, CDCl3): δ 8.32 (s, 1H), 7.79 (s, 1H), 4.38 (q, J = 7.1 Hz, 2H), 1.53 (s, 9H), 1.38 (t, J = 7.1 Hz, 3H). |
|
With dmap; triethylamine In tetrahydrofuran at 20℃; for 12h; |
|
|
With dmap; N,N,N,N,-tetramethylethylenediamine In 1,4-dioxane |
18.A Step A.
Step A. A solution of 2-amino-4-ethoxycarbonylthiazole (1 mmole) in 1,4-dioxane (5 mL) was treated with di-tert-butyl dicarbonate (1.2 mmole), TMEDA (0.1 mmole) and DMAP (0.1 mmole) at room temperature. After the reaction was stirred for 20 h, it was evaporated to dryness. The residue was subjected to extraction to give 2-[N-Boc(amino)]-4-ethoxycarbonyl thiazole as a yellow solid. |
|
With triethylamine In tetrahydrofuran at 0 - 20℃; for 1h; |
5
1.39 g of tert-butyl dicarbonate was added to a solution in which 1.00 g of ethyl 2-amino-l , 3-thiazole-4-carboxylate, 0.88 g of triethylamine, and 0.07 g of DMAP were dissolved in THF, under ice cooling. Then, after the reaction mixture was stirred at room temperature for 1 hr, it was poured into water and-extracted_with_ethyl acetate. The organic layer was washed with an aqueous 2N-hydrochloric acid solution and dried over anhydrous magnesium sulfate. After distilling off the solvent, the obtained crude product was washed with a mixed solvent of hexane and t-butyl methyl ether to obtain 1.5 g of the aimed ethyl 2-( tert-butoxycarbonyl)amino-l , 3-thiazole -4-carboxylate.1H NMR (300MHz, CDCl3) 58.27 (lH,brs), 7.78 (IH s), 4.39 (2H, q, J=7.2Hz), 1.54 ( 9H s), 1.39 (3H, t, J=7.2Hz). |
|
With triethylamine In dichloromethane at 0 - 20℃; for 16h; |
13.A
A solution of di-tert-butyl dicarbonate (11.3 g; 51.83 mmol) in THF (50 mL) was added to a cold (0° C.) solution of ethyl 2-aminothiazole-4-carboxylate (8.59 g; 49.36 mmol) in THF (150 mL). TEA (7.57 mL; 54.30 mmol) was added followed by a catalytic amount of 4-DMAP (30 mg). The reaction mixture was stirred at RT for 16 h, then was concentrated in vacuo. The residue was portioned between EtOAc (300 mL) and 0.5 N aqueous HCl (250 mL). The organic phase was washed with brine (150 mL), dried (MgSO4) and concentrated in vacuo to give Part A compound (12.38 g; yield given below in Part B). The crude product was used in the next step without further purification. |
|
With dmap; N,N,N,N,-tetramethylethylenediamine In 1,4-dioxane at 20℃; for 20h; |
18.A Preparation of Analogs with X Being Methoxycarbonyl, Methylthiocarbonyl, Methylaminocarbonyl and Methylcarbonylamino; Preparations of 4-phosphonomethoxycarbonylthiazoles and 4-phosphonomethoxycarbonyloxazoles
Step A. A solution of 2-amino-4-ethoxycarbonylthiazole (1 mmole) in 1,4-dioxane (5 ML) was treated with di-tert-butyl dicarbonate (1.2 mmole), TMEDA (0.1 mmole) and DMAP (0.1 mmole) at room temperature.. After the reaction was stirred for 20 h, it was evaporated to dryness.. The residue was subjected to extraction to give 2-[N-Boc(amino)]-4-ethoxycarbonyl thiazole as a yellow solid. |
|
With dmap In tetrahydrofuran at 20 - 50℃; |
19.1
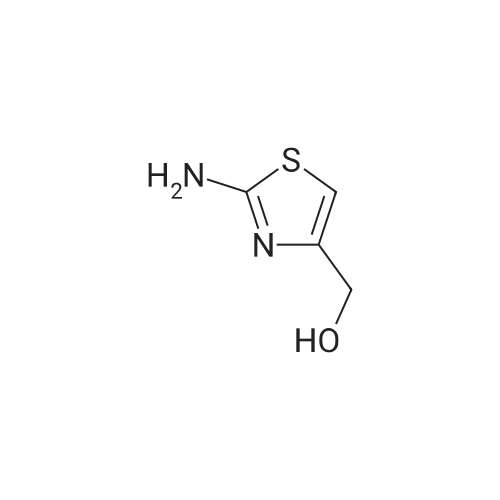
Ethyl 2-aminothiazole-4-carboxylate (55; 10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (BOC20, 12.67 g, 58.1 mmol) along with 10 mg of 4-(dimethyl)aminopyridine (DMAP). The reaction mixture was stirred at 50 0C for 4 h and then at room temperature for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10.5 g of ethyl 2-(tert~ butoxycarbonylamino)thiazole-4-carboxylate 56. This material 56 (10.5 g, 38.5 mmol) was dissolved in 300 mL of anhydrous THF and cooled in Dry Ice-acetonitrile bath. A solution of 1 M Super Hydride in THF (85 mL) was then added over a period of 10 min. The resulting reaction mixture was stirred at -45 0C for 2 h. Another portion of 1 M Super Hydride in THF (35 mL) was then added and the reaction mixture was stirred for an additional 2 h at -45 0C. The reaction was quenched at -45 0C by the addition of 50 mL of brine. Upon warming to room temperature, the reaction mixture was concentrated under reduced pressure. The resulting mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The resulting residue was purified by chromatography to afford 6.39 g oftert-butyl 4- (hydroxymethyl)thiazol-2-ylcarbamate 57 (72%). |
|
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
5.1
Ethyl 2-aminothiazole-4-carboxylate (14; 10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (BOC2O, 12.67 g, 58.1 mmol) along with 10 mg of 4- (dimethyl) aminopyridine (DAP). The reaction mixture was stirred at 50 0C for 4 h and then at room temperature for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10.5 g of ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate 15. |
|
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
2.1
Example 2. Preparation of 2-(4-fluoro-2-(trifluoromethyl)phenyl)-N-(4- (morpholinomethyl)thiazol-2-yl)-3H-imidazo[4,5-b]pyridine-7-carboxamide (Compound 109): Step 1) Synthesis oftert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate (6):5 6Ethyl 2-aminothiazole-4-carboxylate (5; 10 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (BOC2O, 12.67 g, 58.1 mmol) along with 10 mg of 4-(dimethyl)aminopyridine (DMAP). The reaction mixture was stirred at 50 0C for 4 h and then at room temperature for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10.5 g of ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate. This material (10.5 g, 38.5 mmol) was dissolved in 300 mL of anhydrous THF and cooled in Dry Ice- acetonitrile bath. A solution of 1 M Super Hydride in THF (85 mL) was then added over a period of 10 min. The resulting reaction mixture was stirred at -45 0C for 2 h. Another portion of 1 M Super Hydride (lithium triethylborohydride) in THF (35 mL) was then added and the reaction mixture was stirred for an additional 2 h at -45 0C. The reaction was quenched at -45 0C by the addition of 50 mL of brine. Upon warming to room temperature, the reaction mixture was concentrated under reduced pressure. The resulting mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The resulting residue was purified by chromatography to afford 6.39 g of tert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate 6 (72%). MS (ESI) calcd for C9Hi4N2O3S: 230.3; found: 231 [M+H]. |
|
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
16.1
Ethyl 2-aminothiazole-4-carboxylate (59; 10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (BoC2O, 12.67 g, 58.1 mmol) along with 10 mg of 4-(dimethyl)aminopyridine (DMAP). The reaction mixture was stirred at 500C for 4 h and then at room temperature for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10.5 g of ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate 60. This material (60; 10.5 g, 38.5 mmol) was dissolved in 300 mL of anhydrous THF and cooled in Dry Ice-acetonitrile bath. A solution of 1 M Super Hydride in THF (85 mL) was then added over a period of 10 min. The resulting reaction mixture was stirred at -45°C for 2 h. Another portion of 1 M Super Hydride in THF (35 mL) was then added and the reaction mixture was stirred for an additional 2 h at -45°C. The reaction was quenched at -45°C by the addition of 50 mL of brine. Upon warming to room temperature, the reaction mixture was concentrated under reduced pressure. The resulting mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The resulting residue was purified by chromatography to afford 6.39 g of tert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate 61 (72%). MS (ESI) calcd for C9Hi4N2O3S: 230.3; found: 231 [M+H]. |
|
Stage #1: 2-aminothiazole-4-carboxylate; di-<i>tert</i>-butyl dicarbonate With dmap; triethylamine In tetrahydrofuran at 20℃; for 16h;
Stage #2: With hydrogenchloride In water |
A
Intermediate 2[00168] A solution of di-tert-butyl dicarbonate (1 1.3 g; 51.83 mmol) in THF (50 niL) was added to a cold (0°C) solution of ethyl 2-aminothiazole-4-carboxylate (8.59 g; 49.36 mmol) in THF (150 mL). TEA (7.57 mL; 54.30 mmol) was added followed by a catalytic amount of 4-DMAP (30 mg). The reaction mixture was stirred at RT for 16 h, then was concentrated in vacuo. The residue was partitioned between EtOAc (300 mL) and 0.5 N aqueous HC1 (250 mL). The organic phase was washed with brine (150 mL), dried (MgS04) and concentrated in vacuo to give Part A compound (12.38 g; yield given below in Part B). The crude product was used in the next step without further purification. |
|
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
5.1
Step 1. Preparation of tert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate:Ethyl 2-aminothiazole-4-carboxylate (10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (BOC2O, 12.67 g, 58.1 mmol) along with 10 mg of 4-(dimethyl)aminopyridine (DMAP). The reaction mixture was stirred at 50 °C for 4 h and then at room temperature for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10.5 g of ethyl 2-(tert- butoxycarbonylamino)thiazole-4-carboxylate. This material (10.5 g, 38.5 mmol) was dissolved in 300 mL of anhydrous THF and cooled in Dry Ice-acetonitrile bath. A solution of 1 M lithium triethylborohydride (Super Hydride) in THF (85 mL) was then added over a period of 10 min. The resulting reaction mixture was stirred at -45 °C for 2 h. Another portion of 1 M Super Hydride in THF (35 mL) was then added and the reaction mixture was stirred for an additional 2 h at -45 °C. The reaction was quenched at -45 °C by the addition of 50 mL of brine. Upon warming to room temperature, the reaction mixture was concentrated under reduced pressure. The resulting mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried ( a2S04) and concentrated under reduced pressure. The resulting residue was purified by chromatography to afford 6.39 g of tert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate (72%). |
| 2.5 g |
With dmap In dichloromethane at 0 - 20℃; for 16h; |
88.1 Ethyl-2-((tert-butoxycarbonyl)amino)thiazole-4-carboxylate3-nitropyridin-2- amine (1):
Ethyl-2-((tert-butoxycarbonyl)amino)thiazole-4-carboxylate3-nitropyridin-2- amine (1): To a stirred solution of ethyl 2-aminothiazole-4-carboxylate (1 g, 5.8 mmol) in DCM (30 ml) was added DMAP (121 mg, 0.98 mmol) and (Boc)20 (2.78 g, 12.76 mmol) dropwise at 0 °C and stirred at room temperature for 16 h. The reaction mixture was diluted with DCM and washed with 10 % citric acid solution. The organic layer was separated, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography to obtain 1 (2.5 g ) as an off white solid. MS m z (M+H): 272.3 |
| 10.5 g |
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
1
Step 1) Preparation of tert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate: Ethyl 2-aminothiazole-4-carboxylate (10.0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (Boc2O, 12.67 g, 58.1 mmol) along with 10 mg of 4-(dimethyl)aminopyridine (DMAP). The reaction mixture was stirred at 50 °C for 4 h and then at room temperature for 18 h. It was then concentrated under reduced pressure to obtain a thick oil. Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10.5 g of ethyl 2-(tert-butoxycarbonylamino)thiazole-4-carboxylate. This material (10.5 g, 38.5 mmol) was dissolved in 300 mL of anhydrous THF and cooled in Dry Ice-acetonitrile bath. A solution of 1 M Super Hydride in THF (85 mL) was then added over a period of 10 min. The resulting reaction mixture was stirred at -45 °C for 2 h. Another portion of 1 M Super Hydride in THF (35 mL) was then added and the reaction mixture was stirred for an additional 2 h at -45 °C. The reaction was quenched at -45 °C by the addition of 50 mL of brine. Upon warming to room temperature, the reaction mixture was concentrated under reduced pressure. The resulting mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The resulting residue was purified by chromatography to afford 6.39 g of tert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate (72%). The following material was prepared in a similar fashion: |
|
With pyridine; dmap for 24h; Reflux; |
|
|
With dmap; N,N,N,N,-tetramethylethylenediamine In 1,4-dioxane |
18.A Step A.
Step A. A solution of 2-amino-4-ethoxycarbonylthiazole (1 mmole) in 1,4-dioxane (5 mL) was treated with di-tert-butyl dicarbonate (1.2 mmole), TMEDA (0.1 mmole) and DMAP (0.1 mmole) at room temperature. After the reaction was stirred for 20 h, it was evaporated to dryness. The residue was subjected to extraction to give 2-[N-Boc(amino)]-4-ethoxycarbonyl thiazole as a yellow solid. |
|
With dmap In tetrahydrofuran at 20 - 50℃; for 22h; |
9.1
Example 9. Synthesis of N-(4-(morpholinomethyl)thiazol-2-yl)-2-(3- (trifluoromethyl)phenyl)imidazo[l,2-a]pyridine-8-carboxamide (Compound 132): Step 1) Preparation oftert-butyl 4-(hydroxymethyl)thiazol-2-ylcarbamate (35): Ethyl 2-aminothiazole-4-carboxylate (33; 10 0 g, 58.1 mmol) was taken up in 150 mL of anhydrous THF along with di-tert-butyl carbonate (BOC2O, 12 67 g, 58 1 mmol) along with 10 mg of 4-(dimethyl)aminopyridine (DMAP) The reaction mixture was stirred at 50 0C for 4 h and then at room temperature for 18 h It was then concentrated under reduced pressure to obtain a thick oil Pentane was added and the resulting crystalline materials were collected by filtration and dried to afford 10 5 g of ethyl 2-(tert- butoxycarbonylamino)thiazole-4-carboxylate 34 This mateϖal (10 5 g, 38 5 mmol) was dissolved in 300 mL of anhydrous THF and cooled in Dry Ice-acetonitϖle bath. A solution of 1 M Super Hydride in THF (85 mL) was then added over a penod of 10 min The resulting reaction mixture was stirred at -45 0C for 2 h. Another portion of 1 M Super Hydride in THF (35 mL) was then added and the reaction mixture was stirred for an additional 2 h at -45 0C. The reaction was quenched at -45 0C by the addition of 50 mL of brine. Upon warming to room temperature, the reaction mixture was concentrated under reduced pressure. The resulting mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and concentrated under reduced pressure. The resulting residue was purified by chromatography to afford 6.39 g of tert-butyl 4- (hydroxymethyl)thiazol-2-ylcarbamate 35 (72%). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






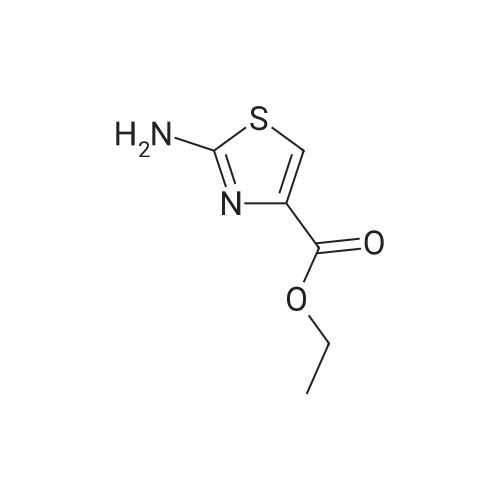

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping