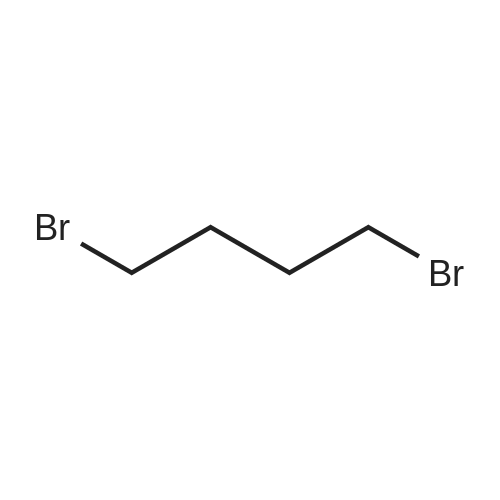
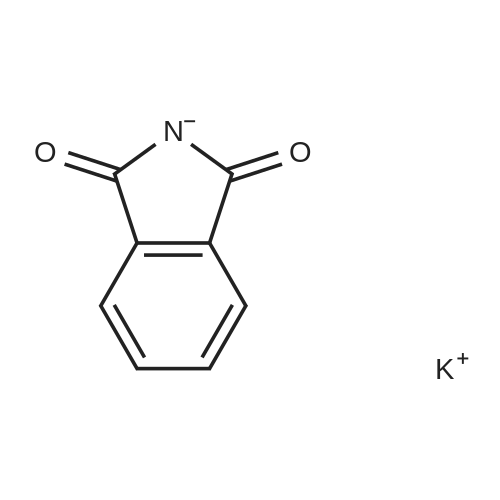
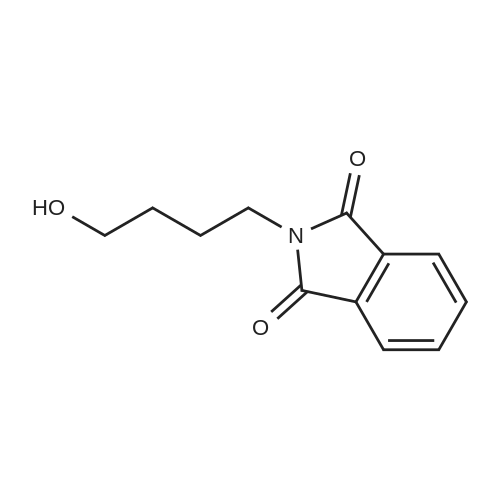
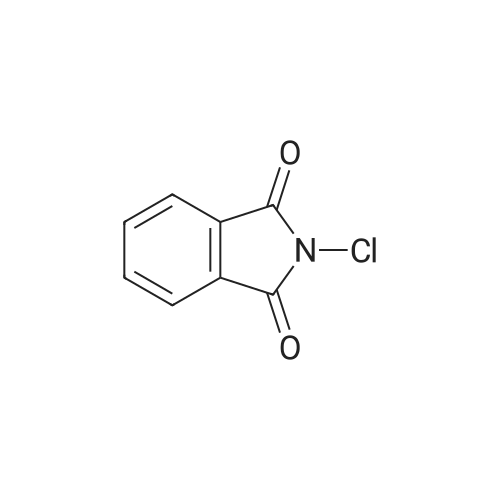
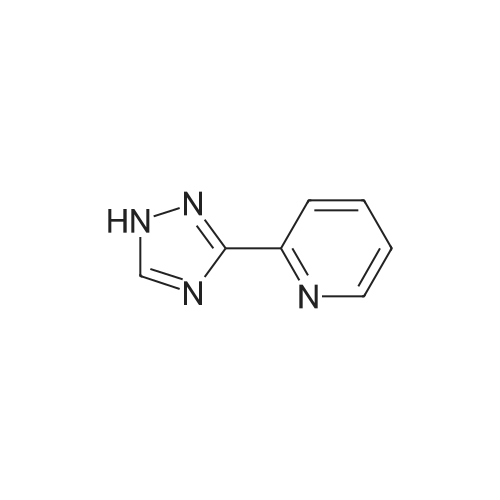
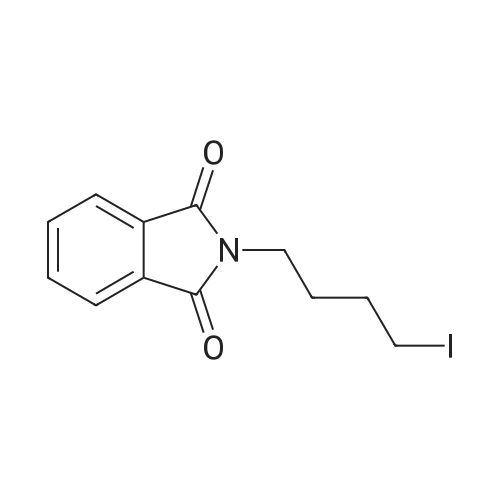
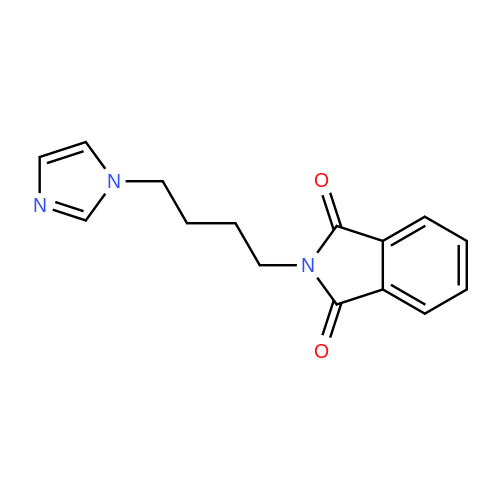
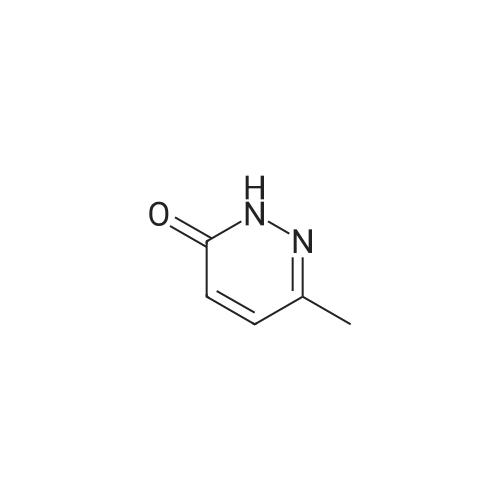
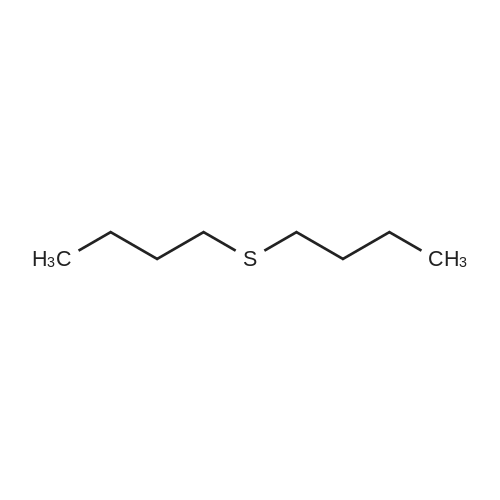
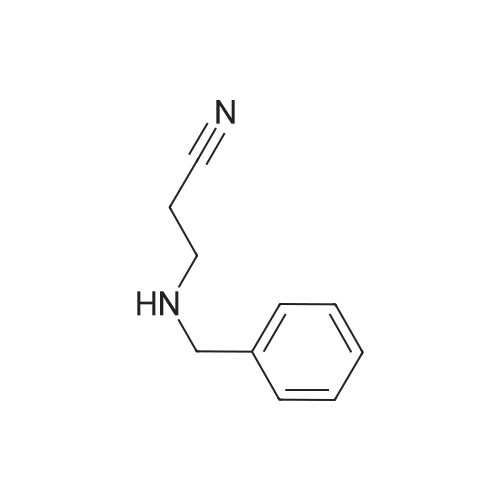
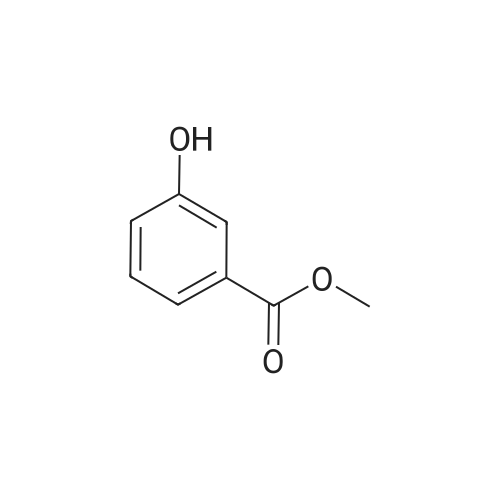
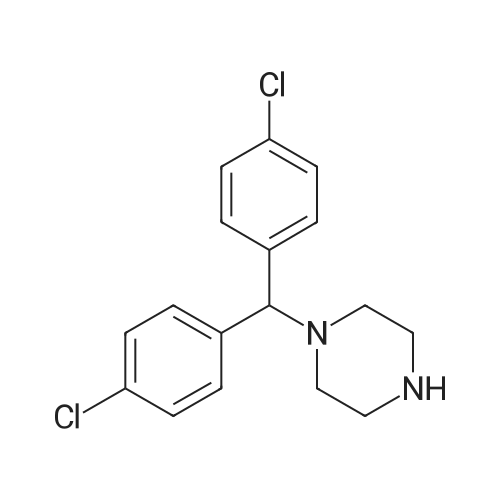
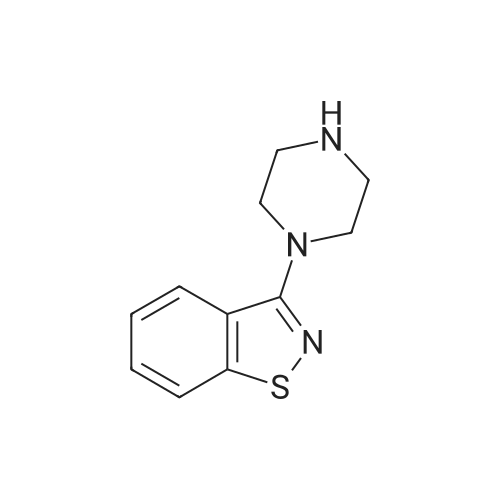
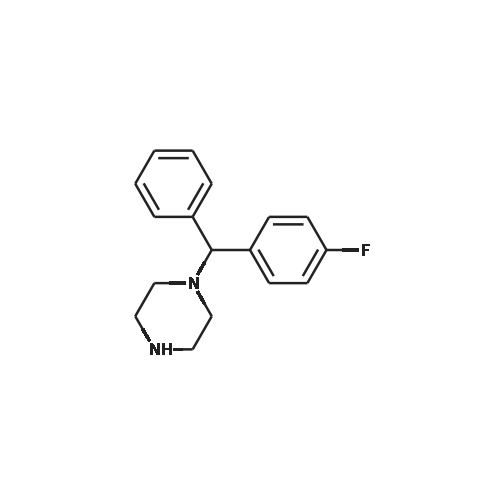
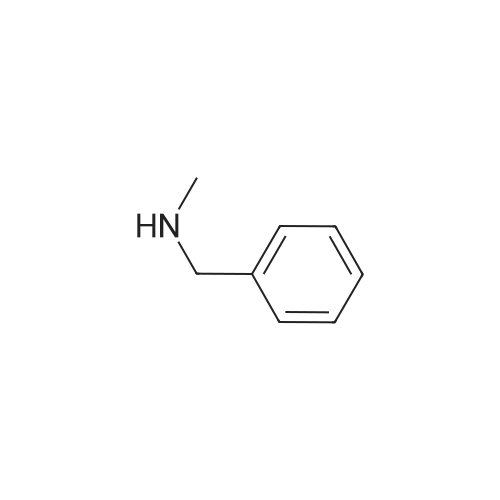
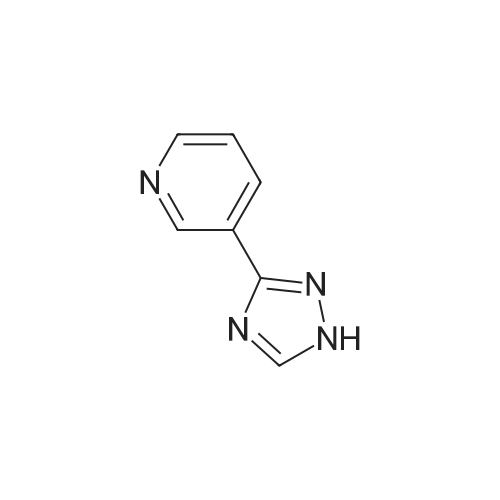
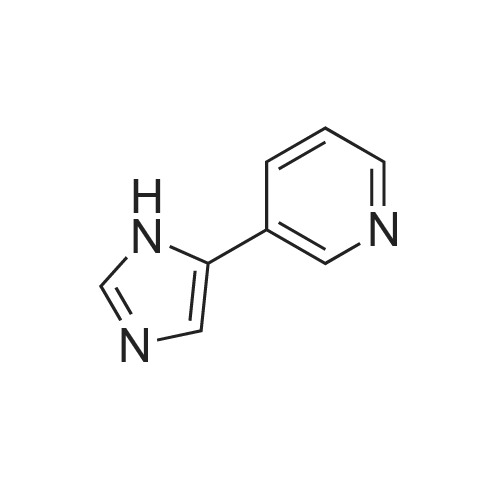
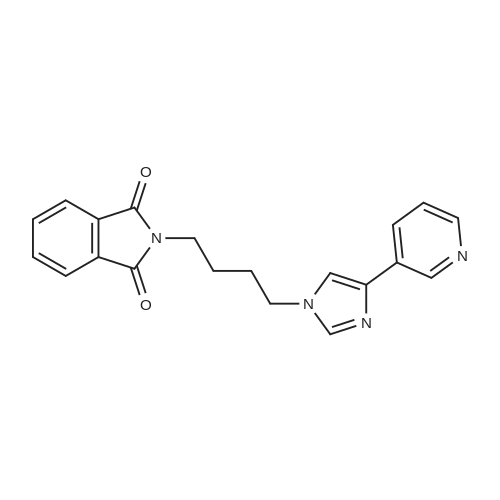
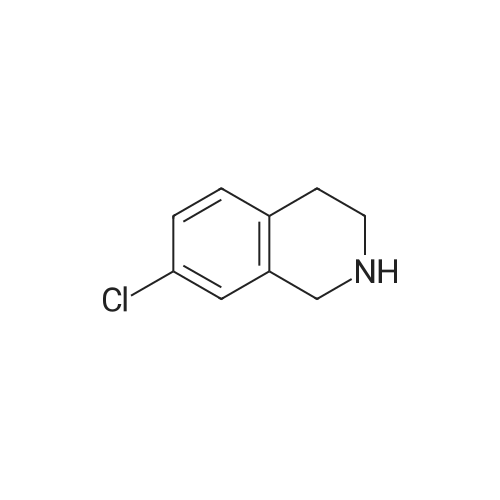
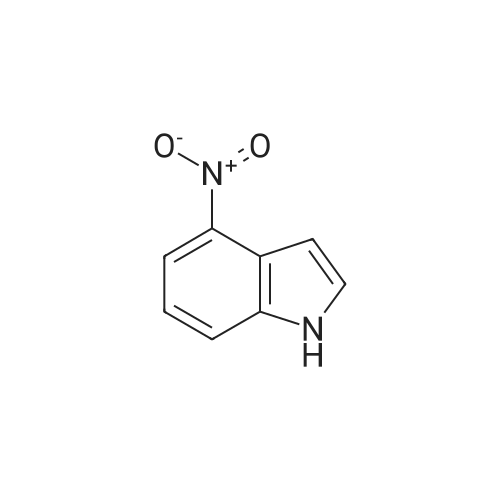
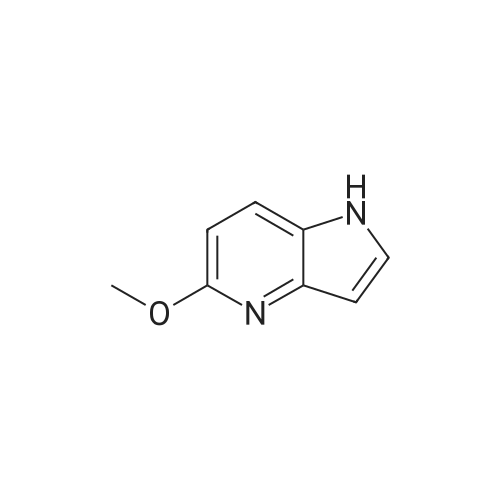
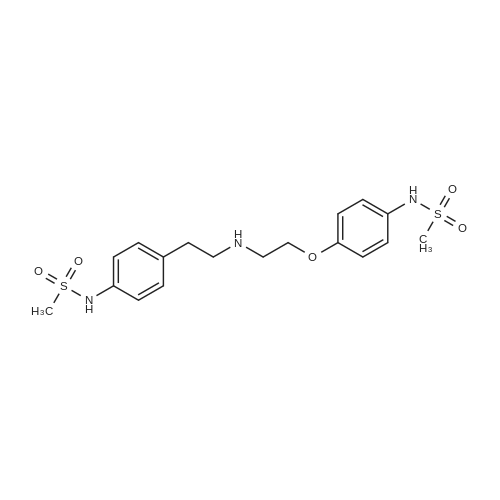
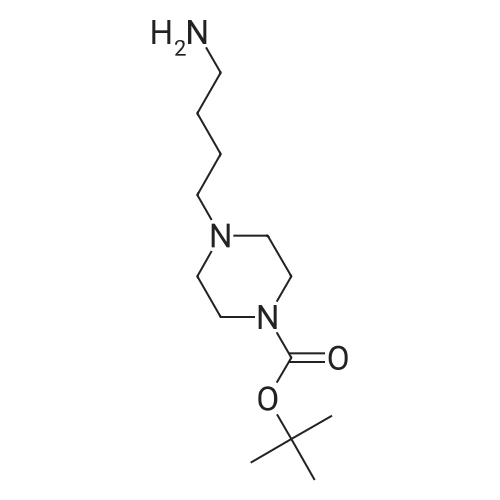
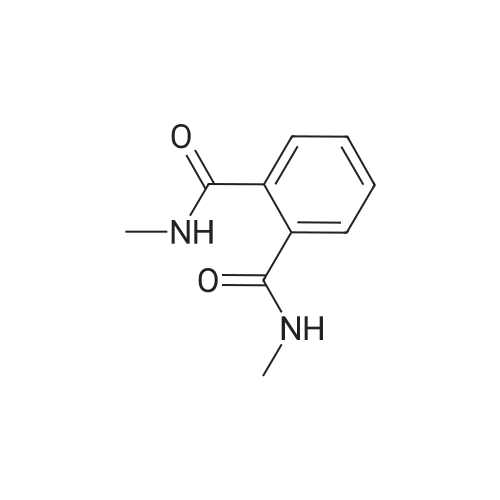
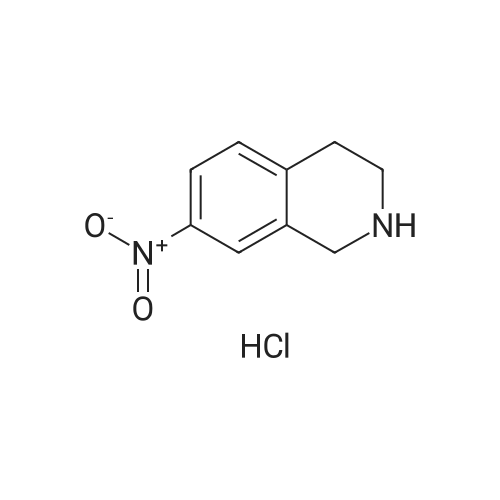
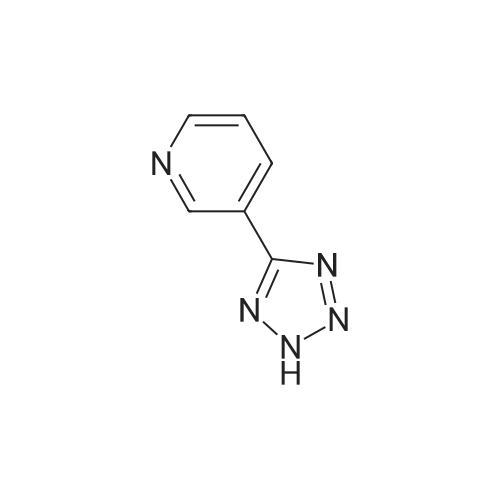
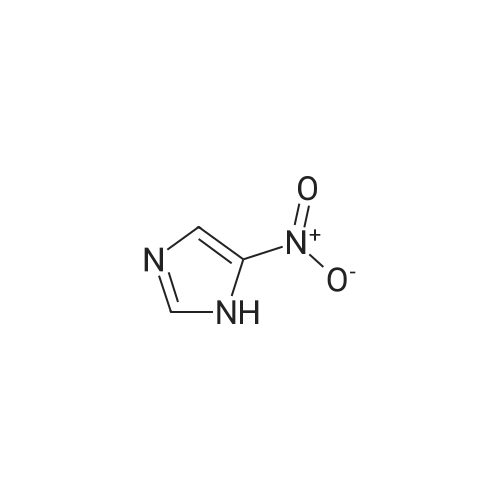
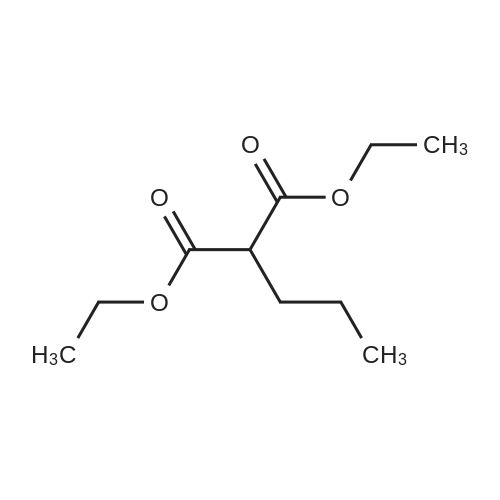
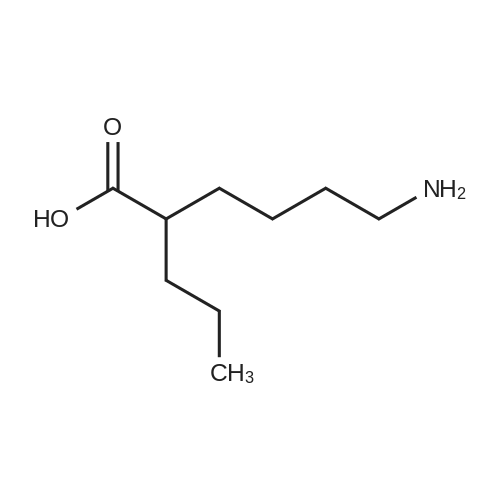
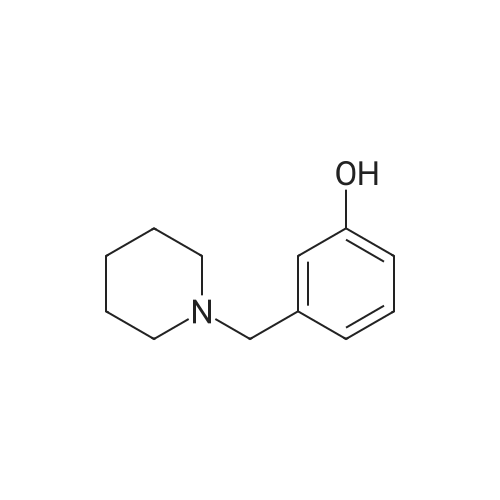
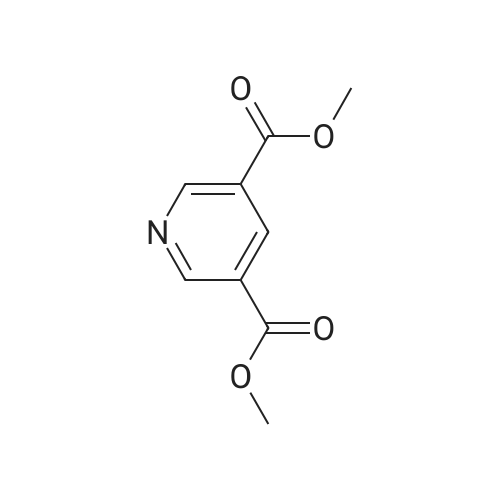
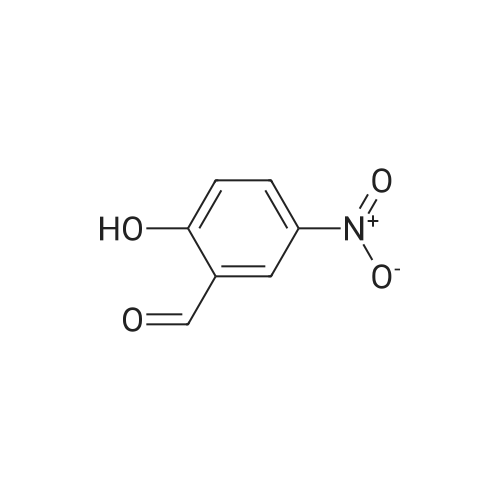
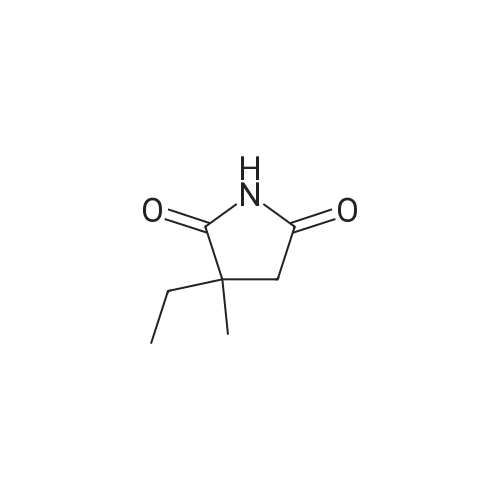
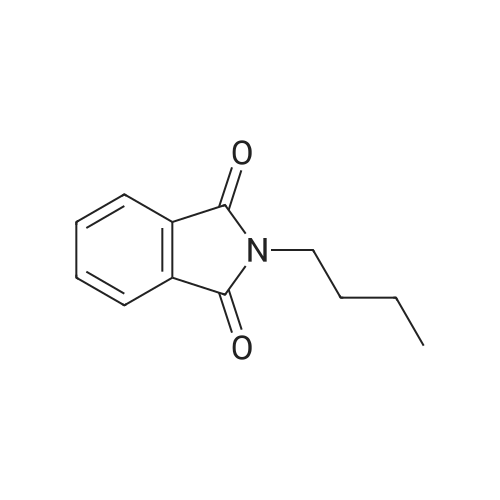
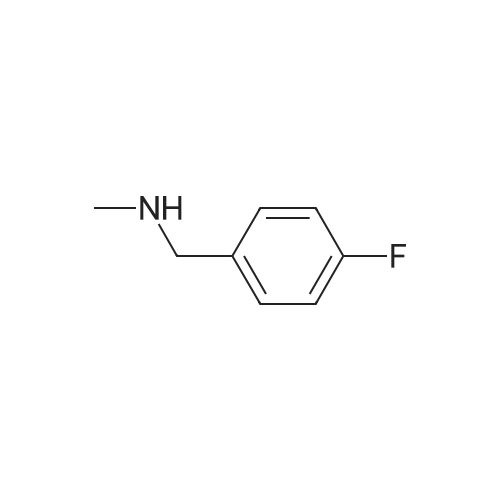
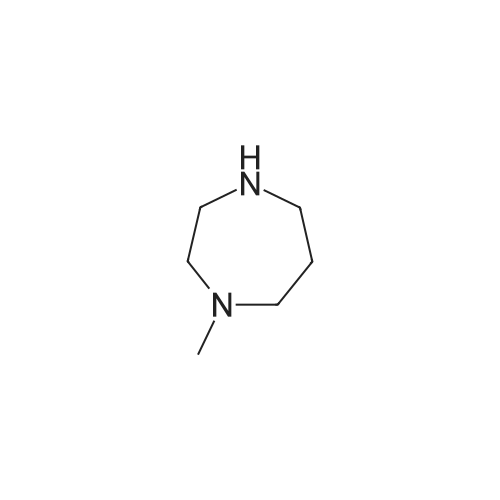
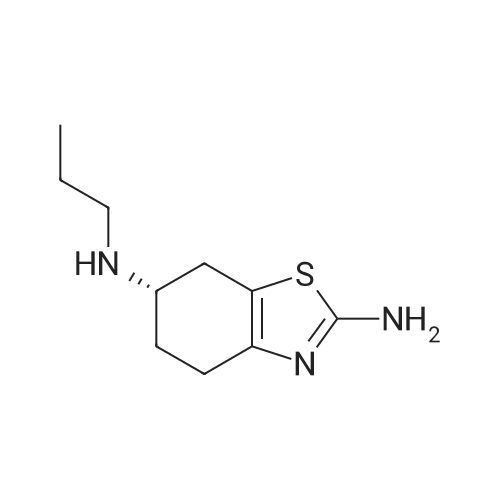
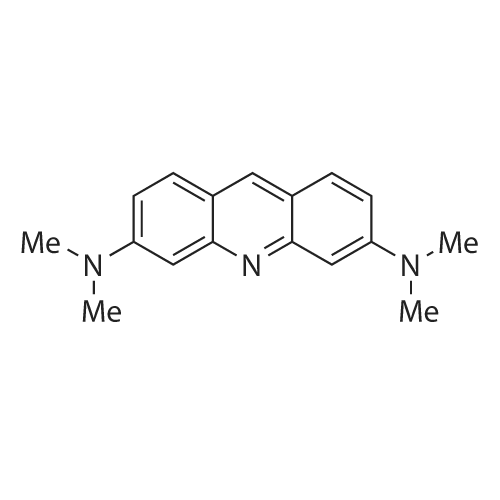
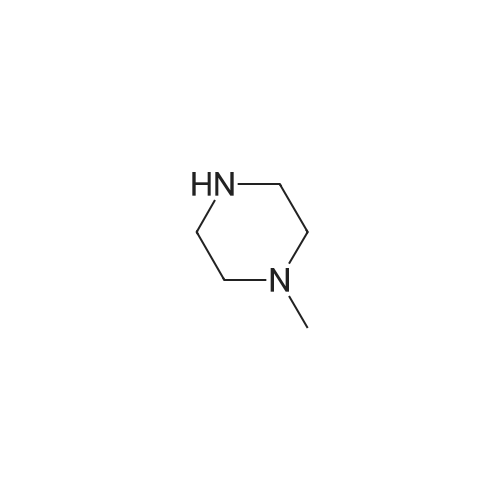
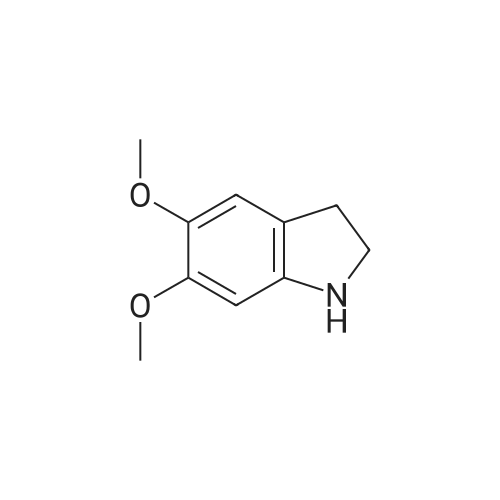
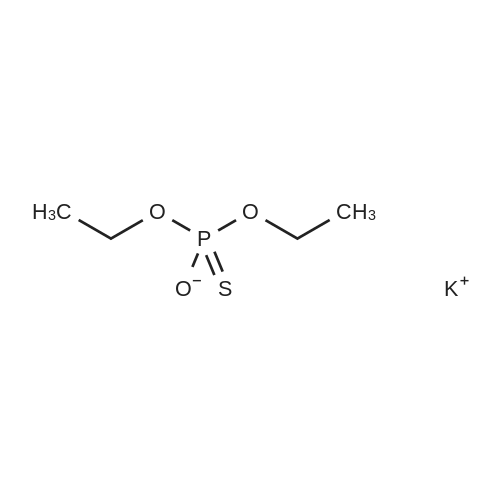
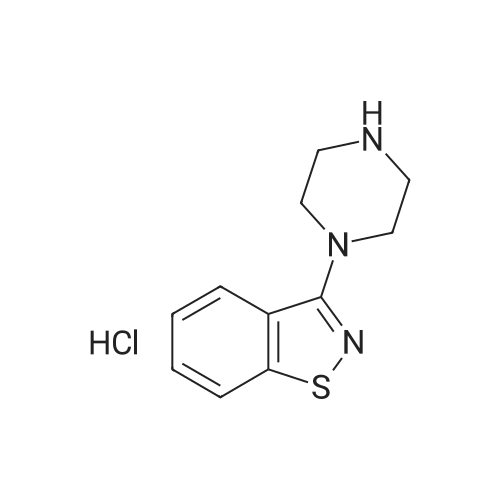
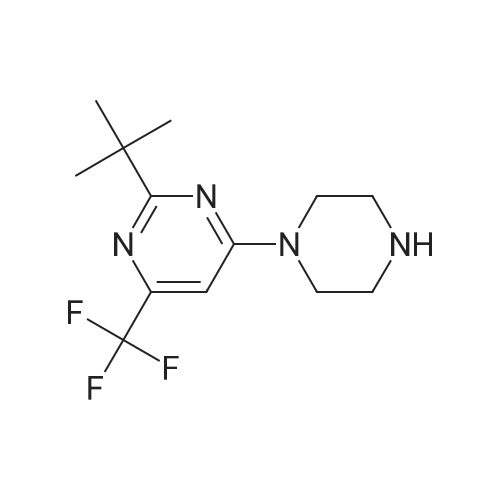
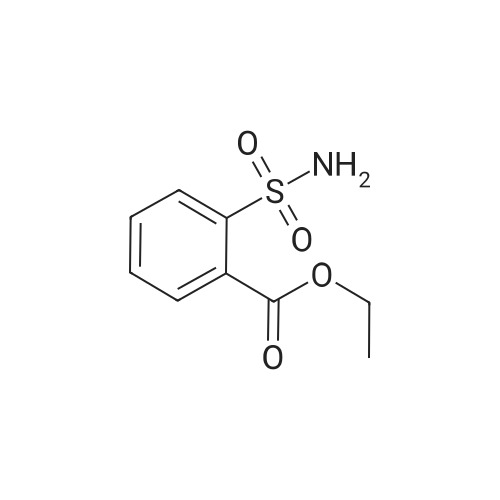
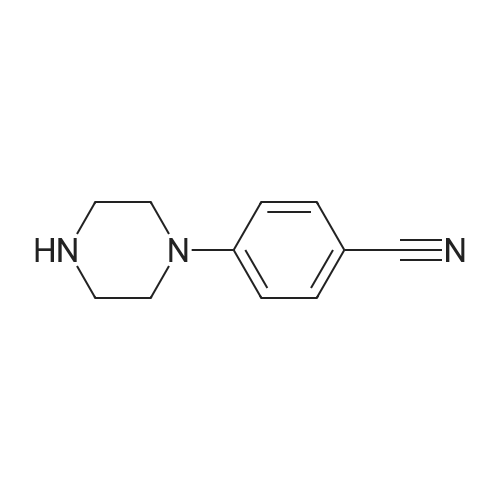
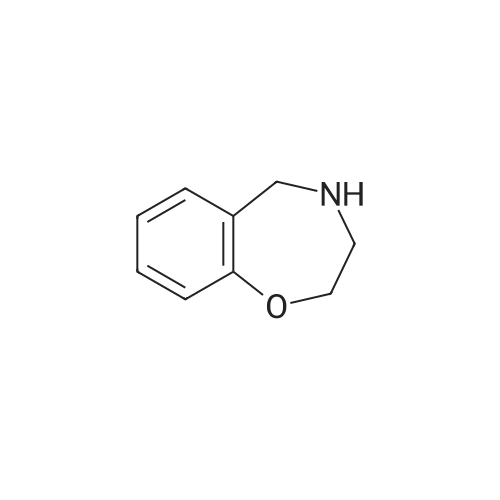
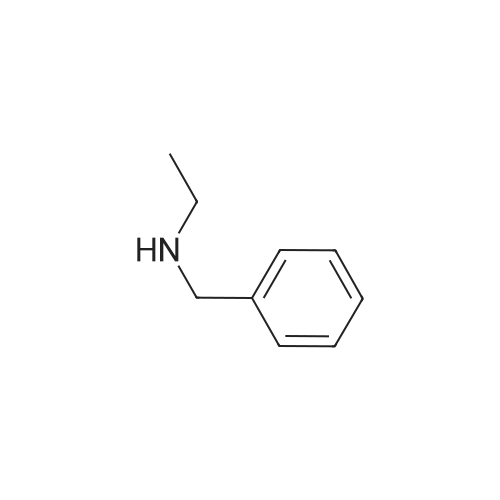
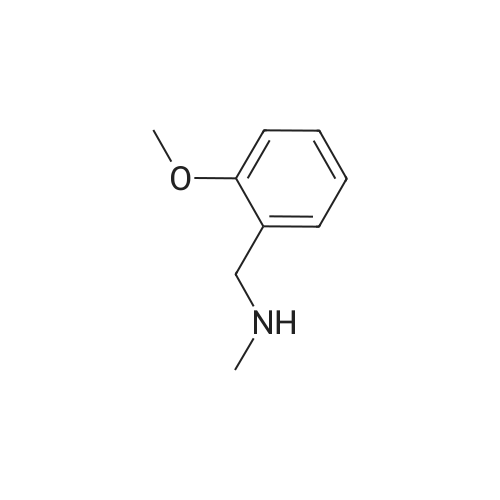
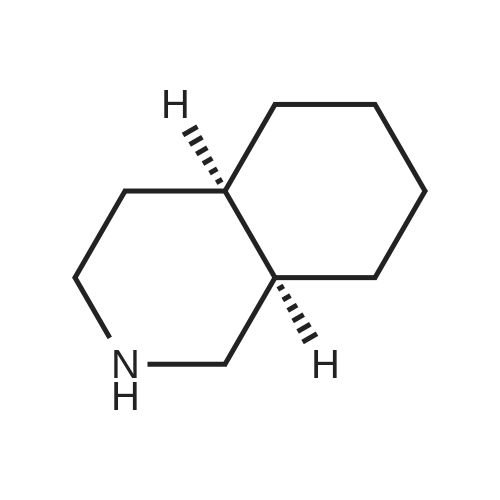
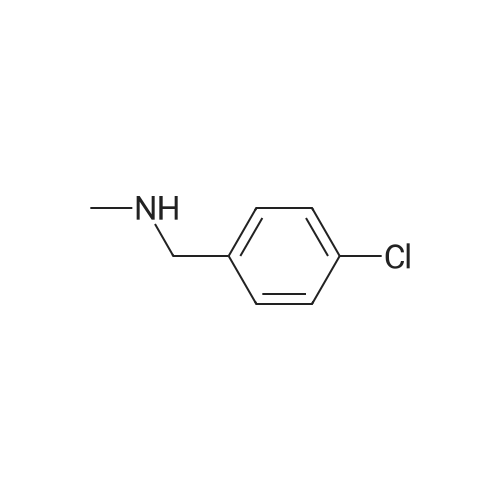
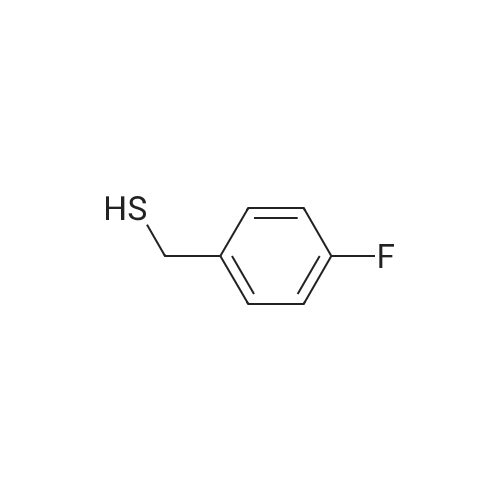
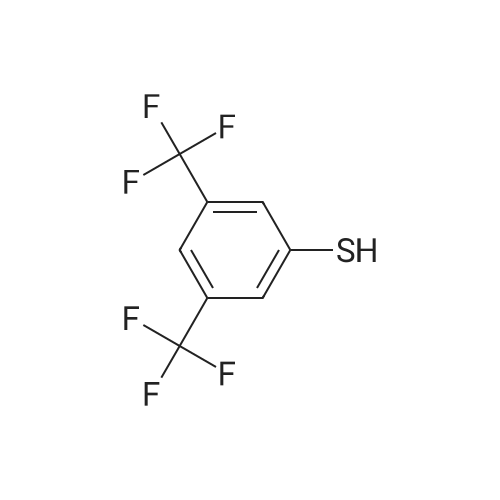
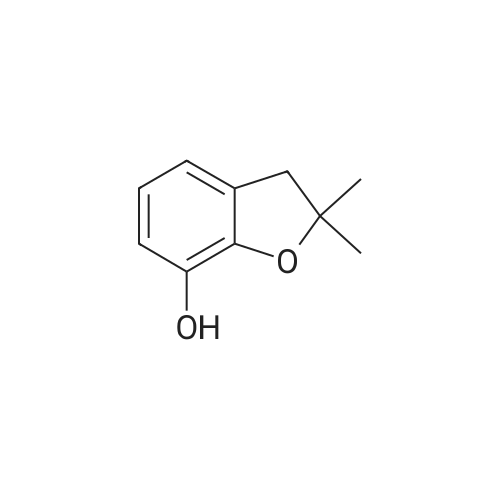
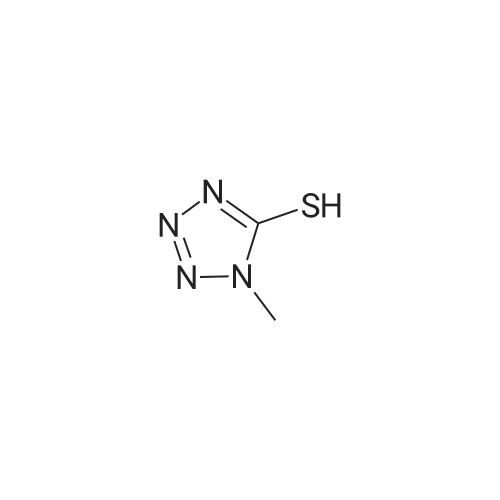
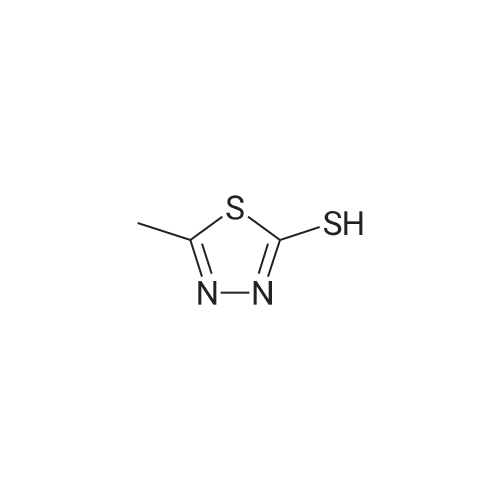
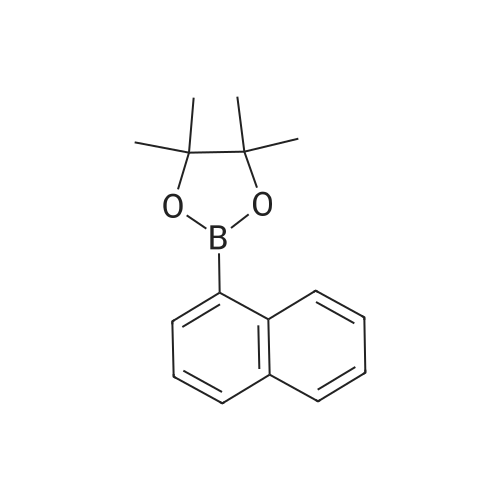
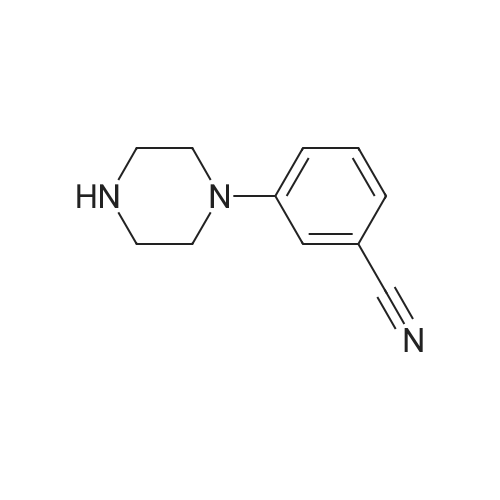
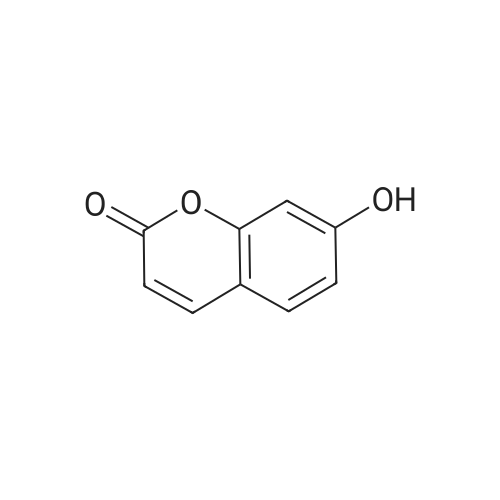
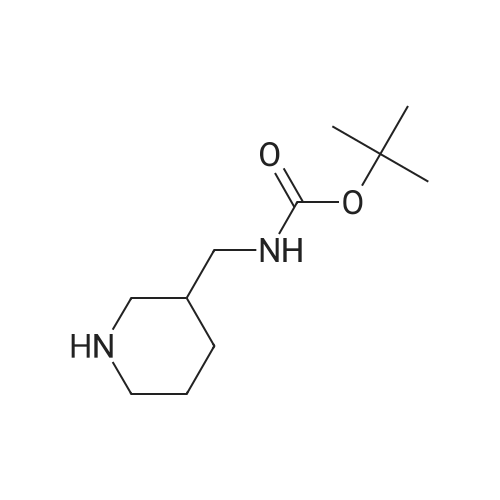
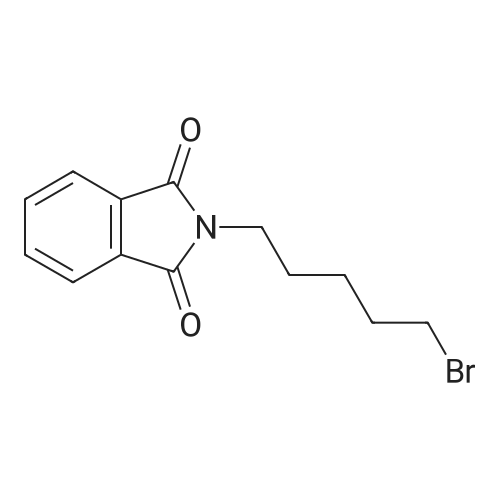
| 87% |
In N,N-dimethyl-formamide at 100℃; for 12h; |
|
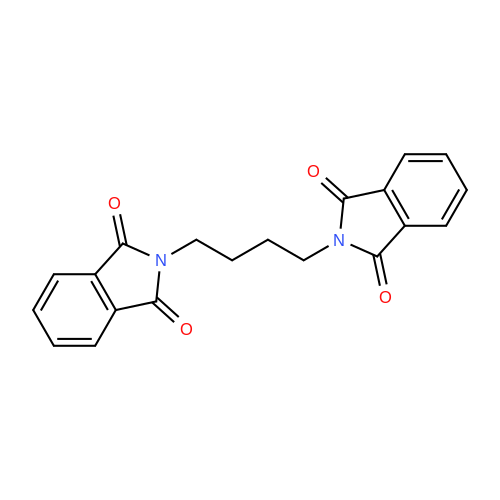
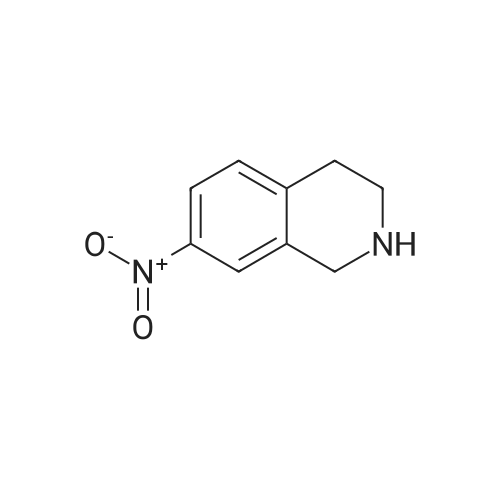
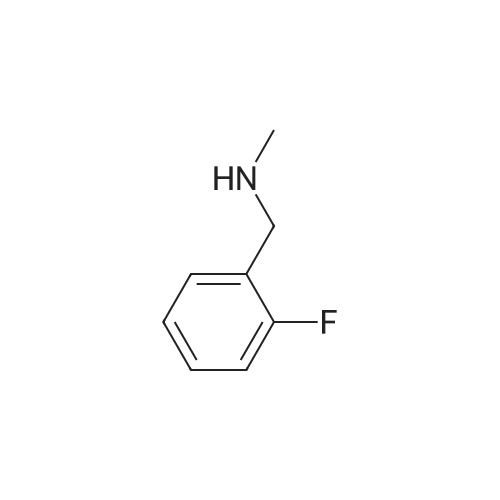
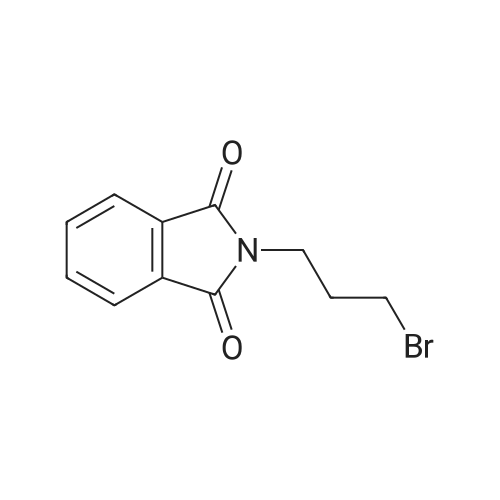
| 85% |
In N,N-dimethyl-formamide at 30℃; |
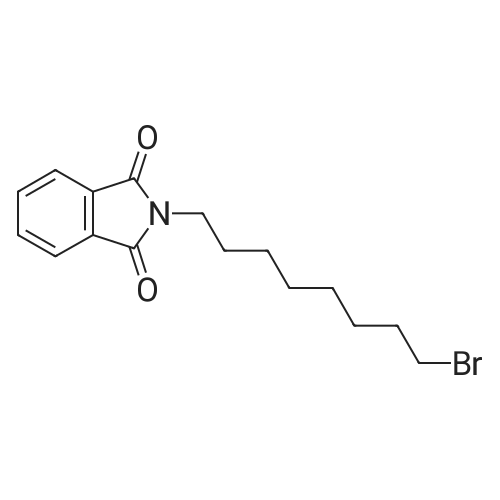
2-(2-Bromoethyl)isoindoline-1,3-dione (g)
General procedure: Potassium phthalimide (0.93 g, 5 mmol) was added to a solution of 1,2-dibromoethane (1.3 mL, 15 mmol) in DMF (8 mL). The mixture was stirred at room temperature overnight and evaporated the solvent in vacuo, the residue dissolved in H2O and extracted with ethyl acetate. The organic layer was washed by brine and dried by MgSO4. Filtered and the solvent evaporated in vacuo, recrystallized from ethyl acetate to give white solid (566 mg, 45 %). |
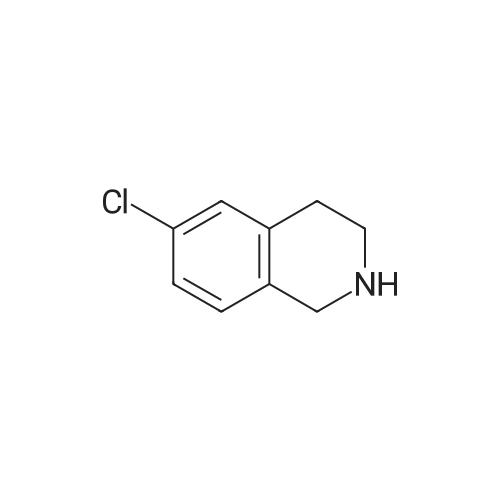
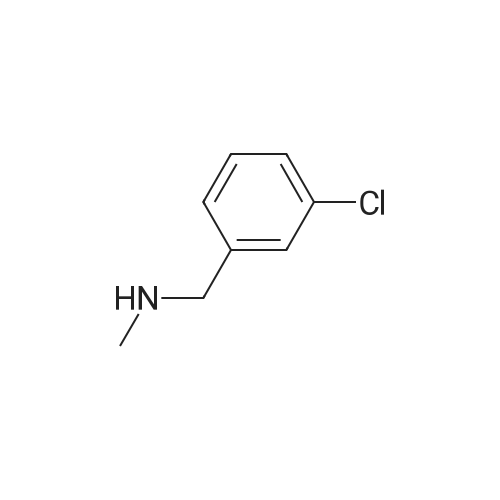
| 85% |
In N,N-dimethyl-formamide at 20℃; for 20h; |
|
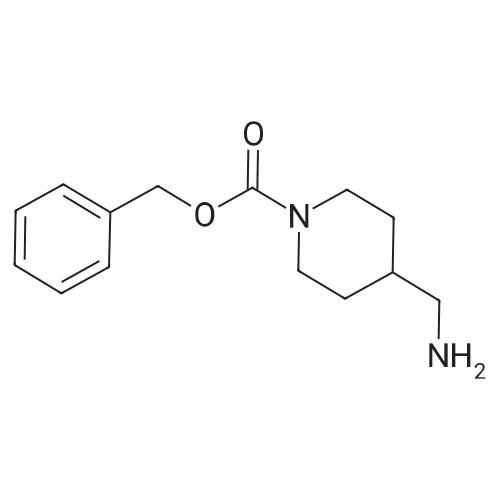
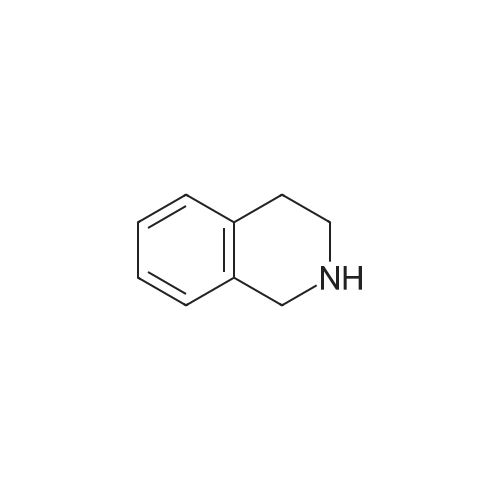
| 85% |
In N,N-dimethyl-formamide at 90℃; for 24h; |
|
| 84% |
With tetrabutylammonium bromide In N,N-dimethyl-formamide at 78℃; for 6h; |
3.1. Procedure for Synthesis of Intermediate Compound 2
Potassium phthalimide (18.62 g, 0.10 mol) and tetrabutylammoniumbromide (TBAB, 5.00 g) were added to a solutionof 1,4-dibromobutane (35.5 mL, 0.30mol) in N, N-Dimethylformamide (DMF, 240 mL) at room temperature and then the mixture was stirred at 78 °C for 6 h. After the completion of the reaction (monitored by TLC analysis), the mixture was filtered, and washed with DMF (20 mL × 2).The filtrate was evaporated under vacuum to removed DMF,the residual liquid was poured into ice-water bath, immediately a large amount of white solid generated. The white solid was collected by filtration, washed on the filter withwater and dried in vacuum. The crude residue was purifiedby recrystallization from methanol to afford pure N-(4-Bromobutyl) phthalimide 2 (23.60 g, 84%, m.p. 79-81oC aswhite solid. 1H NMR (CDCl3, 400 MHz), δ: 1.82-1.95 (m,4H, C13H, C14H), 3.45 (t, J=6.3 Hz, 2H, C15H), 3.73 (t,J=6.6 Hz, 2H, C16H), 7.27-7.74 (m, 2H, C3H, C6H), 7.83-7.86 (m, 2H, C1H, C2H); 13C NMR (CDCl3, 100 MHz), δ:27.3 (C13), 29.9 (C14), 32.8 (C15), 37.0 (C12), 123.3 (C3,C6), 132.1 (C4, C5), 134.0 (C1, C2), 168.4 (C7, C9). KunHu has reported the same compound in 2013 [17], the NMRcharacterization in the literature is described as follows: 1HNMR (CDCl 3, 500 MHz): d 1.84-1.94 (m, 4H), 3.45 (t,2H, J = 6.0 Hz), 3.73 (t, 2H, J = 6.5 Hz), 7.73 (dd, 2H, J =3.0, 5.5 Hz), 7.85 (dd, 2H, J = 3.0, 5.0 Hz); 13 C NMR(CDCl 3, 125 MHz): d 27.27, 29.89, 32.73, 36.99, 123.27,132.11, 133.99, 168.36. |
| 82.5% |
With tetrabutylammonium bromide In N,N-dimethyl-formamide at 70℃; for 2h; |
3 Example 3
50 mL DMF, 0.025 mol potassium salt of phthalimide,0.10 mol 1,4-dibromobutane and 0.5 g TBAB,70 reaction 2.0h;Cool to room temperature,Pour ice waterEthyl acetate extraction,Washed,dry,De-soluble,Stand overnight,5.82 g of N- (4-bromobutyl) phthalimide precipitated as a white solid,m.p. 78-81 ° C, yield 82.5%. |
| 82% |
In acetone for 24h; Reflux; |
General procedure for the synthesis of 11a-h:
General procedure: A mixture of phthalimide10(6.8 mmol) dissolved in EtOH (20 mL) was gently boiled for about 1h. The hot solution was decanted from any solid into 1.25 mL of a specially prepared solution of KOH (15.25 g KOH dissolved in 15 mL of H2O and 45 mL of EtOH). A precipitate of potassium phthalimide separates at once. The mixture was stirred and cooled quickly to room temperature, and the precipitate was filtered under vacuum. To the alcoholic mother liquors a second 1 g portion of phthalimide was added, and the entire process was repeated. The two crops of crystals were united and washed with acetone to remove any unchanged phthalimide. After air-dried pure potassium phthalimidewas obtained as white crystals (yield 30%). In a second step, a mixture of potassium phthalimide (2.2 mmol),the appropriate dibromoalkane derivative (2.9 mmol) and 2.5 mL of acetone was stirred and refluxed for 24h, and then cooled to 15 °C. After filtering off the precipitated potassium bromide, the cake was washed with acetone and the solvent evaporated to give pure compound. |
| 82.5% |
With tetrabutylammonium bromide In N,N-dimethyl-formamide at 70℃; for 2h; |
10 Preparation of N-(4-bromobutyl)phthalimide
50 mL of DMF, 0.025 mol of the potassium salt of phthalimide, 0.10 mol of 1,4-dibromobutane and 0.5 g of TBAB, reaction at 70° C. for 2.0 h; cooling to room temperature, pouring into ice water, extraction with ethyl acetate, After washing with water, drying and desolvation, the mixture was allowed to stand overnight to precipitate 5.82 g of N-(4-bromobutyl)phthalimide as a white solid, mp 78-81°C, yield 82.5%. |
| 81% |
In acetone for 24h; Reflux; |
|
| 80% |
In acetone for 12h; Reflux; |
5.4.4. 2-(4-bromobutyl)isoindoline-1,3-dione (8)
Potassium phthalimide salt 7 (572 mg, 3.09 mmol) was added toa solution of 1,4-dibromobutane (2.0 g, 1.11 ml, 9.26 mmol) inacetone (5 mL). The reaction mixture was refluxed for 12 h; laterthe solid was filtered off and the resulting solution was evaporatedunder reduced pressure. The crude product was purified throughflash chromatography using EP/AcOEt 9:1 as eluting mixture toafford derivative 8 (696.9 mg, 2.47 mmol, 80% yield). 1H NMR(CDCl3):δ 1.83-1.93 (m, 4H, CH2); 3.44 (t, J 6.4 Hz, 2H, CH2Br);3.72 (t, J 6.8 Hz, 2H, CH2N); 7.71-7.73 (m, 2H, Ar); 7.83-7.86 (m,2H, Ar) ppm. |
| 79% |
With tetrabutylammonium bromide In acetonitrile for 20h; Reflux; |
|
| 78.2% |
With potassium carbonate In acetone at 60 - 65℃; Inert atmosphere; |
General procedure for the synthesis of intermediates 3a-3e
General procedure: The appropriate dibromoalkane derivative 2a-2e (11.9 mmol) was added slowly to a mixture of the starting material phthalimide potassium salt (1) (1g, 5.4 mmol) and anhydrous K2CO3 (0.82 g, 5.94 mmol) in acetone (15 mL). The reaction mixture was heated to 60-65 °C and stirred for 6-10 h under an argon atmosphere. After complete reaction, the solvent was evaporated under reduced pressure. Water (50 mL) was added to the residue and the mixture was extracted with dichloromethane (30 mL × 3). The combined organic phases were washed with saturated aqueous NaCl, dried over Na2SO4, and filtered. The solvent was evaporated to dryness under reduced pressure. The crude product was purified on a silica gel chromatography using dichloromethane/acetone (50:1) as eluent to give the intermediates 3a-3e. |
| 77% |
With tetrabutylammonium bromide |
|
| 76% |
With potassium carbonate; potassium iodide In acetone at 100℃; for 6h; |
4.2.6.1 (R,S) 3-(4-bromobutyl)-4-phenyloxazolidin-2-one 23
General procedure: To a solution of 1.6g (10.0mmol) of (R,S) 4-phenyloxazolin-2-one (13), 3.4g (25.0mmol) of K2CO3 and a catalytic amount of KI in acetonitrile, 5.4g (25.0mmol) of 1,4-dibromobutane were slowly added drop by drop stirring at rt. The mixture was then heated under reflux for 6-12h, monitored by TLC (CH2Cl2/EtOH 9:1). The hot solution was filtered and evaporated under reduced pressure. The yellow oil residue of (R,S) 3-(4-bromobutyl)-4-phenyloxazolidin-2-one 23 was used without further purification. (0045) Yield: 96%; |
| 75% |
In acetone for 10.25h; Reflux; |
1 5.1.1 General procedures for preparation of 2-5
General procedure: Acetone (150mL) and dibromoalkyl (30mol) were added to a 250mL three-necked round-bottom flask fitted with a mechanical stirrer and reflux condenser. Potassium phthalimide (11.85g, 10 mol) was added slowly over a 15-min period, and then the reaction mixture was heated under reflux for 10h. The reaction mixture was filtered via suction, and the acetone was removed via rotary evaporation. The crude product was purified by flash chromatography on silica gel (EtOAc:petroleum ether=1:14) to afford 2-5 as white solid. |
| 75% |
In acetone for 24h; Reflux; |
|
| 72.3% |
In N,N-dimethyl-formamide at 90℃; for 18.5h; |
25a.1 10649] Step 1: Synthesis of 2-(4brornobuty)isoindoine-4 ,3-dione (3-j)
To a stirred solution of 1,4-dibromobutane (2j) (9,7 mL, 27,0 mmoi) (100 mL), was added potassIum phthaiate (i-) (5.0 g, 27,0 mrnol) portion-wise over 30 mm at room temperature. After complete addition, the reaction mixture was stirred at 90 °C for 18 h, then quenched with water (300 rnL) and extracted with diethyl ether (150 mL x 2). The combined organic extracts were washed with water (100 mL x 2), followed by brine (50 rnL x 2) and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the crude. The crude product was purified by silica gel column chromatography (60- 120 mesh) using 5.40% EtOAc / hexanes to afford 3j as an off- white solid (5.5 g, 72.3% yield). LCMS: 2.3.9(M+1) |
| 72.3% |
In N,N-dimethyl-formamide at 20 - 90℃; for 18.5h; |
25a.1 Step 1: Synthesis of 2-(4-bromobutyl)isoindoline-l,3-dione (3-j)
To a stirred solution of 1,4-dibromobutane (2-j) (9.7 mL, 27.0 mmol) in DMF (100 mL), was added potassium phthalate (1-j) (5.0 g, 27.0 mmol) portion-wise over 30 min at room temperature. After complete addition, the reaction mixture was stirred at 90 °C for 18 h, then quenched with water (300 mL) and extracted with diethyl ether (150 mL x 2). The combined organic extracts were washed with water (100 mL x 2), followed by brine (50 mL x 2) and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the crude. The crude product was purified by silica gel column chromatography (60- 120 mesh) using 5-10% EtOAc / hexanes to afford 3-j as an off- white solid (5.5 g, 72.3% yield). LCMS: 283.9(M+1). |
| 72.3% |
In N,N-dimethyl-formamide at 20 - 90℃; for 18.5h; |
25a.1 Step 1: Synthesis of 2-(4-bromobutyl)isoindoline-1,3-dione (3-j)
To a stirred solution of 1,4-dibromobutane (2-j) (9.7 mL, 27.0 mmol) in DMF (100 mL), was added potassium phthalate (1-j) (5.0 g, 27.0 mmol) portion-wise over 30 mm at room temperature. After complete addition, the reaction mixture was stirred at 90 °C for 18 h, then quenched with water (300 mL) and extracted with diethyl ether (150 mL x 2). The combined organic extracts were washed with water (100 mL x 2), followed by brine (50 mL x 2) and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the crude. The crude product was purified by silica gel column chromatography (60- 120 mesh) using 5-10% EtOAc / hexanes to afford 3-j as an off- white solid (5.5 g, 72.3% yield). LCMS: 283.9(M+1). |
| 72.3% |
In N,N-dimethyl-formamide at 90℃; for 18h; |
25a.1 Step 1: Synthesis of 2-(4-bromobutyl)isoindoline-l,3-dione (3-j)
To a stirred solution of 1,4-dibromobutane (2-j) (9.7 mL, 27.0 mmol) in DMF (100 mL), was added potassium phthalate (1-j) (5.0 g, 27.0 mmol) portion-wise over 30 min at room temperature. After complete addition, the reaction mixture was stirred at 90 °C for 18 h, then quenched with water (300 mL) and extracted with diethyl ether (150 mL x 2). The combined organic extracts were washed with water (100 mL x 2), followed by brine (50 mL x 2) and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the crude. The crude product was purified by silica gel column chromatography (60- 120 mesh) using 5-10% EtOAc / hexanes to afford 3-j as an off- white solid (5.5 g, 72.3% yield). LCMS: 283.9(M+1). |
| 72.3% |
In N,N-dimethyl-formamide at 20 - 90℃; for 18.5h; |
25a.1 Step 1: Synthesis of 2-(4-bromobutyl)isoindoline-1,3-dione (3-j)
To a stirred solution of 1,4-dibromobutane (2-j) (9.7 mL, 27.0 mmol) in DMF (100 mL), was added potassium phthalate (1-j) (5.0 g, 27.0 mmol) portion-wise over 30 mm at room temperature. After complete addition, the reaction mixture was stirred at 90 °C for 18 h, then quenched with water (300 mL) and extracted with diethyl ether (150 mL x 2). The combined organic extracts were washed with water (100 mL x 2), followed by brine (50 mL x 2) and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the crude. The crude product was purified by silica gel column chromatography (60- 120 mesh) using 5-10% EtOAc / hexanes to afford 3-j as an off- white solid (5.5 g, 72.3% yield). LCMS: 283.9(M+1). |
| 72.3% |
In N,N-dimethyl-formamide at 20 - 90℃; |
25a.1
Production Example 25a: Synthesis of (R)-N-(1-(3-(cyclopropylmethoxy)-4- fluorophenyl) ethyl)-4-(2,4-dioxoimidazolidin-1-yl)butane-1-sulfonamideScheme 11. [0697] Step 1: Synthesis of 2-(4-bromobutyl)isoindoline-1,3-dione (3-j) To a stirred solution of 1,4-dibromobutane (2-j) (9.7 mL, 27.0 mmol) in DMF (100 mL), was added potassium phthalate (1-j) (5.0 g, 27.0 mmol) portion-wise over 30 min at room temperature. After complete addition, the reaction mixture was stirred at 90 °C for 18 h, then quenched with water (300 mL) and extracted with diethyl ether (150 mL x 2). The combined organic extracts were washed with water (100 mL x 2), followed by brine (50 mL x 2) and dried over anhydrous sodium sulfate, filtered and concentrated under reduced pressure to obtain the crude. The crude product was purified by silica gel column chromatography (60- 120 mesh) using 5-10% EtOAc / hexanes to afford 3-j as an off- white solid (5.5 g, 72.3% yield). LCMS: 283.9(M+1). |
| 68.3% |
With potassium carbonate In acetone Reflux; |
General procedure for the preparation of compounds 2-4.
General procedure: A mixture of dibromoalkane (23 mmol) and potassium carbonate (12 mmol)was added in acetone (30 mL), and potassium phthalimide (10 mmol) was added slowly over a 15-min period, and then the reaction was heated under reflux for 8-10 h. The reaction mixture was filtered,and the acetone was evaporated in vacuo. The crude product was purified by column chromatography on silica gel using mixtures of petroleum/acetone as eluent) to obtain the white solid compounds 2-4. |
| 67.7% |
With potassium carbonate In acetone Reflux; |
1 Example 1 Synthesis of Compound 1
0.011mol (2.375g) 1.4-dibromobutane was added to a 50mL reaction flask, 0.005mol (0.926g) potassium phthalimide, 0.006mol (0.829g) anhydrous potassium carbonate and 20mL acetone were added respectively , stirring and warming up to reflux, holding the reaction, TLC tracking the reaction (developing agent: PF/EA=4/1), the reaction was completed, cooled to room temperature, suction filtration, the filter cake was washed with 10 mL of acetone each time, washed twice, and the filtrate was combined. , the solvent was evaporated under reduced pressure, 10 mL of petroleum ether was added to the residue, stirred well, placed in the refrigerator overnight, suction filtered, the filter cake was washed twice with 5 mL of petroleum ether, and dried to obtain 0.956 g of white solid compound 1, with a yield of 67.77% , the purity is 98.75%. |
| 66% |
In N,N-dimethyl-formamide at 0 - 20℃; for 18h; |
Syntheses of D,L-Sulforaphane and Erysolin; Compound 9; 2-(4-bromobutyl)isoindoline-l,3-dione; The following procedure was adapted from that previously reported by Vermeulen, et al (25) 1 ,4-Dibromobutane (4 400 mL, 36 457 mmols) was dissolved in anhydrous DMF (52 mL) and the resulting solution was chilled to 0°C under argon After 15 min, potassium phthahmide (3 459 g, 18 672 mmols) was slowly added to the stirring solution and the reaction was allowed to warm to ambient temperature under argon over 18 h The reaction was concentrated in vacuo and co-stϖpped with anhydrous THF several times Products were dissolved in 1 1 H2O EtOAc (200 mL) and the aqueous phase was extracted with EtOAc (3 x 100 mL) Combined organics were washed with brine, dried over Na2SO4, and filtered through a celite plug prior to concentration in vacuo Silica gel chromatography (3 1 Hexane EtOAc) and subsequent concentration afforded 3 466 g 9 as a white solid (66% yield) 1H NMR (CDCl3) 5 7 85 (dd, J = 5 4, 3 1 Hz, 2H), 7 73 (dd, J = 5 4, 3 0 Hz, 2H), 3 73 (t, J = 6 7 Hz, 2H), 3 45 (t, J = 6 4 Hz, 2H), 1 89 (m, 4H) 13C NMR (CDCl3) δ 168 5, 134 1, 132 2, 123 4, 37 1, 32 9, 30 0, 27 4 HRMS (ESI-EMM) calc'd for [M + Na]+ m/z 303 9949, found 303 9936 |
| 66% |
In N,N-dimethyl-formamide at 0 - 20℃; for 18h; Inert atmosphere; |
Compound 9: 2-(4-bromobutyl)isoindoline-1,3-dione. The following procedure was adapted from that previously reported by Vermeulen, et al. (25). 1,4-Dibromobutane (4.400 mL, 36.457 mmols) was dissolved in anhydrous DMF (52 mL) and the resulting solution was chilled to 0° C. under argon. After 15 min, potassium phthalimide (3.459 g, 18.672 mmols) was slowly added to the stirring solution and the reaction was allowed to warm to ambient temperature under argon over 18 h. The reaction was concentrated in vacuo and co-stripped with anhydrous THF several times. Products were dissolved in 1:1 H2O:EtOAc (200 mL) and the aqueous phase was extracted with EtOAc (3×100 mL). Combined organics were washed with brine, dried over Na2SO4, and filtered through a celite plug prior to concentration in vacuo. Silica gel chromatography (3:1 Hexane:EtOAc) and subsequent concentration afforded 3.466 g 9 as a white solid (66% yield). 1H NMR (CDCl3) δ 7.85 (dd, J=5.4, 3.1 Hz, 2H), 7.73 (dd, J=5.4, 3.0 Hz, 2H), 3.73 (t, J=6.7 Hz, 2H), 3.45 (t, J=6.4 Hz, 2H), 1.89 (m, 4H). 13C NMR (CDCl3) δ 168.5, 134.1, 132.2, 123.4, 37.1, 32.9, 30.0, 27.4. HRMS (ESI-EMM) calc'd for [M+Na]+m/z 303.9949. found 303.9936. |
| 64.6% |
In acetone for 12h; Reflux; |
|
| 61% |
at 115 - 120℃; for 12h; |
1 EXAMPLE 1 Synthesis of N-(4-bromobutyl)phthalimide With a 1,4-dibromobutane/potassium Phthalimide Ratio of 3
Into a Sovirel type reactor mechanically stirred, provided with a condenser, an opening for introduction of solids, a system inerting with nitrogen, and a temperature probe, are loaded 655 g of 1,4-dibromobutane at 99% purity (3 mol) and 187 g of potassium phthalimide at 99% purity (1 mol). The reaction medium is brought under stirring between 115° C. and 120° C. and then kept at this temperature for 12 h. It is verified that the transformation of potassium phthalimide is completed by a measurement of the KBr in the reaction medium and then the excess 1,4-dibromobutane is evaporated under reduced pressure (10 to 18 mmHg). The recovery rate of the 1,4-dibromobutane is near 90% and this latter may be recycled directly into a later operation. [0052] At the end of the distillation, the heterogeneous reaction medium is 120° C.-130° C. Its temperature is lowered to 70° C. and then 320 g of ethanol are introduced. The medium is kept at 70° C. [0053] The contents of the reactor are filtered at 70° C. on a frit kept at this temperature. The moist cake is dried. It essentially contains KBr and diphthalimidobutane. [0054] The ethanolic filtrate is left to cool to 20° C., which causes crystallization of the N-(4-bromobutyl)phthalimide. The ethanolic suspension of the product is filtered on the frit. The moist cake is washed with 2 ethanol fractions and then dried at 50° C. under reduced pressure (20 to 30 mmHg). Thus, 173 g of N-(4-bromobutyl)phthalimide are obtained or an isolated yield of 61% compared with the potassium phthalimide provided. |
| 61% |
for 12h; Heating / reflux; |
1.A
Potassium phthalimide (65 g, 350 mmol, 1.00 eq.) and tetramethylene dibromide (200 g, 930 mmol, 2.66 eq.) were combined (neat) and heated for 12 hours. The excess tetramethylene dibromide was removed via rotary-evaporation (rotovap). The resulting residue was digested with ethanol and filtered. The material that crystallized on standing was filtered, washed with ethanol, and dried in vacuo. A second crop was obtained by concentration of combined filtrate and washings. The combined mass of crops was 60.5 g (61% yield). |
| 52% |
|
|
| 48.6% |
With N,N,N-tributyl-1-butanaminium iodide In acetone for 18h; Reflux; |
1.3
(3) N-(4-bromobutyl)phthalimide 93.6g (0.43mol) 1,4-dibromobutane was weighed, and added to 380mL acetone. The mixture was added with 72.5g (0.39mol) phthalimide potassium salt and 2.1g tetrabutylammonium iodide under stirring, subjected to refluxing for 18h, cooled, filtrated to remove solid, and washed with acetone. All acetone solutions are combined, subjected to recovering the solvent under a reduced pressure, and added with petroleum ether for crystallization while the reaction mixture was hot. A solid was filtered out, washed with with petroleum ether and dried to obtain a product 48.0g(43.6%),mp 75∼78°C. The mother liquor was concentraed to precipitate crystal, cooled in an ice-bath to obtain a solid 5.5g (5.0%), mp 73∼76°C. |
| 48.6% |
With N,N,N-tributyl-1-butanaminium iodide In acetone for 18h; Reflux; |
1.3
(3) N-(4-bromobutyl)phthalimide 93.6 g (0.43 mol) 1,4-dibromobutane was weighed, and added to 380 mL acetone. The mixture was added with 72.5 g (0.39 mol) phthalimide potassium salt and 2.1 g tetrabutylammonium iodide under stirring, subjected to refluxing for 18 h, cooled, filtrated to remove solid, and washed with acetone. All acetone solutions are combined, subjected to recovering the solvent under a reduced pressure, and added with petroleum ether for crystallization while the reaction mixture was hot. A solid was filtered out, washed with petroleum ether and dried to obtain a product 48.0 g (43.6%), mp 75~78° C. The mother liquor was concentrated to precipitate crystal, cooled in an ice-bath to obtain a solid 5.5 g (5.0%), mp 73~76° C. |
| 43.6% |
With N,N,N-tributyl-1-butanaminium iodide In acetone for 18h; Reflux; |
1.3 (3) N-(4-bromobutane)phthalimide:
Weigh 93.6 g (0.43 mol) of 1,4-dibromobutane and add it to 380 mL of acetone.72.5 g (0.39 mol) of phthalimide potassium salt and 2.1 g of tetrabutylammonium iodide were added under stirring, and the reaction was refluxed for 18 hours, cooled, and the solid was filtered, and washed with acetone.The acetone solution is combined, the solvent is recovered under reduced pressure, and petroleum ether crystals are added as it is, and the solid is collected by filtration.Wash with petroleum ether and dry to 48.0g (43.6%). |
| 42.2% |
In N,N-dimethyl-formamide at 20℃; for 24h; Inert atmosphere; |
|
| 40% |
for 12h; Heating; |
|
| 39.5% |
In N,N-dimethyl-formamide at 20℃; for 24h; |
|
| 32% |
In acetone for 24h; Heating; |
|
| 103.0 mmol |
In N,N-dimethyl-formamide |
|
|
for 12h; Heating; |
|
|
In acetone for 30h; Heating; |
|
|
With potassium iodide In acetone at 50℃; for 72h; |
|
|
In acetone for 10h; Reflux; |
|
|
In acetonitrile Reflux; |
|
|
In N,N-dimethyl-formamide at 20℃; for 18h; |
|
| 17.3 g |
In N,N-dimethyl-formamide at 20℃; for 48h; |
23 N-(4-Bromobutyl)phthalimide
A mixture of 1,4-dibromobutane (22 mL, 185 mmol) and potassium phthalimide (11.35 g, 61.4 mmol) in 60 mL of DMF was mixed at room temperature for 1 day. Then, the reaction mixture was extracted with hexane (3x150 mL). The hexane fractions were dried over MgS04, filtered, and concentrated to give 30 g of a 1:2.2 molar mixture of recovered 1,4-dibromobutane and DMF. This mixture was diluted with 30 mL of DMF and retreated with potassium phthalimide (4.80 g, 26 mmol) at room temperature for 1 day. The two reaction mixtures in DMF were partitioned between 1: 1 EA/Hex (3x150 mL) and H20 (2x100 mL), 0.1M HCl (100 mL), and brine (100 mL).The organic phases were dried over MgS04 and concentrated. SPE, eluting with 0% and 10% EA/Hex, gave 17.3 g of colorless solid. R 0.55 (40% EA/Hex); 1H NMR (CDC13) δ 7.86-7.81 (m, 2H), 7.73-7.69 (m, 2H), 3.71 (t, 2H), 3.43 (t, 2H), 1.94-1.80 (m, 4H); 13C NMR (CDC13) δ 168.5, 134.2, 132.3, 123.5, 37.2, 32.9, 30.1, 27.4. |
|
With tetrabutylammonium bromide In acetonitrile for 15h; Reflux; |
|
|
With N,N,N-tributylbutan-1-aminium fluoride In acetonitrile for 15h; Reflux; |
|
|
With potassium carbonate In acetone for 8h; Reflux; |
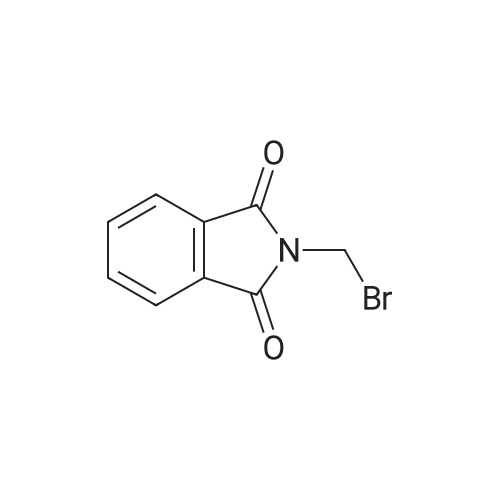
The preparation of N-(ω-bromoalkyl)phthalimide compounds is similar to the following process. N-(2-Bromoethyl) phthalimide (6I)
General procedure: A 500 mL round-bottom flask was charged with4.63 g (0.025 mol) Phthalimide potassium salt, dissolve in 120 mL Anhydrousacetone. 6.9 g (0.05 mol) potassium carbonate and 11.75 g (0.0625 mol) 1,2-Dibromoethanewas added. The solution was heated to reflux for 8 h. The stirring mixture wasallowed to cool, and then was filtered. The solids were washed with 50 mL ofacetone, followed by 50 mL of EtOAc. The filtrate and the washer liquid werepooled and concentrated in a rotary evaporator, then purified by flashchromatography on SiO2 (gradient, 1:6 to 1:4 petroleum ether:EtOAc)to obtain 5.2 g (81.8%) of N-(2-Bromoethyl) phthalimideas a white solid. |
|
With potassium carbonate In acetone Reflux; |
|
| 5.6 g |
In N,N-dimethyl-formamide at 20℃; for 15h; |
7.8ml (6. 5 X 10 2 mol) of 1,4-dibromobutane with 6 g (3. 2 X 10 2 moles) Phthalimide potassium salt dissolved in 100ml of N, N- dimethylformamide andunder room temperatureit was stirred for 15hours.The solvent was distilled off under reduced pressure The residue usingn-hexane: ethyl acetate with a volumeratio of 10: 1 eluent to carry out column chromatography, gradually increasingthe polarity to hexane: ethyl acetate with a volume ratio of 5: 1 to give aproduct of 5.6g (2.0X102mole) a bromo group substituted by Phthalimide. The product with 5. 0g (5. 0X 10 2 mol) ofsodium sulfite, 140ml water and 85ml95% ethanol mixed reaction was heated to 95° C for 18h, the remaining solvent was drained, and the resulting residue was mixed with 73ml ofconcentrated hydrochloric acid then was heated to 110 ° C the for 18H, itwas drained, with water - 95% ethanol on the residue to carry out recrystallization to give 2. 5g of4-amino-1-Butanesulfonic acid (total yield 50%). |
|
In N,N-dimethyl-formamide at 100℃; for 12h; |
1 4.1. synthesis of 2-(4-bromobutyl)isoindoline-1,3-dione, 1b
A mixture of potassium phthalimide 1a (0.93 g, 5 mmol) and1,4-dibromobutane (5.4 g, 25 mmol) was stirred in dry DMF(10 mL) at 100 °C for 12 h. The condenser was then set for distillation, and the excess of 1,4-dibromobutane and DMF was removed under reduced pressure. The crude product obtained was purifiedby column chromatography (silica gel, ethyl acetate/light petroleum1:50) to afford intermediate 1b as a colorless solid. 1H NMR(CDCl3): d 7.87-7.84 (2H, m), 7.74-7.71 (2H, m), 3.73 (2H, t,J = 6.9 Hz), 3.45 (2H, t, J = 6.3 Hz), 1.90-1.88 (4H, m). |
|
In acetone for 20h; Reflux; |
3.2 Preparation of compound b
In a 250 mL round bottom flask was added 97.16 g (0.45 mol) of 1,4-dibromobutane and 100 mL of acetone, 27.75g (0.15mol) potassium phthalimide was added portionwise under stirring, this mixture was stirred at reflux for 20h, cool down. The KBr was filtered off and the acetone was removed by distillation under reduced pressure and excess 1,4-dibromobutane, The remaining solid was recrystallized from absolute ethanol to give a white solid b. |
|
In N,N-dimethyl-formamide at 100℃; for 12h; |
Example 2
Synthesis of 2-(4-Bromobutyl)isoindoline-1,3-dione, 1b (FIG. 1): A mixture of 5 potassium phthalimide 1a (0.93 g, 5 mmol) and 6 1,4-dibromobutane (5.4 g, 25 mmol) was stirred in dry 7 DMF (10 mL) at 100° C. for 12 h. The condenser was then set for distillation, and the excess of 1,4-dibromobutane and DMF was removed under reduced pressure. The crude 8 product obtained was purified by column chromatography (silica gel, ethyl acetate/light petroleum 1:50) to afford intermediate 9 1b (FIG. 1) as a colorless solid. 1H NMR (CDCl3): δ 7.87-7.84 (2H, m), 7.74-7.71 (2H, m), 3.73 (2H, t, J=6.9 Hz), 3.45 (2H, t, J=6.3 Hz), 1.90-1.88 (4H, m). |
|
With potassium iodide In acetone for 10h; Reflux; |
General Procedures for the Synthesis of Compounds 4a-i
General procedure: To a solution of potassium 1,3-dioxoisoindolin-2-ide (1.85 g, 10.0 mmol), potassium iodide (0.08 g, 0.5 mmol) in acetone (25.0 mL), 1,2-dibromoethane dissolved in acetone (5.0 mL) was added by dropping funnel (3.72 g, 20.0 mmol), refluxed for 10 h. Monitored by TLC, and then filtered, concentrated by vacuum. The crude compound was purified by column chromatography on silica gel to give the desired intermediate 4 2-(2-bromoethyl) isoindoline-1,3-dione (1.83 g, 72.5% yield) as a white solid. |
|
With N,N,N-tributyl-1-butanaminium iodide In N,N-dimethyl-formamide at 20℃; for 24h; |
2.3 Synthesis of alkyl bromides (3a-c)
General procedure: In a round bottomed flask (100mL), it was added potassium phthalimide (1.85g, 0.01mol) and desire 1,n-dibromoalkane (0.02mol) in the presence of a catalytic amount of TBAI (0.1g) in anhydrous DMF (10mL). The reaction was stirred for 24h at r.t. After the completion of the reaction (TLC check), the solvent was evaporated in vacuo and the remaining residue was diluted in CHCl3 (150mL) and washed with H2O (3×150mL). The organic phase was evaporated and the remaining residue was purified by a short column chromatography on silica and n-hexane as an eluent. |
|
In acetone at 70℃; |
|
|
With potassium carbonate In acetonitrile at 60 - 65℃; |
|
|
In N,N-dimethyl-formamide at 20℃; for 10h; |
Synthesis of S2-S5.
General procedure: A mixture of potassium phthalimide (3.7 g, 20 mmol, 1 eq.) and 1,3-dibromopropane (8.0 g, 40 mmol, 2 eq.) in DMF (30 mL) was stirred at room temperature for 10 h. The solution was removed under reduced pressure. Then, the residue was purified by silica gel (petroleum ether/EtOAc = 15/1) to afford S2a (4.8 g, 90% yield) as a white solid. To a solution of (3-chloro-4-fluorophenylamino)-7-methoxyquinazolin-6-ol (640 mg, 2 mmol, 1 eq.) and compound S2a (641 mg, 2.4 mmol, 1.2 eq.) in 5 mL DMF, K2CO3 (1106 mg, 8 mmol, 4 eq.) was added, then the mixture was stirred at 80 for 5 h, after cooled to room temperature, 50 mL water was added, and extracted by 50 mL ethyl acetate for three times, the organic layer was combined and washed by brine and dried by Na2SO4, filtered, and concentrated in vacuo, the crude product was purified by silica gel (EtOAc/MeOH = 10/1) to give S2b (840 mg, 83% yield). To the solution S2b (840 mg, 1.66 mmol) in EtOH, hydrazine monohydrate (530 μL) was added, then the mixture was stirred at 78 for 2 h, then the mixture was cooled and filtered to remove the white insoluble solid to gain the crude product, purified by silica gel (EtOAc/MeOH/NH3(aq.) = 1/1/0.05) to give S2 as a bright yellow powder (280 mg, 45% yield). 1H NMR (DMSO-d6, 500 MHz) δH 9.54 (1H, br. s), 8.48 (1H, s), 8.11 (1H, dd, J = 6.9, 2.6 Hz), 7.81 (1H, s), 7.79 (1H, ddd, J = 9.0, 4.3, 2.6 Hz), 7.43 (1H, t, J = 9.0 Hz), 7.19 (1H, s), 4.20 (2H, t, J = 6.3 Hz), 3.93 (3H, s), 2.75 (2H, t, J = 6.7 Hz), 1.89 (2H, p, J = 6.5 Hz); 13C NMR (DMSO-d6, 125 MHz) δC 156.0, 154.5, 153.1, 152.6, 148.5, 146.9, 136.8, 123.5, 122.3, 118.8, 116.5, 108.8, 107.2, 102.4, 66.9, 55.9, 38.4, 32.4. ESI-MS: ([M + H]+): 377. |
|
In N,N-dimethyl-formamide at 20℃; for 10h; |
Synthesis of S2-S5.
General procedure: A mixture of potassium phthalimide (3.7 g, 20 mmol, 1 eq.) and 1,3-dibromopropane (8.0 g, 40 mmol, 2 eq.) in DMF (30 mL) was stirred at room temperature for 10 h. The solution was removed under reduced pressure. Then, the residue was purified by silica gel (petroleum ether/EtOAc = 15/1) to afford S2a (4.8 g, 90% yield) as a white solid. To a solution of (3-chloro-4-fluorophenylamino)-7-methoxyquinazolin-6-ol (640 mg, 2 mmol, 1 eq.) and compound S2a (641 mg, 2.4 mmol, 1.2 eq.) in 5 mL DMF, K2CO3 (1106 mg, 8 mmol, 4 eq.) was added, then the mixture was stirred at 80 for 5 h, after cooled to room temperature, 50 mL water was added, and extracted by 50 mL ethyl acetate for three times, the organic layer was combined and washed by brine and dried by Na2SO4, filtered, and concentrated in vacuo, the crude product was purified by silica gel (EtOAc/MeOH = 10/1) to give S2b (840 mg, 83% yield). To the solution S2b (840 mg, 1.66 mmol) in EtOH, hydrazine monohydrate (530 μL) was added, then the mixture was stirred at 78 for 2 h, then the mixture was cooled and filtered to remove the white insoluble solid to gain the crude product, purified by silica gel (EtOAc/MeOH/NH3(aq.) = 1/1/0.05) to give S2 as a bright yellow powder (280 mg, 45% yield). 1H NMR (DMSO-d6, 500 MHz) δH 9.54 (1H, br. s), 8.48 (1H, s), 8.11 (1H, dd, J = 6.9, 2.6 Hz), 7.81 (1H, s), 7.79 (1H, ddd, J = 9.0, 4.3, 2.6 Hz), 7.43 (1H, t, J = 9.0 Hz), 7.19 (1H, s), 4.20 (2H, t, J = 6.3 Hz), 3.93 (3H, s), 2.75 (2H, t, J = 6.7 Hz), 1.89 (2H, p, J = 6.5 Hz); 13C NMR (DMSO-d6, 125 MHz) δC 156.0, 154.5, 153.1, 152.6, 148.5, 146.9, 136.8, 123.5, 122.3, 118.8, 116.5, 108.8, 107.2, 102.4, 66.9, 55.9, 38.4, 32.4. ESI-MS: ([M + H]+): 377. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping