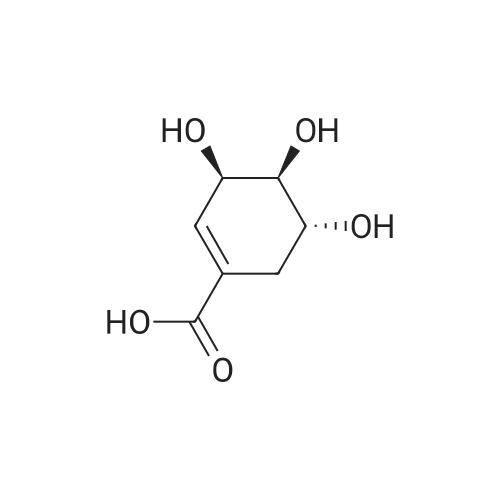
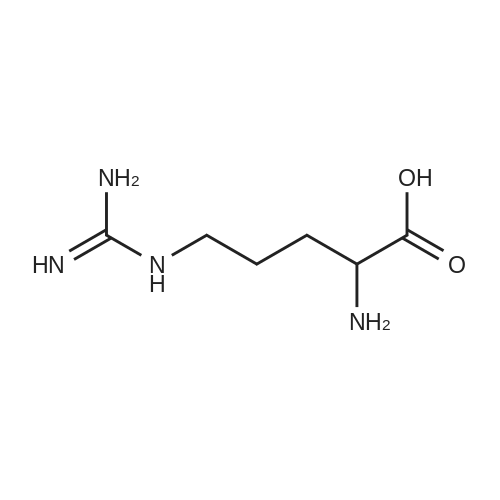
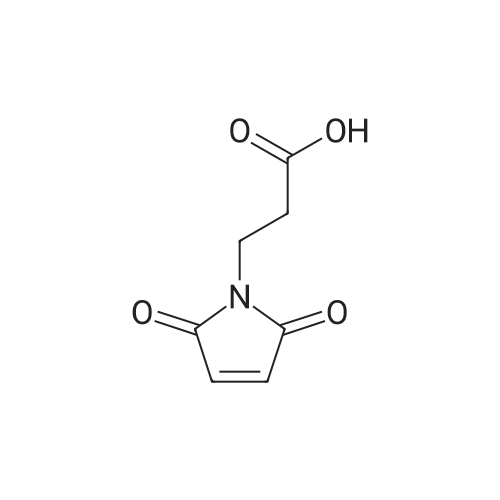
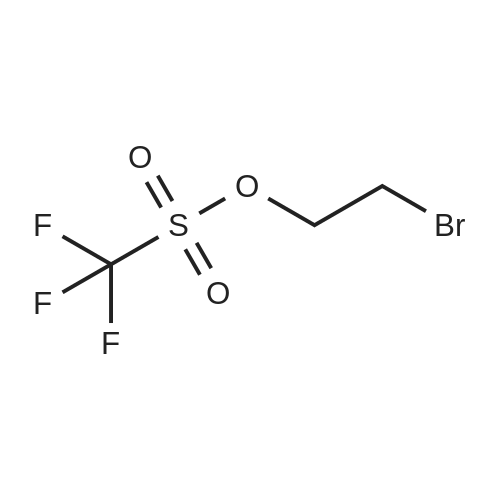
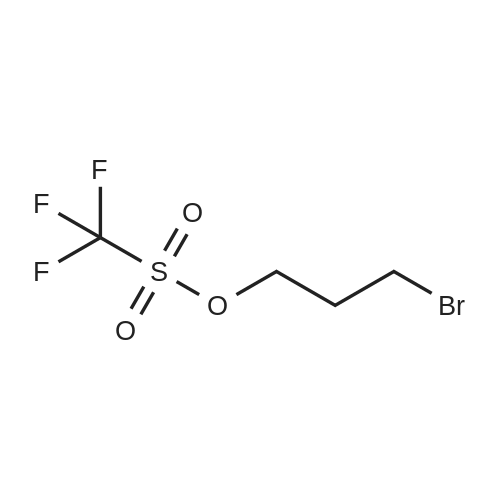
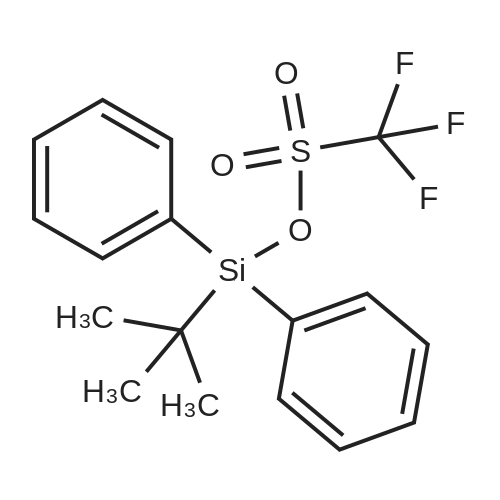
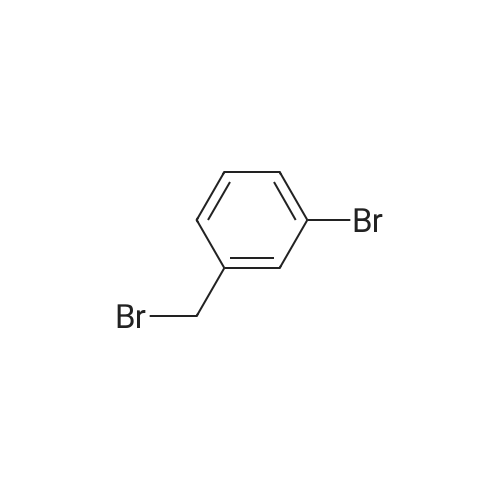
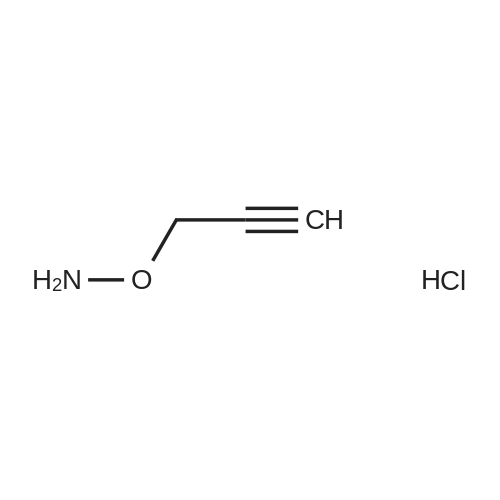
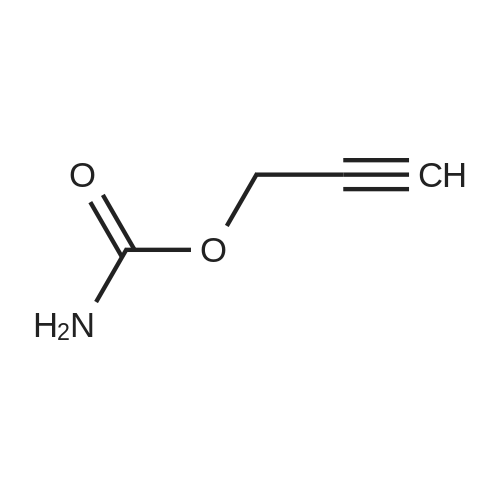
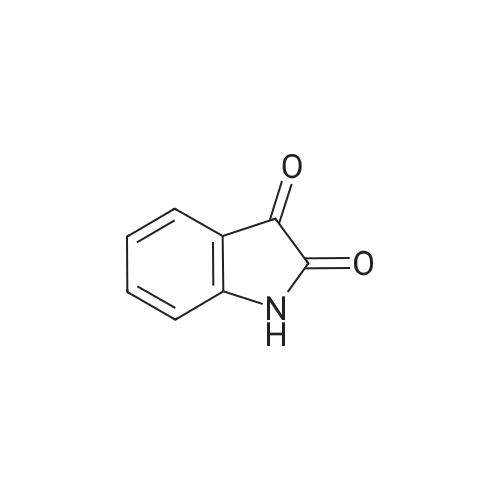
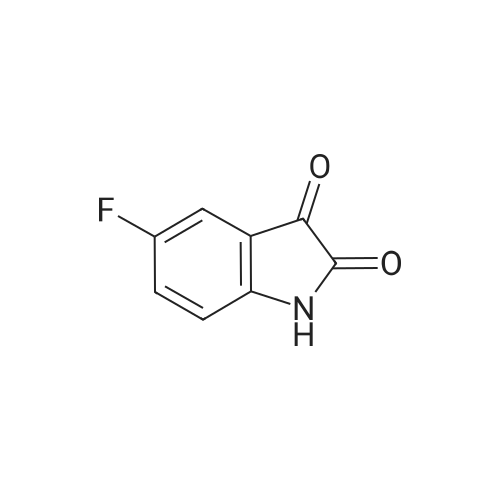
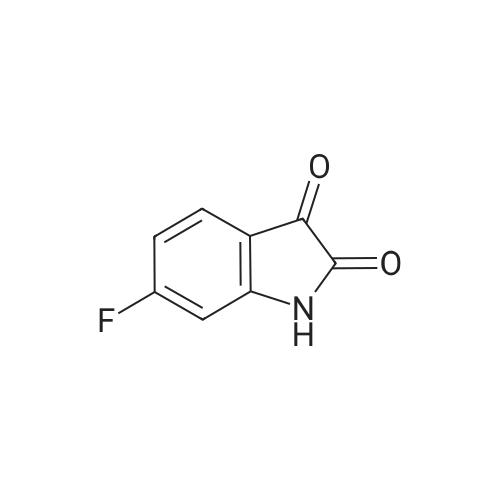
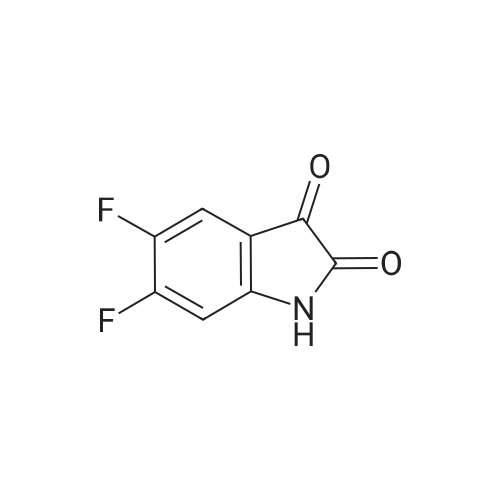
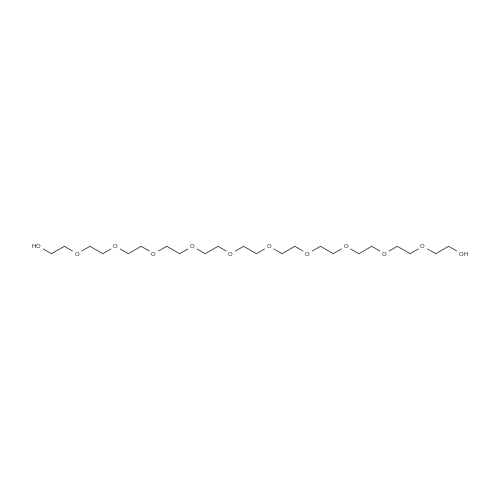
| 1: 8 mg
2: 16 mg |
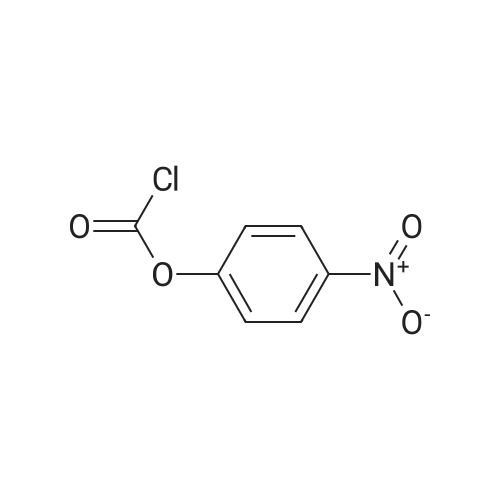
With toluene-4-sulfonic acid In tetrahydrofuran at 0 - 35℃; Inert atmosphere; |
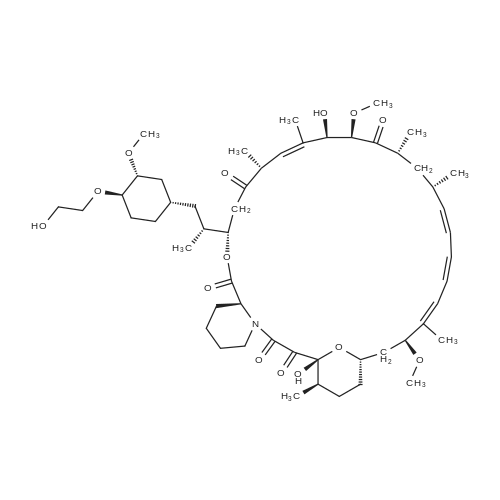
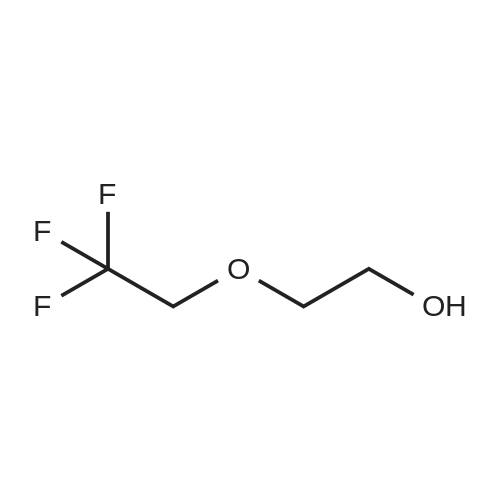
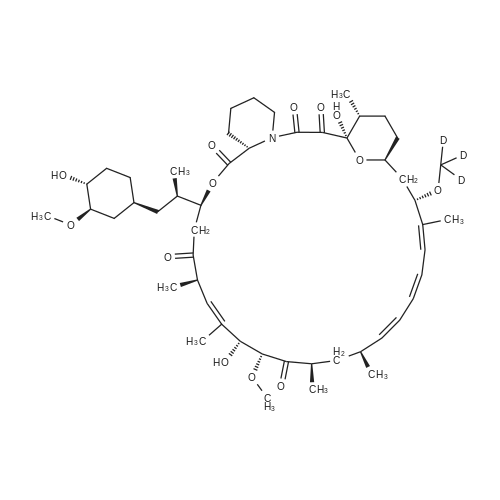
46.1-46.2 Step 1: Synthesis of (21E,23E,25E,26E,33R,34S,35R,36R,38S,40S,43S,44R,45R,54R)- 44,54-dihydroxy-43-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1- methyl-ethyl]-45-methoxy-33,34,35,36,46,47-hexamethyl-42-[2-(2,2,2-trifluoroethoxy) ethoxy]-65,66-dioxa-56-azatricyclohexatriaconta-21,23,25(46),26(47)-tetraene- 48,49,50,51,52-pentone (I-77):
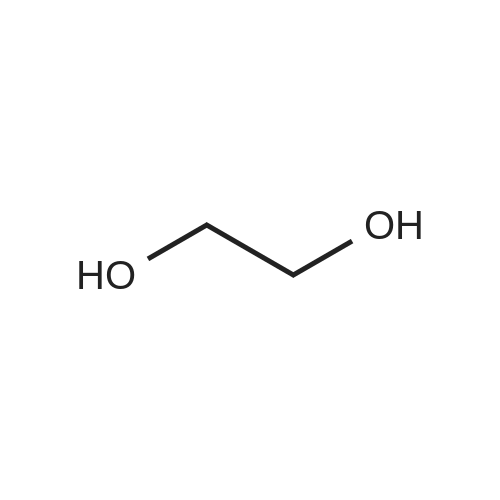
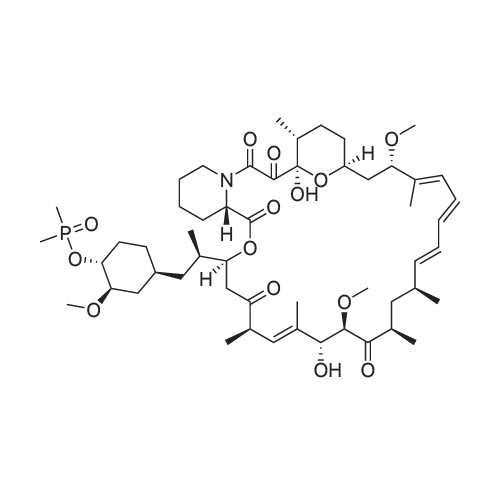
A solution of rapamycin (0.3 g, 0.313 mmol) and 2-(2,2,2-trifluoroethoxy)ethanol (0.9 g, 6.26 mmol) in THF (9 mL) was cooled to 0°C and p-TsOH (0.27 g, 1.57 mmol) was added. The resulting mixture was stirried at 35 °C for 5h under N2 then poured into ice cold NaHCO3 and extracted with EtOAc (40 mL × 3). The combined organic layers were washed with water (30 mL), brine (30 mL), dried, filtered, and concentrated. The residue was purified via reverse phase chromatography (C18, CH3CN:H2O = 57:43) to provide (21E,23E,25E,26E,33R,34S,35R,36R,38S,40S,43S,44R,45R,54R)-44,54-dihydroxy-43- [(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-45- methoxy-33,34,35,36,46,47-hexamethyl-42-[2-(2,2,2-trifluoroethoxy)ethoxy]-65,66-dioxa- 56-azatricyclohexatriaconta-21,23,25(46),26(47)-tetraene-48,49,50,51,52-pentone (I-77: 0.063 g, 19% yield) as a white solid. ESI-MS (EI+, m/z): 1092.3 [M+Na]+. 1H NMR (400 MHz, CDCl3) d 6.55- 5.88 (m, 4H), 5.72- 5.03 (m, 4H), 4.78 (s, 1H), 4.63- 4.34 (m, 1H), 4.32- 4.09 (m, 1H), 4.00- 2.81 (m, 21H), 2.77- 2.43 (m, 3H), 2.41- 2.17 (m, 2H), 2.18- 1.93 (m, 3H), 1.93- 1.54 (m, 18H), 1.54- 1.14 (m, 10H), 1.13- 0.79 (m, 16H), 0.78- 0.63 (m, 1H). 100 mg of (21E,23E,25E,26E,33R,34S,35R,36R,38S,40S,43S,44R,45R,54R)- 44,54-dihydroxy-43-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1- methyl-ethyl]-45-methoxy-33,34,35,36,46,47-hexamethyl-42-[2-(2,2,2-trifluoroethoxy) ethoxy]-65,66-dioxa-56-azatricyclohexatriaconta-21,23,25(46),26(47)-tetraene- 48,49,50,51,52-pentone was purified via prep chiral HPLC and the resulting epimers purified via silica gel chromatography (hexane:DCM:EtOAc:MeOH = 3:3:1:0.3) to provide (21E,23E,25E,26E,33R,34S,35R,36R,38S,40S,42S,43S,44R,45R,54R)-44,54-dihydroxy-43- [(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1-methyl-ethyl]-45- methoxy-33,34,35,36,46,47-hexamethyl-42-[2-(2,2,2-trifluoroethoxy)ethoxy]-65,66-dioxa- 56-azatricyclohexatriaconta-21,23,25(46),26(47)-tetraene-48,49,50,51,52-pentone (I-71: 16 mg, 16% yield) and (21E,23E,25E,26E,33R,34S,35R,36R,38S,40S,42S,43R,44R,45R,54R)- 44,54-dihydroxy-43-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxy-cyclohexyl]-1- methyl-ethyl]-45-methoxy-33,34,35,36,46,47-hexamethyl-42-[2-(2,2,2- trifluoroethoxy)ethoxy]-65,66-dioxa-56-azatricyclohexatriaconta-21,23,25(46),26(47)- tetraene-48,49,50,51,52-pentone (I-70: 8 mg, 8% yield) as a white solid. Chiral separation method: I-71: ESI-MS (EI+, m/z): 1092.4 [M+Na]+.1H NMR (400 MHz, CDCl3) d 6.41- 6.20 (m, 2H), 6.13 (dd, J = 15.1, 9.7 Hz, 1H), 5.93 (dd, J = 22.9, 10.3 Hz, 1H), 5.58- 5.45 (m, 1H), 5.41 (d, J = 9.8 Hz, 1H), 5.27 (d, J = 5.2 Hz, 1H), 5.19- 5.03 (m, 1H), 4.78 (s, 1H), 4.19 (dd, J = 13.9, 5.9 Hz, 1H), 3.95- 3.63 (m, 10H), 3.63- 3.53 (m, 2H), 3.52- 3.25 (m, 11H), 3.24- 3.01 (m, 3H), 2.72 (dd, J = 16.8, 5.8 Hz, 2H), 2.58 (dd, J = 16.8, 6.3 Hz, 1H), 2.31 (t, J = 23.5 Hz, 2H), 2.15- 1.40 (m, 18H), 1.27 (ddd, J = 32.5, 16.2, 6.3 Hz, 8H), 1.15- 0.81 (m, 18H), 0.70 (dt, J = 17.8, 9.0 Hz, 1H). I-70: ESI-MS (EI+, m/z): 1092.4 [M+Na]+. 1H NMR (400 MHz, CDCl3) d 6.43- 5.90 (m, 4H), 5.56- 5.08 (m, 5H), 4.33- 3.99 (m, 3H), 3.95- 3.63 (m, 8H), 3.62- 3.02 (m, 18H), 2.89- 1.97 (m, 12H), 1.76 (dd, J = 31.4, 24.8 Hz, 8H), 1.40 (ddd, J = 39.2, 29.5, 12.0 Hz, 9H), 1.14- 0.79 (m, 18H), 0.76- 0.61 (m, 1H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping