* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Journal of the Chemical Society. Perkin transactions 1, 1968, vol. 5, p. 531 - 540
10
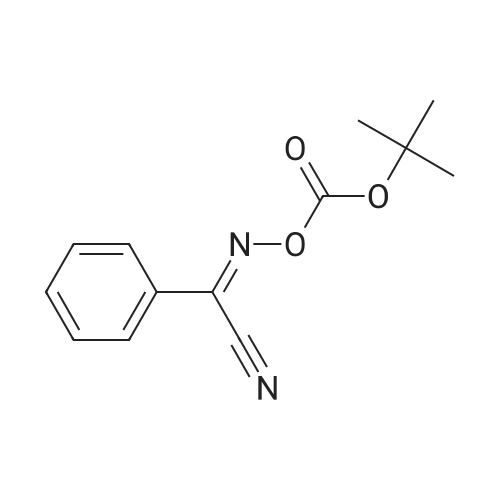
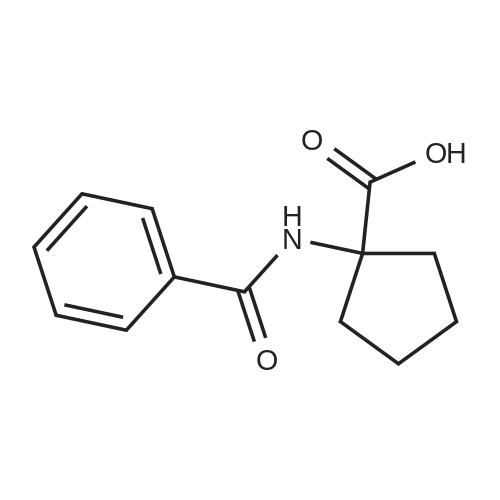
[ 24424-99-5 ]
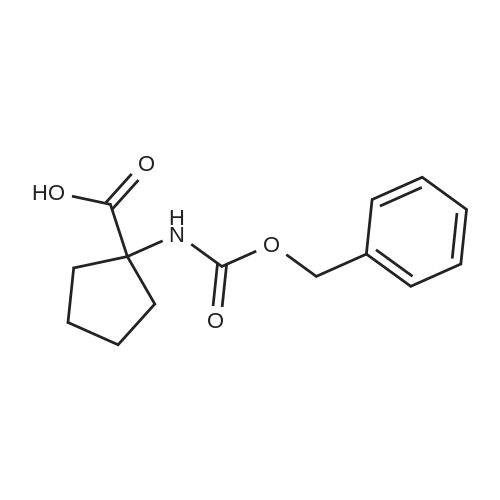
[ 52-52-8 ]
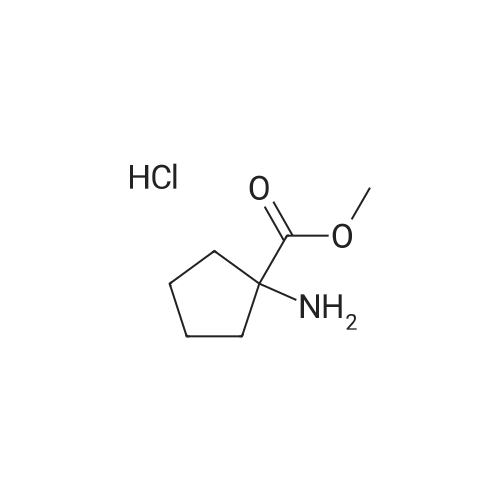
[ 35264-09-6 ]
Yield
Reaction Conditions
Operation in experiment
73%
Stage #1: With sodium hydroxide; triethylamine In water; acetonitrile at 0 - 20℃; for 4 h; Stage #2: With water In ethyl acetate Stage #3: With hydrogenchloride In water
To a 250 mL round bottom flask was added cycloleucine (1. 0g, 7. [74MMOL),] [ET3N] (5.4mL, 38. [7MMOL)] 1M NaOH (7.74mL, 7. [74MMOL)] and [CH3CN] (10mL). The clear solution was cooled down to [0°C] and to it was added [(BOC)] [2O.] The reaction was warmed to room temperature and stirred for four hours, during which time a white precipitate formed. The reaction mixture was concentrated and the residue was dissolved in EtOAc: water (1: 1) [(100ML).] The organic phase was washed with water and the aqueous phases were combined and treated with 10percent HCI and then were extracted with EtOAc three times. The combined organic phase was washed successively with water, brine, dried over [MGS04,] filtered and concentrated to yield the title compound as a white solid (1.29g, 73percent).
63%
With sodium hydrogencarbonate In 1,4-dioxane; water at 0 - 20℃; for 12 h;
To a stirred solution of 1-aminocyclopentane-1-carboxylic acid (6 g, 46 mmol) in 1,4- dioxane: H2O (50: 50 mL), were added NaHCO3 (13 g, 138 mmol) and (Boc)2O (13.5 g, 55.8 mmol) at 0°C and stirred the reaction mixture for about 12 hours at room temperature. After complete conversion of starting material, reaction mixture was washed with EtOAc (50 mL) to remove the impurities, then aqueous layer was acidified with 1N HCl (pH=2-3) and extracted with CH2Cl2 (2x100 mL). The combined organic extracts were washed with water, brine, dried over Na2SO4, filtered and evaporated under reduced pressure. The crude residue was purified by silica gel column chromatography by using 35percent EtOAc: n-Hexane as an eluent to afford the desired product (6.7 g, yield: 63percent) as an off white solid. 1H MR (300 MHz, CDCl3): δ 4.96 (s, 1H), 2.31-2.22 (m, 2H), 1.96-1.90 (m, 2H), 1.85-1.78 (m, 4H), 1.44 (s, 9H); ES Mass: 252.09 [M+Na]+.
56.33%
With sodium hydroxide In 1,4-dioxane at 0 - 20℃;
To a stirred solution of 1-aminocyclopentane-l-carboxylic acid (10 g, 77.51 mmol, 1.0 eq) in 1,4-dioxane (100 ml) at 0 °C was added 2N NaOH solution (100 ml) followed by (Boc)20 (25.34 g, 116.27 mmol, 1.5 eq). The reaction mixture was allowed to stir at room temperature for overnight. TLC indicated starting material was consumed and the desired product was observed. The organic phase was evaporated under reduced pressure, the reaction mixture was diluted with water (50 ml), cooled to 0 °C, pH adjusted to 5 with IN HC1 and then extracted with DCM (3x300 ml). The combined organic extracts were washed with water (300 ml), brine (100 ml) solution, dried over Na2S04, filtered and evaporated under reduced pressure. The residue was stirred with n-hexane (300 ml) at room temperature for about 30 minutes, the obtained solid was filtered and dried under vacuum to obtain the desired product (10.0 g, yield: 56.33percent) as a white solid. 1H NMR (300 MHz, DMSO-d6): δ ppm 12.11 (s, 1H), 7.10-6.86 (m, 1H), 1.94-1.84 (m, 4H), 1.62-1.58 (m, 4H), 1.36 (s, 9H); ESI-MS: m/z 252.02 (M+Na)+.
13%
With dmap; potassium carbonate In tetrahydrofuran; water
1-tert-Butoxycarbonylamino-cyclopentanecarboxylic acid To a suspension of 1-amino-1-cyclopentane carboxylic acid (4.52 g, 35.0 mmol) in THF (50 mL) and water (50 mL) was added potassium carbonate (14.51 g, 105.0 mmol), di-t-butyl-dicarbonate (7.72,g, 35.4 mmol), and DMAP (5 mg, 0.04 mmol). The reaction was stirred at rt. under nitrogen overnight. The THF was removed in vacuo, and the remaining aqueous solution was acidified to pH 5.5 with an aqueous 5N HCl solution. The aqueous solution was extracted with 20percent isopropanol/chloroform (*6), dried over magnesium sulfate, and concentrated to give 1.01 g of the title compound as a white solid, 13percent yield. 1H NMR: consistent with structure. MS (ion spray) 229 (M+).
Reference:
[1] Chemistry - A European Journal, 2011, vol. 17, # 19, p. 5256 - 5260
[2] Tetrahedron Letters, 1996, vol. 37, # 20, p. 3441 - 3444
[3] Organic Process Research and Development, 1998, vol. 2, # 4, p. 238 - 244
[4] Patent: WO2004/22534, 2004, A1, . Location in patent: Page 39
[5] Patent: WO2017/21922, 2017, A1, . Location in patent: Page/Page column 29
[6] Patent: WO2016/178092, 2016, A2, . Location in patent: Page/Page column 55; 56
[7] European Journal of Medicinal Chemistry, 2001, vol. 36, # 3, p. 265 - 286
[8] Patent: US2003/100576, 2003, A1,
[9] Patent: US5936089, 1999, A,
[10] Patent: US5432186, 1995, A,
[11] Patent: US5506244, 1996, A,
[12] Patent: US5506244, 1996, A,
[13] Patent: WO2005/41664, 2005, A1, . Location in patent: Page/Page column 103
[14] Patent: US2010/113546, 2010, A1, . Location in patent: Page/Page column 16-17
[15] Patent: EP1698626, 2006, A1, . Location in patent: Page/Page column 42
a 1-N-t-BOC-amino-1-cyclopentanecarboxylic acid To a solution of 1-amino-1-cyclopentane carboxylic acid (10.0 g, 0.0774 mole) triethylamine (11.75 g, 0.1161 mole), water (45 ml) and 1,4-dioxane (45 ml) was added BOC-ON (20.9 g, 0.085 mole) with stirring for 7 hr at room temperature. The mixture was extracted with ethyl acetate and the ethyl acetate layer was washed with water (50 ml). The aqueous layer was acidified with citric acid to a pH of 3.5 and extracted with ethyl acetate (2*100 ml). The ethyl acetate layer was dried with magnesium sulfate and evaporated to yield the product as an oil, 14 g (79percent).
Reference:
[1] Patent: US5270331, 1993, A,
13
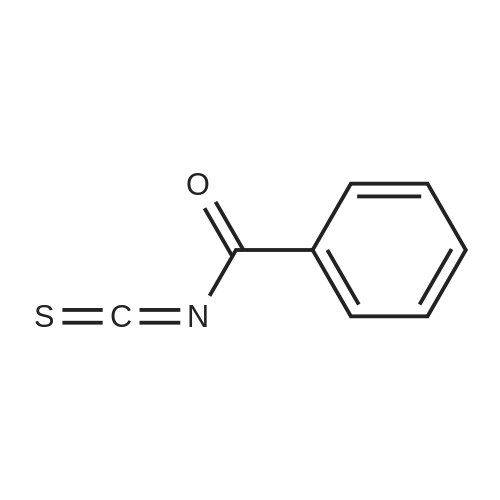
[ 58632-95-4 ]
[ 52-52-8 ]
[ 35264-09-6 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2004, vol. 12, # 16, p. 4477 - 4492
14
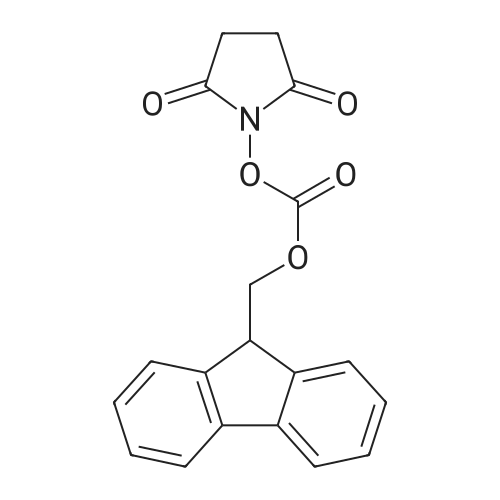
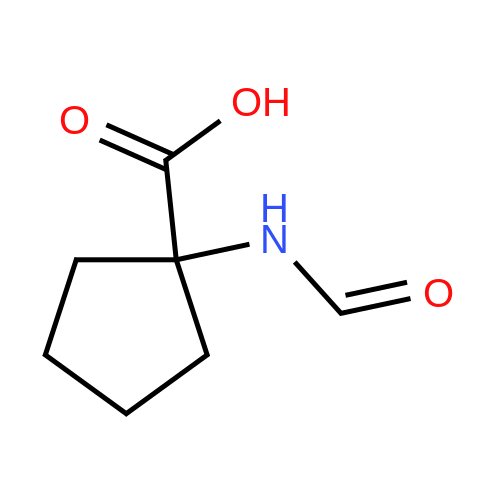
[ 1070-19-5 ]
[ 52-52-8 ]
[ 35264-09-6 ]
Reference:
[1] Journal of medicinal chemistry, 1971, vol. 14, # 10, p. 904 - 906
15
[ 501-53-1 ]
[ 52-52-8 ]
[ 17191-44-5 ]
Yield
Reaction Conditions
Operation in experiment
78%
Stage #1: With sodium carbonate In 1,4-dioxane; water at 20℃; Stage #2: With lithium hydroxide In tetrahydrofuran; water at 20℃; Stage #3: Acidic conditions
To a solution of 1-aminocyclopentanecarboxylic acid (3.0 g, 23.2 mmol) in 1:1 dioxane/water (60 mL), was slowly added Na2CO3 (12.3 g, 116 mmol) followed by benzyl chloroformate (3.6 mL, 25.5 mmol) and the mixture stirred overnight at RT. The reaction mixture was carefully acidified to pH=2 with 1M HCl then extracted with EtOAc (3*30 mL). The combined organic extracts were washed with brine (30 mL), dried (MgSO4), filtered and concentrated in vacuo to leave a pale yellow oil. LCMS and NMR showed the crude product to be a mixture of desired product and corresponding benzyl ester. The crude product was dissolved in 1:1 THF/water (60 mL) and treated with lithium hydroxide (2.67 g, 116 mmol). The mixture was stirred at RT overnight then washed with Et2O (3*30 mL), acidified to pH=2 and extracted with EtOAc (3*30 mL). The combined organic extracts were washed with brine (30 mL), dried (MgSO4), filtered and concentrated under reduced pressure to afford the title compound (4.76 g, 78percent). LCMS: m/z 264 [M+H]+.
78%
Stage #1: With sodium carbonate In 1,4-dioxane; water at 20℃; Stage #2: With hydrogenchloride In 1,4-dioxane; water
To a solution of 1-aminocyclopentanecarboxylic acid (3.0 g, 23.2 mmol) in 1 :1 dioxane / water (60 ml), was slowly added Na2CO3 (12.3 g, 116 mmol) followed by benzyl chloroformate (3.6 ml, 25.5 mmol) and the mixture stirred overnight at RT. The reaction mixture was carefully acidified to pH 2 with 1 M HCI then extracted with EtOAc (3 x 30 ml). The combined organic extracts were washed with brine (30 ml), dried (MgSO4), filtered and concentrated in vacuo to leave a pale yellow oil. LCMS and NMR showed the crude product to be a mixture of desired product and corresponding benzyl ester. The crude product was dissolved in 1 :1 THF / water (60 ml) and treated with lithium hydroxide (2.67 g, 116 mmol). The mixture was stirred at RT over night then washed with Et2O (3 x 30 ml), acidified to pH 2 and extracted with EtOAc (3 x 30 ml). The combined organic extracts were washed with brine (30 ml), dried (MgSO4), filtered and concentrated under reduced pressure to afford the title compound (4.76 g, 78percent). LCMS: m/z 264 [M+H]+.
62%
With sodium carbonate In 1,4-dioxane; water at 0 - 20℃;
A solution of benzyl chloroformate (0.290 g, 1.1 mmol) in dioxane (2.5 cm3) was added dropwise to a solution of 1 -aminocyclopentanecarboxylic acid (Fluka) (0.2 g, 1.54 mmol) and sodium carbonate (0.490 g, 4.64 mmol) in water (5 cm3) at 0 °C. Stirring was continued at room temperature overnight and the reaction mixture washed with ether. The aqueous layer was acidified with 2M hydrochloric acid, extracted with ethyl acetate, dried (Na2SO4), filtered and the solvent removed to afford carbamate 21 (0.253 g, 62percent) as an oil which solidified on standing. Carbamate 21 was shown to be a 70:30 mixture of conformers by 1H NMR analysis (the ratio was estimated from the integration of the resonances at δ 5.31 and 7.29-7.40, assigned to the N-H protons of the major and minor conformers, respectively): mp 70-80 °C (lit.1 82-86 °C, ethyl acetate, petroleum ether); SH (400 MHz; CDCl3; Me4Si) 1.83 (4H, br s, 2 x cyclopentyl-H2), 2.04 (2H, br s, cyclopentyl-H2), 2.20-2.40 (2H, m, cyclopentyl- H2), 5.13 (2H, br s, OCH2Ph), 5.31 (0.7η, br s, N-H) and 7.29-7.40 (5.3H, m, Ph and N-H*); δc (100 MHz; CDCl3) 24.6 (CH2, cyclopentyl-C), 37.5 (CH2, cyclopentyl-C),' denotes resonance assigned to minor conformer. <n="31"/>66.0 (quat., cyclopentyl-C), 66.8 (CH2, OCH2Ph), 128.0 (CH, Ph), 128.1 (CH, Ph), 128.4 (CH, Ph), 136.1 (quat, Ph), 155.8 (quat., NCO2) and 179.5 (quat., CO2H).
62%
With sodium carbonate In 1,4-dioxane; water at 0 - 20℃;
N-Benzyloxycarbonyl-1-aminocyclopentane-1-carboxylic acid 21 A solution of benzyl CHLOROFORMATE (0.290 g, 1.1 mmol) in dioxane (2.5 CM3) was added dropwise to a solution of L-AMINOCYCLOPENTANECARBOXYLIC acid (Fluka) (0.2 g, 1.54 mmol) and sodium carbonate (0.490 g, 4.64 mmol) in water (5 CM3) at 0 °C. Stirring was continued at room temperature overnight and the reaction mixture washed with ether. The aqueous layer was acidified with 2M hydrochloric acid, extracted with ethyl acetate, dried (NA2S04), filtered and the solvent removed to afford carbamate 21 (0.253 g, 62percent) as an oil which solidified on standing. Carbamate 21 was shown to be a 70: 30 mixture of conformers by 1H NMR analysis (the ratio was estimated from the integration of the resonances at No. 5.31 and 7.29-7. 40, assigned to the N-H protons of the major and minor conformers, respectively): mp 70-80 °C (lit. l 82-86 °C, ethyl acetate, petroleum ether); IH (400 MHz; CDC13 ; Me4Si) 1.83 (4H, br s, 2 x CYCLOPENTYL-H2), 2.04 (2H, br s, cyclopentyl-H2), 2.20-2. 40 (2H, M, cyclopentyl- H2), 5.13 (2H, br s, OCH2Ph), 5.31 (0.7H, br s, N-H) and 7.29-7. 40 (5.3H, M, Ph and N-H*) ; DC (100 MHz; CDC13) 24.6 (CH2, cyclopentyl-C), 37.5 (CH2, cyclopentyl-C), 66.0 (quat. , cyclopentyl-C), 66.8 (CH2, OCH2PH), 128.0 (CH, Ph), 128.1 (CH, Ph), 128.4 (CH, Ph), 136.1 (quat, Ph), 155.8 (quat. , NC02) and 179.5 (quat. , CO2H).
62%
With sodium carbonate In 1,4-dioxane; water at 0 - 20℃;
N-Benzyloxycarbonyl-l-aminocyclopentane-l-carboxylic acid 21 A solution of benzyl chloroformate (0.290 g, 1.1 mmol) in dioxane (2.5 cm3) was added dropwise to a solution of l-aminocyclopentanecarboxylic acid (Fluka) (0.2 g, 1.54 mmol) and sodium carbonate (0.490 g, 4.64 mmol) in water (5 cm3) at 0 °C. Stirring was continued at room temperature overnight and the reaction mixture washed with ether. The aqueous layer was acidified with 2M hydrochloric acid, extracted with ethyl acetate, dried (Na2SC>4), filtered and the solvent removed to afford carbamate 21 (0.253 g, 62percent) as an oil which solidified on standing. Carbamate 21 was shown to be a 70:30 mixture of conformers by NMR analysis (the ratio was estimated from the integration of the resonances at 6 5.31 and 7.29-7.40, assigned to the N-H protons of the major and minor conformers, respectively): mp 70-80 °C (lit.1 82-86 °C, ethyl acetate, petroleum ether); < (400 MHz; CDC; Me4Si) 1.83 (4H, br s, 2 x cyclopentyl-H2), 2.04 (2H, br s, cyclopentyl-H2), 2.20-2.40 (2H, m, cyclopenty[-H2), 5.13 (2H, br s, OCiPh), 5.31 (0.7H, br s, N-H) and 7.29-7.40 (5.3H, m, Ph and N-H*); <5fc (100 MHz; CDC13) 24.6 (CH2, cyclopentyl-C), 37.5 (CH2, cyclopentyl-C), 66.0 (quat., cyclopentyl-C), 66.8 (CH2) OCH2Ph), 128.0 (CH, Ph), 128.1 (CH, Ph), 128.4 (CH, Ph), 136.1 (quat, Ph), 155.8 (quat, NC02) and 179.5 (quat., C02H).
Reference:
[1] Organic Process Research and Development, 1998, vol. 2, # 4, p. 238 - 244
[2] Patent: US2010/317865, 2010, A1, . Location in patent: Page/Page column 14; 15
[3] Patent: WO2009/106844, 2009, A1, . Location in patent: Page/Page column 39
[4] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 2001, vol. 40, # 1, p. 70 - 74
[5] Bioorganic and Medicinal Chemistry, 2005, vol. 13, # 2, p. 533 - 548
[6] Patent: WO2008/63311, 2008, A2, . Location in patent: Page/Page column 29-30
[7] Patent: WO2005/23815, 2005, A2, . Location in patent: Page/Page column 42-43
[8] Patent: WO2015/13397, 2015, A2, . Location in patent: Page/Page column 45-46
[9] Journal of the Chemical Society, 1962, p. 4601 - 4607
[10] Journal of the Chemical Society. Perkin transactions 1, 1968, vol. 5, p. 531 - 540
16
[ 67-56-1 ]
[ 52-52-8 ]
[ 60421-23-0 ]
Yield
Reaction Conditions
Operation in experiment
100%
With thionyl chloride In methanol at -15 - 20℃;
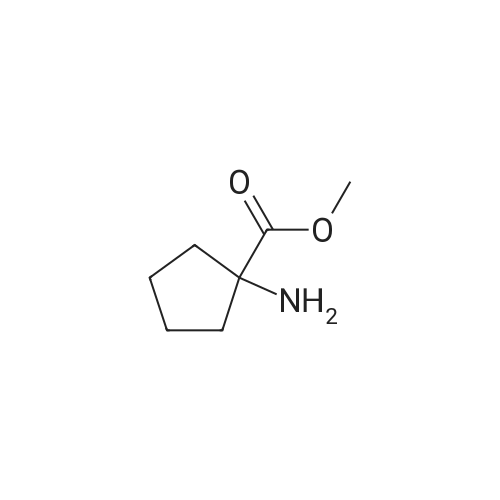
To a suspension of 1-aminocyclopentanecarboxylic acid, (675 g, 5.23 mol, 1.0 equiv.) in MeOH (6.5 L) held at -15° C. with an ice/MeOH bath was added SOCl2 (687 mL, 9.4 mol, 1.8 equiv.), dropwise at such a rate that the reaction temp. did not exceed 7° C. After the addition was complete, cooling was removed, the reaction was allowed to stir at room temp. overnight, then was concentrated under reduced pressure. The residue was treated with CH2Cl2 (1 L) and concentrated under reduced pressure to afford methyl 1-aminocyclopentanecarboxylate HCl salt as a white solid (938 g, 100percent): 1H NMR (CD3OD) d 1.87-1.94 (m, 8H), 3.83 (s, 3H); NMR (DMSO-d6) δ1.67-1.71 (m, 2H), 1.83-1.98 (m, 4H), 2.06-2.14 (m, 2H), 3.73 (s, 3H), 8.81 (br s 3). This material was used in the next step without further purification
98%
at 20℃; for 3 h; Cooling with ice
1-aminocyclopentanecarboxylic acid (3.5 g, 26.9 mmol) and methanol (100 ml) were charged. An ice bath was set and thionyl chloride (3.9 ml, 53.9 mmol) was slowly added thereto. Then, the ice bath was removed, and the mixture was stirred at room temperature for 3 hours. The resultant product was vacuum-distilled to remove a solvent, and dried in a 60 oven so as to obtain methyl-1-aminocyclopentanecarboxylate hydrochloride (4.74 g, 26.4 mmol, 98 percent).[823] 1H NMR (400 MHz, DMSO-d6) δ 8.76 (br, 3H), 3.75 (s, 3H), 2.12 (m, 2H), 1.88 (m, 4H), 1.72 (m, 2H).
98%
at 20℃; for 3 h; Inert atmosphere
Preparation Example 1 Synthesis of N-4-(1-(3-chloro-2-methylphenylsulfonamido)cyclopentanecarboxyamido)adamantane-1-carboxyamide (compound 167) 1-aminocyclopentanecarboxylic acid (3.5 g, 26.9 mmol) and methanol (100 ml) were charged. An ice bath was set and thionyl chloride (3.9 ml, 53.9 mmol) was slowly added thereto. Then, the ice bath was removed, and the mixture was stirred at room temperature for 3 hours. The resultant product was vacuum-distilled to remove a solvent, and dried in a 60° C. oven so as to obtain methyl-1-aminocyclopentanecarboxylate hydrochloride (4.74 g, 26.4 mmol, 98percent). 1H NMR (400 MHz, DMSO-d6) δ 8.76 (br, 3H), 3.75 (s, 3H), 2.12 (m, 2H), 1.88 (m, 4H), 1.72 (m, 2H).
97%
at 20℃;
1-Amino-l-cyclopentanecarboxylic acid (5.00 g, 38.8 mmol) was dissolved in methanol (100 mL) and then thionyl chloride (9.25 g, 77.7 mmol) was added dropwise with stirring. The resulting mixture was stirred overnight at room temperature and then concentrated in vacuo which left a white solid. The solid was tritruated in ethyl ether, filtered, and dried to give 6.78 g (97 percent) of methyl 1-amino- 1-cyclopentanecarboxylate hydrochloride as a white solid. 1H NMR was consistent with product. ESMS (M+1) 144.2
97%
for 18 h;
1-Aminocyclopentanecarboxylic acid (1.00 g, 7.74 mmol) was added to a solution of acetyl chloride (0.60 mL, 8.44 mmol) in methanol (10 mL). The reaction mixture was allowed to stir for EPO <DP n="37"/>18 h and then concentrated under reduced pressure. This yielded 1.35 g (97percent) of the title compound. 1H NMR (400 MHz, CD3OD) δ 1.80-2.00 (m, 6H), 2.34-2.39 (m, 2H), 3.82 (s, 3H).
91%
Reflux
Example 2.1 1-aminocyclopentanecarboxylic acid methyl ester hydrochloride Prepared as a white powder following the general procedure previously described using cycloleucine and methanol. Yield: 91percent Rf (dichloromethane/methanol 9/1): 0.5 MP: 157-159° C. IR: νCO: 1742 cm-1 NMR 1H (DMSO-d6): 1.68-1.84 (m, 6H); 2.04 (m, 2H); 3.71 (s, 3H). Example 2General Procedure for the Preparation of Amino Acid EstersAminocarboxylic acid (1eq) was added at 0° C. to the appropriate alcohol (methanol or ethanol) and the mixture was saturated with anhydrous hydrochloric acid. Thionyl chloride was then added drop by drop. The reaction mixture was stirred at reflux for 12 hours. The reaction mixture was concentrated under reduced pressure and diethyl ether was added to the crude residue. The resulting powder was filtered and washed with diethyl ether.
89%
Stage #1: at 0℃; for 1 h; Stage #2: for 6 - 16 h; Heating / reflux
To a stirring solution of anhydrous alcohol (10 mol eq. ) was added thionyl chloride (2 mol eq. ) at 0° C, and the resulting solution stirred for 1 hr. After warming to room temperature, the appropriate amino acid (1 mol eq) was added and the reaction heated at reflux for 6-16 hrs. Removal of solvent and recrystallisation from methanol/ether gave the amino ester hydrochloride salts.; This was synthesised according to Standard Procedure 1, using 1-AMINO-1- cyclopentanecarboxylic acid (3.876 g, 30 mmol) with thionyl chloride (4.44 mL, 45 mmol, ) and anhydrous methanol (15.5 mL). The product was isolated as a white solid (4.81 g, yield 89percent). 'H-NMR (CDC13 ; 300 MHz): 8 9.1 (3H, bs, NH3+Cl), 3.85 (3H, s, OCH3), 2.3-2. 2 (4H, m, 4H cyclopentane), 2. 15 (2H, 2H cyclopentane), 1.95 (2H, m, 2H cyclopentane). 13C-NMR (CDCl3 ; 75 MHz): 8 26.6 (2CH2 cyclopent), 38.1 (2CH2 cyclopent), 54.8 (CH30), 66.6 (cyclopentane), 174.1 (COOMe).
87%
at 0℃;
General procedure: Thionyl chloride (10 mL) was slowly added to a cold suspension solution of the appropriate aminoacid (50 mmol) in methanol (50 mL) at 0 °C. The reaction mixture was stirred for 8–10 h and thenconcentrated on a rotary evaporator. The white precipitate formed was washed with anhydrous etherand then dried under vacuum. All data agreed with the reported data [43,44].
77%
for 1 h; Heating / reflux
Example 10; Methyl 1-[(1-(2,4-dichlorophenyl)-4-methyl-5-{4[(propylsulfonyl)oxy]phenyl}-1H- pyrazol-3-yl) carbonvl1ammo} cvclopentanecarboxylate; Step A Methyl 1-aminocyclopentanecarboxylate hydrochloride; Thionyl chloride (1.5 ml) was dissolved in methanol (15 ml) and poured over 1- aminocyclopentanecarboxylic acid (100 mg, 0.774 mmol). The mixture was refluxed 1 hour. The solvent was evaporated to give the product (107 mg, 77percent). 'H NMR (399.964 MHz) 8 9.00-8. 60 (br, 3H), 3.79 (s, 3H), 2.23 (s, 4H), 2.14-2. 00 (m, 2H), 1.90-1. 76 (m, 2H).
77%
for 1 h; Heating / reflux
Thionyl chloride (1.5 ml) was dissolved in methanol (15 ml) and poured over 1- Q aminocyclopentanecarboxylic acid (100 mg, 0.774 mmol). The mixture was refluxed 1 hour. The solvent was evaporated to give the product (107 mg, 77percent).1H NMR (399.964 MHz) δ 9.00-8.60 (br, 3H), 3.79 (s, 3H), 2.23 (s, 4H), 2.14-2.00 (m,2H), 1.90-1.76 (m, 2H).
Reference:
[1] Patent: US6353006, 2002, B1, . Location in patent: Page column 31
[2] Patent: WO2013/19091, 2013, A2, . Location in patent: Paragraph 821-823
[3] Patent: US2014/206875, 2014, A1, . Location in patent: Paragraph 0449-0451
[4] Patent: WO2003/87069, 2003, A2, . Location in patent: Page/Page column 103-104
[5] Patent: WO2006/44775, 2006, A2, . Location in patent: Page/Page column 35-36
[6] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2006, vol. 45, # 8, p. 1942 - 1944
[7] Patent: US2010/4159, 2010, A1, . Location in patent: Page/Page column 36
[8] Patent: WO2005/12327, 2005, A2, . Location in patent: Page/Page column 18; 20
[9] Molecules, 2013, vol. 18, # 12, p. 14747 - 14759
[10] Patent: WO2005/80343, 2005, A2, . Location in patent: Page/Page column 48
[11] Patent: WO2007/20388, 2007, A1, . Location in patent: Page/Page column 46
[12] Journal of Medicinal Chemistry, 1984, vol. 27, # 12, p. 1663 - 1668
[13] Heterocycles, 1991, vol. 32, # 10, p. 1879 - 1895
[14] J. Med. Chem., 1996, vol. 39, # 3, p. 773 - 780
[15] Journal of the American Chemical Society, 1997, vol. 119, # 49, p. 11807 - 11816
[16] Patent: US2007/265225, 2007, A1, . Location in patent: Page/Page column 49
[17] Patent: US2012/270881, 2012, A1, . Location in patent: Page/Page column 116
[18] Journal of the American Chemical Society, 2016, vol. 138, # 25, p. 7939 - 7945
[19] Patent: WO2009/120652, 2009, A2, . Location in patent: Page/Page column 71
Stage #1: With sodium hydrogencarbonate In water; acetonitrile at 20℃; Stage #2: With hydrogenchloride In water; acetonitrile
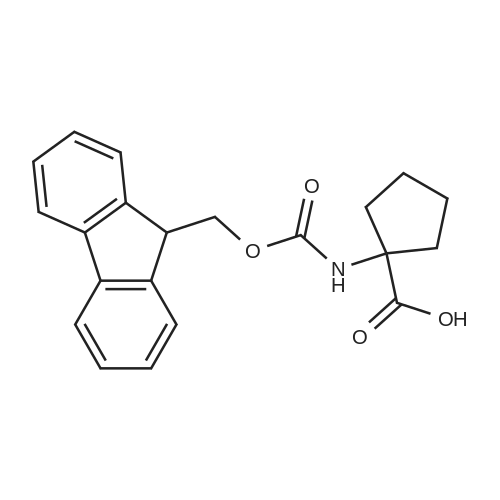
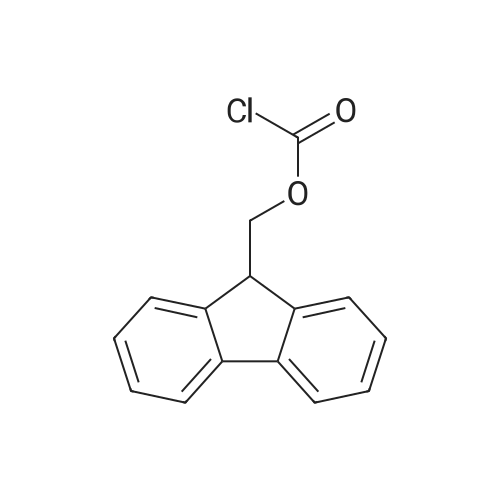
1- (((9H-Fluoren-9-yl)methoxy)carbonylamino)cyclopentanecarboxylic acid(compound 4 of example A): Fmoc-OSu (3.13 g, 9.3 mmol) was added to a solution of 1- aminocyclo- pentanecarboxylic acid (1.0 g, 7.8 mmol) and NaHC03 (1.63 g, 19.4 mmol) in acetonitrile/water (100 mL, 1 : 1). The reaction mixture was stirred at room temperature overnight. Most of the solvent was removed under reduced pressure and the resulting mixture was adjusted to pH = 2 with 2 N HCI and extracted with DCM. The combined extracts were washed with brine, dried over anhydrous Na2S04 and concentrated. The residue was purified by flash column chromatography on silica gel (PE/EA = 20: 1) to afford compound 4 of example A (0.75 g, 28percent yield) as a white solid.
Reference:
[1] Journal of the American Chemical Society, 2000, vol. 122, # 37, p. 8898 - 8909
[2] Patent: WO2012/119941, 2012, A1, . Location in patent: Page/Page column 110
[3] Journal of the American Chemical Society, 2003, vol. 125, # 23, p. 6852 - 6853
[4] Bioorganic and Medicinal Chemistry, 2013, vol. 21, # 18, p. 5694 - 5706
21
[ 497-19-8 ]
[ 52-52-8 ]
[ 117322-30-2 ]
Reference:
[1] Patent: US5385910, 1995, A,
22
[ 28920-43-6 ]
[ 52-52-8 ]
[ 117322-30-2 ]
Reference:
[1] Indian Journal of Chemistry, Section B: Organic Chemistry Including Medicinal Chemistry, 2001, vol. 40, # 1, p. 70 - 74
Cycloleucine was N-sulfonated with toluenesulfonyl chloride using the procedure described in Method 1. The title compound was then prepared as a white solid by following Method 3 using the methyl ester of phenylalanine which was subsequently hydrolyzed in a 1:1 solution of MeOH:H2O containing NaOH to provide for the title compound.NMR data was as follows:1H NMR (300 MHZ, DMSO-d6): delta=8.19 (s, 1H), 7.84 (d, 2H, J=8.46 Hz), 7.64 (d, 1H, J=8.25 Hz), 7.52 (d, 2H, J=8.25 Hz), 7.42 (m, 2H), 7.36 (m, 2H), 4.50 (m, 1H), 3.62 (m, 1H), 3.15 (m, 2H), 2.54 (s, 3H), 1.98 (m, 4H), 1.60 (m, 2H), 1.35 (m, 2H).13C NMR (300 MHZ, CD3OD): delta=176.35, 174.52, 145.17, 141.77, 138.37, 131.23, 130.02, 128.45, 122.08, 109.47, 71.29, 55.72, 39.03, 37.96, 24.68, 22.00.Mass Spectroscopy: (FAB) 431 (M+H).; Method 1N-Tosylation ProcedureN-Tosylation of the appropriate amino acid was conducted via the method of Cupps, Boutin and Rapoport J. Org. Chem. 1985, 50, 3972.
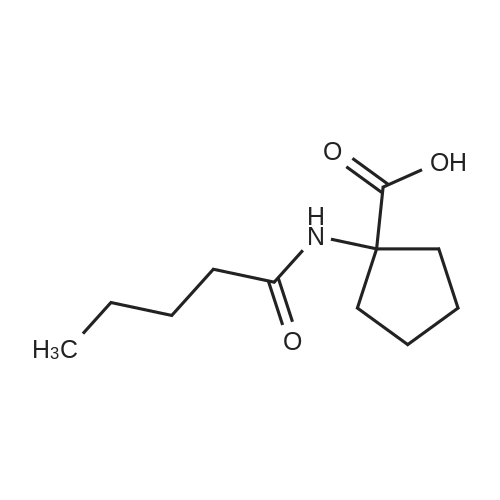
EXAMPLE 1; In a process for the preparation of n-pentanoyl cycloleucine of Formula IV, sodium hydroxide (68.2 grams, 1.7 moles) was added to water (275 ml) and cooled the solution cooled to 0-5C, then there was added slowly a solution of cycloleucine (55 grams, 0.426 moles) and valeryl chloride (77 grams, 0.639 moles) in toluene (55 ml) over about 2-3 hours at 0-10C. The reaction mass was maintained at 0-10C for about 2-3 hours. Water (275 ml) and toluene (55 ml) were added to the reaction mass and the mixture was stirred for about 15 minutes. The aqueous layer was separated and washed with toluene (55 ml), then the aqueous layer pH was adjusted to 2.0-2.5 with 8% aqueous hydrochloric acid (95 ml) and stirred for 15 minutes as a solid formed. The solid was separated by filtration and washed with water (45 ml), then was dried at 70-80C to get the desired compound of Formula IV, in an amount of 50 grams.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping