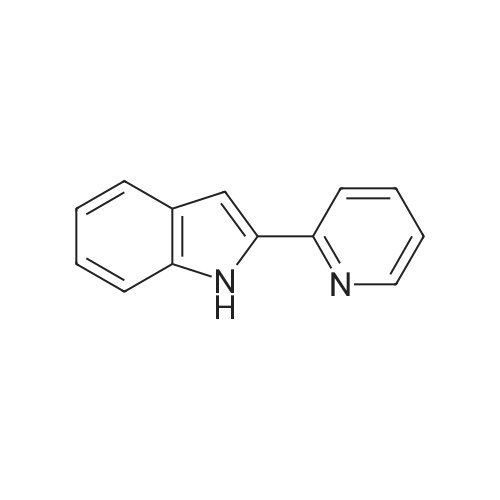
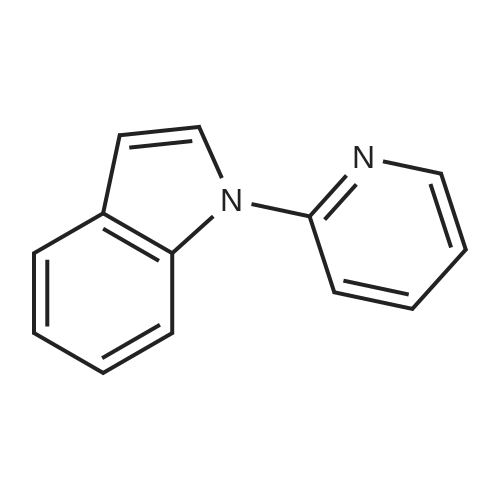
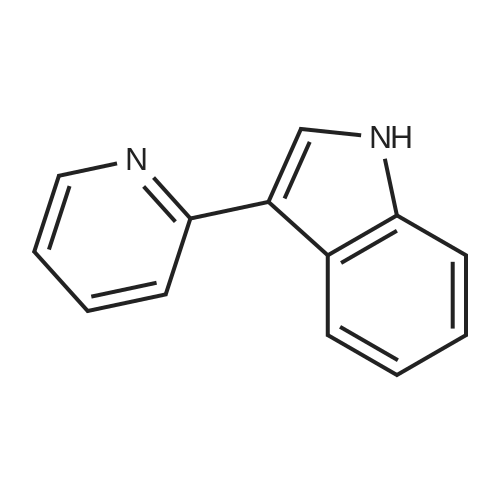
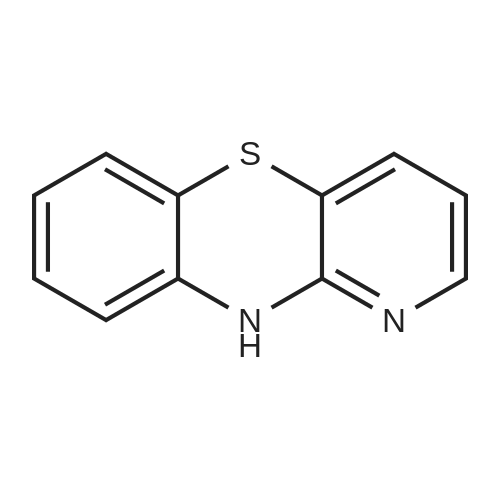
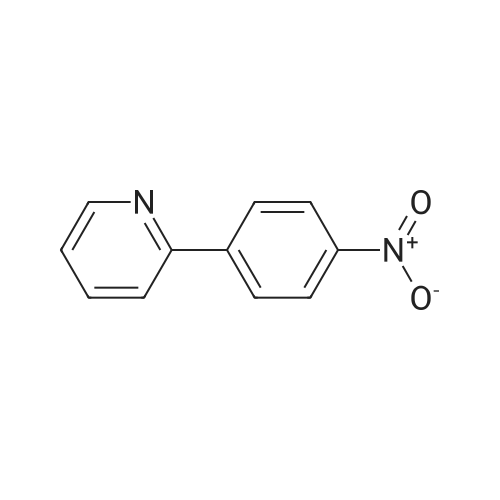
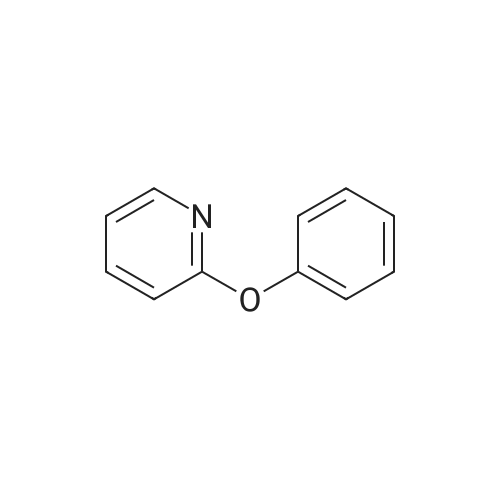
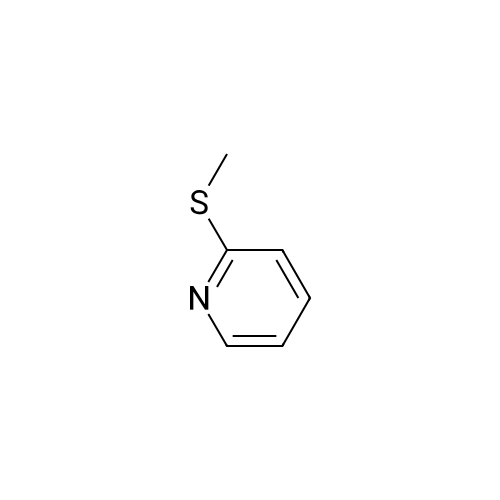
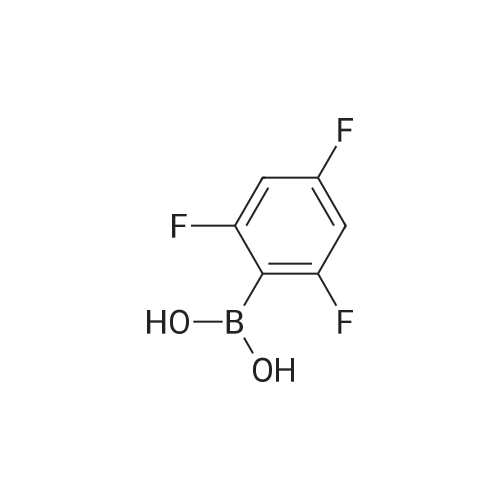
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: at 20℃; for 2 h; Inert atmosphere Stage #2: at 50℃; for 24 h; Inert atmosphere
General procedure: To the suspension of zinc powder (without activation, 65.4 mg, 1.0 mmol, Aldrich 99.995percent purity) in DMPU (0.5 mL), trifluoromethyl iodide (ca. 2.5 mmol, sufficiently dissolved in the solution) was added at room temperature under argon atmosphere. After the solution was stirred for 2 h at room temperature, CuI (1.9 mg, 0.01 mmol, 2 mol percent), 1.10-phenanthroline (1.8 mg, 0.01 mmol, 2 mol percent), and then aryl iodide 1a (138.0 mg, 0.5 mmol) were added. The reaction mixture was stirred at 50 °C for 24 h. After cooling to room temperature, the yield of product 2a was determined by 19F NMR analysis by using benzotrifluoride (BTF) as an internal standard. Except for 2c, all trifluoromethylated products 2 exhibited the same 1H, 13C, and 19F NMR spectra as reported before [14, 17, 29, 31-36].
Reference:
[1] Beilstein Journal of Organic Chemistry, 2013, vol. 9, p. 2404 - 2409
6
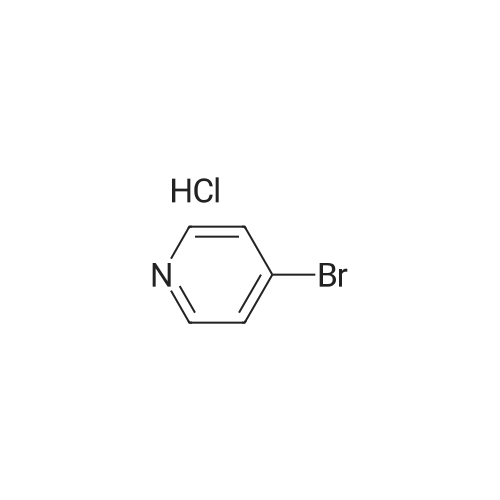
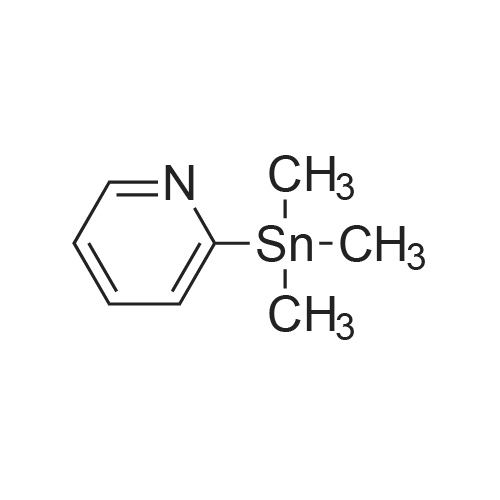
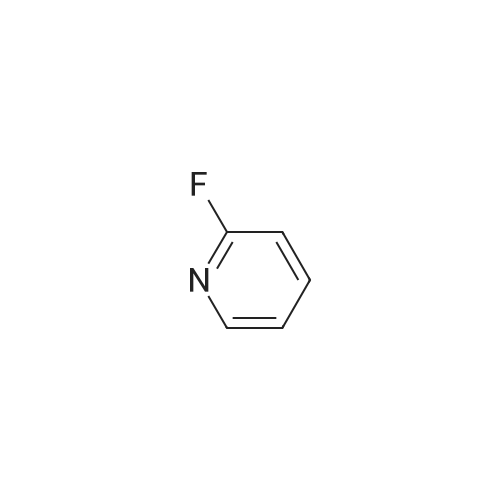
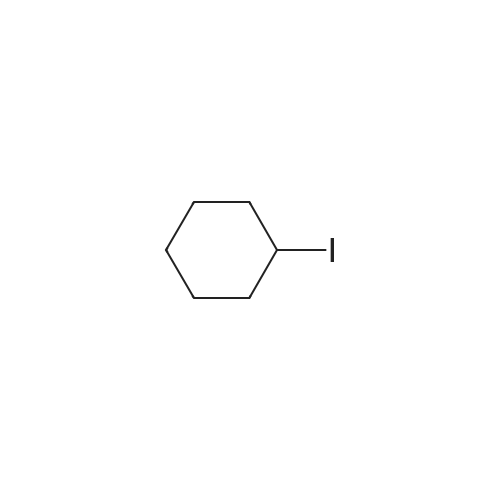
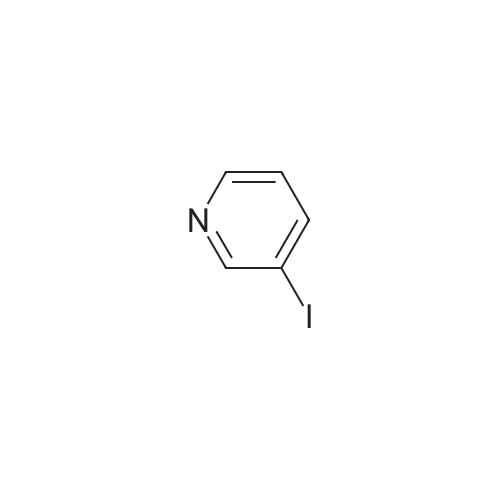
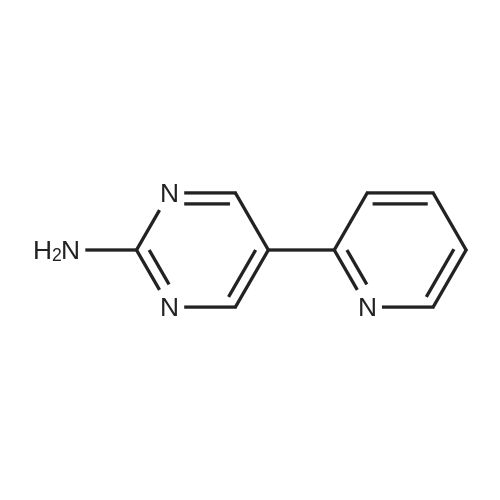
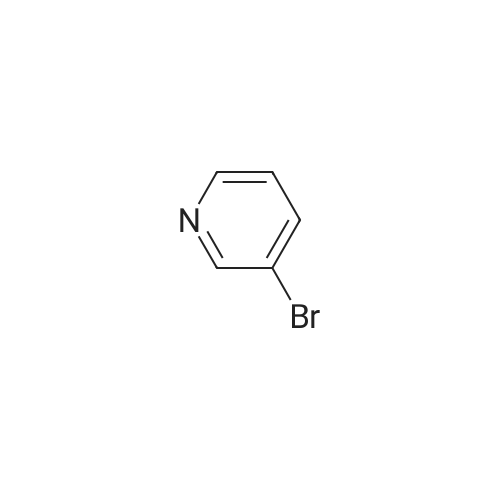
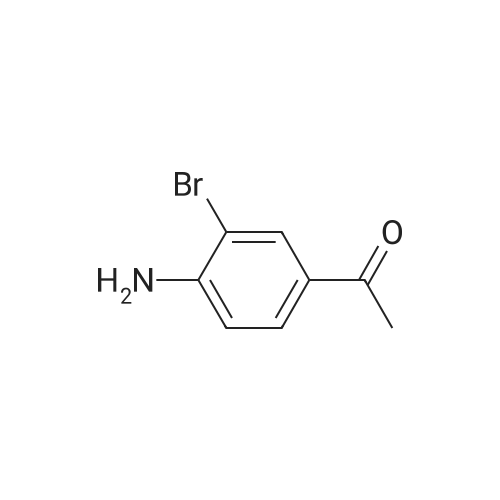
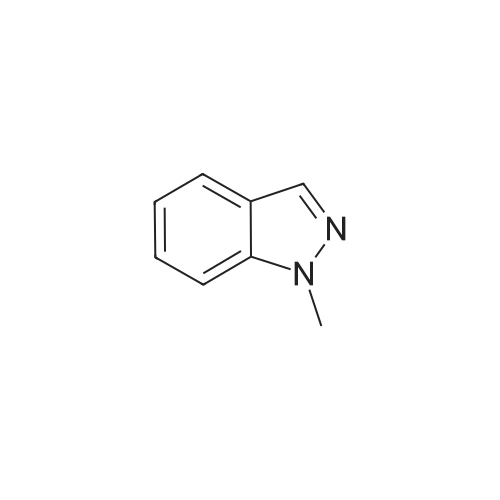
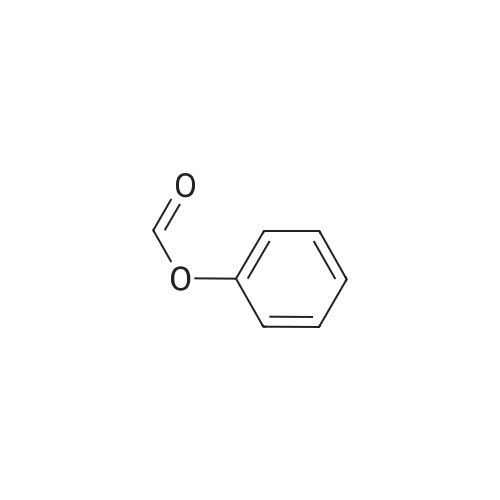
[ 5029-67-4 ]
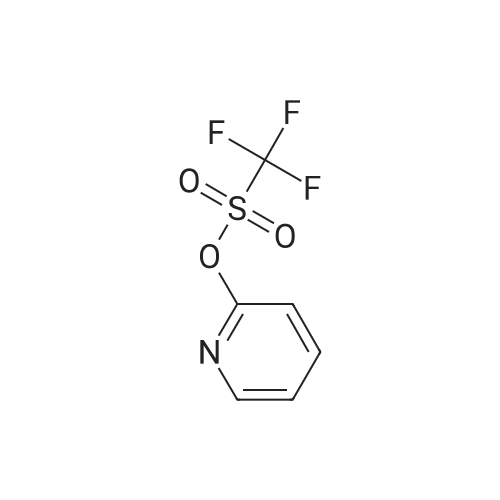
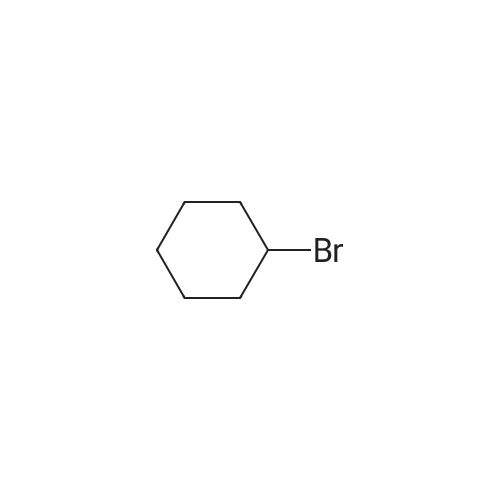
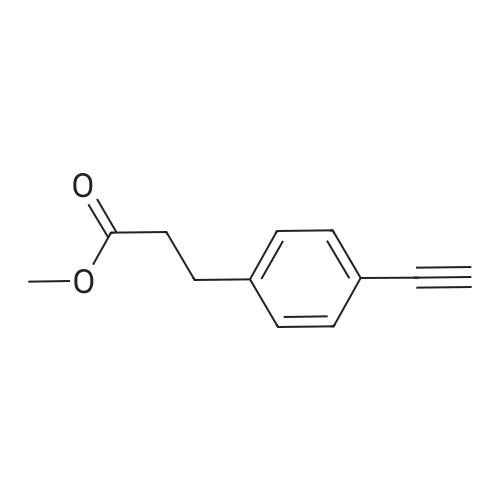
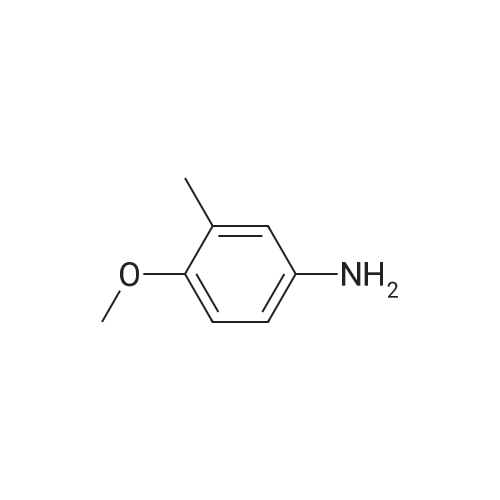
[ 371-76-6 ]
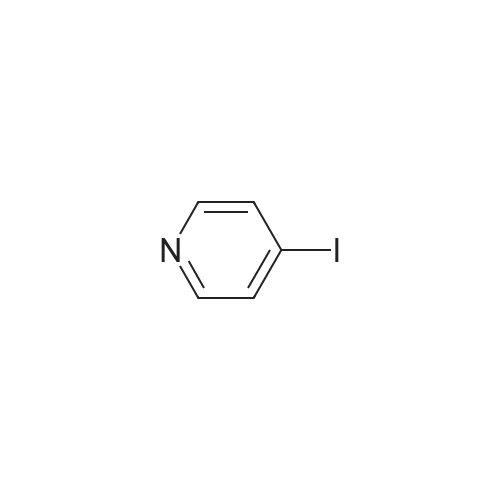
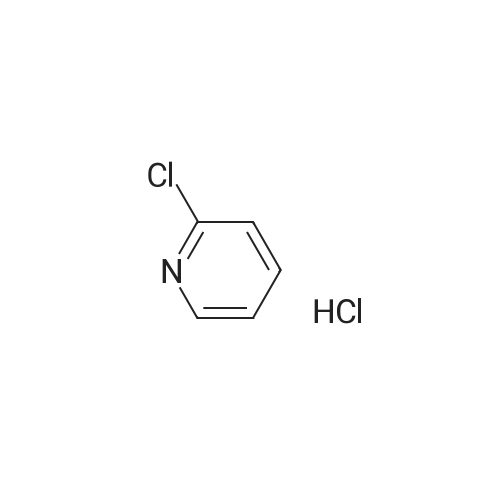
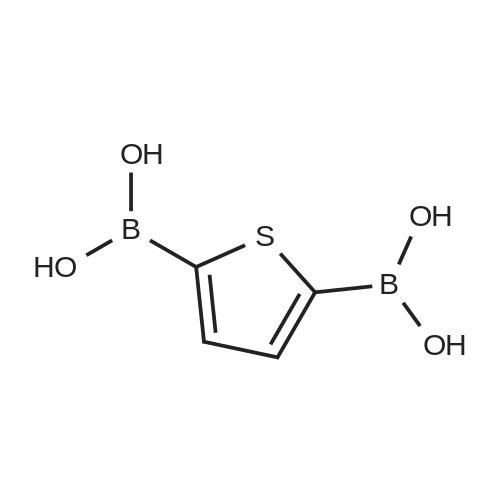
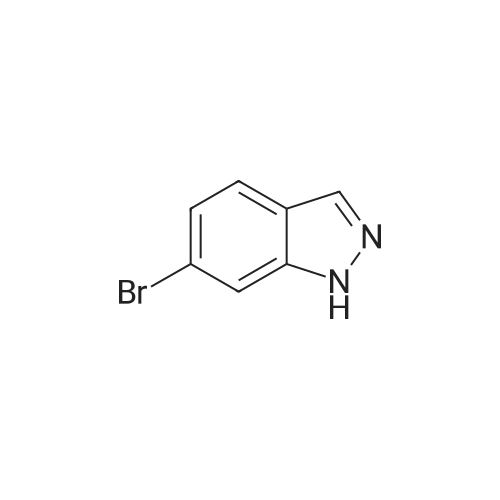
[ 368-48-9 ]
Reference:
[1] Synthesis, 1980, # 11, p. 932 - 933
7
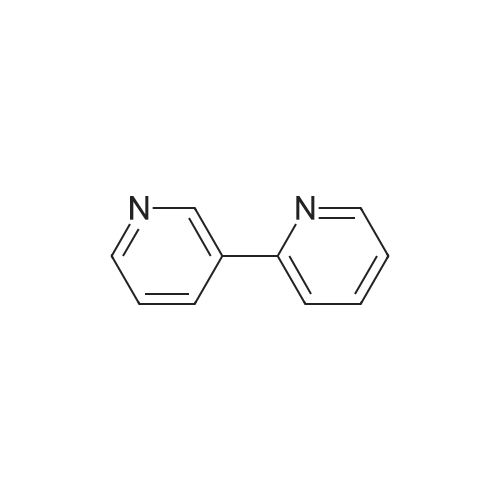
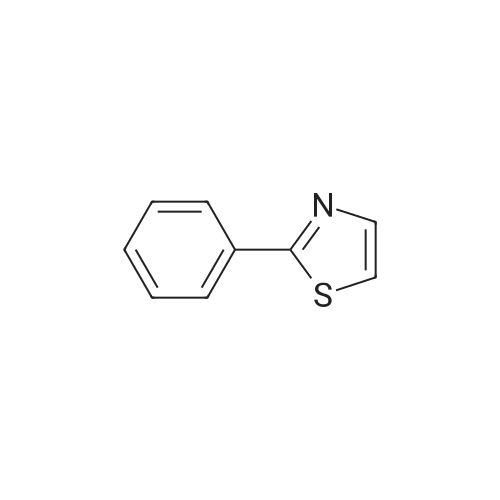
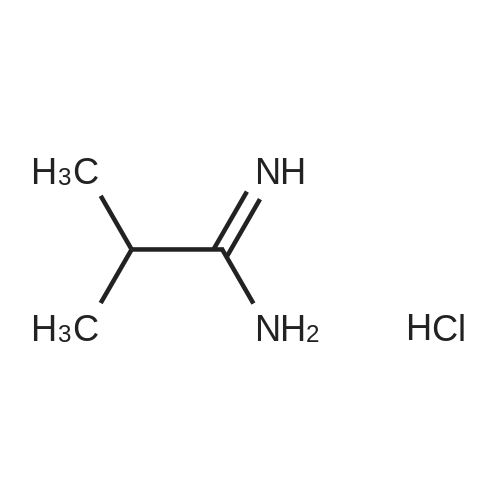
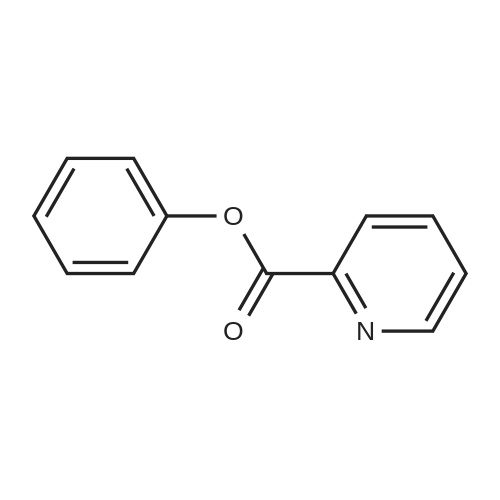
[ 5029-67-4 ]
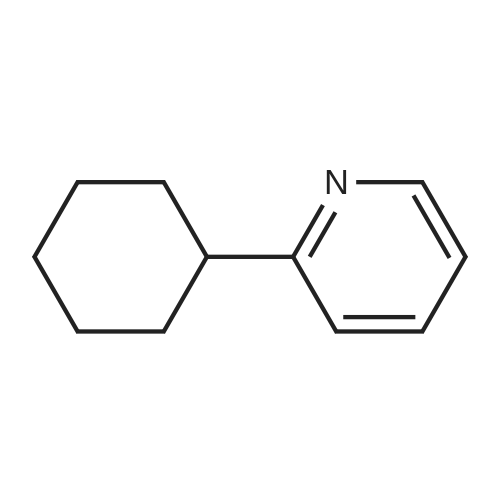
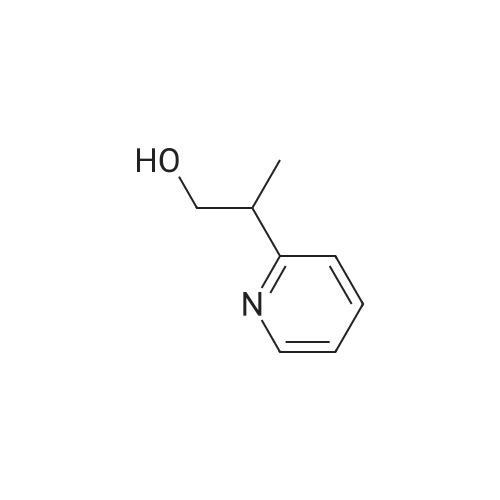
[ 325810-07-9 ]
[ 368-48-9 ]
Reference:
[1] Angewandte Chemie - International Edition, 2011, vol. 50, # 33, p. 7655 - 7659
8
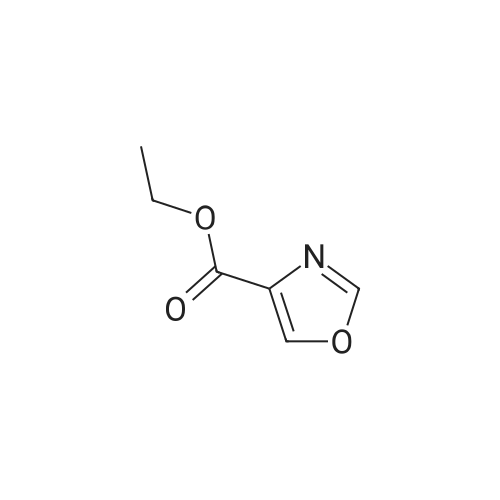
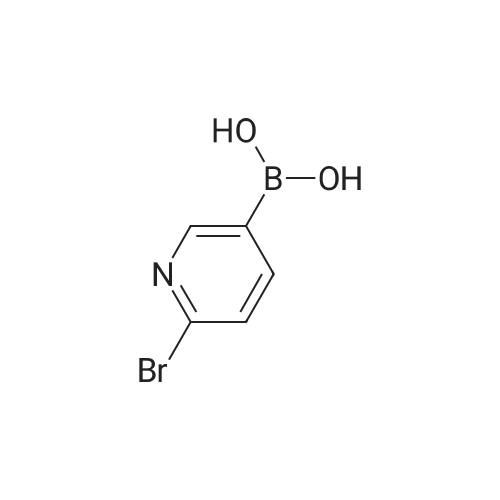
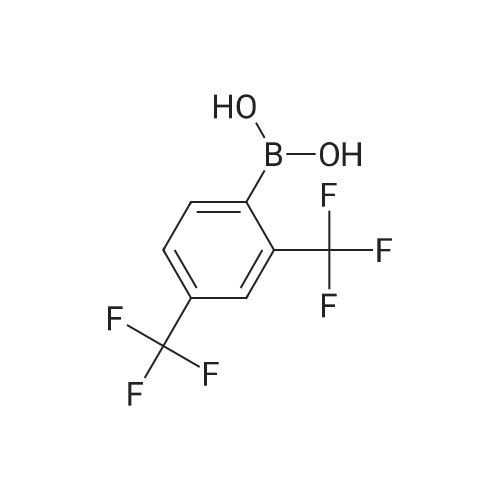
[ 5029-67-4 ]
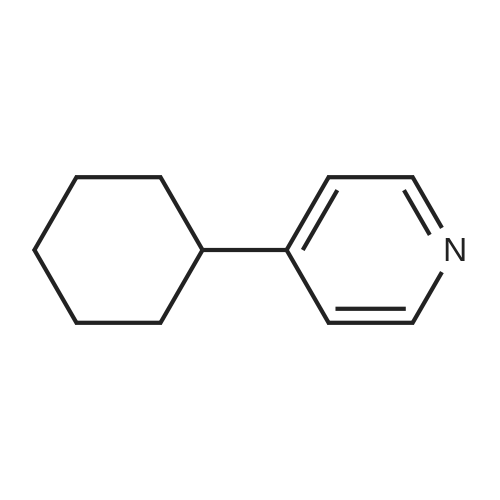
[ 75-46-7 ]
[ 368-48-9 ]
Reference:
[1] Journal of the American Chemical Society, 2011, vol. 133, # 51, p. 20901 - 20913
9
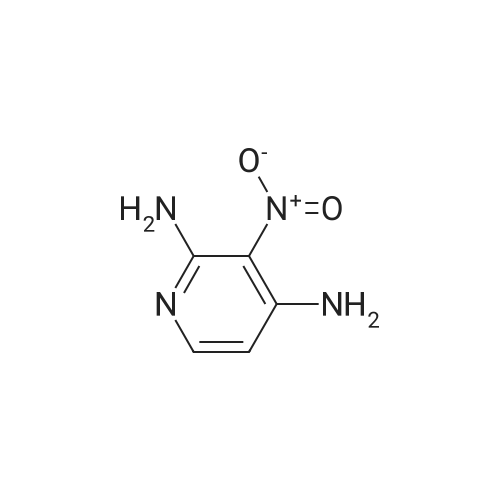
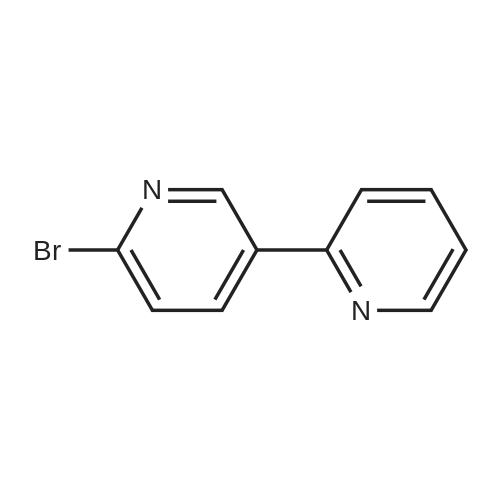
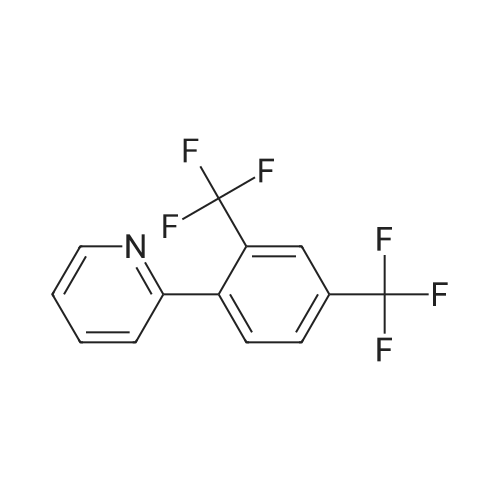
[ 5029-67-4 ]
[ 368-48-9 ]
Reference:
[1] Chemistry--A European Journal, 2015, vol. 21, # 1, p. 96 - 100
10
[ 5029-67-4 ]
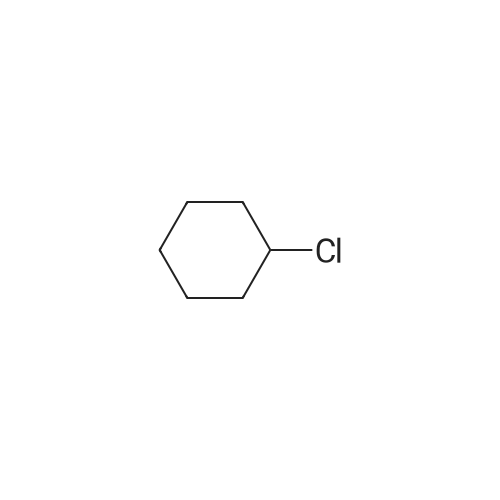
[ 77152-08-0 ]
[ 368-48-9 ]
Reference:
[1] European Journal of Organic Chemistry, 2016, vol. 2016, # 23, p. 4099 - 4104
11
[ 5029-67-4 ]
[ 1744-46-3 ]
[ 368-48-9 ]
Reference:
[1] Journal of Fluorine Chemistry, 2010, vol. 131, # 2, p. 212 - 216
12
[ 5029-67-4 ]
[ 368-48-9 ]
Reference:
[1] Angewandte Chemie - International Edition, 2011, vol. 50, # 33, p. 7655 - 7659
13
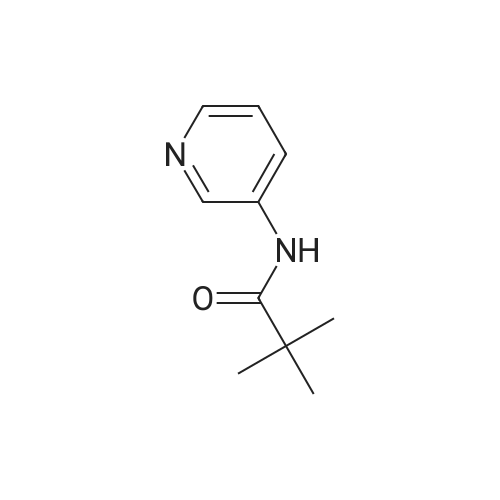
[ 109-04-6 ]
[ 5029-67-4 ]
Yield
Reaction Conditions
Operation in experiment
92%
With copper(l) iodide; sodium iodide; N,N`-dimethylethylenediamine In 1,4-dioxane at 110℃; for 18 h; Inert atmosphere; Schlenk technique
Aromatic Finkelstein Reaction; General ProcedureThe reaction was carried out under argon using standard Schlenktechniques due to the moisture and oxygen sensitivity of the copper(I) iodide. A two-neck pear-shaped flask equipped with a refluxcondenser was charged with the (het)aryl bromide starting material,NaI (2 equiv per bromine to exchange), and CuI (5 molpercent per bromineto exchange). N,N′-Dimethylethylenediamine (L1) or N,N′-dimethyl-1,2-cyclohexanediamine (L2) (10 molpercent per bromine toexchange) and anhydrous 1,4-dioxane (0.5 mL per 1 mmol NaI)were added. The resulting suspension was heated to 110 °C for 18h. After cooling to r.t., the mixture was poured into aq 25percent NH3 solution.The blue solution was diluted to a doubled volume with H2Oand was extracted three times with CH2Cl2. In the case of the 2,2′-bipyridines, the combined organic phases were additionally washedwith aq EDTA solution. Otherwise, the combined organic phaseswere solely washed with brine and dried with MgSO4. The solventwas removed under reduced pressure to give the desired product inpure form. If needed, the crude product can be purified by columnchromatography or recrystallization.
88 %Chromat.
With copper(I) oxide; <i>L</i>-proline; potassium iodide In ethanol at 110℃; for 30 h; Schlenk technique; Inert atmosphere; Sealed tube
General procedure: A Schlenk tube was charged with Cu2O (7.2 mg, 10 molpercent), l-proline (11.5 mg, 20 molpercent), aryl (or heteroaryl) bromide (1 or 3,0.50 mmol), potassium iodide (KI) (249 mg, 0.75 mmol), and EtOH(1.5 mL) under nitrogen atmosphere. The Schlenk tube was sealedwith a teflon valve, and then the reaction mixture was stirred at110C for a period (the reaction progress was monitored by GCanalysis). After the reaction was completed, GC yield of high volatileproduct was determined using an appropriate internal standard(chlorobenzene or 1-chloro-4-methylbenzene) or the solvent wasremoved under reduced pressure. The residue obtained was puri-fied via silica gel chromatography (eluent: petroleum ether/ethylacetate = 10/1) to afford aryl iodides 2a–2o or heteroaryl iodides4a–4g.
Reference:
[1] Tetrahedron Letters, 1999, vol. 40, # 23, p. 4339 - 4342
[2] Tetrahedron, 2000, vol. 56, # 10, p. 1349 - 1360
[3] Synthesis (Germany), 2014, vol. 46, # 8, p. 1085 - 1090
[4] Phosphorus and Sulfur and the Related Elements, 1984, vol. 21, p. 197 - 204
[5] Chemistry - A European Journal, 2010, vol. 16, # 41, p. 12425 - 12433
[6] European Journal of Organic Chemistry, 2002, # 24, p. 4181 - 4184
[7] Chemistry - A European Journal, 2011, vol. 17, # 47, p. 13284 - 13297
[8] Tetrahedron Letters, 1990, vol. 31, # 47, p. 6757 - 6758
[9] Tetrahedron Letters, 1990, vol. 31, # 47, p. 6757 - 6758
[10] Journal of the Chemical Society, 1951, p. 83,86
[11] Journal of Organic Chemistry, 2005, vol. 70, # 17, p. 6904 - 6906
[12] Journal of the Chemical Society. Perkin Transactions 2, 2001, # 9, p. 1620 - 1630
[13] Journal of the Chemical Society - Perkin Transactions 1, 1996, # 15, p. 1781 - 1782
[14] Patent: US2013/109876, 2013, A1, . Location in patent: Page/Page column 5
[15] Journal of Organic Chemistry, 1955, vol. 20, p. 118,128
[16] European Journal of Organic Chemistry, 2014, vol. 2014, # 27, p. 5986 - 5997
[17] Catalysis Today, 2016, vol. 274, p. 129 - 132
[18] Catalysis Science and Technology, 2017, vol. 7, # 10, p. 2110 - 2117
14
[ 109-09-1 ]
[ 5029-67-4 ]
Reference:
[1] Journal of Organic Chemistry, 2009, vol. 74, # 13, p. 4893 - 4895
[2] European Journal of Organic Chemistry, 2002, # 24, p. 4181 - 4184
[3] Tetrahedron Letters, 1990, vol. 31, # 47, p. 6757 - 6758
[4] Chemical & Pharmaceutical Bulletin, 1982, vol. 30, # 5, p. 1731 - 1737
[5] Heterocycles, 1994, vol. 37, # 3, p. 1467 - 1468
[6] Journal of the Chemical Society - Perkin Transactions 1, 1996, # 15, p. 1781 - 1782
15
[ 181647-39-2 ]
[ 5029-67-4 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 2001, vol. 38, # 2, p. 481 - 484
[2] Journal of the Chemical Society - Perkin Transactions 1, 1996, # 15, p. 1781 - 1782
16
[ 20485-44-3 ]
[ 70298-88-3 ]
[ 5029-67-4 ]
[ 113975-32-9 ]
Yield
Reaction Conditions
Operation in experiment
23%
With n-butyllithium; iodine In tetrahydrofuran
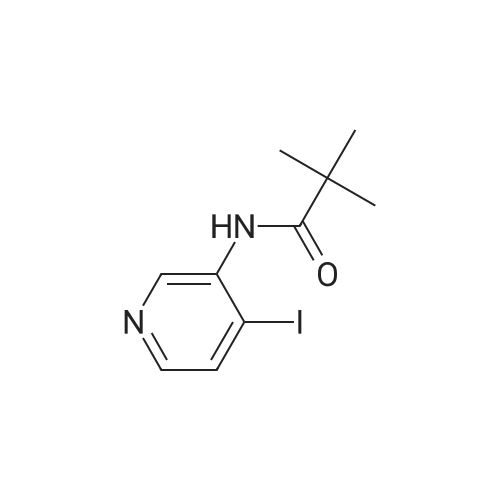
EXAMPLE 10 Compounds of the following general formula II-10 may be made, for example by the following general scheme. Iodopyridine 16. The 3-(Pivaloylamino)pyridine 15 (1.9 g, 11 mmol) and tetramethylethylene-diamine (4.0 mL, 26 mmol) were dissolved in dry THF (60 mL) and cooled to -78° C. While maintaining the temperature between -78° C. and -65° C., nBuLi (2.5 M solution in hexanes, 10.6 mL, 26.5 mmol) was added dropwise. The reaction was allowed to warm to -10° C. for 2 h, and then cooled back down to -78° C. Iodine (6.73 g, 26.5 mmol) dissolved in dry THF (20 mL) was added slowly. After stirring for 2 h at -78° C., the reaction was quenched with ice. Excess iodine was destroyed with addition of saturated potassium thiosulfate solution. The product was extracted with CH2Cl2, and the organic layers were washed with brine. The mixture was concentrated in vacuo to a black oil which was chromatographed (1:1 EtOAc/Hexanes; 2:1 EtOAc/Hexanes) to give 700 mg (23percent) of 2,2-dimethyl-N-(4-iodo-3-pyridinyl)propanamide as a yellow solid. 1H-NMR (DMSO-d6 300 MHz) δ 9.24 (s, 1H), 8.35 (s, 1H), 8.04 (d, 1H), 7.95 (d, 1H), 1.26 (s, 9H). MS (ES+)=305.
Reference:
[1] Journal of Organic Chemistry, 2009, vol. 74, # 14, p. 5111 - 5114
20
[ 110-86-1 ]
[ 5029-67-4 ]
Reference:
[1] Journal of Organic Chemistry, 1984, vol. 49, # 20, p. 3857 - 3859
[2] Journal of the American Chemical Society, 1999, vol. 121, # 14, p. 3539 - 3540
21
[ 36316-71-9 ]
[ 5029-67-4 ]
Reference:
[1] Synlett, 2003, # 12, p. 1801 - 1804
22
[ 19524-06-2 ]
[ 5029-67-4 ]
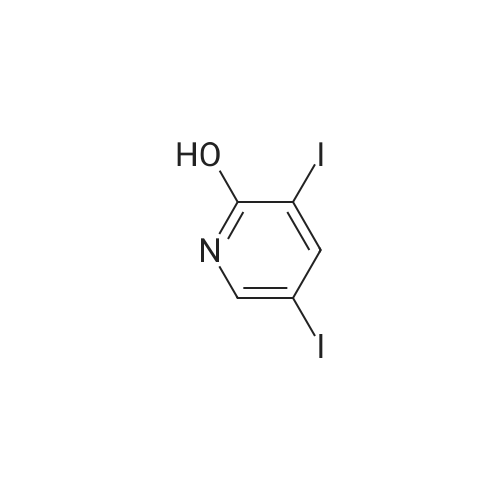
[ 15854-87-2 ]
Reference:
[1] Patent: US6358971, 2002, B1,
23
[ 504-29-0 ]
[ 5029-67-4 ]
Reference:
[1] Zhurnal Russkago Fiziko-Khimicheskago Obshchestva, 1915, vol. 47, p. 1575[2] Chem. Zentralbl., 1916, vol. 87, # II, p. 228
24
[ 7553-56-2 ]
[ 5029-67-4 ]
Reference:
[1] Journal of the American Chemical Society, 1965, vol. 87, p. 5361 - 5366
[2] , Gmelin Handbook: Co: Org.Verb.1, 1.1.3.2.1, page 66 - 68,
25
[ 1054449-81-8 ]
[ 5029-67-4 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 2008, vol. 45, # 4, p. 1167 - 1170
26
[ 107263-95-6 ]
[ 5029-67-4 ]
[ 65007-00-3 ]
[ 372-48-5 ]
Reference:
[1] Journal of Fluorine Chemistry, 1996, vol. 77, # 2, p. 161 - 168
With potassium acetate In N,N-dimethyl acetamide at 130℃; for 3 h; Schlenk technique; Inert atmosphere
General procedure: MCM-41-3N-Pd(0) (21mg, 0.01mmol), KOAc (1.5mmol) and aryl iodide 1 (1.0mmol) (if solid) were placed in an oven-dried 20mL Schlenk tube, the reaction vessel was evacuated and filled with argon for three times. Then aryl iodide 1 (1.0mmol) (if liquid), diphenylphosphine (1.2mmol) and DMAc (1mL) were added with a syringe under a counter flow of argon. The reaction mixture was stirred at 130°C for 3h. After completion of the reaction, the mixture was cooled to room temperature and diluted with CH2Cl2 (20mL) and filtered. The MCM-41-3N-Pd(0) catalyst was washed with distilled water (2×5mL) and ethanol (2×5mL), and reused in the next run. The filtrate was concentrated in vacuo and the residue was purified by flash column chromatography on silica gel to provide the product 2.
With copper(I) oxide; caesium carbonate In dimethyl sulfoxide at 100℃; for 24 h; Inert atmosphere
General procedure: The N-nucleophile (0.735mmol), Cu2O (0.0735mmol), Cs2CO3 (1.47mmol), DMSO (0.3mL) and heteroaryl halide (1.103mmol) were added to a reaction vial and a screw cap was fitted to it. The reaction mixture was stirred under air in a closed system at 100°C for 24h. After cooling to room temperature, the mixture was diluted with dichloromethane and filtered through a pad of Celite. The combined organic extracts were dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by silica-gel column chromatography to afford the N-arylated product. The identity and purity of the products was confirmed by 1H, 13C NMR spectroscopic analysis and elemental analysis or mass spectroscopy.
Reference:
[1] Advanced Synthesis and Catalysis, 2017, vol. 359, # 10, p. 1631 - 1636
[2] Tetrahedron Letters, 2009, vol. 50, # 42, p. 5868 - 5871
[3] Tetrahedron, 2013, vol. 69, # 35, p. 7279 - 7284
[4] Green Chemistry, 2011, vol. 13, # 1, p. 42 - 45
Reference:
[1] Chemical Communications, 2012, vol. 48, # 15, p. 2080 - 2082
[2] Synlett, 2013, vol. 24, # 9, p. 1101 - 1104
[3] RSC Advances, 2015, vol. 5, # 92, p. 75263 - 75267
[4] Organic and Biomolecular Chemistry, 2016, vol. 14, # 6, p. 2127 - 2133
41
[ 5029-67-4 ]
[ 927-74-2 ]
[ 395652-44-5 ]
Yield
Reaction Conditions
Operation in experiment
67%
With potassium phosphate In water; isopropyl alcohol at 80℃; for 20 h; Sealed tube
General procedure: In a sealed tube, aryl iodide (1 mmol, 1 equiv.), K3PO4(2 mmol,2 equiv.), catalyst (2 molpercent Pd) were suspended in i-PrOH (3 mL)and H2O (3 mL). The acetylene derivative (1.2 mmol, 1.2 equiv.)was added and the resulting mixture was stirred at 80C for 20 h.After cooling to room temperature, EtOAc (20 mL) and H2O (20 mL)were added and the mixture was filtered over a pad of Celite®.The aqueous layer was extracted twice with EtOAc (2 × 20 mL). Thecollected organics extracts were washed by brine (60 mL), driedon MgSO4, filtered and concentrated under reduced pressure. Thecrude product was purified by flash chromatography.
Reference:
[1] Applied Catalysis A: General, 2014, vol. 482, p. 157 - 162
42
[ 5029-67-4 ]
[ 34091-51-5 ]
[ 938066-21-8 ]
Reference:
[1] Journal of Organic Chemistry, 2007, vol. 72, # 9, p. 3589 - 3591
With potassium carbonate; N,N`-dimethylethylenediamine;copper(l) iodide; at 95℃;
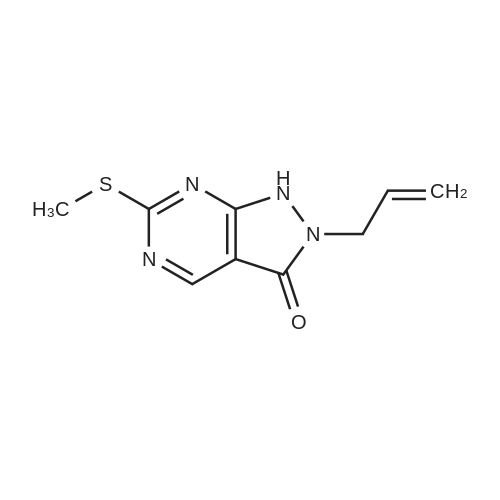
1) Production of 2-allyl-6-(methylthio)-1-pyridin-2-yl-3H-pyrazolo[3,4-d]pyrimidin-3-one 2.4 mL of N,N'-dimethylethylenediamine was added to 1,4-dioxane (50 mL) solution of 4.44 g of <strong>[955368-90-8]2-allyl-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one</strong>, 3.80 g of copper(I) iodide, 5.33 g of 2-iodopyridine and 3.80 g of potassium carbonate, and stirred overnight at 95 C. The reaction liquid was cooled, aqueous ammonia was added thereto and extracted with ethyl acetate, washed with saturated saline water and dried with anhydrous magnesium sulfate. The solvent was evaporated away under reduced pressure, and crystallized with ethyl acetate to obtain 5.15 g of the entitled compound as a white solid. 1H-NMR (400 MHz, CDCl3) delta: 8.94 (1H, s), 8.52 (1H, d, J=5.1 Hz), 7.90 (2H, d, J=3.5 Hz), 7.29-7.25 (1H, m), 5.68 (1H, ddt, J=17.0, 10.2, 6.3 Hz), 5.05 (1H, d, J=10.2 Hz), 4.91 (1H, d, J=17.0 Hz), 4.85 (1H, d, J=6.3 Hz), 2.58 (3H, s). ESI-MS Found: m/z[M+H]+ 300.
With copper(l) iodide; potassium carbonate; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 95℃;
Reference Example 1 :Production of 2-allyl-6-fmethylthio)-l-pyridin-2-yl-3H-pyrazolo[3,4-d1pyrimidin-3-one2.4 mL of N,N'-dimethyl ethyl enediamine was added to 1,4-dioxane (50 mL) solution of 4.44 g of 2-allyl-6-(methylthio)-l,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one, 3,80 <n="30"/>g of copper(I) iodide, 5.33 g of 2-iodopyridine and 3.8O g of potassium carbonate, and stirred overnight at 95C. The reaction liquid was cooled, aqueous ammonia was added thereto and extracted with ethyl acetate, washed with saturated saline water and dried with anhydrous magnesium sulfate. The solvent was evaporated away under reduced pressure, and crystallized with ethyl acetate to obtain 5.15 g of the entitled compound as a white solid. 1 H-NMR (400 MHz, CDCI3) delta: 8.94 (IH, s), 8.52 (IH, d, J=5.1 Hz), 7.90 (2H, d, J=3.5 Hz),7.29-7.25 (IH, m), 5.68 (IH, ddt, J=I 7.0, 10.2, 6.3 Hz), 5.05 (IH, d, J=10.2 Hz), 4.91 (IH, d, J=I 7.0 Hz), 4.85 (IH, d, J=6.3 Hz), 2.58 (3H, s). ESI-MS Found: m/z[M+H]+ 300.
With potassium carbonate;copper(l) iodide; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 95℃;
1) Production of 2-allyl-6-(methylthio)-1-pyridin-2-yl-3H-pyrazolo[3,4-d]pyrimidin-3-one: N,N'-dimethylethylenediamine (2.4 mL) was added to a 1,4-dioxane (50 mL) solution of <strong>[955368-90-8]2-allyl-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one</strong> (4.44 g), copper(I) iodide (3.80 g), 2-iodopyridine (5.33 g) and potassium carbonate (3.80 g), and stirred overnight at 95C. The reaction liquid was cooled, then aqueous ammonia was added thereto and extracted with ethyl acetate, washed with saturated saline water, and dried with anhydrous magnesium sulfate. The solvent was evaporated away under reduced pressure, and the residue was crystallized with ethyl acetate to give the entitled compound as a white solid (5.15 g). 1H-NMR (400 MHz, CDCl3) delta: 8.94 (1H, s), 8.52 (1H, d, J = 5.1 Hz), 7.90 (2H, d, J = 3.5 Hz), 7.29-7.25 (1H, m), 5.68 (1H, ddt, J = 17.0, 10.2, 6.3 Hz), 5.05 (1H, d, J = 10.2 Hz), 4.91 (1H, d, J = 17.0 Hz), 4.85 (2H, d, J = 6.3 Hz), 2.58 (3H, s). ESI-MS Found: m/z[M+H]+ 300.
With copper(l) iodide; potassium carbonate; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 95℃;
Production of 2-allyl-6-(methylthio)- 1 -pyridin-2-yl-3 H-pyrazolo [3 ,4-d]pyrimidin-3 -one : 2.4 mL of Nu,Nu'-dimethylethylenediamine was added to 1,4-dioxane (50 mL) solution of 4.44 g of 2-allyl-6-(memyltWo)-l,2-dmydro-3H-pyrazolo[3,4-d]pyrimidin-3-one, 3,80 g of copper(I) iodide, 5.33 g of 2-iodopyridine and 3.80 g of potassium carbonate, and stirred overnight at 95 C. The reaction liquid was cooled, aqueous ammonia was added thereto and extracted with ethyl acetate, washed with saturated saline water and dried with anhydrous magnesium sulfate. The solvent was evaporated away under reduced pressure, and crystallized with ethyl acetate to obtain 5.15 g of the entitled compound as a white solid. iH-NMR (400 MHz, CDCI3) 5: 8.94 (IH, s), 8.52 (IH, d, J=5.1 Hz), 7.90 (2H, d, J=3.5 Hz), 7.29-7.25 (IH, m), 5.68 (IH, ddt, J=17.0, 10.2, 6.3 Hz), 5.05 (IH, d, J=10.2 Hz), 4.91 (IH, d, J=17.0 Hz), 4.85 (IH, d, J=6.3 Hz), 2.58 (3H, s).
5.15 g
With copper(l) iodide; potassium carbonate; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 95℃;
Preparative Example 1-1 Production of 2-allyl-6-(methylthio)-l-pyridin-2-yl-3H-pyrazolo[3,4-d]pyrimidin-3-one: 2.4 mL of Nu,Nu'-dimethylethylenediamine was added to 1,4-dioxane (50 mL) solution of 4.44 g of 2-allyl-6-(methylthio)-l,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one, 3.80 g of copper(I) iodide, 5.33 g of 2-iodopyridine and 3.80 g of potassium carbonate, and stirred overnight at 95C. The reaction liquid was cooled, aqueous ammonia was added thereto and extracted with ethyl acetate, washed with saturated saline water and dried with anhydrous magnesium sulfate. The solvent was evaporated away under reduced pressure, and crystallized with ethyl acetate to obtain 5.15 g of the entitled compound as a white solid. lH-NMR (400 MHz, CDCI3) delta: 8.94 (IH, s), 8.52 (IH, d, J=5.1 Hz), 7.90 (2H, d, J=3.5 Hz), 7.29-7.25 (IH, m), 5.68 (IH, ddt, J=17.0, 10.2, 6.3 Hz), 5.05 (IH, d, J=10.2 Hz), 4.91 (IH, d, J=17.0 Hz), 4.85 (IH, d, J=6.3 Hz), 2.58 (3H, s).
With copper(l) iodide; potassium carbonate; N,N`-dimethylethylenediamine; In 1,4-dioxane; at 95℃;
2.4 mL of N,N'-dimethylethylenediamine was added to 1,4-dioxane (50 mL) solution of 4.44 g of <strong>[955368-90-8]2-allyl-6-(methylthio)-1,2-dihydro-3H-pyrazolo[3,4-d]pyrimidin-3-one</strong>, 3.80 g of copper(I) iodide, 5.33 g of 2-iodopyridine and 3.80 g of potassium carbonate, and stirred overnight at 95 C. The reaction liquid was cooled, aqueous ammonia was added thereto and extracted with ethyl acetate, washed with saturated saline water and dried with anhydrous magnesium sulfate. The solvent was evaporated away under reduced pressure, and crystallized with ethyl acetate to obtain the entitled compound as a white solid. 1H-NMR (400 MHz, CDCl3) delta: 8.94 (1H, s), 8.52 (1H, d, J=5.1 Hz), 7.90 (2H, d, J=3.5 Hz), 7.29-7.25 (1H, m), 5.68 (1H, ddt, J=17.0, 10.2, 6.3 Hz), 5.05 (1H, d, J=10.2 Hz), 4.91 (1H, d, J=17.0 Hz), 4.85 (1H, d, J=6.3 Hz), 2.58 (3H, s).
EXAMPLE 10 Compounds of the following general formula II-10 may be made, for example by the following general scheme. Iodopyridine 16. The 3-(Pivaloylamino)pyridine 15 (1.9 g, 11 mmol) and tetramethylethylene-diamine (4.0 mL, 26 mmol) were dissolved in dry THF (60 mL) and cooled to -78 C. While maintaining the temperature between -78 C. and -65 C., nBuLi (2.5 M solution in hexanes, 10.6 mL, 26.5 mmol) was added dropwise. The reaction was allowed to warm to -10 C. for 2 h, and then cooled back down to -78 C. Iodine (6.73 g, 26.5 mmol) dissolved in dry THF (20 mL) was added slowly. After stirring for 2 h at -78 C., the reaction was quenched with ice. Excess iodine was destroyed with addition of saturated potassium thiosulfate solution. The product was extracted with CH2Cl2, and the organic layers were washed with brine. The mixture was concentrated in vacuo to a black oil which was chromatographed (1:1 EtOAc/Hexanes; 2:1 EtOAc/Hexanes) to give 700 mg (23%) of 2,2-dimethyl-N-(4-iodo-3-pyridinyl)propanamide as a yellow solid. 1H-NMR (DMSO-d6 300 MHz) delta 9.24 (s, 1H), 8.35 (s, 1H), 8.04 (d, 1H), 7.95 (d, 1H), 1.26 (s, 9H). MS (ES+)=305.
With copper(I) oxide; caesium carbonate; In dimethyl sulfoxide; at 100℃; for 24h;Inert atmosphere;
General procedure: The N-nucleophile (0.735mmol), Cu2O (0.0735mmol), Cs2CO3 (1.47mmol), DMSO (0.3mL) and heteroaryl halide (1.103mmol) were added to a reaction vial and a screw cap was fitted to it. The reaction mixture was stirred under air in a closed system at 100C for 24h. After cooling to room temperature, the mixture was diluted with dichloromethane and filtered through a pad of Celite. The combined organic extracts were dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by silica-gel column chromatography to afford the N-arylated product. The identity and purity of the products was confirmed by 1H, 13C NMR spectroscopic analysis and elemental analysis or mass spectroscopy.
2-Iodopyridine (Tokyo Chemical Industry Co., Ltd.) (200 mg, 0.976 mmoles), 2,5-thiophene diborate (Wako Pure Chemical Industries, Ltd.) (420 mg, 2.44 mmoles), sodium carbonate (Wako Pure Chemical Industries, Ltd.) (311 mg, 2.93 mmoles) and tetrakis (triphenylphosphine) palladium (0) (Wako Pure Chemical Industries, Ltd.) (33.9 mg, 0.0293 mmoles) were dissolved in a mixed solvent containing 5 ml of toluene and 5 ml of methanol. Air was removed to create a vacuum and the space was purged with nitrogen three times after which the reaction mixture was agitated for thirty minutes at 80°C. After letting the reaction solution stand until cooled, Compound 1 obtained in the manner described above was added. The air was removed to create a vacuum and the space was purged with nitrogen three times again. after which the reaction mixture was agitated for seven hours at 80°C. After allowing the reaction solution to cool, the reaction solution was diluted with ethyl acetate, washed once using de-ionized water, and once using an aqueous saturated sodium chloride solution. The organic layer was dried using anhydrous sodium sulfate. The solvent was removed by distillation, and the product was purified using a medium pressure fractionating liquid chromatography to obtain yellow solids (Compound 4) (99.2 mg, 0.236 mmoles, 24percent). The analytical results for the Compound 4 synthesized {N,N-dihexyl-4-[5-(pyridine-2-yl) thiophene-2-yl] aniline} are shown below. 1H-NMR (400 MHz, CDCl3, TMS, rt) delta 8.55 (1H, brd, J = 4.5 Hz), 7.67-7.61 (2H, m), 7.53-7.50 (3H, m), 7.14-7.08 (2H, m), 6.64 (2H, d, J = 8.6 Hz) 3.29 (4H, t, J = 7.5 Hz), 1.60 (4H, brs), 1.33 (12H, brs), 0.91 (6H, t, J = 6.3 Hz)
With caesium carbonate;copper(I) oxide; 8-quinolinol; In dimethyl sulfoxide; at 120℃; for 2h;microwave irradiation;
A mixture of (R)-l-(5,6-dichloro-lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3-yl)- piperidine-4-carboxamide (100 mg, 0.261 mmol) dimethylsulfoxide (2 mL), caesium carbonate (0.392 mmol), 2-iodopyridine (0.783 mmol), 8-hydroxyquinoline (0.104 mmol), polyethylene glycol (0.511 mmol) and copper(I) oxide (0.0522 mmol) was subjected to microwave conditions for two hour at 120C. The reaction mixture was filtered and subjected to preparative hplc(performed on a Gilson-Finnigan ThermoQuest AQA system equipped with a Zorbax SB-C8 (5 muetaiota, 21.2 x 150 mm) column, using methanol/water (0.05 % formic acid) gradients at a flow rate of 15 mL/min with UV (214 or 254 nm) and MS (ESI) detection) to give 30 mg (25 % yield) of (R)- 1 -(5,6-dichloro- 1 -(pyridin-2-yl)- lH-benzo[d]imidazol-2-yl)-N-(tetrahydrofuran-3 - yl)piperidine-4-carboxamide as a pale yellow solid. LC-MS (m/z) 460.3 (M+l).
With copper(I) oxide; caesium carbonate; In dimethyl sulfoxide; at 100℃; for 24h;Inert atmosphere;
General procedure: The N-nucleophile (0.735mmol), Cu2O (0.0735mmol), Cs2CO3 (1.47mmol), DMSO (0.3mL) and heteroaryl halide (1.103mmol) were added to a reaction vial and a screw cap was fitted to it. The reaction mixture was stirred under air in a closed system at 100C for 24h. After cooling to room temperature, the mixture was diluted with dichloromethane and filtered through a pad of Celite. The combined organic extracts were dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by silica-gel column chromatography to afford the N-arylated product. The identity and purity of the products was confirmed by 1H, 13C NMR spectroscopic analysis and elemental analysis or mass spectroscopy.
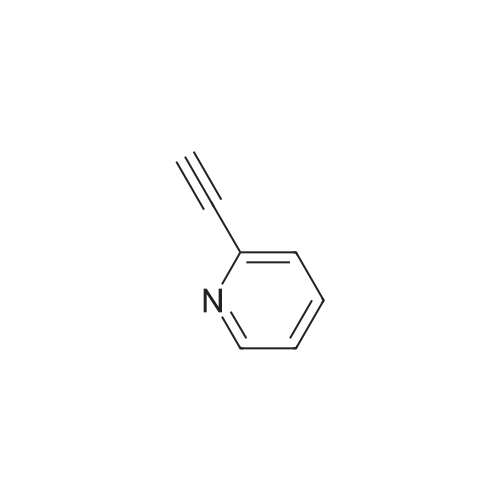
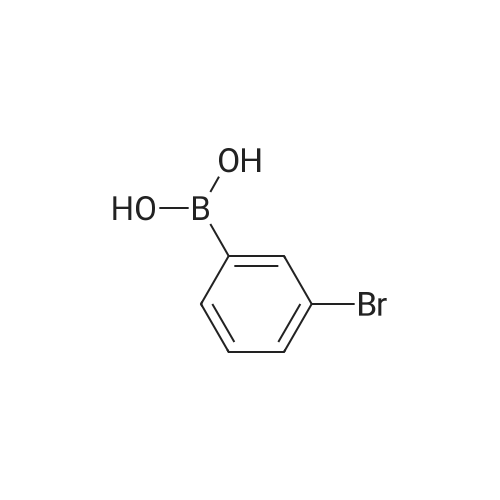
General procedure: To the suspension of zinc powder (without activation, 65.4 mg, 1.0 mmol, Aldrich 99.995% purity) in DMPU (0.5 mL), trifluoromethyl iodide (ca. 2.5 mmol, sufficiently dissolved in the solution) was added at room temperature under argon atmosphere. After the solution was stirred for 2 h at room temperature, CuI (1.9 mg, 0.01 mmol, 2 mol %), 1.10-phenanthroline (1.8 mg, 0.01 mmol, 2 mol %), and then aryl iodide 1a (138.0 mg, 0.5 mmol) were added. The reaction mixture was stirred at 50 C for 24 h. After cooling to room temperature, the yield of product 2a was determined by 19F NMR analysis by using benzotrifluoride (BTF) as an internal standard. Except for 2c, all trifluoromethylated products 2 exhibited the same 1H, 13C, and 19F NMR spectra as reported before [14, 17, 29, 31-36].
With potassium phosphate; In water; isopropyl alcohol; at 80℃; for 20.0h;Sealed tube;
General procedure: In a sealed tube, aryl iodide (1 mmol, 1 equiv.), K3PO4(2 mmol,2 equiv.), catalyst (2 mol% Pd) were suspended in i-PrOH (3 mL)and H2O (3 mL). The acetylene derivative (1.2 mmol, 1.2 equiv.)was added and the resulting mixture was stirred at 80C for 20 h.After cooling to room temperature, EtOAc (20 mL) and H2O (20 mL)were added and the mixture was filtered over a pad of Celite.The aqueous layer was extracted twice with EtOAc (2 × 20 mL). Thecollected organics extracts were washed by brine (60 mL), driedon MgSO4, filtered and concentrated under reduced pressure. Thecrude product was purified by flash chromatography.
With copper(l) iodide; 8-quinolinol; caesium carbonate; In tert-butyl alcohol; at 90℃; for 18h;Inert atmosphere;
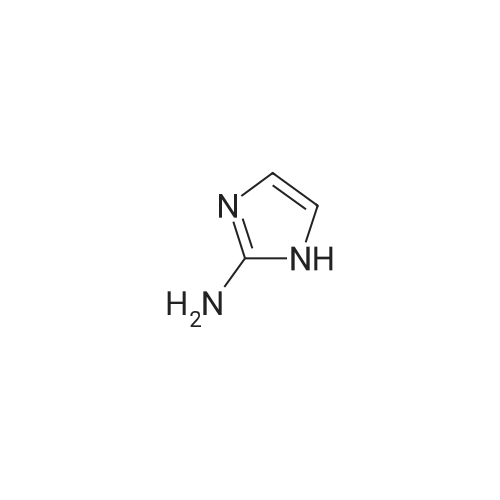
General procedure: To a stirred solution of iodopyridine (1.0 equivalent) and 2-amino imidazole (1.1 equivalents) in tert-butanol ( 15 volumes) was added cesium carbonate ( 1.5 equivalents). The reaction mixture was degassed by purging with nitrogen continuously for 15 minutes. 8-Hydroxy- quinoline (0.15 equivalents) and copper(I) iodide (0.1 equivalents) were added, and the reaction mixture was heated to 90 C for 18 hours. The reaction mixture was cooled to room temperature and quenched with water and extracted with ethyl acetate (3x). The combined organic extracts were washed with water and a saturated brine solution, dried over sodium sulfate, and concentrated. Purification by preparative HPLC provided the title molecule.XH NMR(400 MHz, DMSO-d6) delta 12.79 (br s, IH), 8.77 (s, IH), 8.57- 8.56 (m, IH), 8.12-8.11 (m, IH), 7.85 (d, J = 8.36 Hz, IH), 7.77 (d, J = 1.84 Hz, IH), 7.49-7.22 (m, IH), 7.21 (s, IH); ESIMS m/z 161 ([M+H]+).
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; potassium carbonate; In water; N,N-dimethyl-formamide; at 90℃; for 3h;
2-iodopyridine (3. lg, 15. 0mmol), 3-(4,4,5,5- tetramethyl _1,3,2_ dioxolan boron2-yl) phenol (5. (^, 23.0_1), [1,1'-bis (diphenylphosphino) ferrocene] dichloropalladium (11) (1.88,2.00mmol) and potassium carbonate (9.3g, 68.0mmol) was suspended in DMF (llOmL) / water (60mL) mixed solvent at 90 C underStirred for 3 hours. Thereafter, the reaction solution was filtered under reduced pressure and the insoluble matter was removed, the solvent was distilled off under reduced pressure. The residueChloroform was added (lOOmL), washed with water (100 ml x 3). Dried over magnesium sulfate, and purified by column chromatography (baby gel, chloroform / AAlcohol = 99/1 (nu / nu)) to give compound 6 (2. Lg, 82% yield).
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; tris-(dibenzylideneacetone)dipalladium(0); potassium carbonate; In toluene; at 120℃; for 18h;
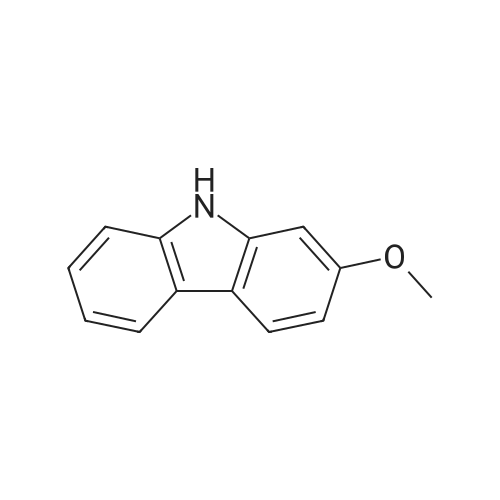
<strong>[6933-49-9]2-methoxy-9H-carbazole</strong> (1.0 eq), 2-iodopyridine (1.2 eq), Pd2(dba)3 (0.05 eq), SPhos (0.10 eq), and K2CO3 (1.0 eq) were dissolved in toluene (0.1 M) and stirred at a temperature of 120 C. for 18 hours. After the reaction mixture was cooled to room temperature, an organic layer was extracted therefrom three times using dichloromethane and water. The extracted organic layer was dried over magnesium sulfate and concentrated, and column chromatography was performed thereon to obtain Intermediate [1-A] (yield: 91%).
60%
With (1,1'-bis(diphenylphosphino)ferrocene)palladium(II) dichloride; tris-(dibenzylideneacetone)dipalladium(0); sodium t-butanolate; In toluene; for 18h;Inert atmosphere; Reflux;
A 1 L three-neck round-bottom flask was charged with <strong>[6933-49-9]2-methoxy-9H-carbazole</strong> (4.6 g, 23.2 mmol), 2-iodopyridine (3.1 ml, 29.0 mmol), 1,1?-bis(diphenylphosphino)ferrocene (0.514 g, 0.927 mmol), Pd2(dba)3 (0.424 g, 0.463 mmol), sodium tert-butoxide (3.12 g, 32.4 mmol) and toluene (150 mL). The reaction mixture was degassed for 20 minutes and heated to reflux for 18 hours. The reaction mixture was cooled to room temperature and diluted with water. The aqueous layer was extracted three times with EtOAc and the combined organic layers were concentrated. The crude material was chromatographed on silica with 85/15 hexane/EtOAc (v/v) to 70/30 hexane/EtOAc (v/v) to yield 5.5 g (60%) of 2-methoxy-9-(pyridin-2-yl)-9H-carbazole as an off-white solid. The product was confirmed by GC/MS and NMR.
2-methoxy-10-(pyridin-2-yl)-phenothiazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
52%
With dicyclohexyl-(2',6'-dimethoxybiphenyl-2-yl)-phosphane; tris-(dibenzylideneacetone)dipalladium(0); sodium t-butanolate; In toluene; at 130℃; for 14h;Inert atmosphere;
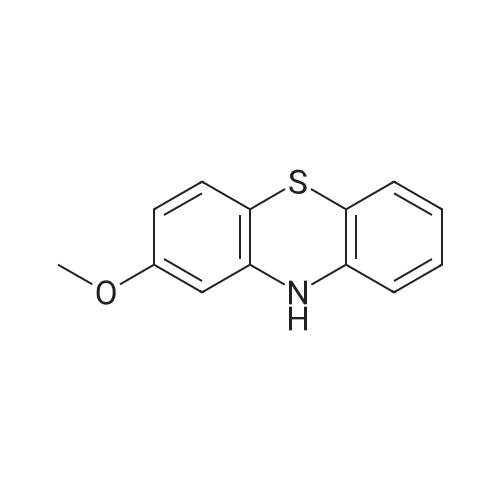
Step 1: Synthesis of 2-methoxy-10-(pyridin-2-yl)-phenothiazine First, 3.1 g of <strong>[1771-18-2]2-methoxy phenothiazine</strong>, 3.1 g of 2-iodopyridine, and 2.0 g of sodium-t-butoxide were put into a three-neck flask equipped with a reflux pipe, and the air in the flask was replaced with nitrogen. Then, 97 mL of toluene, 0.56 g of 2-dicyclohexylphosphino-2',6'-dimethoxybiphenyl (product name: SPhos), and 0.62 g of tris(dibenzylideneacetone)dipalladium(0) (abbreviation: Pd2(dba)3) were added to this mixture, and the mixture was heated and stirred at 130 C. for 14 hours. Water was added to the obtained mixture, and an organic layer was extracted with ethyl acetate. The solution of the extract was washed with saturated brine. Then, magnesium sulfate was added and filtration was performed. The solvent of the filtrate was distilled off and the obtained residue was purified by flash column chromatography using ethyl acetate and hexane in a ratio of 1:5 as a developing solvent, so that the desired substance was obtained as 2.2 g of a brown oily substance in a yield of 52%.
With potassium phosphate; copper(l) iodide; glycine; In N,N-dimethyl-formamide; for 24h;Inert atmosphere; Reflux;
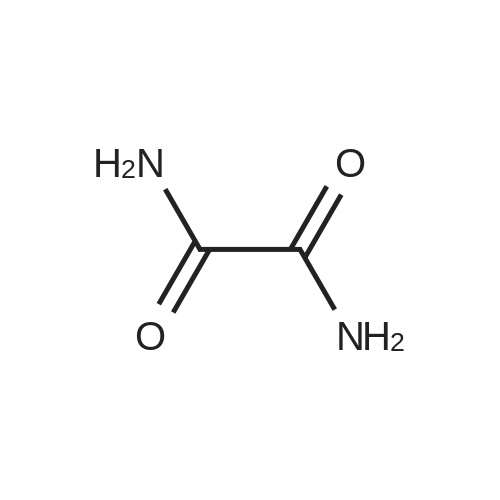
A 1000 mL three-neck reaction flask fitted with a thermowell, a condenser and a stirrer was charged with 2-iodopyridine (244.8 g, 1.2 mol)<strong>[471-46-5]Oxamid</strong>e (44.0 g, 0.5 mol),Glycine (9.4 g, 25 mol%, 0.125 mol),CuI (9.6 g, 10 mol%, 0.05 mol),K3PO4 (212.0 g, 1.0 mol) was added and 600 mL of dimethylformamide was added as a solvent. The mixture was purged with nitrogen and the mixture was stirred and the reaction was refluxed for 24 hours. The reaction was cooled to completion, the solvent was distilled off under reduced pressure, and absolute ethanol was added to the residual solid. The filter cake was washed with water and cold ether to give 105.3 g of N, N'-bis (2-pyridyl) oxamide Rate of 87%,
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In ethanol; benzene; for 16h;Reflux; Inert atmosphere;
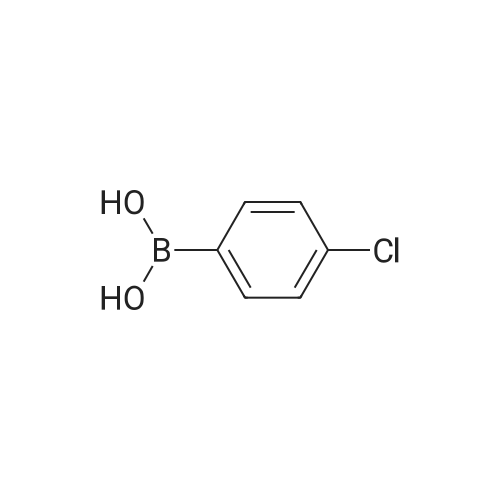
In accordance with the following formula, a C-N ligand (3) was obtained by reacting <strong>[153254-09-2]2,4-bis(trifluoromethyl)phenylboronic acid</strong> with 2-iodopyridine. A mixture of <strong>[153254-09-2]2,4-bis(trifluoromethyl)phenylboronic acid</strong> (2.31 g, 8.96 mmol), 2-iodopyridine (1.16 g, 5.66 mmol), benzene (25 mL), ethanol (10 mL), K2CO3 (7.31 g, 52.9 mmol) and PdCl2(PPh3)2 (0.383 g, 0.546 mmol) was heated and refluxed for 16 hours under a nitrogen atmosphere. After being allowed to cool, the reaction mixture was transferred to a separating funnel. After diluting with an appropriate amount of chloroform, the mixture was washed with water and a saturated saline, and the organic layer was dried on anhydrous magnesium sulfate. After removal of the magnesium sulfate by filtration, the solvent was distilled off with a rotary evaporator. The C-N ligand (3) was obtained in a yield of 59% (0.978 g, 3.36 mmol) by purifying the residue with a silica gel chromatography (development solvent; chloroform). The 1H NMR property of the thus synthesized compound was as follows. 1H NMR (CDCl3): delta7.33 (ddd, J=7.8, 7.7 and 1.1 Hz, 1H), 7.42 (d, J=7.8 Hz, 1H), 7.64 (d, J=7.7 Hz, 1H), 7.76 (ddd, J=7.7, 7.7 and 1.9 Hz, 1H), 7.86 (d, J=8.3 Hz, 1H), 8.01 (s, 1H)
With bis-triphenylphosphine-palladium(II) chloride; potassium carbonate; In ethanol; water; benzene; for 18h;Reflux; Inert atmosphere;
In accordance with the following formula, a C-N ligand (4) was obtained by reacting <strong>[871940-31-7]2,4-difluoro-3-cyanophenylboronic acid</strong> with 2-iodopyridine. A mixture of 2,4-difluoropyridineboronic acid (0.976 g, 5.34 mmol), 2-iodopyridine (0.733 g, 3.58 mmol), benzene (15 mL), ethanol (6 mL), water (15 mL), K2CO3 (4.56 g, 33.0 mmol) and Pd(PPh3)2 Cl2 (0.215 g, 0.306 mmol) was heated and refluxed for 18 hours under a nitrogen atmosphere. After being allowed to cool, the mixture was concentrated to approximately in solution volume by a rotary evaporator, and then the obtained mixture was transferred to a separating funnel. After diluting with an appropriate amount of chloroform, the mixture was washed with water and a saturated saline, and the organic layer was dried on anhydrous magnesium sulfate. After removal of the magnesium sulfate by filtration, the solvent of the filtrate was distilled off with a rotary evaporator. The C-N ligand (4) was obtained in a yield of 80% (0.620 g, 2.87 mmol) by purifying the residue with a silica gel chromatography (development solvent; chloroform). The 1H NMR property of the thus synthesized compound was as follows. 1H NMR (CDCl3): delta7.18 (ddd, J=1.4, 7.8 and 9.2 Hz, 1H), 7.33 (ddd, J=1.4, 5.0 and 7.3 Hz, 1H), 7.76-7.84 (m, 2H), 7.86 (td, J=6.4 and 8.7 Hz, 1H), 8.72 (m, 1H)
With iron(III) chloride; tetrabutyl-ammonium chloride; In water; dimethyl sulfoxide; at 140℃; for 20h;Inert atmosphere;
General procedure: The sulfinate salt 1 (2 mmol, 1.0 equiv) and halopyridine 2 (1.2 equiv)in DMSO-H2O (3:1, 2 M) were added to a round-bottom flask containing nBu4NCl (0.3 equiv) and FeCl3 (0.01 equiv). The mixture was degassed for 30 min in a sonicator under argon atmosphere, and then heated to 140 C and stirred for 20 h. The resulting mixture was cooled to r.t. and neutralized with 2 M aq NaOH solution, and then extracted with EtOAc (3×). The combined organic phase was washed with distilled H2O, followed by a brine solution, and then was dried over Na2SO4 and filtered. The solvent was then removed under reduced pressure. The residue was purified by column chromatography(silica gel, EtOAc-hexanes-CH2Cl2, 5:45:55) to give the desired sulfonylated pyridine product 3.
With palladium diacetate; caesium carbonate; tris-(o-tolyl)phosphine; In toluene; at 110℃; for 18h;
[1397] Cs2CO3 (4.6 g, 14.2 mmol) was added to a mixture of compound 314b (1.0 g, 7.1 mmol) and 2-iodopyridine (2.9 g, 14.2 mmol) in toluene (20.00 ml). Then p(o-tolyl)3 (216 mg, 0.71 mmol) and pd(oac)2 (80 mg, 0.35 mmol) was added. The mixture was de-gassed for 3 times. Then the mixture was heated to 110 C and stirred for 18h. The mixture was filtered through celite; the cake was washed with ea (15 ml x 2). The combined filtrates were concentrated. The residue was purified by flash column chromatography (pe/ea = 10/1 to 1/1) to afford compound 314c (1.0 g, yield 64.6%) as pale yellow solid. 1H NMR (DMSO-d6, 400 mhz) delta 8.69 - 8.67 (m, 1h), 8.08 - 8.06 (m, 1h), 7.96 -7.91 (m, 1h), 7.48 - 7.45 (m, 1h), 3.77 (s, 3h), 2.50 (s, 3h).
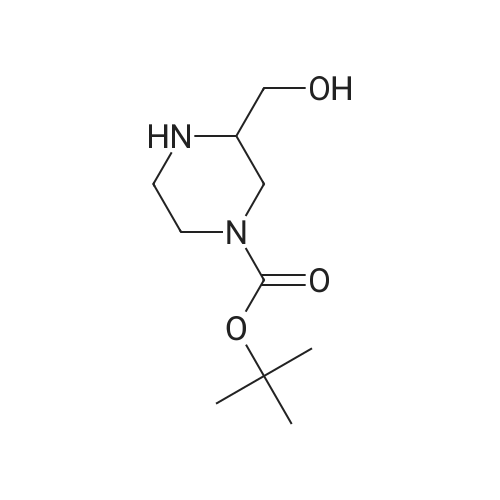
tert-butyl 3-(hydroxymethyl)-4-(pyridin-2-yl)piperazine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium phosphate; copper(l) iodide; In isopropyl alcohol; at 90℃;
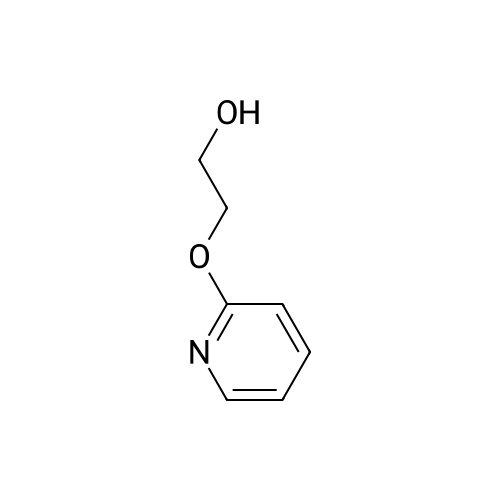
To a pressure tube; <strong>[301673-16-5]tert-butyl 3-(hydroxymethyl)piperazine-1-carboxylate</strong> (681 mg, 3.15 mmol), 2-iodopyridine (615 mg, 3 mmol), ethylene glycol (372 mg, 6 mmol) and potassium phosphate (1.28 g, 6 mmol) in isopropanol (6 ml) purged with nitrogen 5 mins were added then CuI (60 mg, 0.3 mmol) was added. The reaction was stirred and heated to 90° C. overnight. After cooling to ambient temperature MBTE (4 ml) and water (2 ml) were then added to the reaction mixture. The organic layer was extracted with MBTE (2×4 ml). The combined organic phases were washed with brine and dried over Na2SO4. The solvent was concentrated in vacuo giving 918 mg of a green oil. Purified by flash column chromatography on silica gel using gradient 0percent to 100percent MBTE in DCM. The product containing fractions were combined and reduced in vacuo to yield the title compound as a colorless gum (143 mg, 9percent). (0477) Found by 1H NMR to be a 3:5 mixture of the title product and 2-(pyridine-2-yloxy)ethan-1-ol. Used the crude product for the next stage. 1H NMR (500 MHz, Chloroform-d) delta 8.20-8.10 (m, 1.6H), 7.68-7.58 (m, 1H), 7.57-7.47 (m, 0.6H), 6.97-6.89 (m, 1H), 6.87-6.80 (m, 1H), 6.72-6.61 (m, 1.2H), 4.72-4.35 (m, 2.6H), 4.32-3.62 (m, 7H), 3.24 (d, J=7.7 Hz, 2H), 1.51 (s, 5.4H). LCMS Method 2?Tr=0.98 min, (ES+) (M+H+) 294.
ethyl 4-((pyridin-2-ylsulfonyl)methyl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
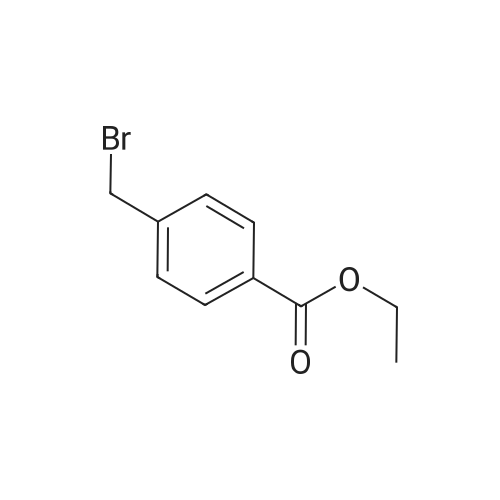
Add in the reaction tube2-iodopyridine (0.5 mmol, 1.0 equivalent)And sodium dithionite (0.55mmol, 1.1 equivalent),Replace the air in the test tube with high purity nitrogen.Add 3 mL of N,N-dimethylformamide as solvent.The reaction was stirred for 16 hours while heating to 90 C.After the reaction was cooled, tetrabutylammonium iodide (0.05 mmol, 0.1 equivalent) and potassium iodide (0.6 mmol, 1.2 equivalent) were added to the reaction mixture.And 4-carboxyethyl benzyl bromide (1.0 mmol, 2.0 eq.),Replace the air in the test tube with high purity nitrogen.Stir at room temperature for 10 hours.The reaction was quenched with saturated brine.Extracted with ethyl acetate,Combine the organic phase,dry,Concentrate and separate by column chromatography.The target product Ie (purity: 98%) was obtained in 85% yield.
81%
(1) Add 0.5 mmol of 2-iodopyridine to the test tube,1.0 mmol of thiourea dioxide,2.0mmol sodium hydroxide,0.05 mmol of fluorescein,Replace the air in the test tube with high purity nitrogen.3 mL of dimethyl sulfoxide was added, and the reaction was stirred for 16 hours under irradiation with a 25 W fluorescent lamp; (2) After cooling the mixed solution obtained in the step (1), 0.05 mmol of tetrabutylammonium iodide is added to the reaction solution.0.6mmol potassium iodide and 1.0mmol 4-ethyl benzyl bromide,Then, the air in the test tube was replaced with high-purity nitrogen, and stirred at room temperature for 10 hours; (3) quenching the reaction of the step (2) with saturated brine, extracting the obtained reaction solution with ethyl acetate, and extracting the organic phase obtained by drying, concentration, column chromatography and solvent removal to obtain a mixture. Cyclic sulfoneThe developing solvent for the column chromatography separation was a mixture obtained by mixing ethyl acetate and petroleum ether in a volume ratio of 1:4.The yield of this example was calculated to be 81%.The product was found to have a purity of 98% by HPLC.
Add in the reaction tube2-iodopyridine (0.5 mmol, 1.0 equivalent)And sodium dithionite (0.55mmol, 1.1 equivalent),Replace the air in the test tube with high purity nitrogen.Add 3 mL of N,N-dimethylformamide as solvent.The reaction was stirred for 16 hours while heating to 90 C.After the reaction is cooled, it is added to the reaction liquid.Tetrafluoroborate diphenyl iodide(0.75mmol, 1.5 equivalents),Replace the air in the test tube with high purity nitrogen.Stir at 90 C for 10 hours.The reaction was quenched with saturated brine.Extracted with ethyl acetate,Combine the organic phase,dry,Concentrate and separate by column chromatography.The target product IIa (purity: 98%) was obtained in a yield of 65%.
63%
(1) Add 0.5 mmol of 2-iodopyridine to the test tube,1.0 mmol of thiourea dioxide,2.0mmol sodium hydroxide,0.05mmol of fluorescein, the air in the test tube was replaced with high-purity nitrogen, and 3mL of dimethyl sulfoxide was added.The reaction was stirred for 16 hours under irradiation of a 25 W fluorescent lamp;(2) After cooling the mixed solution obtained in the step (1), 0.75 mmol of diphenyliodanium tetrafluoroborate is added to the reaction solution.Then replace the air in the test tube with high purity nitrogen.Stir at room temperature for 10 hours;(3) quenching the reaction of the step (2) with saturated brine,The obtained reaction liquid is extracted with ethyl acetate, and the organic phase extracted is sequentially dried, concentrated,Column chromatography separation and solvent removal to obtain a heterocyclic sulfone;The developing solvent for the column chromatography separation was a mixture obtained by mixing ethyl acetate and petroleum ether in a volume ratio of 1:4.The yield of this example was calculated to be 63%.The product was found to have a purity of 98% by HPLC.
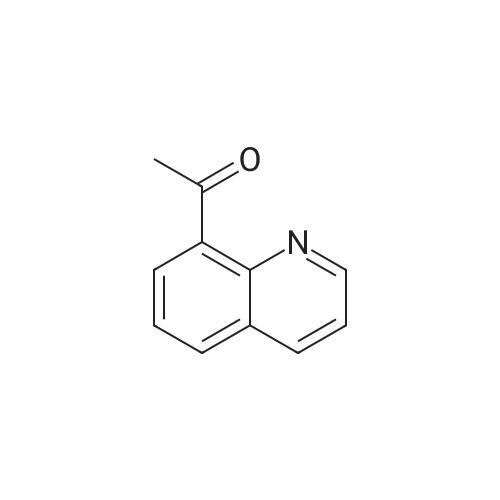
N-methyl-2-phenyl-2-trifluoromethylbenzimidazoline[ No CAS ]
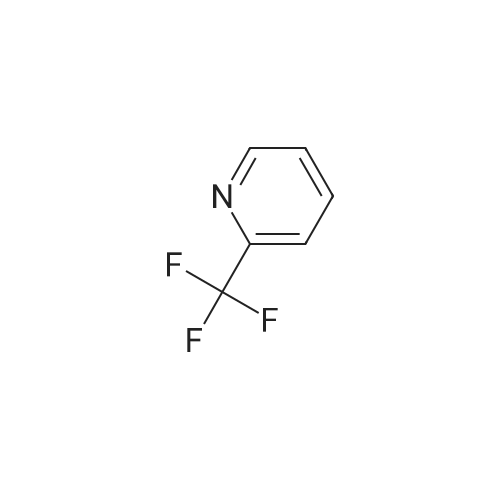
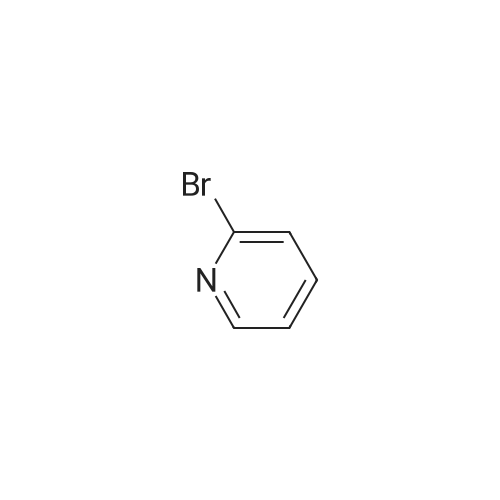
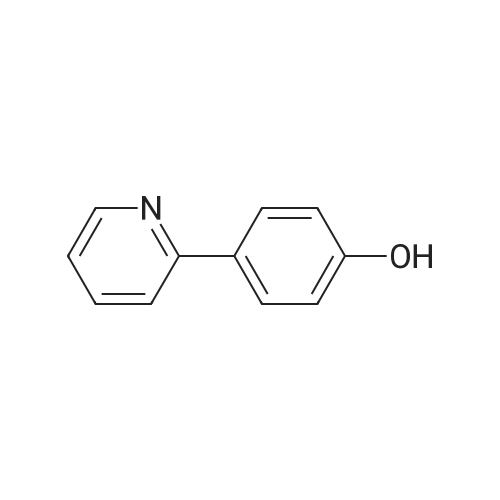
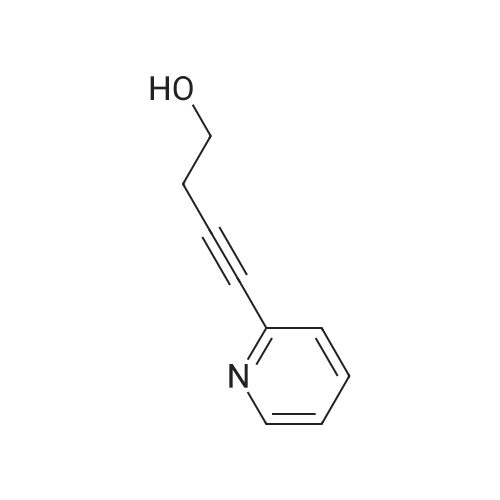
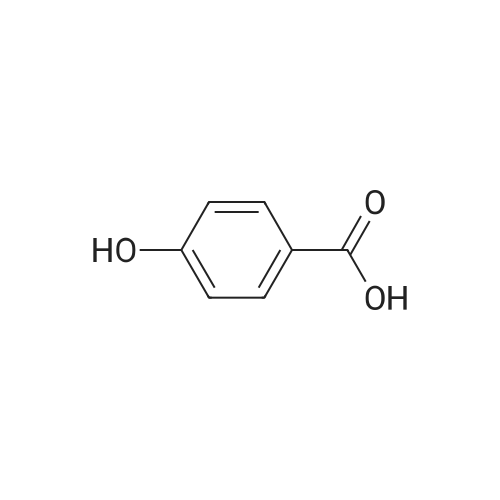
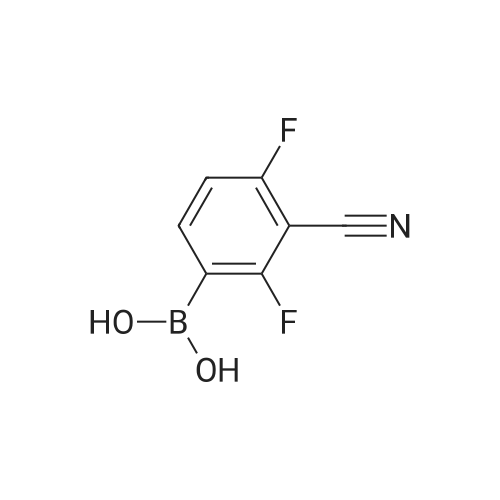
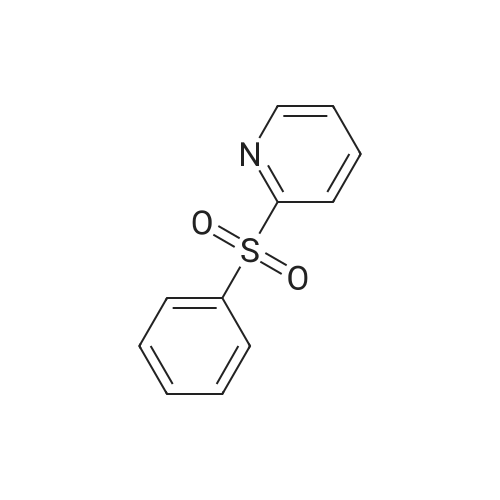
[ 368-48-9 ]
Yield
Reaction Conditions
Operation in experiment
57%
With potassium carbonate; copper(l) chloride; at 60℃; for 48h;Inert atmosphere;
General procedure: Under a nitrogen atmosphere, a mixture of iodoarene 1a (0.10 mmol), 5a (0.20 mmol, 2.0 equiv), copper(I) chloride (0.30 mmol, 3.0 equiv), and potassium carbonate (0.40 mmol, 4.0 equiv) in propionitrile (1.0 mL) was stirred at 60 C for 2 d, and the reaction was monitored by TLC. After completion of the reaction, supernatant of the reaction mixture was purified by preparative TLC to give trifluorotoluene 2a (88%).
With sodium sulfite; In water; for 0.333333h;Sonication; Green chemistry;
In a 50 mL round bottom flask, 2.05 g of 2-iodopyridine, 1.94 g of benzenesulfonyl chloride, 1.51 g of sodium sulfite, 15 ml of water, and a 30 W/100 KHz ultrasonic reaction apparatus were ultrasonically reacted for 20 minutes. The crude product of 2-phenylsulfonylpyridine was filtered, and the crude product was washed with 95% ethanol to give 2.14 g of 2-benzenesulfonylpyridine as a pure product, yield 98%.
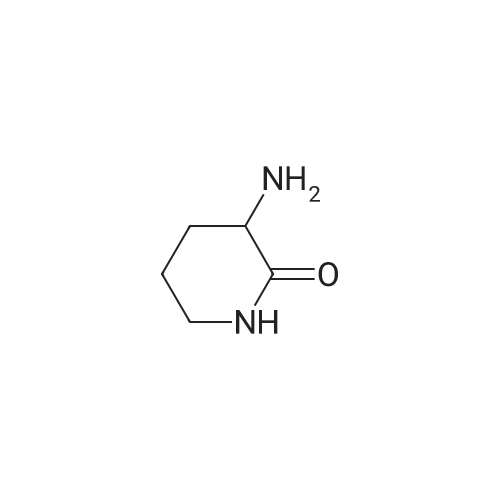
With palladium diacetate; triethylamine; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In N,N-dimethyl-formamide; at 50℃; under 750.075 Torr; for 2h;
General procedure: In a typical experiment Pd(OAc)2 (2.81 mg, 0.0125 mmol), triphenylphosphine (6.55 mg, 0.025 mmol) or Xantphos (7.23 mg, 0.0125 mmol), iodoalkene (1-5) or iodo(hetero)arene (6-11) substrates (0.5 mmol), and 3-aminolactams (3-amino-azepan-2-one (a), <strong>[1892-22-4]3-amino-piperidin-2-one</strong> (b), 3-amino-pyrrolidin-2-one (c)) (0.55 mmol) and triethylamine (0.25 mL) were dissolved in DMF (5 mL) under argon in a 100 mL three-necked flask equipped with reflux condenser connected to a balloon filled with argon. The atmosphere was changed to carbon monoxide. The reaction was conducted for the given reaction time upon stirring at 50 C and analyzed by Gc and GC-MS. The cooled reaction mixture was then distilled to dryness under reduced pressure. The residue was dissolved in chloroform (15 mL) and washed twice with water (15 mL). The organic phase was dried over Na2SO4, filtered and evaporated under reduced pressure to a solid material. All compounds (except 10a, 10b) were subjected to column chromatography (Silicagel 60 (Sigma), 0.063-0.200 mm) or Aluminum oxide (Sigma), activated, neutral, Brockmann activity I), CHCl3/MeOH or CHCl3/EtOH eluent mixtures (the exact ratios are specified in Characterization (3.4) for each compound). In the case of 10a and 10b chloroform (10 mL) was added to the residue and the insoluble material (product) was filtered and dried.
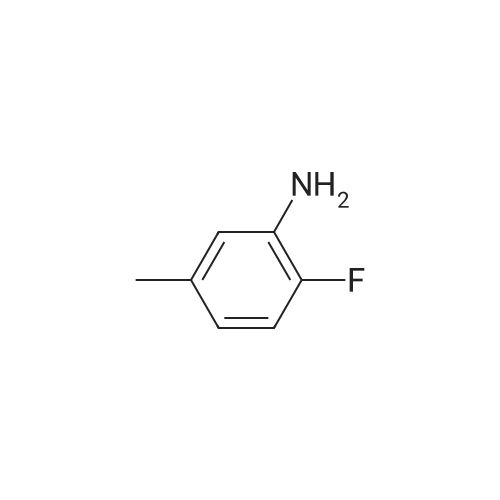
N-(2-fluoro-5-methylphenyl)pyridin-2-amine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
With potassium hexamethylsilazane; at 100℃; for 16h;Inert atmosphere;
General procedure: Under the N2 atmosphere,2x (0.5 mmol) was added to the reaction tube in turn.3b (0.75mmol) and dissolved in 1.0M in advanceThe mixture obtained from KHMDS (0.75 mmol) of THF was heated to 100 C, and the reaction was stirred for about 16 hours until the conversion of the starting material was completed, and the temperature was lowered to room temperature.Diluted with THF (3 ml) to the reaction mixture.Filter through silica gel or diatomaceous earth, wash with THF,The crude product was concentrated in vacuo and subjected to silica gel column chromatography to give the corresponding product 1xb.As shown in the following equation, where, lists the yield of 1xb isolated using different 2x as raw materials.For example, when using 2f?,The yield of the product 1f'b was 88%.
With palladium diacetate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; sodium t-butanolate; In toluene; at 130℃; for 17h;Inert atmosphere;
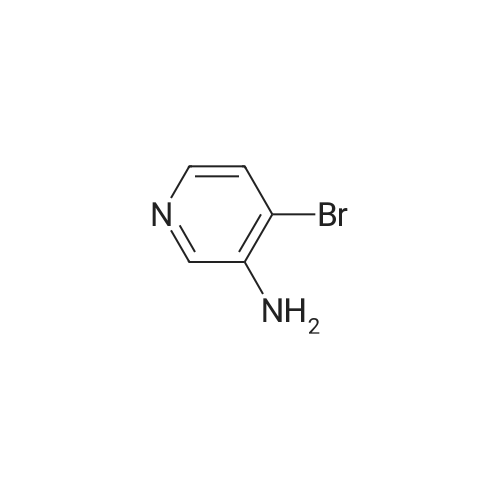
A mixture of <strong>[239137-39-4]4-bromopyridin-3-amine</strong> (1 g, 5.78 mmol), 2-iodopyridine (1.4 g, 6.94 mmol), palladium acetate (0.13 g, 0.58 mmol), xantphos (0.34 g, 0.58 mmol) and sodium tert-butoxide (0.83 g, 8.67 mmol) in toluene (20 mL) was degassed with nitrogen, heated to 130 C. and stirred for 17 hours under nitrogen atmosphere. The reaction was cooled to r.t and filtered. The filtrate was concentrated in vacuum. The residue was purified by column chromatography (Petroleum ether/EtOAc=2:1) to give N-(4-bromopyridin-3-yl)pyridin-2-amine (1.0 g, 69.4% yield) as a yellow solid. LC/MS (ESI, m/z): [M+1]+=251.1.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping