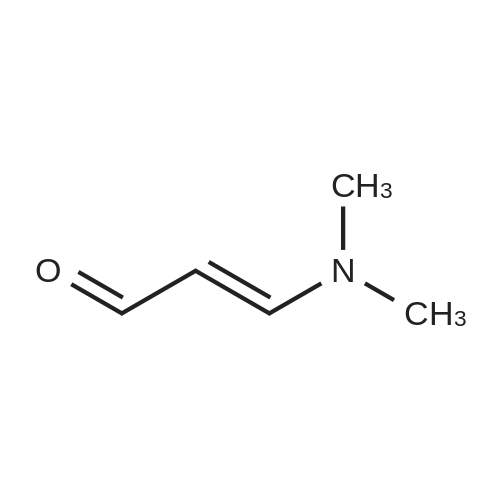
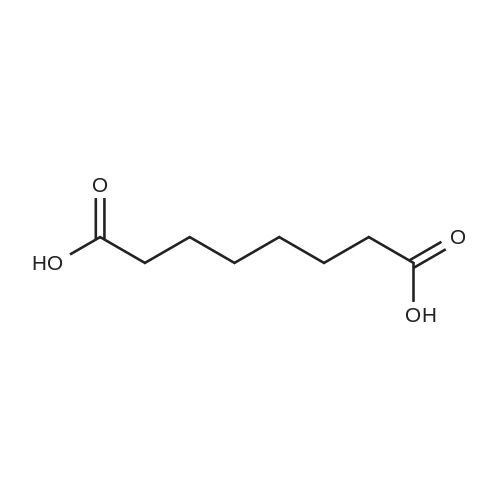
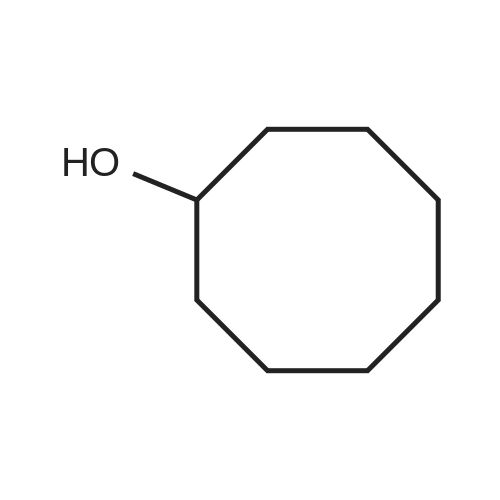
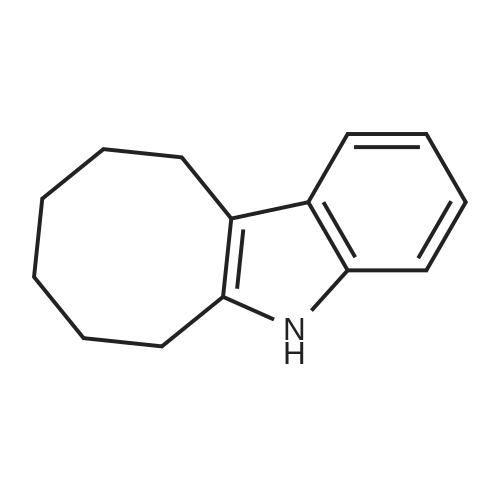
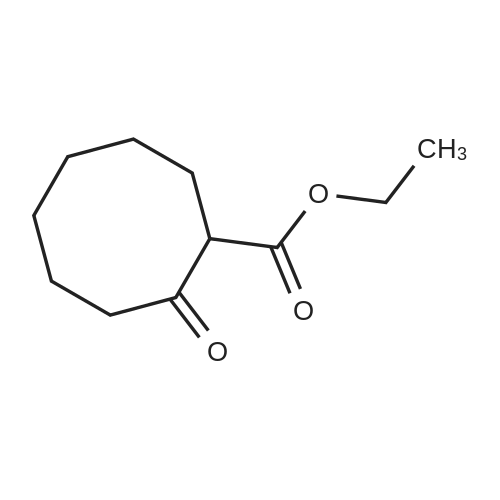
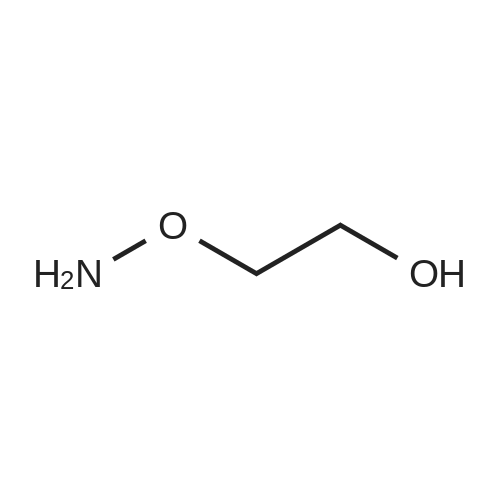
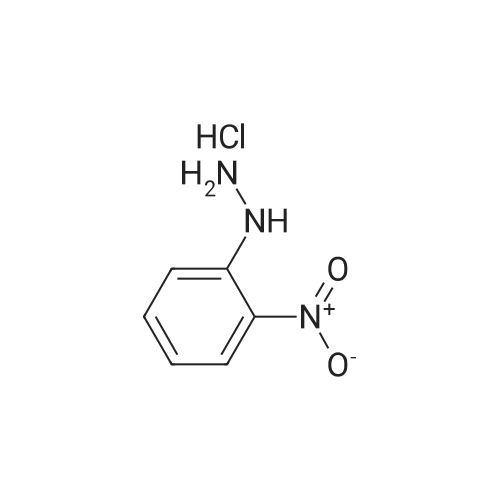
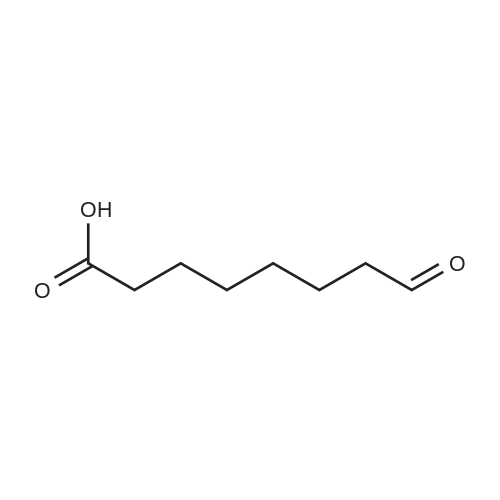
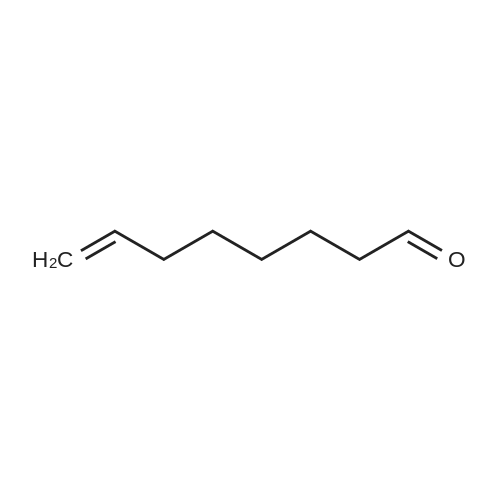
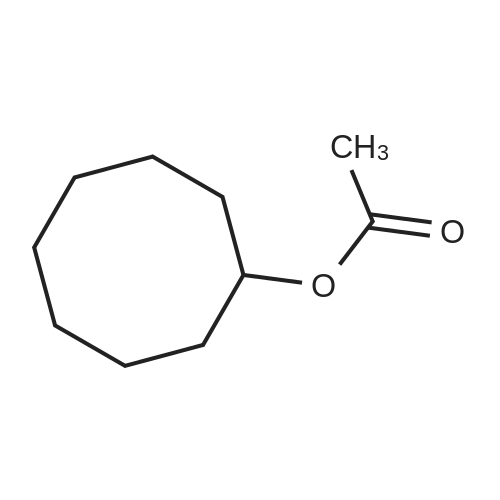
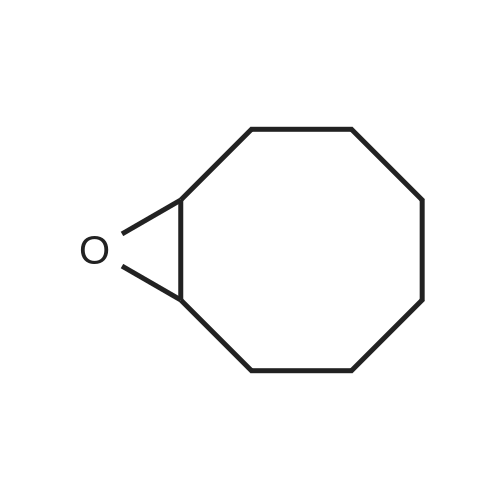
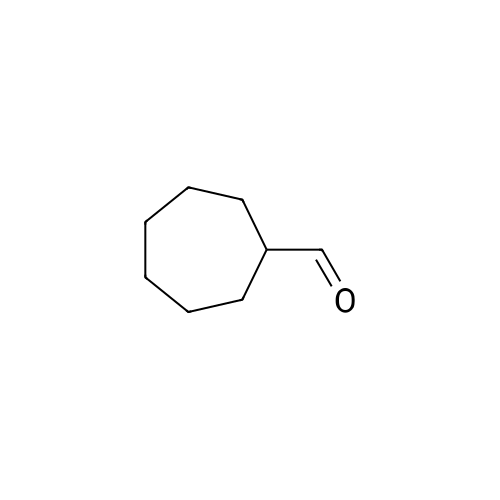
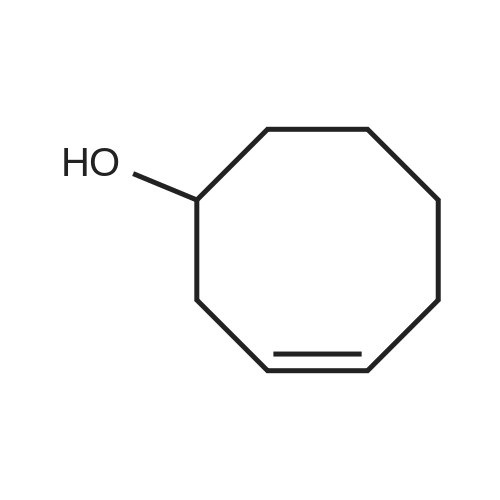
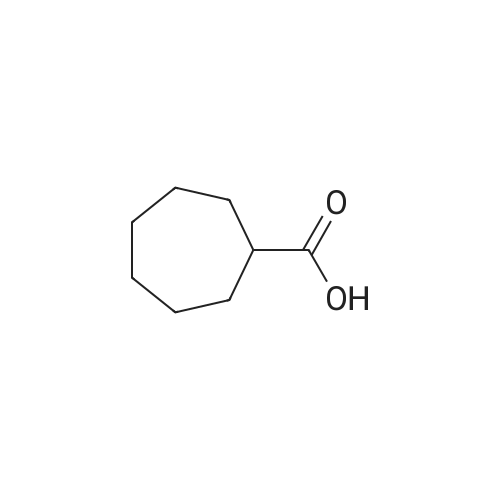
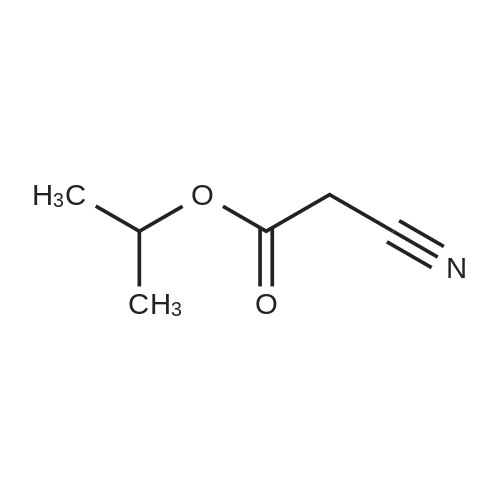
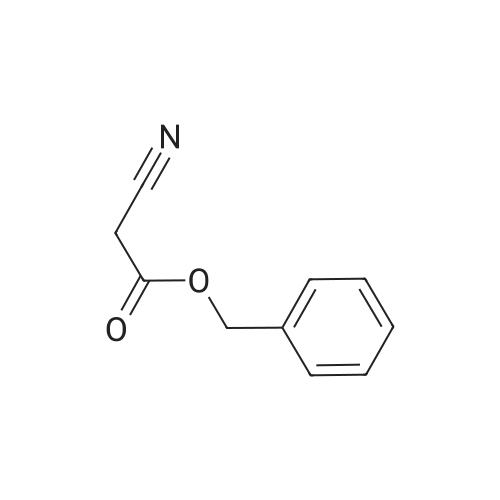
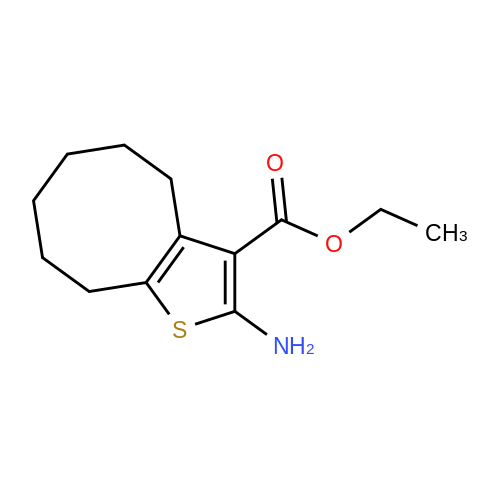
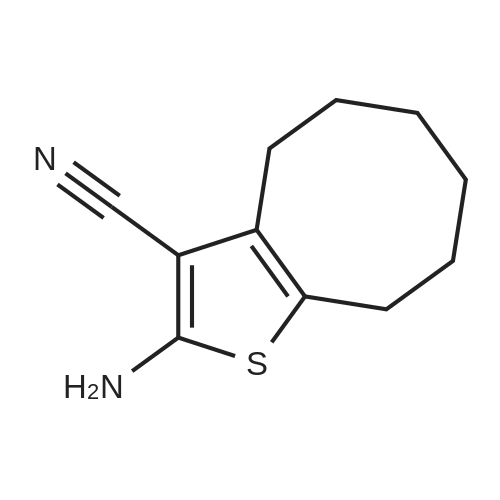
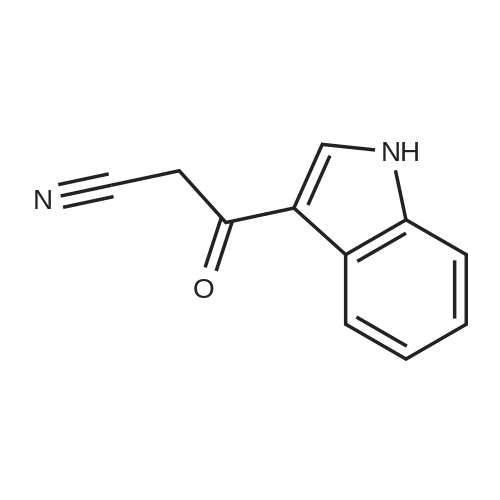
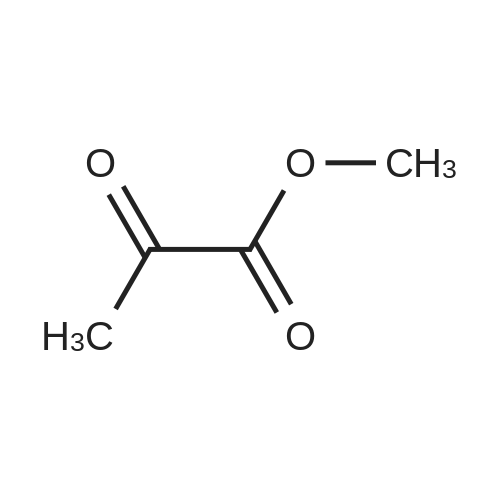
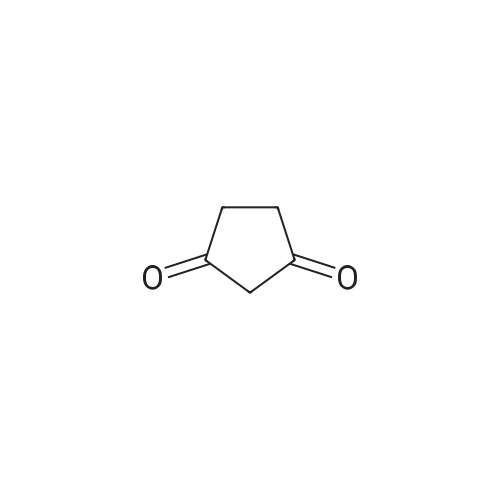
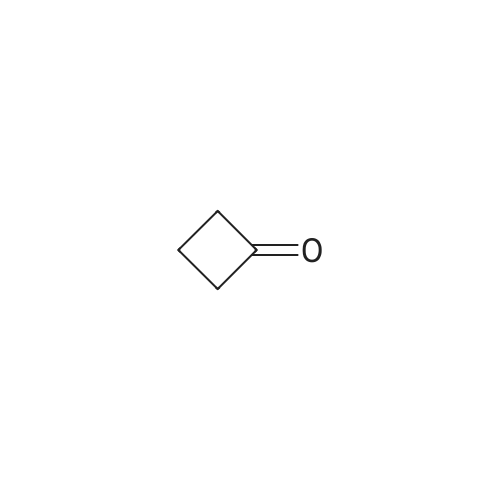
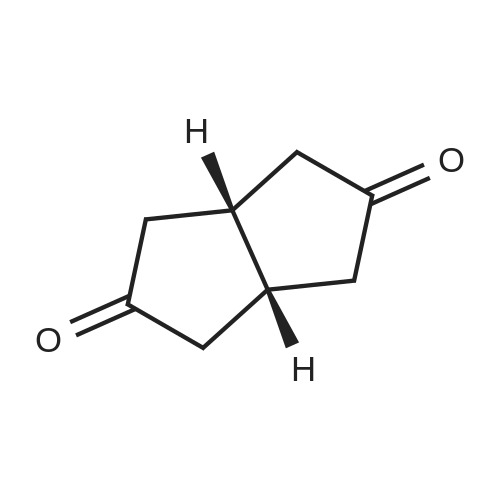
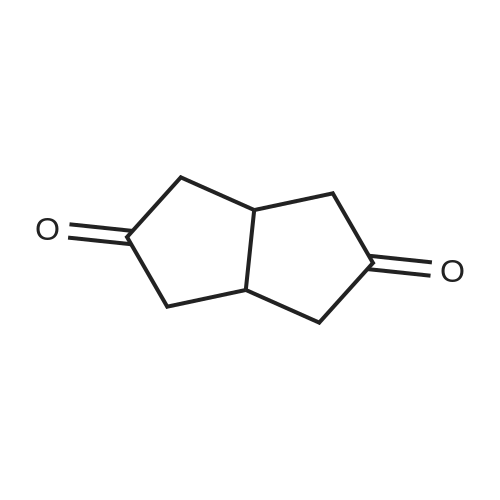
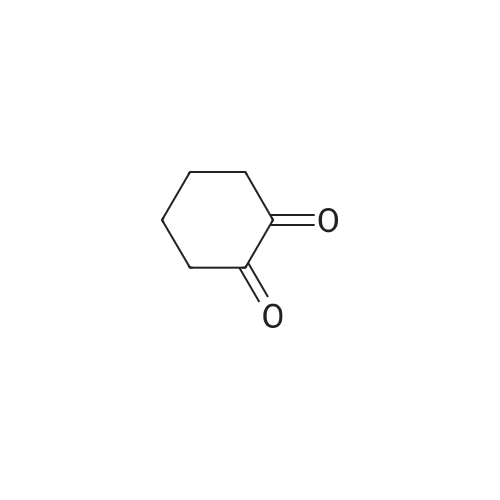
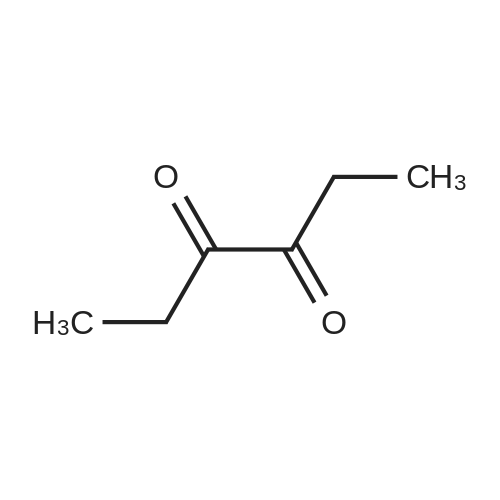
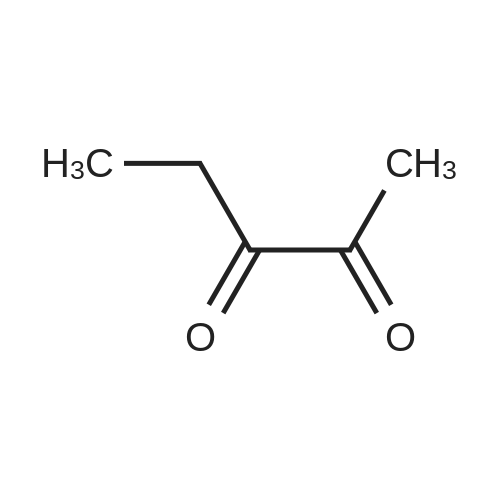
| 1: 71%
2: 10.8% |
With (5,10,15,20-tetrakis(pentafluorophenyl)porphyrinato)iron(III) chloride; oxygen In benzene at 120℃; for 6h; |
2.3. Catalytic experiments
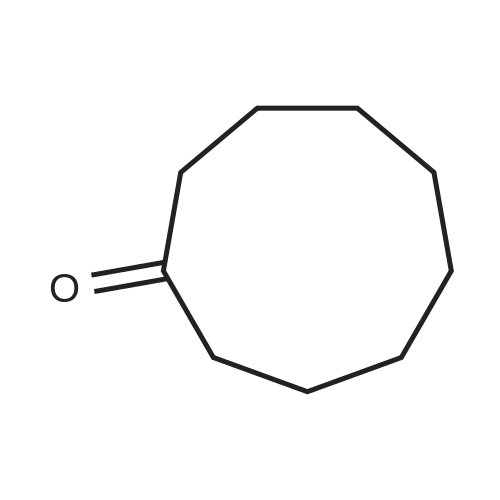
General procedure: Oxidation of cycloalkanes by molecular oxygen was carried outin the stainless steel batch reactor at 393 K and under the air pressureof 10 atm. Cycloalkane to oxygen molar ratio was set at 6.5.The amount of iron μ-oxo or monomeric iron porphyrin givingthe final concentration 3.3 x 10-5 M of the catalyst in the reactionmixture was dissolved in 1 ml of benzene and added to 60 ml ofcycloalkane. Reactor filled with substrate and the catalyst waspre-treated under the argon flow to remove air and to provide aninert atmosphere. Then the rector was heated to 393 K and airwas introduced. After 6 h of reaction time the oxidation wasstopped by immersing the hot reactor in a cold water bath. Yieldsof products were calculated based on the oxygen quantity in thebatch reactor for all catalytic tests. Reaction products were analyzedusing Agilent Technologies 6890N chromatograph equippedwith Innovax chromatography column (30 m). The yield valueswere verified by an addition of internal standard, chlorobenzene,at the end of the reaction. Cycloalcohol and cycloketone were theonly oxygen-containing products, together with traces of cycloalkanehydroperoxide. The blank experiment confirmed that thecycloalkane was not oxidized by O2 in the absence of catalyst. |
| 1: 68.8%
2: 14.2% |
With [5,10,15,20-tetrakis(2,6-dichlorophenyl)porphinato]cobalt(II); oxygen at 120℃; for 6h; |
|
| 1: 66%
2: 34% |
With [PPh4]2[MnV(N)(CN)4]; tetrabutylammonium periodite; acetic acid In 2,2,2-trifluoroethanol at 23℃; Inert atmosphere; |
|
| 1: 63%
2: 34% |
With [PPh4]2[MnV(N)(CN)4]; dihydrogen peroxide; acetic acid In 2,2,2-trifluoroethanol at 23℃; for 5h; Inert atmosphere; |
|
| 1: 57%
2: 15% |
With ammonium acetate; chloro(meso-tetrakis(2,6-dichlorophenyl)porphyrinato)manganese(III); dihydrogen peroxide In dichloromethane; acetonitrile for 2h; Ambient temperature; |
|
| 1: 53%
2: 23% |
With dihydrogen peroxide; oxalic acid In water; acetonitrile at 60℃; for 8h; Green chemistry; regioselective reaction; |
|
| 1: 52.3%
2: 6.8% |
With C48H12F68MnN2O2; oxygen at 119.84℃; for 6h; Autoclave; Green chemistry; |
2.3 General procedure for the oxidation of cycloalkanes
General procedure: The oxidation of cyclooctane (cyclopentane or cyclohexane) was performed in a stainless steel batch reactor system at 393 K and under the air pressure of 10 atm, with the substrate to oxygen molar ratio set at 6.5. In a typical experiment, 60 mL of substrate containing the amount of catalyst providing a concentration of 3.3×10-4 M was introduced in the deaerated autoclave and the whole system was heated until a temperature of 393 K was reached. Air was then introduced and after 6 h the oxidation products were analyzed by GC Agilent 6890 N equipped with an Innowax (30 m) column. The yield values were verified by addition of an internal standard, chlorobenzene, at the end of the reaction. |
| 1: 51%
2: 6% |
With tert.-butylhydroperoxide; manganese(II,III) oxide In acetonitrile at 70℃; for 3h; chemoselective reaction; |
|
| 1: 47%
2: 30% |
With tert.-butylhydroperoxide In water; acetonitrile at 50℃; for 5h; |
General procedure for the catalytic oxidation reaction
General procedure: In a typical experiment, a mixture of 2 mmol of tert-butyl hydroperoxide (TBHP, 70% aqueous solution) oxidizing agent and 1 mmol of alkene or alkane and 0.028 mmol (100 mg) of γ-Fe2O3[VO(salenac-OH)] in 5 mL of CH3CN was preparedin a test tube. A magnetic hotplate stirrer was used to stirring the reaction mixture at 50 °C, and the reaction progress was supervised using thin layer chromatography (TLC) or gas chromatography (GC). Having the reaction completed, 20 mL of CH2Cl2 was added to the reaction mixture to dilute it and an external magnet was used to remove the catalyst. Using CH2Cl2, the catalyst was completely washed and the combined washings were passed through a silica gel column to purify the product. |
| 1: 46%
2: 29% |
With 1H-imidazole; sodium periodate; Mn(TDCPP)OAc In dichloromethane for 24h; Ambient temperature; |
|
| 1: 43%
2: 9% |
With 1H-imidazole; dihydrogen peroxide In dichloromethane; acetonitrile for 2h; Ambient temperature; |
|
| 1: 3%
2: 43% |
With 1H-imidazole; dihydrogen peroxide In dichloromethane; acetonitrile for 2h; Ambient temperature; |
|
| 1: 9%
2: 43% |
With 1H-imidazole; dihydrogen peroxide In dichloromethane; acetonitrile at 20℃; |
|
| 1: 42%
2: 31% |
With dihydrogen peroxide In water; acetonitrile at 60℃; for 3h; |
|
| 1: 35%
2: 21 % Chromat. |
With 1H-imidazole; sodium periodate; MnTPPS-Ad In water; acetonitrile for 8h; Ambient temperature; |
|
| 1: 25.8%
2: 34.6% |
With chloro[N,N'-bis(salicylaldehyde)cyclohexanodiminate]iron(III); dihydrogen peroxide; nitric acid In acetonitrile at 20℃; for 3h; |
2.4. Catalytic experiments
General procedure: The homogeneous phase catalytic studies were performed using 5.00 mmol of cyclohexane, 0.05 mmol of homogeneous catalyst(1 mol% relative to cyclohexane), 0.5 mmol of nitric acid and25 mmol of hydrogen peroxide as sustainable oxidant in 20.00 mlof acetonitrile in batch reactors at room temperature, atmospheric pressure and under constant stirring (1300 rpm) (Scheme 2).Therefore, the ratio of cyclohexane/catalyst/HNO3/H2O2 was,respectively, 100/1/10/500 [18]. The reactions were performed at least twice |
| 1: 33%
2: 12% |
With 1H-imidazole; dihydrogen peroxide In dichloromethane; acetonitrile at 20℃; |
|
| 1: 31%
2: 19% |
With C22H16N4O8V2; dihydrogen peroxide In acetonitrile at 60℃; for 4h; |
General oxidation procedure
General procedure: The liquid phase catalytic oxidations were carried out under air(atmospheric pressure) in a 25 mL round bottom flask equippedwith a magnetic stirrer and immersed in a thermostated oil bathat 80 or 60 C. In a typical experiment, a mixture of 30% H2O2(3 mmol), solvent (3 mL), NaHCO3 (0.25 mmol), chlorobenzene(0.1 g) as internal standard and cis-cyclooctene (1 mmol) wasadded to a flask containing the catalyst 1-8 (1.70 lmol). Thecourse of the reaction was monitored using a gas chromatographequipped with a capillary column and a flame ionization detector.The oxidation products were identified by comparing their retentiontimes with those of authentic samples or alternatively by 1HNMR and GC-Mass analyses. Yields based on the added substratewere determined by a calibration curve. Control reactions werecarried out in the absence of catalyst, H2O2 and NaHCO3 underthe same conditions as the catalytic runs verifying that no products,or only trace yields, were detected. |
| 1: 26%
2: 8.1% |
With perchloric acid; C13H30N4*Fe(3+)*CF3O3S(1-)*C2H2F3O(1-)*C6H5IO In 2,2,2-trifluoroethanol; acetone at -40℃; for 0.166667h; Inert atmosphere; Schlenk technique; Further stages; |
|
| 1: 3.7%
2: 21.2% |
With tert.-butylhydroperoxide; 4 A molecular sieve In benzene at 60℃; for 48h; |
|
| 1: 21%
2: 35 % Chromat. |
With 1H-imidazole; sodium periodate; MnTPPS-Ad In water; acetonitrile for 8h; Ambient temperature; |
|
| 1: 13.6%
2: 11.4% |
With dihydrogen peroxide; C52H45Cu6Ge8O24*6C3H7NO; trifluoroacetic acid In water; acetonitrile at 50℃; for 3h; Overall yield = 25 percent; |
|
| 1: 9%
2: 12% |
Stage #1: Cyclooctan In water-d2 at 20℃; for 0.166667h;
Stage #2: In water-d2 at 20℃; for 3h; UV-irradiation; |
|
| 1: 3%
2: 11% |
With BaFeO(2.8-x); oxygen at 89.84℃; for 72h; |
|
| 1: 11.4%
2: 5.5% |
With dihydrogen peroxide; C16H36Cu6O24Si8*5C3H7NO*0.5H2O; trifluoroacetic acid In water; acetonitrile at 50℃; for 3h; Overall yield = 16.9 percent; |
|
| 1: 7.9%
2: 5.4% |
With C11H14FeN3O11S*3H2O; dihydrogen peroxide; nitric acid In water; acetonitrile at 25℃; for 6h; Overall yield = 13.3 %; |
|
| 1: 29 % Chromat.
2: 15 % Chromat. |
With cetyldimethylbenzylammonium chloride In dichloromethane at 20℃; for 0.05h; |
|
|
With lithium perchlorate; acetic acid In water; acetonitrile electrolysis; Yield given. Yields of byproduct given; |
|
|
With pyridine; tert.-butylhydroperoxide; oxygen; acetic acid at 60℃; |
|
| 1: 1 % Chromat.
2: 39 % Chromat. |
With tert.-butylhydroperoxide In benzene for 2h; Ambient temperature; |
|
|
With pyridine; oxygen; trifluoroacetic acid at 20 - 30℃; electrolyse: i = 14 mA/cm2, anode: platinum, cathode: mercury; Yield given. Yields of byproduct given; |
|
|
With 2-Picolinic acid; tert.-butylhydroperoxide; ferric nitrate In pyridine; acetic acid for 10h; Ambient temperature; Yield given. Yields of byproduct given; |
|
|
With iodosylbenzene In dichloromethane; acetonitrile at 20℃; for 2h; Yield given. Yields of byproduct given; |
|
|
With 1-methyl-1H-imidazole; (5,10,15,20-tetraphenylporphyrinato)manganese(III) chloride; oxygen; acetic acid; zinc In dichloromethane; acetonitrile for 0.5h; Yield given; |
|
|
With peracetic acid In ethyl acetate for 2h; Ambient temperature; Yield given. Yields of byproduct given; |
|
|
With ferric picolinate complexes; dihydrogen peroxide In pyridine; water; acetic acid at 0℃; for 18h; Yield given. Yields of byproduct given; |
|
| 1: 33 % Turnov.
2: 67 % Turnov. |
With oxygen; isobutyraldehyde In acetonitrile Ambient temperature; |
|
|
With 18-crown-6 ether; oxygen; acetaldehyde; copper dichloride 1.) CH2Cl2, 20 min, 2.) CH2Cl2, 1 atm, 24 h, 70 deg C; Yield given. Multistep reaction. Yields of byproduct given; |
|
| 1: 10 % Chromat.
2: 12 % Chromat. |
With sodium periodate In dichloromethane; water at 80℃; for 15h; Title compound not separated from byproducts; |
|
| 1: 33 % Turnov.
2: 67 % Turnov. |
With <bis(salicylidene-N-methyl 3-hydroxypropionate)>cobalt; oxygen; isobutyraldehyde In acetonitrile Ambient temperature; |
|
|
With tert.-butylhydroperoxide; di-tert-butyl peroxide; oxygen In various solvent(s) at 20℃; for 24h; Yield given; |
|
| 1: 12 % Chromat.
2: 31 % Chromat. |
With sodium periodate In dichloromethane; water at 80℃; for 15h; Title compound not separated from byproducts; |
|
| 1: 53 % Chromat.
2: 6 % Chromat. |
With iodosylbenzene; chloro<meso-tetra(4-N-methylpyridinio)porphyrinato>manganese(III) tetrachloride (Mn(tmpyp)Cl) on montmorillonite (ClaypMn) In dichloromethane; acetonitrile at 20℃; for 1h; other cyclo- and linear alkanes; various reagents; |
|
|
With 4-tert-butylpyridine; Mn(tclpp)Cl; dihydrogen peroxide; benzoic acid In dichloromethane at 0℃; for 0.333333h; pH 4.5 - 5.0; other alkanes; various Mn(III)-porphyrins and times; |
|
|
With 5,10,15,20-tetra(2',6'-dichlorophenyl)porphyrinatoiron(III) chloride; oxygen at 22℃; Irradiation; other iron-porphyrin complexes; |
|
| 1: 0.95 mmol
2: 0.27 mmol |
With tert.-butylhydroperoxide; oxygen In acetonitrile at 60℃; for 48h; other hydrocarbons, other solvent, time, temperature; presence of 18O2; |
|
|
With ferric picolinate complexes; dihydrogen peroxide In pyridine at 0℃; for 18h; functionalization with other catalyst; |
|
| 1: 20 % Chromat.
2: 36 % Chromat. |
With 1H-imidazole; 4-vinylpyridine; sodium periodate; manganese(III) 5,10,15,20-tetra(sulfonato)porphyrin In acetonitrile for 8h; Ambient temperature; other alkanes; |
|
|
With air; tris(μ-oxo)di[(1,4,7-trimethyl-1,4,7-triazanonane)manganese(IV)] hexafluorophosphate; cyclohexanecarbaldehyde In acetonitrile at 60℃; for 8h; reaction course with time, other temperature and reaction time; |
|
|
With cis-Cyclooctene; 3-chloro-benzenecarboperoxoic acid In dichloromethane at -15℃; for 0.0833333h; |
|
|
With dihydrogen peroxide In acetonitrile at 25℃; for 1.5h; |
|
| 1: 8 % Chromat.
2: 77 % Chromat. |
With tert.-butylhydroperoxide; cis-<Ru(6,6-Cl2bpy)2(OH2)2>(CF3SO3)2 In acetone at 20℃; for 24h; |
|
|
With peracetic acid In ethyl acetate at 20℃; for 0.25h; Title compound not separated from byproducts; |
|
| 1: 28 % Chromat.
2: 17 % Chromat. |
With 1H-imidazole; sodium periodate; Mn(III)meso-(p-sulfonato-Ph)4-β-Br8-porphyrin*Amberl.IRA400 In water; acetonitrile at 20℃; for 10h; |
|
| 1: 9 % Chromat.
2: 3 % Chromat. |
With 1H-imidazole; air; tetra-n-butylammonium hydrogen monopersulfate In dichloromethane at 20℃; for 0.05h; |
|
|
With tert.-butylhydroperoxide; acetic acid In acetonitrile at 30℃; |
|
| 1: 33 % Chromat.
2: 17 % Chromat. |
With 1H-imidazole; sodium periodate; Mn(III) tetrakis(4-aminophenyl)porphyrin on polystyrene In water; acetonitrile at 20℃; for 8h; |
|
| 1: 23 % Chromat.
2: 19 % Chromat. |
With 1H-imidazole; sodium periodate; Mn(III)TPPS-PSMP In acetonitrile at 20℃; for 8h; |
|
|
With chromium(VI) oxide; dihydrogen peroxide In acetonitrile at 60℃; for 0.833333h; |
|
| 1: 51 % Chromat.
2: 5 % Chromat. |
With 1H-imidazole; [bis(acetoxy)iodo]benzene; [bmim]PF6 In dichloromethane at 20℃; |
|
| 1: 13 % Chromat.
2: 3 % Chromat. |
With [bis(acetoxy)iodo]benzene In dichloromethane at 20℃; |
|
| 1: 65 % Chromat.
2: 19 % Chromat. |
With 2,6-dichloropyridine N-oxide; tetra-n-butylammonium nitridoosmate(VIII); iron(III) chloride In acetic acid; 1,2-dichloro-ethane at 60℃; for 0.5h; |
|
| 44 % Chromat. |
With dihydrogen peroxide; acetic acid In acetonitrile at 20℃; for 5h; |
|
|
With dihydrogen peroxide In acetonitrile at 70℃; for 4h; |
|
| 1: 4 % Chromat.
2: 76 % Chromat. |
With potassium permanganate; boron trifluoride acetonitrile complex In acetonitrile at 24.85℃; |
|
| 1: 3 %Chromat.
2: 19 %Chromat. |
With oxygen; acetaldehyde; acetonitrile at 70℃; for 24h; |
|
| 1: 3 %Chromat.
2: 33 %Chromat. |
With oxygen; acetaldehyde; acetonitrile In dichloromethane at 25℃; for 48h; |
|
| 1: 38 % Chromat.
2: 36 % Chromat. |
With 1H-imidazole; sodium periodate In water; acetonitrile at 20℃; for 0.916667h; |
|
|
With dihydrogen peroxide In acetonitrile at 25℃; for 0.166667h; Title compound not separated from byproducts.; |
|
| 1: 68 %Chromat.
2: 22 %Chromat. |
With tert.-butylhydroperoxide; iron(III) chloride; {tetra-n-butylammonium}{osmiumnitrido(chloro)4} In dichloromethane; acetic acid at 23℃; for 0.0833333h; |
|
|
Stage #1: Cyclooctan With aluminum(III) nitrate nonahydrate; dihydrogen peroxide In water; acetonitrile at 70℃;
Stage #2: With triphenylphosphine In water; acetonitrile |
|
|
With oxygen at 19.84℃; Neat (no solvent); UV-irradiation; |
|
|
With tert.-butylhydroperoxide; [(pymox-Me2)2RuCl2]+BF4- In water at 20℃; for 24h; |
|
| 1: 32 %Chromat.
2: 22 %Chromat. |
With 1H-imidazole; sodium periodate In water; acetonitrile at 20℃; for 8h; |
|
| 1: 40 %Chromat.
2: 31 %Chromat. |
With 1H-imidazole; sodium periodate In water; acetonitrile at 20℃; for 0.5h; Sonication; |
|
| 1: 31 %Chromat.
2: 6 %Chromat. |
With 1H-imidazole; manganese(III) meso-tetraphenylporphyrin acetate; dihydrogen peroxide; sodium hydrogencarbonate In water; acetonitrile at 25℃; for 4h; |
|
| 1: 71 %Chromat.
2: 29 %Chromat. |
With C8H9N4S(1-)*O2V(1+); dihydrogen peroxide; chlorobenzene In acetonitrile at 80℃; for 5h; chemoselective reaction; |
|
|
With C42H22FeN9; dihydrogen peroxide In dichloromethane; water; acetonitrile at 20℃; for 5h; chemoselective reaction; |
|
|
Stage #1: Cyclooctan With tert.-butylhydroperoxide; (OC2H4)(OHC2H4)NC2H4N(C2H4OH)Cu(thiocyanate) In water; acetonitrile at 50℃; for 11h;
Stage #2: With triphenylphosphine In water; acetonitrile |
|
| 1: 52 %Chromat.
2: 26 %Chromat. |
With dihydrogen peroxide; sodium hydrogencarbonate In water; acetonitrile at 80℃; for 5h; |
|
|
With dihydrogen peroxide In water at 80℃; for 8h; |
|
| 1: 42 %Chromat.
2: 28 %Chromat. |
With sodium periodate; water In acetonitrile for 6h; |
|
| 1: 54 %Chromat.
2: 6 %Chromat. |
With Fe(triflate)2(1-(6-methyl-2-pyridylmethyl)-4,7-dimethyl-1,4,7-triazacyclononane); dihydrogen peroxide; acetic acid In water; acetonitrile at 0℃; for 0.666667h; |
|
| 1: 62 %Chromat.
2: 33 %Chromat. |
With 1H-imidazole; sodium periodate; 5,10,15,20-tetrakis-(4-aminophenyl) manganese(III) porphyrin chloride In water; acetonitrile at 25℃; for 2h; |
General procedure for oxidation of alkanes with NaIO4 catalyzed by [Mn(TNH2PP)Cl(at)MWCNT]
General procedure: To a mixture of alkane (1 mmol), [Mn(TNH2PP)Cl(at)MWCNT] (350 mg, 0.05 mmol) and imidazole (0.2 mmol) in CH3CN (10 mL) was added a solution of NaIO4 (2 mmol) in H2O (10 mL). The reaction mixture was stirred magnetically at room temperature. The progress of the reaction was monitored by GC. At the end of the reaction, the reaction mixture was diluted with Et2O (20 mL) and filtered. The catalyst was thoroughly washed with Et2O and combined washings and filtrates were purified on silica-gel plates or with a silica-gel column. IR and 1H NMR spectral data confirmed the identities of the products. |
| 1: 33.9 %Chromat.
2: 18.5 %Chromat. |
With C12H8Mo2N2O16(2-)*2C6H6NO2(1+)*2H2O; oxygen at 119.84℃; for 6h; Autoclave; |
2.5 Catalytic activity
The oxidation of cyclooctane was performed in a stainless steel batch reactor system at 393K and under the air pressure of 10atm, with the cyclooctane-to-oxygen molar ratio set at 6.5. In a typical experiment, 60ml of substrate containing the amount of catalyst providing a concentration of 3.3×10-4M was introduced in the deaerated autoclave and the whole system was heated until a temperature of 393K was reached. Air was then introduced and after 6h the oxidation products were analyzed by an Agilent 6890N Gas Chromatograph equipped with an Innowax (30m) column. The yield values were verified by addition of an internal standard, chlorobenzene, at the end of the reaction. |
|
With Oxone; [FeIII(1,4,7-trimethyl-1,4,7-triazacyclononane)(acac)Cl]ClO4; sodium hydrogencarbonate In water; acetone at 20℃; for 0.0833333h; |
|
|
With 1-(2-methyl-1-benzimidazoyl)methyl-4,7-dimethyl-1,4,7-triazacyclononane; water; dihydrogen peroxide In acetonitrile at 20℃; Overall yield = 6.8 %; |
|
|
With cis-(Cl,Cl)-[Re(p-NC6H4CH3)Cl2(indazole-3-carboxylate)(P(phenyl)3)] methanol solvate; dihydrogen peroxide In water; acetonitrile |
|
| 1: 45 %Chromat.
2: 30 %Chromat. |
With sodium periodate In water; acetonitrile at 20℃; for 4h; |
2.3 Catalytic experiments
General procedure: All of the reactions were carried out at room temperature with magnetic stirring. To a mixture of alkene or alkane (1 mmol), Mn(TPP)ClIm-MIL-101(100 mg) in CH3CN (10 ml), a solution of NaIO4 (2 mmol) in H2O (10 ml) was added. The reaction mixture was stirred at room temperature. The progress of the reaction was monitored by GC. At the end of the reaction, the mixture was diluted with Et2O (20 ml) and filtered. The catalyst was thoroughly washed with Et2O and the combined washings and filtrates were purified on a silica gel plates or a silica gel column to afford the product. |
|
With dihydrogen peroxide In acetonitrile |
|
|
With Cr2O4(2-)*Cu(2+); dihydrogen peroxide In acetonitrile at 50℃; for 10h; |
|
| 1: 25 %Chromat.
2: 7 %Chromat. |
With [Fe(N,N'-dimethyl-N,N'-bis(2-pyridylmethyl)-1,2-diaminoethane)(μ-O)FeCl3]; 3-chloro-benzenecarboperoxoic acid In acetonitrile at 20℃; for 1h; Inert atmosphere; |
|
|
With dihydrogen peroxide In acetonitrile at 40℃; for 1.5h; |
|
|
With rac-tris(1,10-phenanthroline)copper(II); dihydrogen peroxide In water at 70℃; for 3h; Autoclave; |
Catalytic reaction
General procedure: The liquid-phase oxidation of cycloalkane with H2O2 (30% in aqueous solution) was carried out under a stirring condition in a sealed autoclave. A typical reaction mixture is as follows: 0.05 g catalyst, 10 mL solvent, 9.5 mmol substrate, and 38 mmol H2O2 (30% in aqueous solution). Unless otherwise stated, the reaction temperature is 70 °C and time is 3 h. After reaction, the liquid product was separated by centrifugation and analyzed by a GC-7890F gas chromatograph equipped with a polyethylene glycol packed column and a flame ionization detector with benzyl chloride as an internal standard. |
|
With bismuth (III) nitrate pentahydrate; dihydrogen peroxide; nitric acid In water; acetonitrile at 60℃; |
|
| 1: 8.9 %Chromat.
2: 7.1 %Chromat. |
With [Cu4(μ4-(N,N,N',N'-tetrakis(2-hydroxyethyl)ethylenediamine(-2H)))(μ5-(N,N,N',N'-tetrakis(2-hydroxyethyl)ethylenediamine(-2H)))(salicylic acid(-2H))2]*10H2O; dihydrogen peroxide; trifluoroacetic acid In water; acetonitrile at 50℃; for 3h; Overall yield = 16 %Chromat.; |
|
| 1: 12.3 %Chromat.
2: 7 %Chromat. |
With [Cu4(μ4-(N,N,N',N'-tetrakis(2-hydroxyethyl)ethylenediamine(-H)))2(phenylmalonic acid(-H))2(H2O)]*7.5H2O; dihydrogen peroxide; trifluoroacetic acid In water; acetonitrile at 50℃; for 3h; Overall yield = 19.3 %Chromat.; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






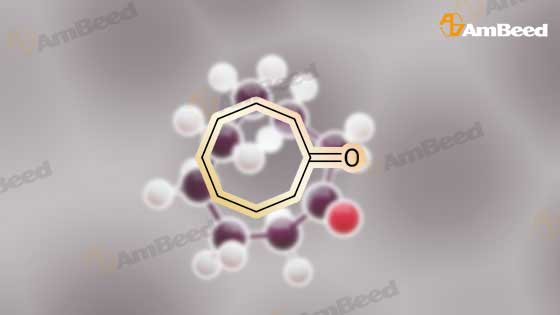
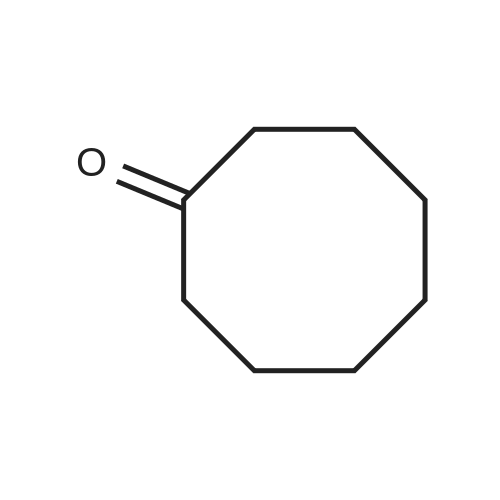

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping