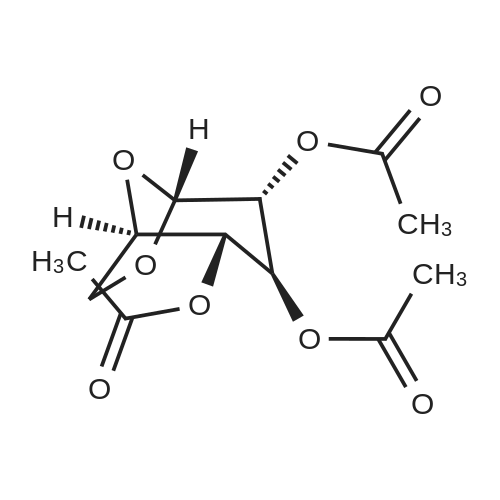
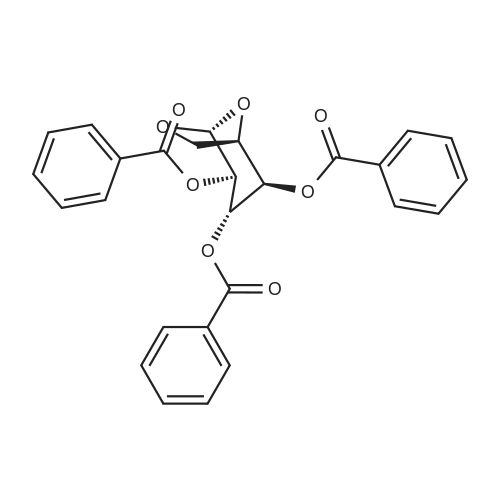
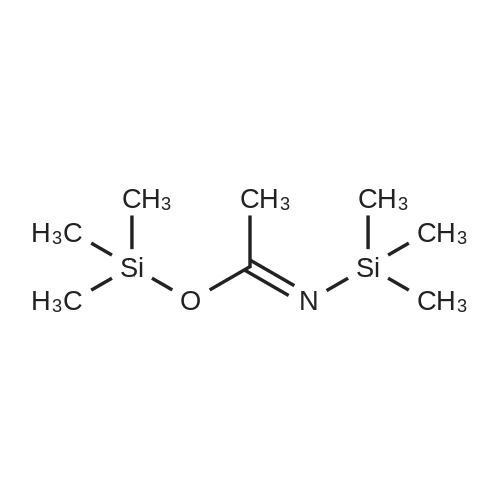
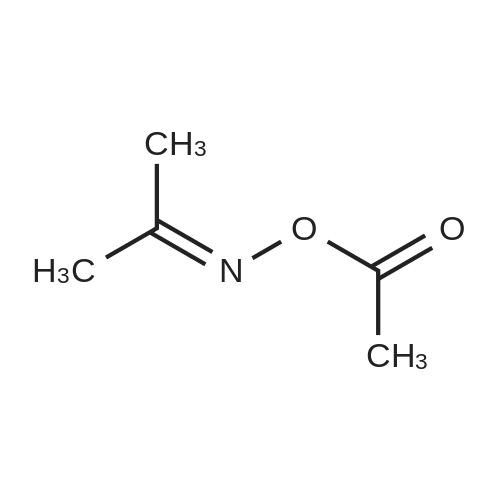
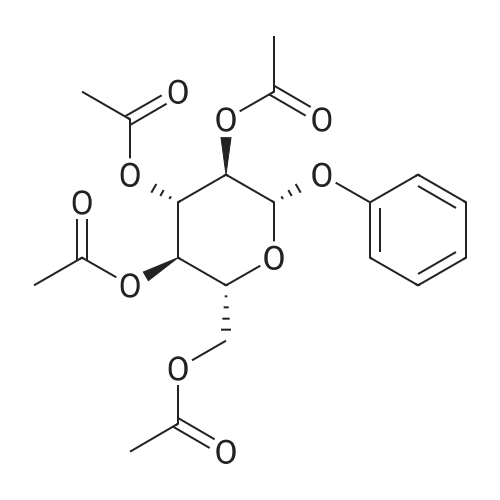
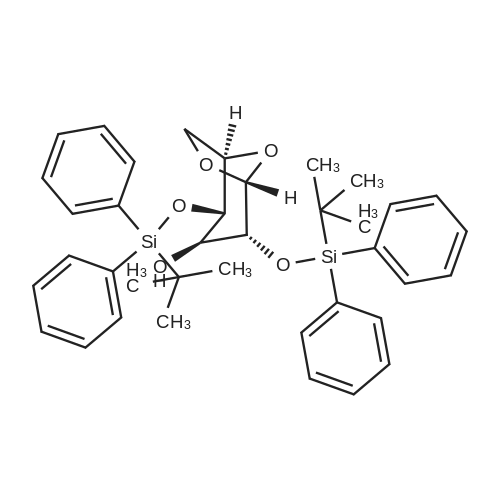
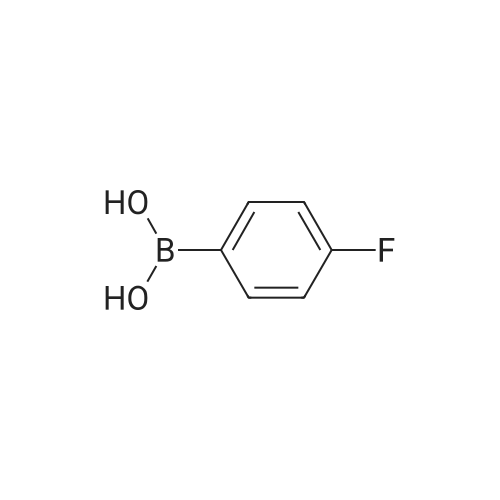
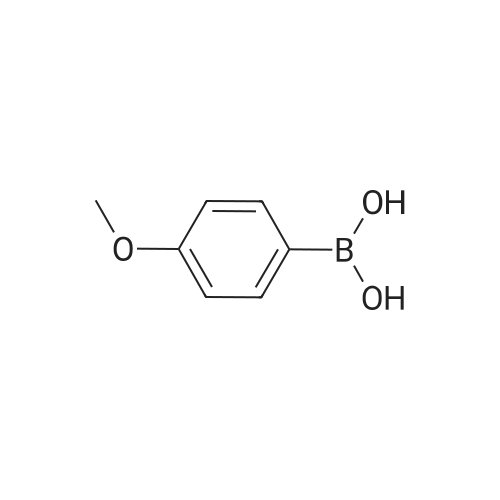
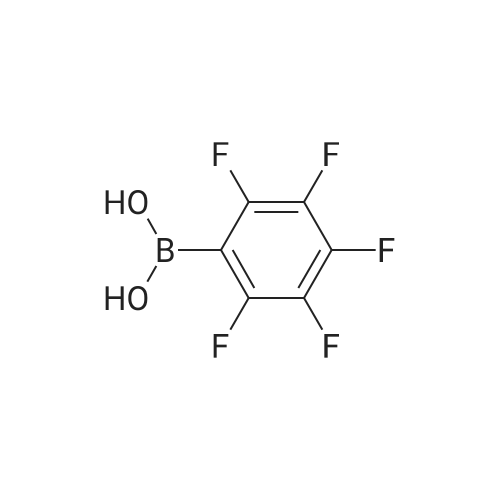
| 95% |
Stage #1: levoglucosan With sodium hydride In N,N-dimethyl-formamide; mineral oil at -20 - 0℃; for 1h;
Stage #2: allyl bromide In N,N-dimethyl-formamide; mineral oil at -20 - 20℃; |
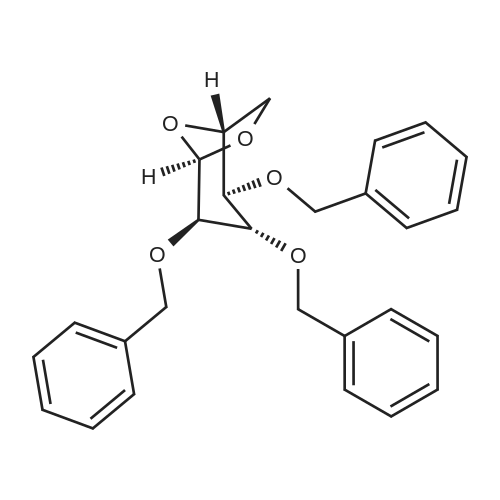
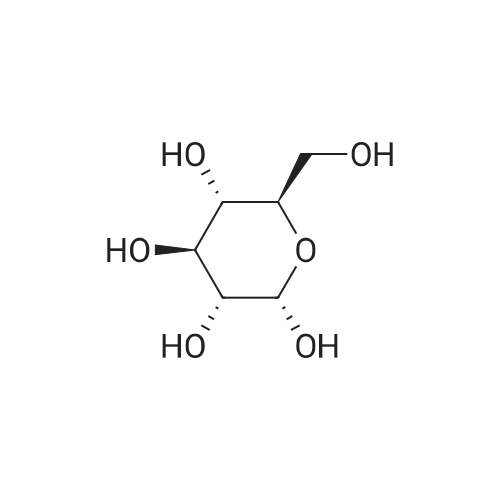
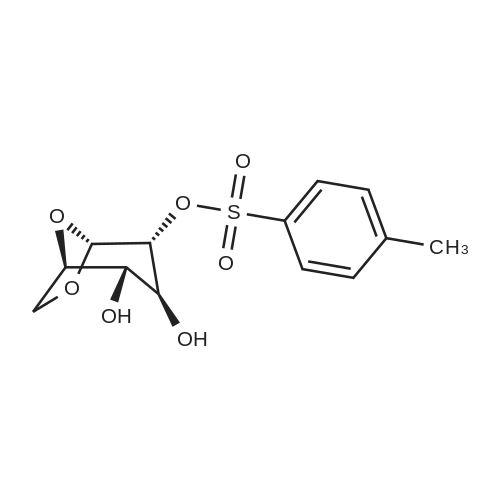
1,6-Anhydro-2,3,4-tri-O-allyl-β-D-glucopyranose (5b).
To a solution of levoglucosan (1 equiv., 1 g,6.11 mmol) in DMF (45 mL) at -20 °C was added sodium hydride (60% in oil, 4 equiv., 0.98 g,24.4 mmol) portionwise. The cooling bath was allowed to reach 0°C within 1 h, then it was cooledagain to -20 °C and allyl bromide (4.5 equiv., 2.4 mL, 27.6 mmol) was added under vigorous stirring.The reaction mixture was stirred overnight at r.t. The mixture was diluted with water and extractedwith diethyl ether (3 x 100 mL). The organic phases are combined and washed 5 times with water,dried with Na2SO4, filtered, and concentrated to afford a yellow oil which was purified by flashchromatography (Petroleum Ether/EtOAc, 9/1 to 1/1), to give 5b (1.63 g, 5.77 mmol) as a yellow oil in95% yield. 1H NMR (400 MHz, CDCl3) δ (ppm) 5.92-5.79 (m, 3H, OCH2CHCH2), 5.37 (s, 1H, H-1),5.26-5.12 (m, 6H, OCH2CHCH2), 4.51 (m, 1H, H-5), 4.08-4.02 (m, 6H, OCH2CHCH2), 3.85 (dd, J = 7.1,0.8 Hz, 1H, H-6), 3.64 (dd, J = 6.8, 6.0 Hz, 1H, H-6), 3.45 (m, 1H, H-3), 3.24 (m, 1H, H-4), 3.21 (m, 1H,H-2). The analyses are in good agreement with the experimental data reported in literature [21]. |
| 90% |
Stage #1: levoglucosan With sodium hydride In N,N-dimethyl-formamide; mineral oil at -20 - 0℃; for 1h; Inert atmosphere;
Stage #2: allyl bromide In N,N-dimethyl-formamide; mineral oil at -20 - 20℃; Inert atmosphere; |
2,3,4-Tri-O-allyl-1,6-anhydro-β-d-glucopyranose
Sodium hydride (60% in oil, 500mg, 12.5mmol) was added to a solution of 1,6-anhydro-β-d-glucopyranose (530mg, 3.27mmol) in anhydrous N,N-dimethylformamide (15mL) under cooling (-20°C) and stirring. The cooling bath temperature was allowed to reach 0°C within 1h, then it was cooled again to-20°C and allyl bromide (1.3mL, 15mmol) was added under vigorous stirring. The reaction mixture was stirred overnight at room temperature. It was then diluted with water, extracted with diethyl ether, the organic extract was dried (Na2SO4) and concentrated to afford crude product (1.12g). Chromatography in ethyl acetate-light petroleum 1:5 yielded syrupy 2,3,4-tri-O-allyl-1,6-anhydro-β-d-glucopyranose (835mg, 90%). The product was unstable and slowly polymerized at room temperature. It could be stored at-30°C for a few weeks, the purity being >95% according to NMR. 1H NMR [300MHz, CDCl3]: δ (ppm) 3.25 (dd, 3J(H1,2)=1.3Hz and 3J(H2,3)=1.3Hz, 1H, H2), 3.28 (dd, 3J(H4,5)=1.3Hz and 3J(H3,4)=3.9Hz, 1H, H4), 3.49 (dddd, 4J(H1,3)=1.3Hz, 4J(H3,5)=1.3Hz, 3J(H2,3)=1.3Hz and 3J(H3,4)=3.9Hz, 1H, H3), 3.68 (dd, 2J(H6,6′)=7.1Hz and 3J(H6′,5)=5.8Hz, 1H, H6′), 3.89 (dd, 2J(H6,6′)=7.1Hz and 3J(H6,5)=1.1Hz, 1H, H6), 4.09 (m, 3× 2H, OCH2CH), 4.55 (dddd, 4J(H3,5)=1.3Hz, 3J(H4,5)=1.3Hz, 3J(H6,5)=1.1Hz and 3J(H6′,5)=5.8Hz, 1H, H5), 5.31 (m, 3× 2H, OCH2CHCH2), 5.41 (dd, 3J(H1,2)=1.3Hz and 4J(H1,3)=1.3Hz, 1H, H1), 5.90 (m, 3× 1H, OCH2CHCH2). 13C {1H} NMR [300MHz, CDCl3]: δ (ppm) 65.43 (C6), 70.40, 70.97 and 71.19 (OCH2CHCH2), 74.30 (C5), 76.62 (C2 or C3), 76.65 (C2 or C3), 77.28 (C4), 100.75 (C1), 117.37, 117.66 and 117.80 (OCH2CHCH2), 134.79, 134.83 and 134.94(OCH2CHCH2). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping