| 50% |
With pyridine hydrochloride; |
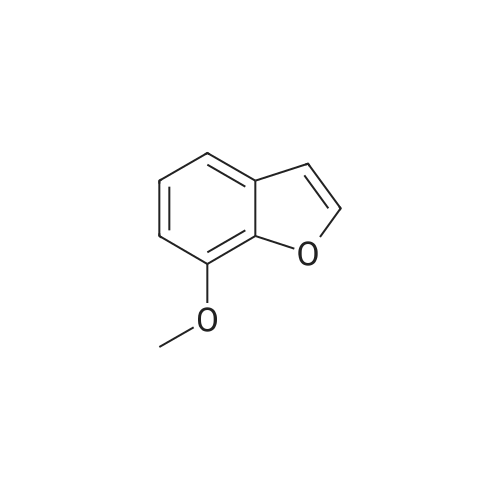
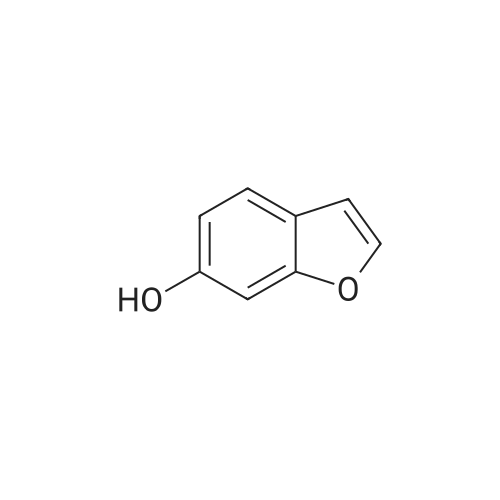
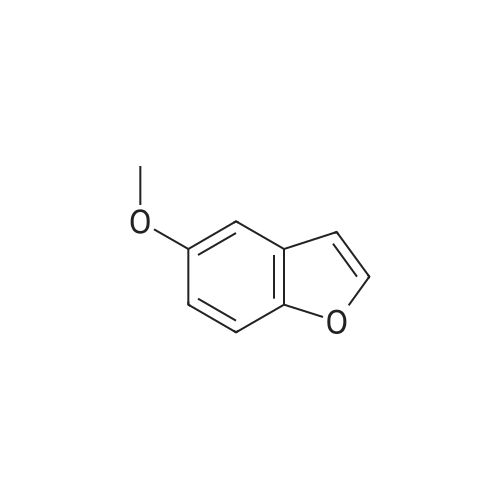
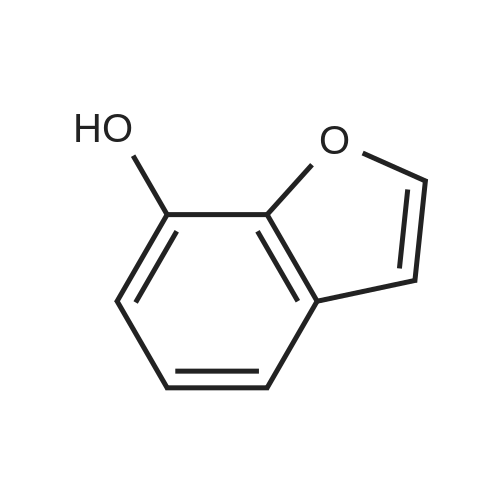
EXAMPLE 1C 7-Hydroxybenzofuran A mixture of 7methoxybenzofuran (1.8 g.) and pyridine hydrochloride (4.6 g.) was heated at 220 C. for 21/2 hours. The reaction was cooled and diluted with methylene chloride. The solution was washed with 5% aqueous hydrochloric acid (six times); dried (MgSO4) and concentrated to an oil, 0.8 g. (50% yield). |
| 42% |
With boron tribromide; In dichloromethane; at -78 - 20℃;Inert atmosphere; |
To a stirred solution of <strong>[7168-85-6]7-methoxybenzofuran</strong> (3.3 mmol)) in dry DCM (5.0 mL) at -78 C. under N2 atmosphere was added BBr3 (3.0 mmol) (1.0M solution in DCM) and then the reaction was allowed to stir overnight at room temperature. The crude reaction was quenched with ice water and extracted with DCM (3×5.0 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The product was purified by column chromatography to give 51. [0380] benzofuran-7-ol (51) (42) 1H NMR (500 MHz, CDCl3) delta 7.60 (d, J=2.0 Hz, 1H), 7.16 (d, J=7 Hz, 1H), 7.10 (t, J=7.5 Hz, 1H), 6.83 (d, J=7.5 Hz, 1H), 6.77 (d, J=2.0 Hz, 1H), 5.43 (s, 1H). GC-MS (ES) for C8H6O2 [M]+ 134. |
|
With hydrogenchloride; lithium iodide; In 2,3,5-trimethyl-pyridine; |
Reference Example 214 To a solution of <strong>[7168-85-6]7-methoxybenzofuran</strong> (8.0 g) in collidine (80 ml) was added lithium iodide (14.5 g), and the mixture was refluxed under argon atmosphere for 1 day and cooled. To the mixture was added 1N hydrochloric acid, and the mixture was extracted with ethyl acetate (twice). The organic layer was washed with 1N hydrochloric acid (twice), water and saturated brine, and dried with magnesium sulfate. Under reduced pressure, the solvent was evaporated, and the residue was purified with silica gel column chromatography to give dark brown oil of 7-hydroxybenzofuran (7.0 g). 1H-NMR (200 MHz, CDCl3) delta 5.45 (br, 1H), 6.78 (d, 1H, J=2.2 Hz), 6.84 (dd, 1H, J=7.0, 1.4 Hz), 7.09 (d, 1H, J=7.4 Hz), 7.17 (dd, IH, J=7.8, 1.8 Hz), 7.61 (d, 1H, J=2.2 Hz). |
|
With boron trichloride; tetra-(n-butyl)ammonium iodide; In dichloromethane; at -78℃; for 6h; |
Preparation 13 Benzofuran-7-ol To 7-methoxy benzofuran (6.3 g, 42.5 mmol) in a 0.5 M solution of CH2Cl2 at -78 C. is added tetrabutylammonium iodide (18.9 g, 51 mmol). To this stirred solution, BCl3 (100 mL of a 1.0 M solution in CH2Cl2, 100 mmol) was added dropwise via addition funnel. The reaction mixture was stirred at -78 C. for 6 h. Water is added dropwise very slowly to quench. The resulting mixture was stirred overnight at rt, basified with 6 N NaOH (pH=10), stirred for 1 h, neutralized with 2 N HCl (pH=7), and extracted with CH2Cl2 (5*). The combined extracts were dried (MgSO4), filtered, concentrated, and chromatographed (10% EtOAc in hexanes) to provide 5.3 grams of benzofuran-7-ol as an oil. 1H NMR (400 MHz, CDCl3): delta 7.61 (d, 1, J=2.1), 7.19 (d, 1, J=7.7), 7.13 (t, 1, J=7.7), 6.88 (d, 1, J=7.7), 6.79 (d, 1, J=2.1), 6.04 (bs, 1). Previously prepared but no data reported: Musser, J. H.; Chakraborty, U.; Bailey, K.; Sciortino, S.; Whyzmuzis, C.; Amin, D.; Sutherland, C. A. J. Med. Chem. 1987, 30, 62-67. |
|
|
prepared from <strong>[7168-85-6]7-methoxybenzo[b]furan</strong> according to a similar demethylation method as described in WO 04/043904) |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping