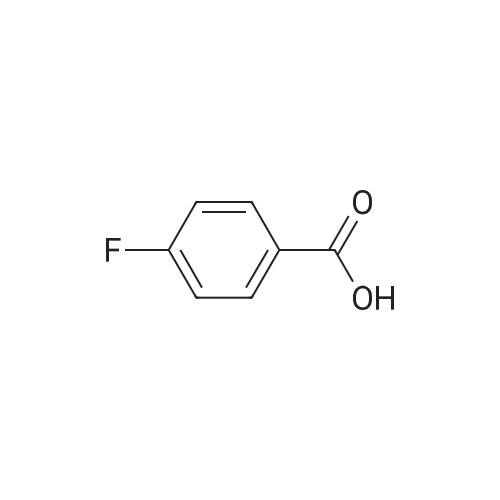
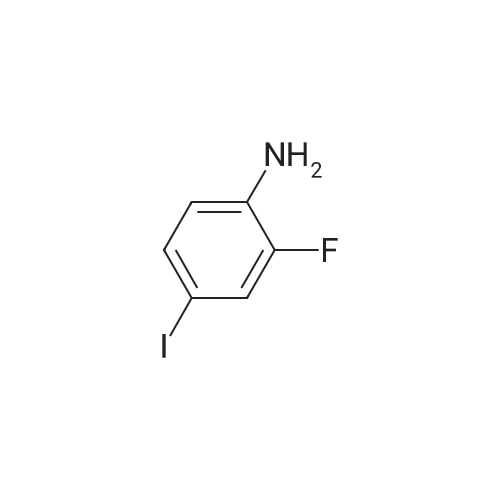
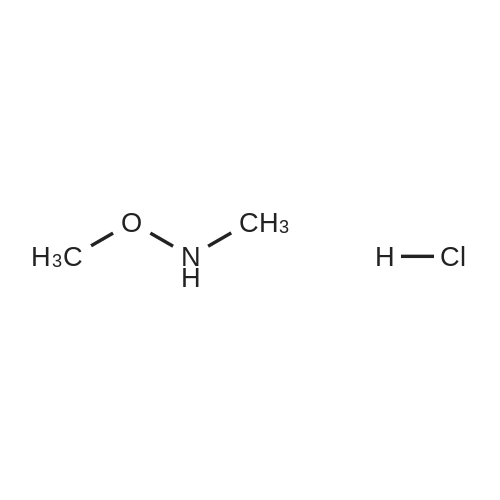
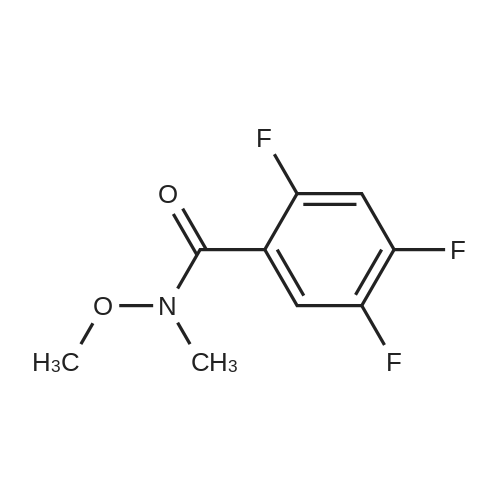
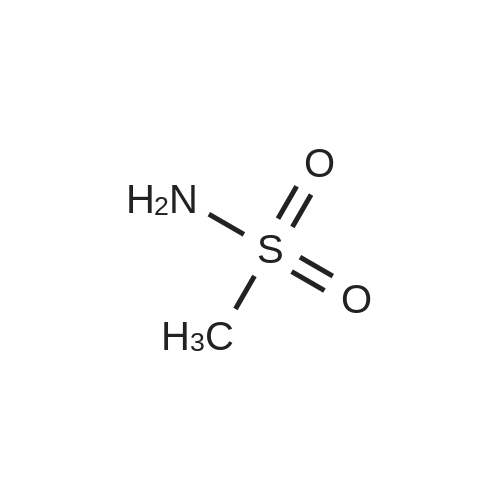
|
With thionyl chloride; In toluene;Heating / reflux; |
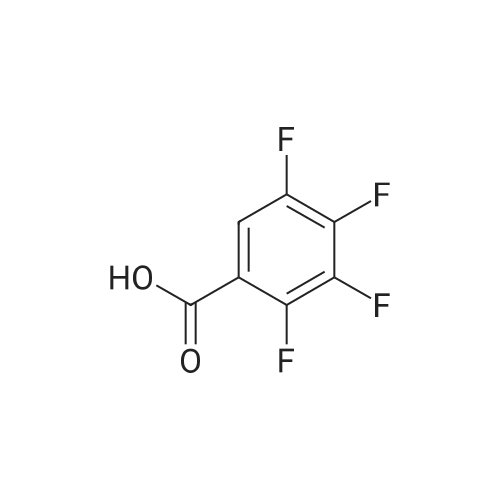
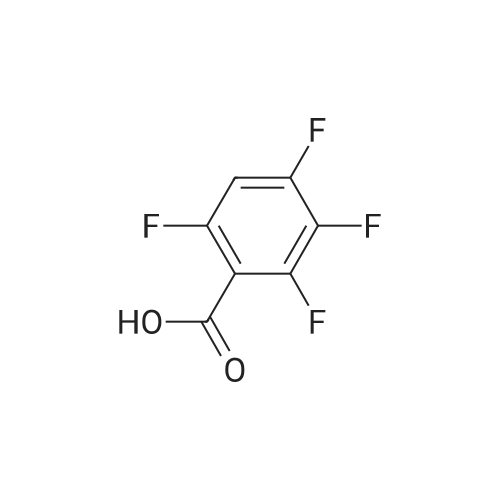
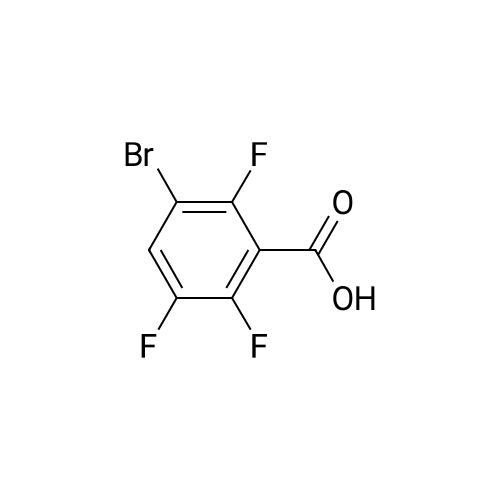
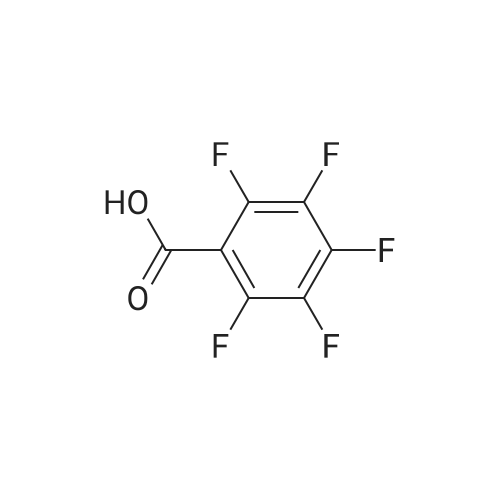
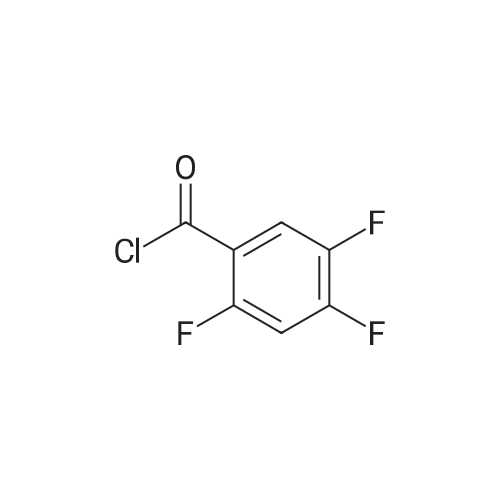
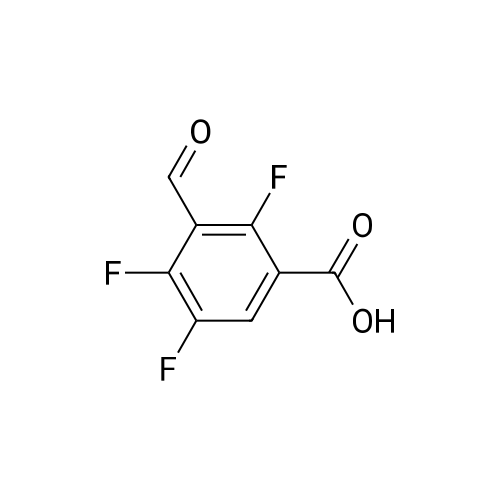
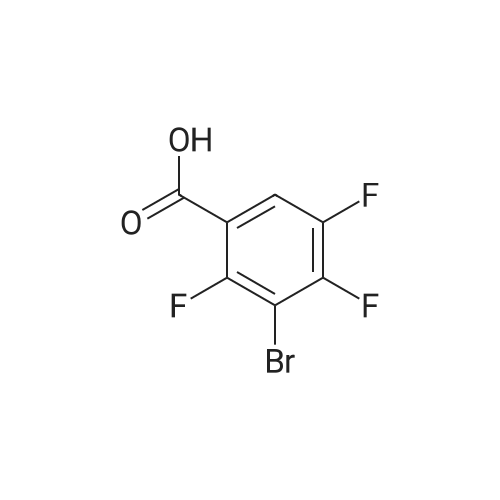
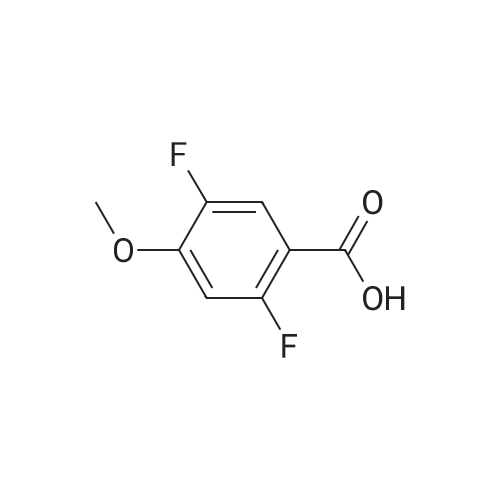
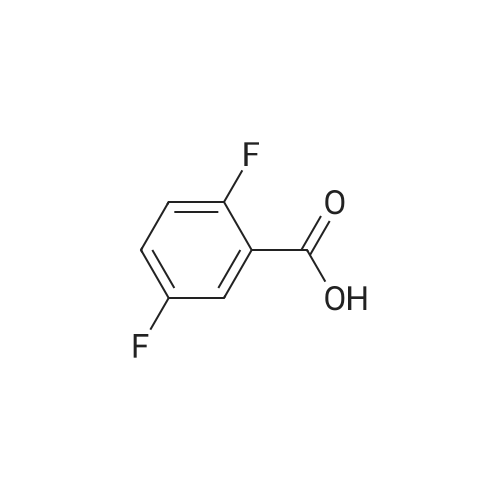
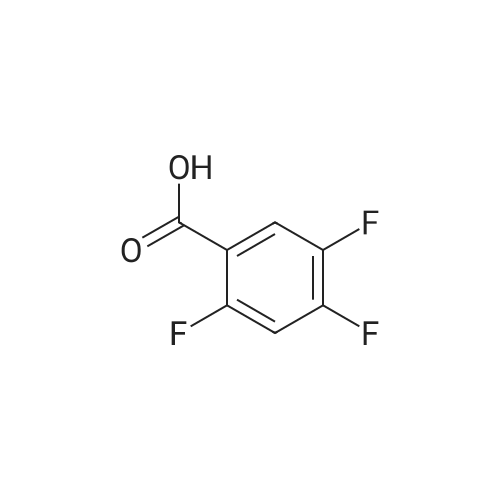
Example 3; 1 - ( 3 - F2-oxo-3 -( 1 H-p yrrol-2- ylmethylene)-2.3 -dihvdro- 1 H-indol-6- ylaminol phenyl 1-3-(2A5-trifluorophenyl)urea; [00107] Synthesis of 2,4,5-trifluorobenzoyl azide (10); [00108] To a solution of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (10.0 g, 0.057 mol) in toluene(150 mL) is added SOCl2 (12.5 mL, 0.171 mol). The mixture is heated at reflux overnight. All the solvent is removed and the crude is dried in vacuo for an hour to remove residual SOCl2. The crude is then dissolved in acetone (100 ml) and cooled to 00C. A solution of NaN3 (4.5 g, 0.0684 mol) in water (20 mL) is added slowly. The resulting mixture is warmed to room temperature and stirred for 2 hr. Acetone is removed. The mixture is extracted with EtOAc (3x150 mL). The combined organic layer is dried over Na2SO4, filtered and concentrated. The crude is purified by column chromatography (EtOAc/Hexane, gradient, 0-20%) to give the desired product as oil. 1H NMR (400 MHz, CDCl3) delta 7.90-7.70 (m, 1 H), 7.10-7.00 (m, 1 H). |
|
With oxalyl dichloride; |
<strong>[446-17-3]2, 4, 5-trifluorobenzoic acid</strong> (20 g, 113.6 mmol) was dissolved in thionyl chloride (15ml, 170.4 mmol) at room temperature under nitrogen atmosphere. The reaction mixture was heated to reflux for 4 h and then thionyl chloride was removed under reduced pressure to provide 2, 4, 5-trifluorobenzoyl chloride 2b.2 (20 g, 103.8 mmol). The acid chloride 2b.2 was dissolved in DCM, cooled to 00C, and triethyl amine(17.2 ml, 126.2mmol) followed by 3- dimethyl amino acrylonitrile 2b.3 (1 1.3 ml, 103.8 mmol) were added. The ice bath was removed and the reaction mixture was stirred at room temperature overnight. The reaction mixture was portioned between water and DCM. The organic layer was separated and dried over Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography eluting with 30% ethyl acetate in n-hexane to provide compound 2b.4 (12.2 g, 46.4%). 1H-NMR (DMSO-ct°delta 7.81 (IH, s, -CH), 7.78-7.56 (2H, m, Ar-H), 3.31 (3H, s, - CH3), 3.27 (3H, s, -CH3). EIMS (m/z): 255.21 (M + H)+ |
|
With thionyl chloride; for 4h;Heating / reflux; |
2,4,5-trifluorobenzoyl chloride (2) is prepared from <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> as described previously. [Reuman, M.; et. al, J. Med. Chem. (1995) 38, 2531-2540]. Thionyl chloride (8 ml) is added to a solid 2,4,5-trifluoro-3-methoxy-benzoic acid (154 mg, 0.75 mmole ). The reaction mixture is refluxed for 4 hours. It is distilled to remove excess thionyl chloride and further dried under vacuum at rt for the further reaction. |
|
With thionyl chloride; at 55℃; for 18h; |
Thionyl chloride (50 mL, 680 mmol) was added to <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (10 g, 57 mmol) and the mixture stirred at 55 C for 18 hours. After cooling, the excess thionyl chloride was removed in vacuo. The resulting crude oil was azeotroped twice with DCM (30 mL) and toluene (20 mL) and the residue redissolved in DCM (50 mL), then cooled to 0 C. A mixture of 4-methylphenol (6.4 g, 59 mmol) and triethylamine (10 mL, 71 mmol) in DCM (20 mL) was added over 30 minutes. The reaction was allowed to warm up to room temperature over 1 hour. The crude reaction mixture was partitioned between EtOAc (200 mL) and saturated sodium bicarbonate solution (70 mL). The aqueous layer was further extracted with EtOAc (100 mL). The combined organic extracts were combined, washed with saturated sodium bicarbonate solution (70 mL) and water (100 mL). The organic layer was dried over magnesium sulfate and concentrated to provide a crude solid, which was purified by silica gel chromatography eluting with 5% EtOAc in heptane to provide the title compound (10.08 g, 66%) as a white solid. |
|
With oxalyl dichloride; In N,N-dimethyl-formamide; acetonitrile; for 1.5h; |
To a solution of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (5.00 g, 28.4 mmol) in acetonitrile (50 mL) was added N,N-dimethylformamide (40 muL) followed by addition of oxalyl chloride (3.60 mL, 42.6 mmol). After 90 min, the volatiles were removed under reduced pressure. The residue was co-evaporated with acetonitrile (50 mL). The residue was then dissolved in methylene chloride (50 mL). This solution was added drop-wise into a cooled (ice bath) mixture of (2S)-1,1,1-trifluoropropan-2-amine hydrochloride (5.52 g, 36.9 mmol) (from Synquest, 98% ee) in toluene (100 mL) and 0.5 M sodium hydroxide aqueous solution (142 mL, 71.0 mmol). After addition, the ice bath was removed, and the reaction was allowed to warm to rt. The reaction was stirred overnight. The organic layer was separated. The aqueous layer was extracted with methylene chloride (50 mL). The combined organic layers were washed with 20% brine (75 mL) and water (2×75 mL), dried over MgSO4, filtered and concentrated under reduced pressure to afford the desired product (6.49 g, 84%) which was directly used in the next step without further purification. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In acetonitrile; for 1.5h; |
Step 1: 2,4, 5-Trifluoro-N-[(JS)-2, 2, 2-trifluoro-1-methylethylJbenzamideTo a solution of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (5.00 g, 28.4 mmol) in acetonitrile (50 mL) was added N,N-dimethylformamide (40 .iL) followed by addition of oxalyl chloride (3.60 mL, 42.6 mmol). After 90 mm, the volatiles were removed under reduced pressure. The residue was co-evaporated with acetonitrile (50 mL). The residue was then dissolved in methylene chloride (50 mL). This solution was addeddrop-wise into a cooled (ice bath) mixture of (2S)-i, 1,1 -trifluoropropan-2-amine hydrochloride (5.52 g, 36.9 mmol) (from Synquest, 98% ee) in toluene (100 mL) and 0.5 M sodium hydroxide aqueous solution (142 mL, 71.0 mmol). After addition, the ice bath was removed, and the reaction was allowed to warm to rt. The reaction was stirred overnight. The organic layer was separated. The aqueous layer was extracted with methylene chloride (50 mL). The combined organic layers were washed with 20% brine (75 mL) and water (2 x 75 mL), dried over MgSO4, filtered and concentrated under reduced pressure to afford the desired product (6.49 g, 84%) which was directly used in the next step without further purification. ?H NMR (300MHz, DMSO-d6) oe 9.01 (d, J= 7.6 Hz, 1H), 7.92-7.50 (m, 2H), 4.76 (m, 1H), 1.31 (d, J= 7.0 Hz, 3H) ppm. LCMS cacid. for C,0H8F6N0 (M+1): mlz = 272.0; Found:272.0. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In acetonitrile; for 1.5h; |
To a solution of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (5.00 g, 28.4 mmol) in acetonitrile (50 mL) was added N,N-dimethylformamide (40 followed by addition of oxalyl chloride (3.60 mL, 42.6 mmol). After 90 min, the volatiles were removed under reduced pressure. The residue was co-evaporated with acetonitrile (50 mL). The residue was then dissolved in methylene chloride (50 mL). This solution was added drop-wise into a cooled (ice bath) mixture of (2S)-l,l,l-trifluoropropan-2-amine hydrochloride (5.52 g, 36.9 mmol) (from Synquest, 98%> ee) in toluene (100 mL) and 0.5 M sodium hydroxide aqueous solution (142 mL, 71.0 mmol). After addition, the ice bath was removed, and the reaction was allowed to warm to rt. The reaction was stirred overnight. The organic layer was separated. The aqueous layer was extracted with methylene chloride (50 mL). The combined organic layers were washed with 20% brine (75 mL) and water (2 x 75 mL), dried over MgS04, filtered and concentrated under reduced pressure to afford the desired product (6.49 g, 84%) which was directly used in the next step without further purification. lU NMR (300 MHz, DMSO-de) delta 9.01 (d, J= 7.6 Hz, 1H), 7.92 - 7.50 (m, 2H), 4.76 (m, 1H), 1.31 (d, J = 7.0 Hz, 3H) ppm. LCMS cacld. for CioHsFeNO (M+l)+: m/z = 272.0; Found: 272.0. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; |
(a) 2,4,5-trifluorobenzoyl chloride A suspension of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (5 g, 28.4 mmol) in DCM (60 mL) was cooled to0CC. Oxalyl chloride (3.72 mL, 42.59 mmol) was added followed by 3 drops of DMF and thereaction allowed to warm to room temperature. Effervescence commenced on warming.The mixture was stirred at room temperature for 2 h then evaporated (co-evaporated fromDCM x 3) and used without further purification |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In acetonitrile; for 1.5h; |
To a solution of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (5.00 g, 28.4 mmol) in acetonitrile (50 mL) was added N,N-dimethylformamide (40 muL) followed by addition of oxalyl chloride (3.60 mL, 42.6 mmol). After 90 min, the volatiles were removed under reduced pressure. The residue was co-evaporated with acetonitrile (50 mL). The residue was then dissolved in methylene chloride (50 mL). This solution was added drop-wise into a cooled (ice bath) mixture of (2S)-1,1,1-trifluoropropan-2-amine hydrochloride (5.52 g, 36.9 mmol) (from Synquest, 98% ee) in toluene (100 mL) and 0.5 M sodium hydroxide aqueous solution (142 mL, 71.0 mmol). After addition, the ice bath was removed, and the reaction was allowed to warm to rt. The reaction was stirred overnight. The organic layer was separated. The aqueous layer was extracted with methylene chloride (50 mL). The combined organic layers were washed with 20% brine (75 mL) and water (2×75 mL), dried over MgSO4, filtered and concentrated under reduced pressure to afford the desired product (6.49 g, 84%) which was directly used in the next step without further purification. 1H NMR (300 MHz, DMSO-d6) delta 9.01 (d, J=7.6 Hz, 1H), 7.92-7.50 (m, 2H), 4.76 (m, 1H), 1.31 (d, J=7.0 Hz, 3H) ppm. LCMS cacld. for C10H8F6NO (M+1)+: m/z=272.0. Found: 272.0. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In dichloromethane; at 0 - 20℃; for 2h; |
A suspension of <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (5 g, 28.4 mmol) in DCM (60 mL) was cooled to 0C. Oxalyl chloride (3.72 mL, 42.59 mmol) was added followed by 3 drops of DMF and the reaction allowed to warm to room temperature. Effervescence commenced on warming. The mixture was stirred at room temperature for 2 h then evaporated (co-evaporated from DCM x 3) and used without further purification. |
|
With thionyl chloride; In chloroform; at 20 - 70℃; |
General procedure: In a 100 mL round bottom flask equipped with mechanical stirrer and a water bath, taken 2-methyl-3-nitrobenzoic acid (15 g, 0.00 mmol), chloroform (200 mL) and then freshly distilled thionyl chloride (30 mL) was added drop wise at room temperature. The reaction mass stirred for 10 min at ambient temperature and slowly heated to 70 C,maintain the same temperature for 1-2 h. After completion ofthe TLC removed the excess thionyl chloride and once stripped off with chloroform to get desired substituted benzoyl chlorides. |
|
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃; for 2h;Inert atmosphere; |
General procedure: Commercially available <strong>[446-17-3]2,4,5-trifluorobenzoic acid</strong> (1a) (10.1 g,57.4 mmol) was dissolved in 100 mL of distilled DCM and stirred underargon atmosphere. Oxalyl chloride (5.0 mL, 58.300 mmol) was thenadded to the stirring mixture, the solution was allowed to stir for 5 minat room temperature. Anhydrous DMF (0.45 mL, 5.8 mmol) was added,dropwise, to the stirring solution over 35 s, the resulting solution wasallowed to stir at room temperature for 2 h. The DCM was removedfrom the reaction mixture by rotary evaporation, the resulting oil wasreconstituted in 100 mL of DCM and placed in an ice bath to coolto ~ 0 C. Ammonium hydroxide (40 mL, 28-30% ammonia) wasadded, dropwise, to the cool stirring solution over 2 min, the resultingmixture was allowed to stir for 2.5 h, gradually warming up to roomtemperature. The product was extracted by washing the aqueous layerwith 100 mL of DCM and then 150 mL of ethyl acetate. The organiclayers were pooled and concentrated by rotary evaporation, the resultingsolid was triturated with 9:1 hexanes:ethyl acetate and subjectedto vacuum filtration, which yielded UIAA-I-061 (2a) as a pureyellow solid. 84% yield. 1H NMR (300 MHz, CDCl3) delta = 8.01 (ddd,J = 7.17, 8.94, 10.62 Hz, 1H), 7.05 (ddd, J = 6.05, 9.51, 10.83 Hz,1H), 6.66 (bs, 1H), 6.03 (bs, 1H). 19F NMR (282 MHz, CDCl3)delta = -113.67 (m, 1F), -125.90 (ddd, J = 9.08, 17.12, 21.57 Hz, 1F),-140.15 (m, 1F). HRMS (ESI) calculated for (M + H+) 176.0318,found 176.0323. Retention time (analytical HPLC) = 12.3 min |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping