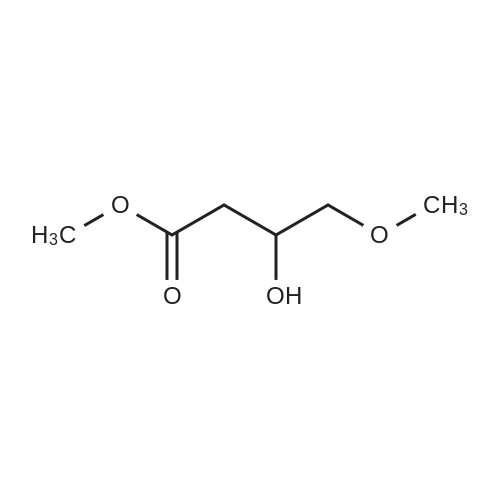
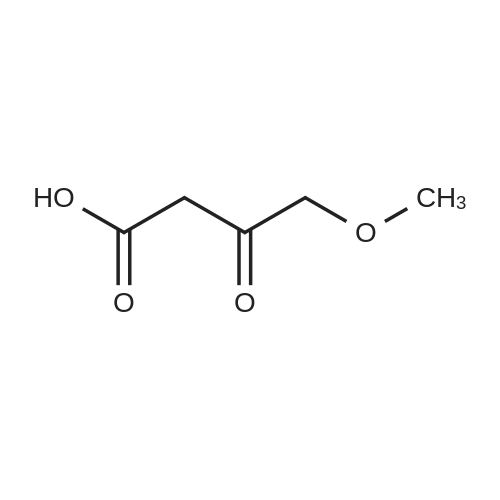
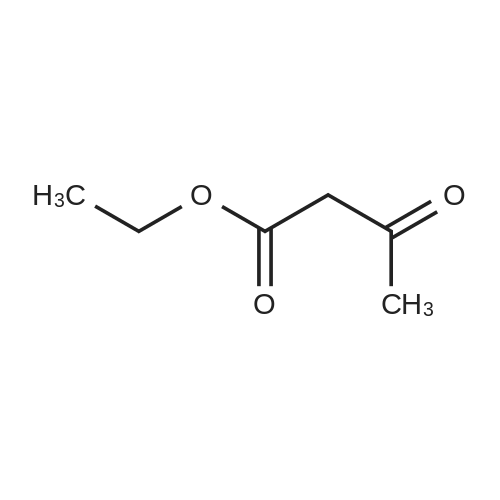
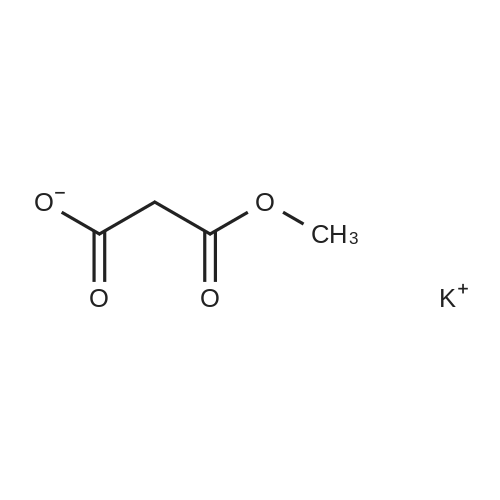
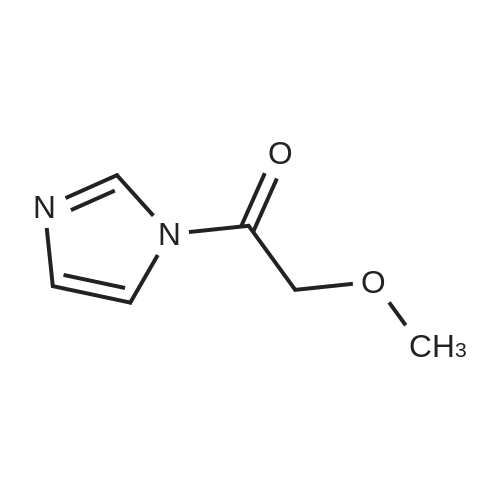
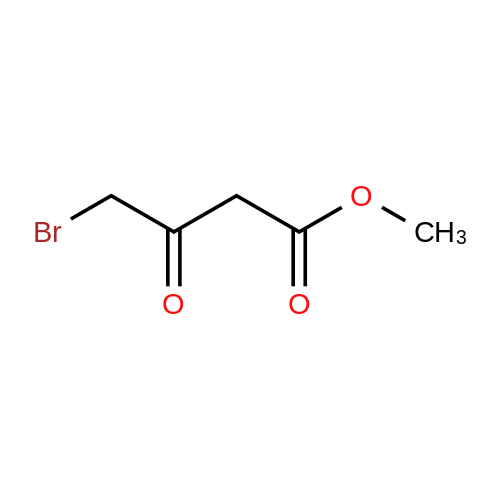
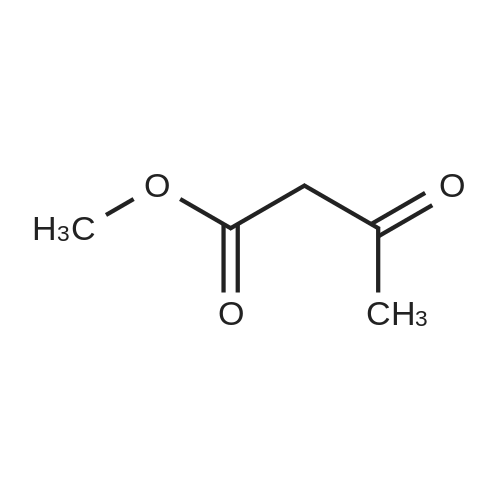
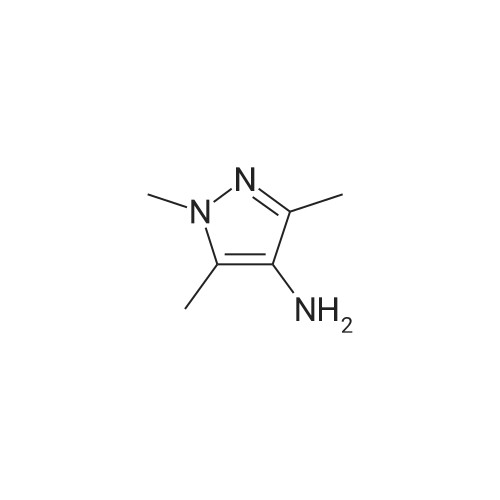
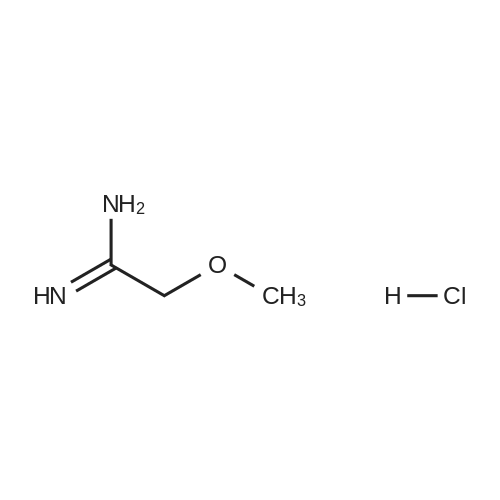
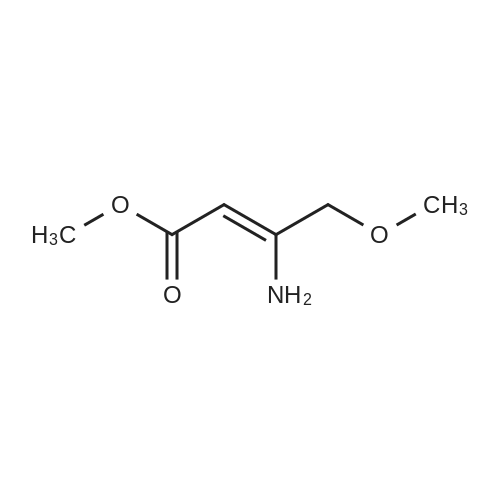
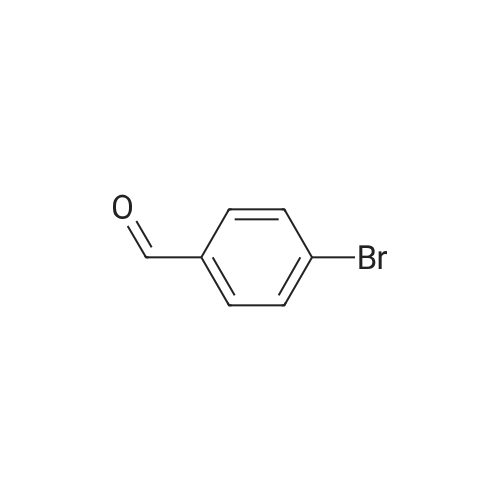
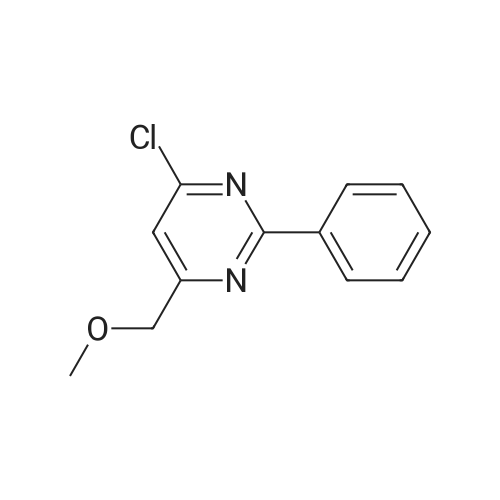
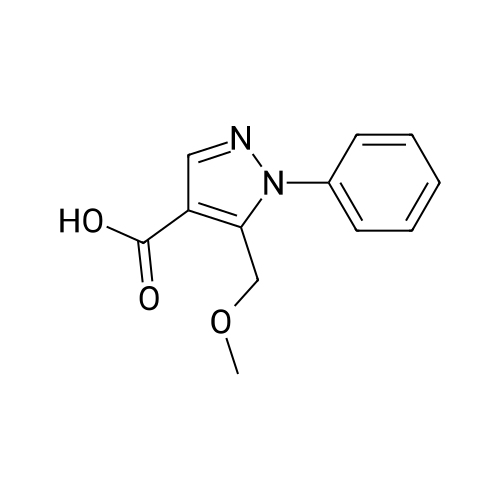
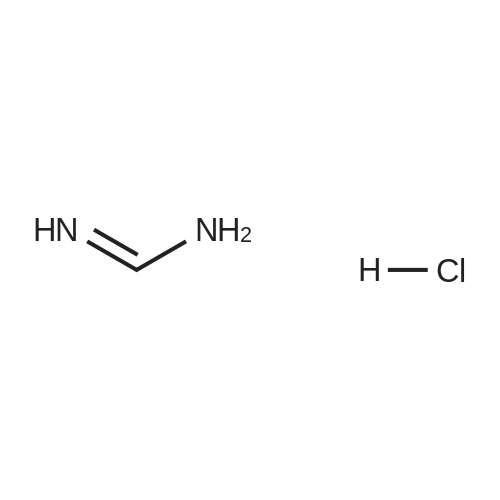
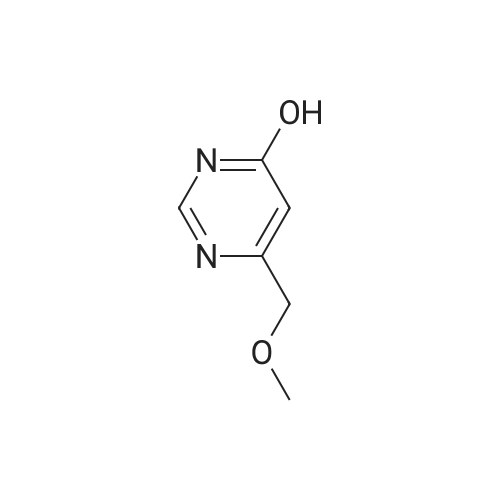
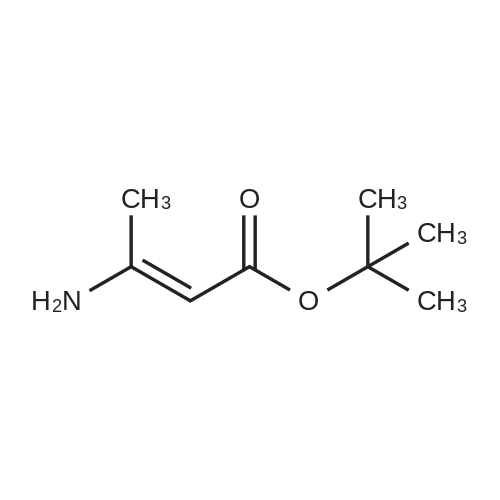
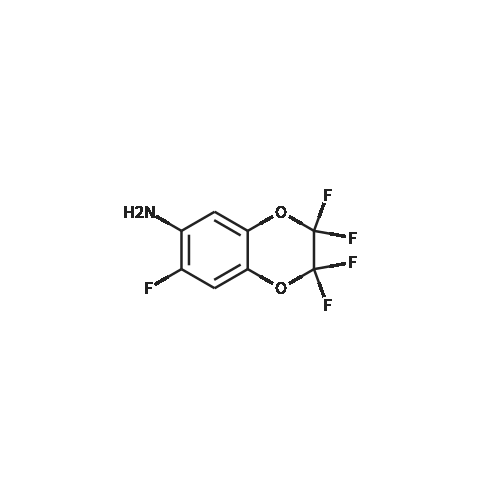
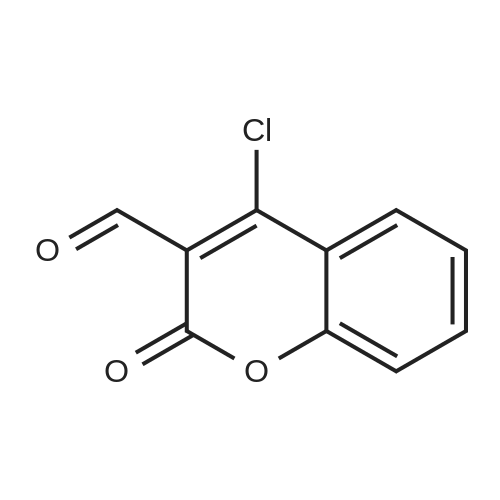
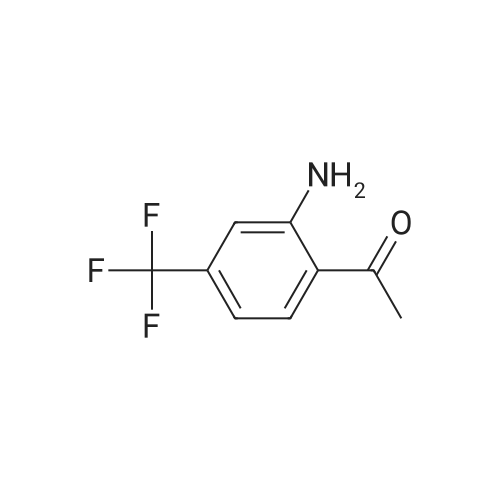
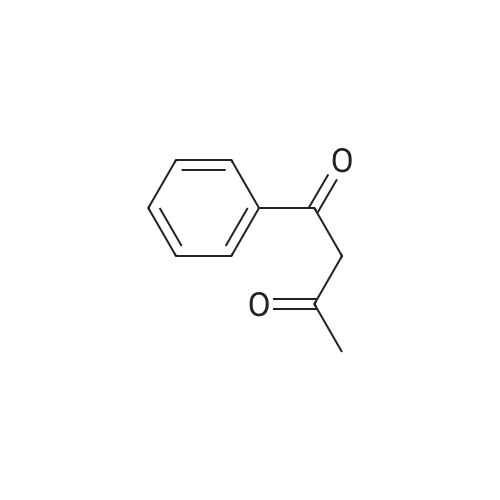
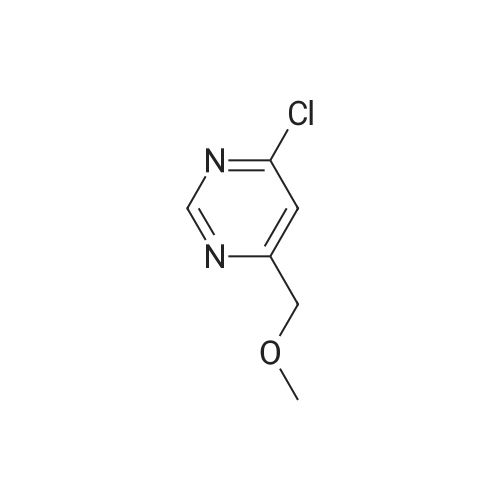
| 50% |
at 20 - 90℃; for 20h; |
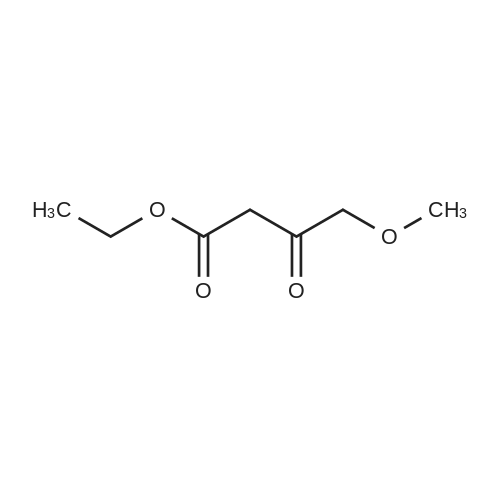
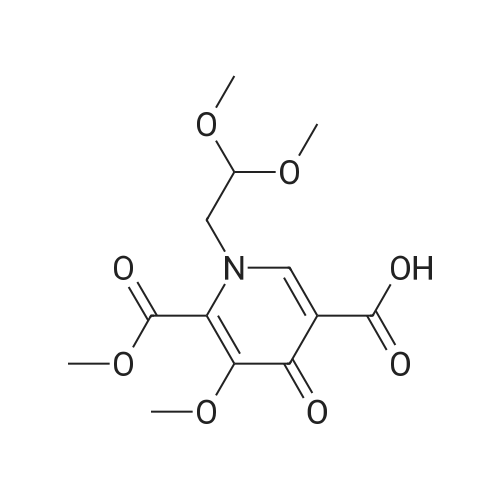
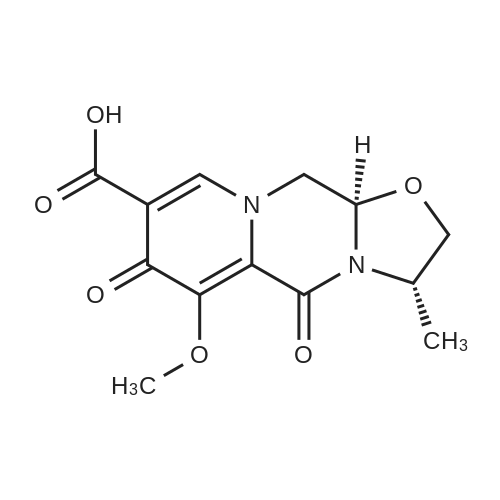
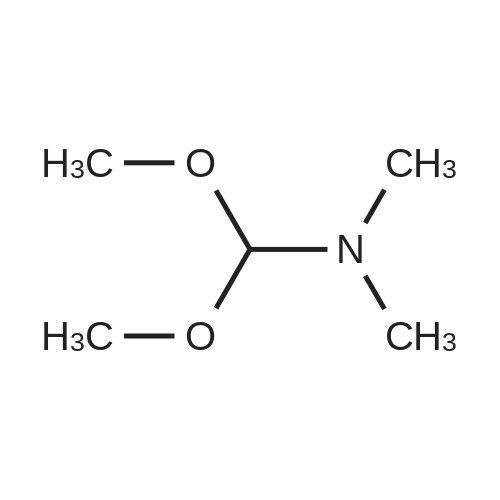
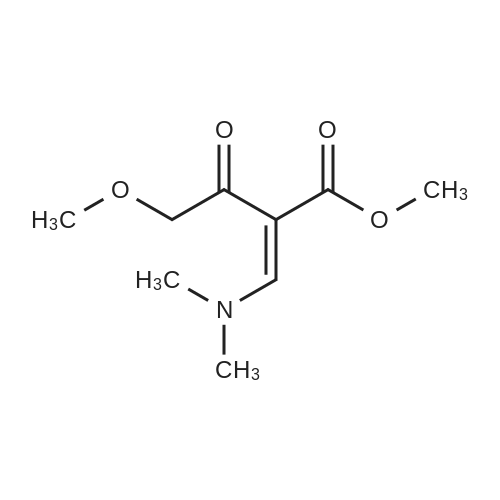
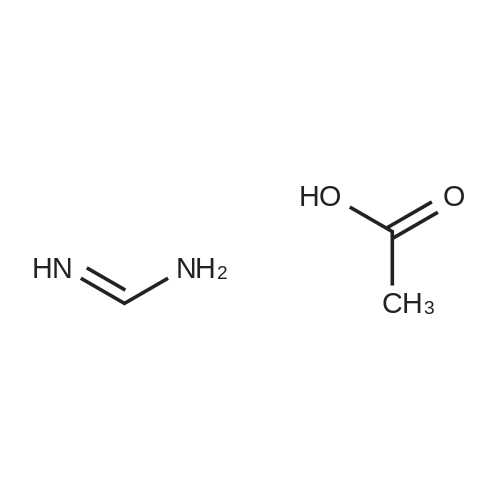
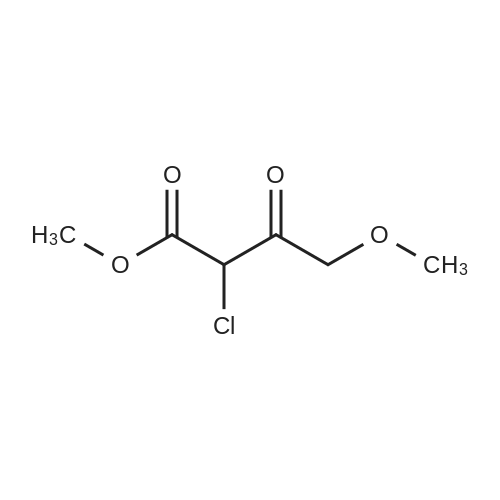
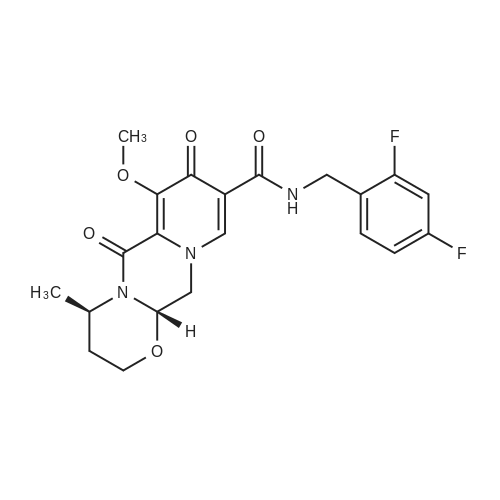
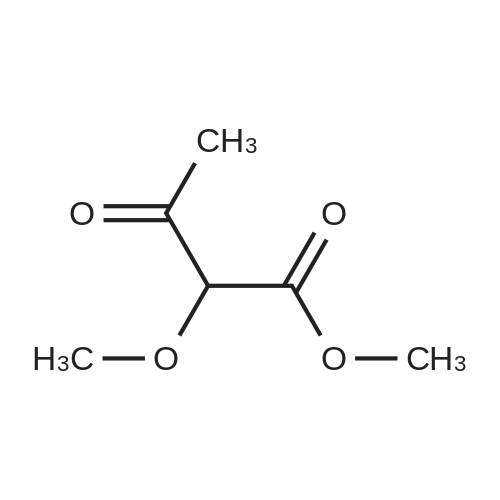
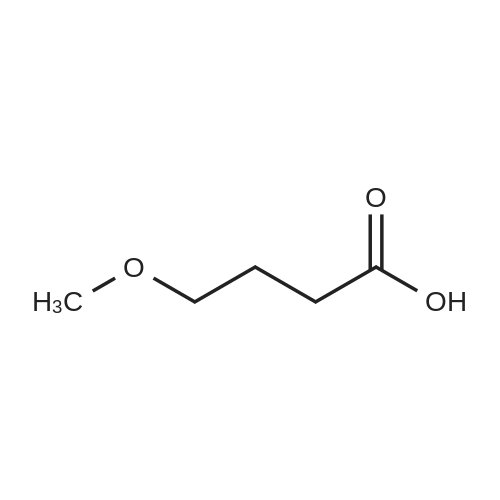
Preparation 13Methyl 2-f(dimethylamino)methylene1-4-methoxy-3-oxobutanoateMethyl-4-methoxyacetoacetate (9 ml, 70 mmol) was added to dimethylformamide dimethylacetal (18.8 ml, 139 mmol) and the reaction stirred at 9O0C for 2 hours before cooling to room temperature and stirring for 18 hours. The reaction was concentrated <n="62"/>in vacuo and purified by silica gel column chromatography, eluting with 70:30 to 100:0 ethyl acetate: heptane to afford the title compound as an oil (7.68 g, 50% yield).1HNMR (CDCI3): 2.87 (br s, 3H), 3.25 (br s, 3H), 3.39 (s, 3H), 3.72 (s, 3H), 4.37 (s, 2H), 7.74 (s, 1H) |
|
In toluene; for 2.16667h;Reflux; |
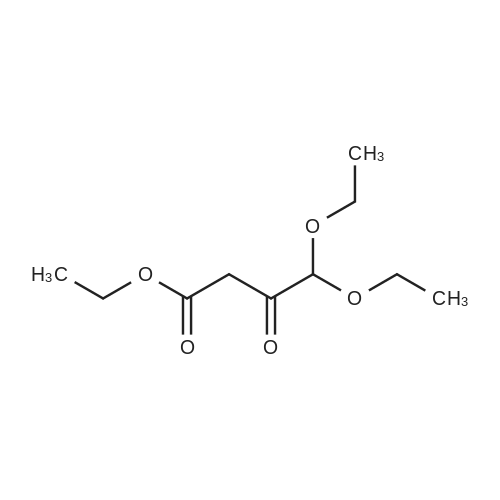
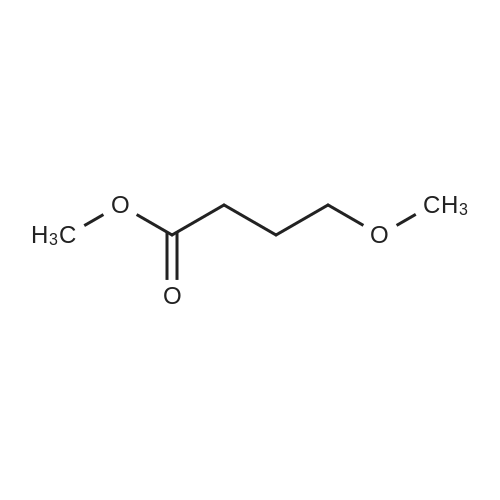
To a solution of Compound II-24(4-methoxy-3-oxobutanoic acid methyl ester, 9.87g) in toluene(99ml) was added dropwise 1,1-dimethoxy-N,N-dimethylmethanamine(26.9ml) over 10min, then the reaction mixture was refluxed for 2hrs. After termination of the reaction, the mixture was concentrated. The obtained product was used for the next reaction. According to the above procedure, the obtained residue was dissolved in acetonitrile soln.(136ml), and 80% hydrazine ethanol(6.58g) was added to the resulting solution. The reaction mixture was stirred at 50 C for one hour. After termination of the reaction, the mixture was concentrated. 1N aqueous HCl was added to the residue and extracted with ethyl acetate. The organic layer was washed with saturated aqueous NaHCO3 and brine successively, and dried over magnesium sulfate and concentrated. The residue was purified by silicagel columnchromatography to give Compound II-26(9.74g). |
|
at 20℃; for 1.5h; |
A. l-[2,2-Bis(methyloxy)ethyl]-5-(methyloxy)-6-[(methyIoxy)carbonyl]-4-oxo-l,4-dihydro- 3-pyridinecarboxylic acidA mixture of methyl 4-methoxyacetoacetate (20 mL) and DMFDMA (24 mL) was stirred at room temperature for 1.5 h. The reaction mixture was diluted with MeOH (50 mL) and aminoacetaldehyde dimethyl acetal (16.7 mL) was added. The mixture was stirred for 1 h at room temperature, concentrated, and then diluted with MeOH (113 mL). Dimethyl oxalate (45.66 g) was charged followed by portion-wise addition of LiH (2.15 g) while maintaining the reaction temperature below 25 C. The reaction content was heated to 40 C for 14 h. The reaction mixture was cooled to -5 C and LiOH (14.82 g) was added while maintaining the reaction temperature below 5 C. When addition was complete, the mixture was stirred for a further 2 h at 3-5 C for 1 h. The reaction mixture was quenched with aqueous HC1 (2 N, 367 mL), maintaining the reaction temperature below 5 C. When addition was complete, EtOAc (450 mL) was added and the mixture was warmed to 20 C. The reaction mixture was filtered and the aqueous layer discarded. Water (225 mL) was added and the organic layer was removed under reduced pressure. The product was collected by filtration and dried in a vacuum oven overnight at 50 C. The product was obtained as a solid. |
|
In toluene; at 20℃; for 11h; |
Example 5 First StepCompound 5A (598 mg, 4.09 mmol) and N,N-dimethylformamide dimethyl acetal (488 mg, 4.09 mmol) were dissolved in toluene (1 ml), and the mixture was stirred at room temperature for 11 hours. The solvent was distilled off from the reaction solution under reduced pressure, and the resulting residue (containing Compound 5B) was used in Second step without purification. |
|
In toluene; at 20℃; for 11h; |
Compound 5A (598 mg, 4.09 mmol) and N,N-dimethylformamide dimethyl acetal (488 mg, 4.09 mmol) were dissolved in toluene (1 ml), and the solution was stirred at room temperature for 11 hours. The solvent in the reaction solution was distilled off under reduced pressure, and the obtained residue (containing compound 5B) was used in Step 2 without being purified. |
|
at 20 - 27℃; for 8h; |
Methyl-4-methoxyacetoacetate (100 gm) was charged followed by slow addition of 1,1-dimethoxy dimethylethanamine (DMF-DMA) (98 gm) at a temperature of 20C. The reaction content was warmed to 27C and maintained the reaction mass for 8 hours at the same temperature. The reaction mixture was diluted with methanol (200 mL) and aminoacetaldehyde dimethyl acetal (78.8 gm) was added. The mixture was stirred for 2 hours at a temperature of 20C, distilled of the solvent and extracted with methylene dichloride. Lithium hexamethyldisilazane (LiHDMS) in THF (tetrahydrofuran) solution (1685 mL) was added slowly followed by gradual addition of dimethyloxalate (202 gm) while maintained the reaction temperature below 4C. The reaction content was heated to a temperature of 45C, stirred for 26 hours and then cooled to a temperature of 27C. The pH of the reaction mixture was adjusted to 3-4 with 2N HC1 solution (615 mL) and the product was extracted with ethyl acetate (1500 mL). The obtained crude product was purified by flash chromatography to get a purified compound of Formula 18 (65 gm). |
|
at 25 - 35℃; |
N,N-Dimethyl-l, l-bis(methyloxy)methanamine (196 g) was added to methyl-4-methoxy acetoacetate (formula 8a, 200 g) at 0-5 C. The temperature of the reaction mass was raised to 25-35 C and stirred at the same temperature until complete consumption of starting material. The reaction mass was then diluted with methanol and cooled to 15-20 C. Allylamine (86 g) was added and stirring was continued at 25-35 C until completion of reaction, as monitored by TLC. (NMR data of compound 6a: 1H NMR (CDC13): delta 3.61 (s, 3H), 4.40 (s, 2H), 3.29 (s, 3H), 8.02 (d, 1H, J=14.1Hz), 4.05 (t, 2H, J=5.7Hz), 5.87-6.00 (m, 1H), 5.16-5.22 (m, 2H), 10.80- 10.84 (broad, 1H)) Thereafter, the solution was concentrated under reduced pressure and then diluted with methanol (700 mL). Dimethyl oxalate (404 g) was then added and the solution was warmed to 40-45 C. Next, a sodium methoxide solution (530 g of sodium methoxide dissolved in 1.4 L of methanol) was added and the solution was stirred until complete consumption of the starting material, as monitored by TLC. The reaction mass was poured into a mixture of water (3.5 L) and methylene dichloride (2.4 L). The pH adjusted to 4-5 using acetic acid and the solution was filtered. The organic layer was separated and concentrated to yield a crude residue of dimethyl l-allyl-3-methoxy-4-oxo-l,4-dihydropyridine-2,5-dicarboxylate (NMR data of compound 5b: 1H NMR (CDC13): delta 8.39 (s, 1H), 4.63 (d, 2H, J=5.7Hz), 5.86-5.99 (m, 1H), 5.15-5.21 (dd, 1H, J=17.1Hz, 1.2Hz), 5.26-5.30 (dd, 1H, J=10.2Hz, 0.9Hz), 3.77 (s, 3H), 3.74 (s, 3H), 3.88 (s, 3H)) Boric acid (85 g) was added in portions to acetic anhydride (560 g) and heated to 70 C. The boroacetate solution was heated further to 90 C and maintained at that temperature for an hour. The crude residue of dimethyl l-allyl-3-methoxy-4-oxo-l,4- dihydropyridine-2,5-dicarboxylate (formula 5b) diluted in acetic acid (440 mL) was then added to the boroacetate solution at 70-75 C and stirring was maintained until complete conversion of starting material. The reaction mass was cooled to 2-5 C, water was added, and the solution was filtered to yield formula 5c (NMR data of compound of formula 5c: 1H NMR (CDCI3): delta 9.22 (s, 1H), 5.10 (d, 2H, J=6.3Hz), 6.00-6.13 (m, 1H), 5.33-5.43 (m, 2H), 3.84 (s, 3H), 4.00 (s, 3H), 1.92 (s, 6H)). Formula 5c was then hydrolyzed with methanolic HC1 (150 mL) at 25-35 C to give l-allyl-5-methoxy-6-(methoxycarbonyl)-4-oxo-l,4-dihydropyridine-3-carboxylic acid as white solid (formula 4a, 239 g, 1H NMR (DMSO-d6): delta 8.75 (s, 1H), 4.83 (d, 2H, J=5.7Hz), 5.89-6.02 (m, 1H), 5.22-5.28 (dd, 1H, J=17.1, 0.9Hz), 5.30-5.34 (dd, 1H, J=10.2, 0.9Hz), 3.89 (s, 3H), 3.93 (s, 3H), 15.28 (s, 1H)). |
|
at 20℃; for 1.5h; |
<strong>[41051-15-4]Methyl 4-methoxyacetoacetate</strong> (8 ml, 61.8 mmol),9.6 ml of N,N-dimethylformamide dimethyl acetal (DMF-DMA, 72.26 mmol) was stirred in a 150 ml round bottom flask for 1.5 h at room temperature.The solution gradually turns from yellow to brownish yellow, and the reaction gives compound 1(Rf = 0.14, TLC developing solvent: AcOEt/petroleum ether = 5/1).The reaction solution was diluted with 20 ml of methanol, and 6.68 ml (61.31 mmol) of aminoacetaldehyde dimethylacetal was added, and the mixture was stirred at room temperature for 1 h.The solution turns into wine red and the reaction gives compound 2(Rf = 0.56, TLC developing agent: AcOEt).The reaction solution was concentrated under reduced pressure to give a red brown oil,45.2 ml of methanol and dimethyl oxalate (18.34 g, 155.34 mmol) were added.After the dimethyl oxalate was completely dissolved, LiH (0.86 g, 108.67 mmol) was added in portions under the control of ice water bath at 25 C.The solution is a brown-red suspension. After the addition is completed, the reaction solution is placed at a bath temperature of 40 C for 14 hours.The solution changes from a suspension to a brick red solution, and the reaction gives compound 3(Rf = 0.23, TLC developing solvent: AcOEt).The reaction solution was cooled to 3-5 C and anhydrous LiOH (5.94 g, 123.8 mmol) was added to continue the reaction for 2 h.The reaction mixture was changed from brick red to orange yellow suspension, and the reaction gave Compound 3 (Rf=0.12, TLC developing solvent: AcOEt).The reaction was quenched by the addition of 2N HCl (146.8 mL).180 ml of ethyl acetate was added to the obtained reaction solution, and the solid was discarded by filtration.The liquid phase was collected and liquid-separated, 90 ml of water was added to the organic phase, and the organic solvent was evaporated under reduced pressure to give a large white solid.Filter by suction and wash the filter cake with a small amount of water.Obtained after vacuum drying at 50 C6.93g of compound 4(Rf = 0.44, TLC developing solvent: CH2Cl2 / MeOH = 40/1 + 0.5% HOAc),It was a colorless solid with a total yield of 36%. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping