| 150.4 g (89.5%) |
With thionyl chloride; In N,N-dimethyl-formamide; toluene; |
Step A 4-Fluorophenylacetyl chloride A solution of 150 g (0.974 mol) of 4-fluorophenylacetic acid an 1 mL of N,N-dimethylformamide in 500 mL of toluene at 40 C. was treated with 20 mL of thionyl chloride and heated to 400 C. An additional 61.2 mL of thionyl chloride was added dropwise over 1.5 hours. After the addition, the solution was heated at 50 C. for 1 hour, the solvent was removed in vacuo and the residual oil was distilled at reduced pressure (1.5 mmHg) to afford 150.4 g (89.5%) of the title compound, bp=68-70 C. |
| 150.4 g (89.5%) |
With thionyl chloride; In N,N-dimethyl-formamide; toluene; |
Step A' 4-Fluorophenylacetyl chloride A solution of 150 g (0.974 mol) of 4-fluorophenylacetic acid an 1 mL of N,N-dimethylformamide in 500 mL of toluene at 40 C. was treated with 20 mL of thionyl chloride and heated to 40 C. An additional 61.2 mL of thionyl chloride was added dropwise over 1.5 hours. After the addition, the solution was heated at 50 C. for 1 hour, the solvent was removed in vacuo and the residual oil was distilled at reduced pressure (1.5 mmHg) to afford 150.4 g (89.5%) of the title compound, bp=68-70 C. |
| 150.4 g (89.5%) |
With thionyl chloride; In N,N-dimethyl-formamide; toluene; |
Step A': 4-fluorophenylacetyl chloride A solution of 150 g (0.974 mol) of 4-fluorophenylacetic acid an 1 mL of N,N-dimethylformamide in 500 mL of toluene at 40 C. was treated with 20 mL of thionyl chloride and heated to 40 C. An additional 61.2 mL of thionyl chloride was added dropwise over 1.5 hours. After the addition, the solution was heated at 50 C. for 1 hour, the solvent was removed in vacuo and the residual oil was distilled at reduced pressure (1.5 mmHg) to afford 150.4 g (89.5%) of the title compound, bp=68-70 C. |
| 150.4 g (89.5%) |
With thionyl chloride; In N,N-dimethyl-formamide; toluene; |
Step A' 4-Fluorophenylacetyl chloride A solution of 150 g (0.974 mol) of 4-fluorophenylacetic acid an 1 mL of N,N-dimethylformamide in 500 mL of toluene at 40 C. was treated with 20 mL of thionyl chloride and heated to 40 C. An additional 61.2 mL of thionyl chloride was added dropwise over 1.5 hours. After the addition, the solution was heated at 50 C. for 1 hour, the solvent was removed in vacuo and the residual oil was distilled at reduced pressure (1.5 mmHg) to afford 150.4 g (89.5%) of the title compound, bp=68-70 C. |
| 150.4 g (89.5%) |
With thionyl chloride; In N,N-dimethyl-formamide; toluene; |
Step A': 4-Fluorophenylacetyl chloride A solution of 150 g (0.974 mol) of 4-fluorophenylacetic acid an 1 mL of N,N-dimethylformamide in 500 mL of toluene at 40 C. was treated with 20 mL of thionyl chloride and heated to 40 C. An additional 61.2 mL of thionyl chloride was added dropwise over 1.5 hours. After the addition, the solution was heated at 50 C. for 1 hour, the solvent was removed in vacuo and the residual oil was distilled at reduced pressure (1.5 mmHg) to afford 150.4 g (89.5%) of the title compound, bp=68-70 C. |
|
With thionyl chloride; In toluene; |
(a) p-Fluorophenylacetic acid (20 g) was stirred with thionyl chloride (30 ml) at room temperature for 5 hours. The excess of thionyl chloride was then removed under reduced pressure. The remaining oil was dissolved in toluene, and the toluene then removed under reduced pressure. The remaining oil was distilled (b.p. 41-42/0.1 mm) to give p-fluorophenylacetyl chloride (19.74 g). |
|
With thionyl chloride; N,N-dimethyl-formamide; In toluene; at 6 - 70℃; for 3.5h; |
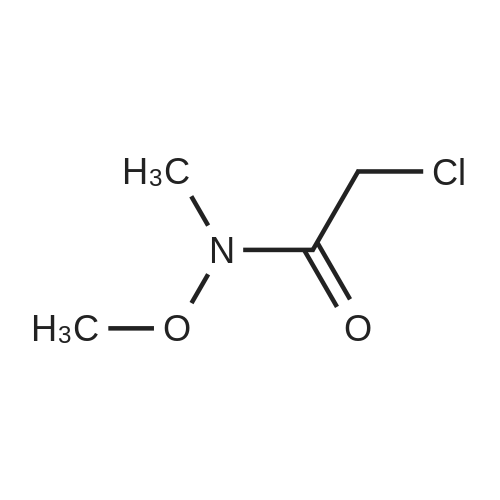
EXAMPLE 1 Process to Ketone Step 1: 2-(4-Fluorophenyl)-N-methoxy-N-methylacetamide (2) Summary: This reaction gives consistently high yield and high purity of material. No major side products have been identified. The final product is an oil (typically clear or slightly yellow) and is isolated with the above purity profile from the crude work up. Procedure: FW: Amt. Moles Equiv. 4-Fluorophenylacetic acid (1) 154 5.0 kg 32.47 mol 1.0 eq. DMF 73.1 48 mL 0.65 mol 0.02 eq. SOCl2 119 2.84 L 38.96 mol 1.2 eq. Weinreb amine-HCl 97.5 4.75 kg 48.70 mol 1.5 eq. NaOH 4.0 M 32.47 L 129.87 mol 4.0 eq. Toluene - 49.19 L - Brine - 64.92 L - A 100 L extractor equipped with a reflux condenser, and a base scrubber was charged with toluene (49.2 L, KF100 ppm) and 4-fluorophenylacetic acid (1) was added (5.0 kg). This solution was heated to 70 C. Once 70 C. was reached the DMF (48 mL, KF150 ppm) was added and thionyl chloride (2.8 L) was slowly added over 3 hours. Batch temperature will decrease while thionyl chloride is added. Typical temperature changes range from 6-10 C. When all thionyl chloride has been added and off-gassing has ceased (typically 30 min. after addition is complete) an aliquot of the batch was quenched into excess methanol for HPLC analysis as the methyl ester. Reaction is done when acid 1 is at <0.5 LCAP. Next the reaction was cooled to 5-10 C. The Weinreb amine-HCl (4.75 kg) was added to the batch at this point. Slow addition of NaOH (32.5 L) was begun at this point. This base was added at a rate that maintained the batch temperature at or below 10 C. with a typical addition time of 3 hours. Once this addition was done an aliquot of the batch was quenched into MeOH and assayed by HPLC to check for complete consumption of the acid chloride. Complete consumption of the acid chloride (in the form of the methyl ester after this quench) should be seen. Additional base can be added if the acid chloride is still present. The biphasic solution was separated at between 5 C. and room temperature and the organic phase was washed with 15 wt. % NaCl (aq) (2×32.5 L). Typical assay yield of the organic phase was 96%. The organic phase was concentrated to a 50 wt. % solution (typical KF500 ppm). |
|
With thionyl chloride; In toluene; at 105℃; for 16h; |
Step B: I-[(4-FluorophenyI)acetyl]piperidine4-FluorophenyIacetic acid (280g, 1.82mol) was suspended in 1.9L toluene followed by the careful addition of 185mL thionyl chloride (303g, 2.545mol). The reaction was heated to 1050C for 16hr (overnight). The reaction was allowed to cool to room temperature and the volatiles were removed in vacuo. The crude acid chloride was dissolved in 1.9L THF, cooled to 00C, and 0.72L piperidine (618g, 7.27mol) was added. The reaction vessel was allowed to warm to ambient temperature for 18hr. The mixture was quenched with a saturated solution of aq. NaHCθ3 and extracted several times withEtOAc. The combined organic extracts were washed with brine, dried over Na2SO^ and filtered on a <n="21"/>fritted funnel. The volatiles were removed in vacuo and the crude residue was purified on silica gel and eluted with a mixture of EtOAc/heptanes (0-75% gradient elution). This furnished the title compound. 1H-NMR (CDCI3): δ 1.32-1.42 (m, 2H), 1.48-1.64 (m, 4H), 3.34-3.42 (m, 2H), 3.54-3.60 (m, 2H), 3.69(s, 2H), 6.90-7.05 (m, 2H), 7.18-7.24 (m, 2H) ppm. |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In tetrahydrofuran; at 20℃; for 1h; |
To a solution of 4-fluorophenylacetic acid (9.9 g) in THF (100 ml) was added DMF (5 drops) and then oxalyl chloride (9.0 ml) was added at room temperature, and the mixture was stirred for 1 hr. The mixture was concentrated in vacuo to give 4-fluorophenylacetyl chloride. Aluminum chloride (16.0 g) was added to a suspension of 2H-1, 4-benzoxazin-3 (4H) -one (8.0 g) in 1, 2-dichloroethane (100 ml) under ice-cooling and then 4-fluorophenylacetyl chloride obtained above was added. The reaction mixture was allowed to warm to room temperature and stirred for 12 hr, then poured into ice-cooled water (200 ml) and the resulting crystals were collected by filtration. The crystals were suspended in methanol and the mixture was refluxed for 1 hr. After cooling the mixture, the resulting crystals were collected by filtration. The title compound was obtained as crystals (5.45 g) . |
|
With thionyl chloride; at 60℃; for 1h; |
4-Fluorophenylacetic acid [starting compound B] (15 g) was dissolved in thionyl chloride (15 ml) to prepare a solution which was then heated at 60C for one hr. Excess thionyl chloride was removed by evaporation under the reduced pressure to give 4-fluorophenylacetyl chloride. |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 1 - 2.1h;Product distribution / selectivity; |
Scheme 1; 2-(4-Fluorophenyl)acetyl isothiocyanate (2); Method A (For the Method B see scheme 20); To a solution of 4-fluorophenylacetic acid (1) (25 g, 162 mmol) in DCM (75 mL) was added oxalyl chloride (28.4 mL, 324 mmol) and 3-4 drops of DMF. The mixture was stirred at r.t. for Ih - 2h and concentrated to produce 2-(4-fluorophenyl)acetyl chloride (Ia) (yellow oil) <n="95"/>that was re-dissolved in toluene (100 mL). To this solution was added lead(II) thiocyanate (55.0 g, 170 mmol). The mixture was heated to reflux for 1.5h - 2h, cooled down, filtered and the filtrate was concentrated. The residue was applied onto a silica gel pad (20 cm) and eluted with EtOAc / hexanes (1/9), to afford after evaporation of the solvents title compound 2 (31 g, 98% yield) as a yellow oil. MS (m/z): 228.1 (M+H+MeOH). 1H NMR (400 MHz, DMSOd6) δ (ppm): 7.26-7.22 (m, 2H), 7.09-7.07 (m, 2H), 3.84 (s, 2H).; Step 5: tert-butyl (6-(7-(2-fluoro-4-(3-(2-(4-fluorophenyl)acetyl)thioureido)phenoxy)thieno[3,2- ]pyridin-2-yl)pyridin-3-yl)methyl(2-methoxyethyl)carbamate (14); To a solution of 4-fluorophenylacetic acid (1, scheme 1) (26.5 g, 172 mmol) in DCM (93 mL) was added oxalyl chloride (2 eq., 30.1 mL, 344 mmol) over 5 min and DMF (0.005 eq., 0.067 mL, 0.86 mmol) over 1 min. The reaction mixture was stirred at r.t for 2h then concentrated. The residual DCM was removed as an azeotrope with toluene (2x 20 mL) to afford intermediate 2-(4-fluorophenyl)acetyl chloride (Ia) (30.85 g, 179 mmol, assumed quantitative yield) as yellow oil. Part of that material was used as is in the next step. |
|
With thionyl chloride; In N,N-dimethyl-formamide; toluene; at 70℃; for 3h; |
A lOO L extractor equipped with a reflux condenser, and a base scrubber was charged with toluene (49.2 L, KF< 100 ppm) and 4-fluorophenylacetic acid (1) was added (5.0 kg). This solution was heated to 70 C. Once 70 "C was reached the DMF (48 mL, KF< 150ppm) was added and thionyl chloride (2.8 L) was slowly added over 3 hours. EPO <DP n="15"/>Batch temperature will decrease while thionyl chloride is added. Typical temperature changes range from 6-10 C.When all thionyl chloride has been added and off-gassing has ceased (typically 30 min. after addition is complete) an aliquot of the batch was quenched into excess methanol for HPLC analysis as the methyl ester.Reaction is done when acid 1 is at <0.5 LCAP.Next the reaction was cooled to 5-10 "C. The Weinreb amine-HCl (4.75 kg) was added to the batch at this point. Slow addition of NaOH (32.5 L) was begun at this point. This base was added at a rate that maintained the batch temperature at or below 10 "C with a typical addition time of 3 hours. Once this addition was done an aliquot of the batch was quenched into MeOH and assayed by HPLC to check for complete consumption of the acid chloride.Complete consumption of the acid chloride (in the form of the methyl ester after this quench) should be seen. Additional base can be added if the acid chloride is still present.The biphasic solution was separated at between 5 C and room temperature and the organic phase was washed with 15 wt. % NaCl (aq) (2 x 32.5 L).Typical assay yield of the organic phase was 96%.The organic phase was concentrated to a 50 wt. % solution (typical KF < 500 ppm). |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In tert-butyl methyl ether; at 20℃; for 0.833333h; |
4-(4-Fluoro-phenyl)-2-methyl-buta-2,3-dienoic acid ethyl ester To a solution of (4-fluoro-phenyl)-acetic acid (22.33 g, 144.9 mmol) in 100 mL of methyl tert-butyl ether and 250 μL of DMF was added 13.02 mL (146.3 mmol) of oxalyl chloride at room temperature dropwise over 30 minutes. The resulting mixture was stirred at room temperature for an additional 20 minutes (HPLC indicated completed reaction), and then the entire solution was added dropwise over 1 hour to a solution of N,N-diisopropylethylamine (50.48 mL, 289.8 mmol) and ethyl 2-(triphenylphosphoranylidene)propionate (50.0 g, 138.0 mmol) in 100 mL of methyl tert-butyl ether, while maintaining the internal temperature between 0-15 C. After the addition was complete, the reaction mixture was stirred for an additional 10 minutes at 0-10 C., when HPLC indicated a completed reaction. The reaction mixture was then diluted with 100 mL of heptane, and stirred for 30 minutes at 0-10 C. The resulting solid was filtered and washed with 2*100 mL of 1:1 methyl tert-butyl ether:heptane. |
|
With oxalyl dichloride;N,N-dimethyl-formamide; In tert-butyl methyl ether; at 20℃; for 0.833333h; |
To a solution of (4-fluoro-phenyl)-acetic acid (22.33 g, 144.9 mmol) in 100 ml_ of methyl terf-butyl ether and 250 μl_ of DMF was added 13.02 ml_ (146.3 mmol) of oxalyl chloride at room temperature dropwise over 30 minutes. The resulting mixture was stirred at room temperature for an additional 20 minutes (HPLC indicated completed reaction), and then the entire solution was added dropwise over 1 hour to a solution of Λ/,Λ/-diisopropylethylamine (50.48 ml_, 289.8 mmol) and ethyl 2- (thphenylphosphoranylidene)propionate (50.0 g, 138.0 mmol) in 100 ml_ of methyl terf-butyl ether, while maintaining the internal temperature between 0-15 C. After the addition was complete, the reaction mixture was stirred for an additional 10 minutes at 0-10 C, when HPLC indicated a completed reaction. The reaction mixture was then diluted with 100 mL of heptane, and stirred for 30 minutes at 0- 10 C. The resulting solid was filtered and washed with 2x 100 mL of 1 :1 methyl tert- butyl etheϖheptane. The filtrate and the washings were combined and washed with 100 mL of water, 100 mL of 1 M citric acid, 2x100 mL of water, then concentrated azeotropically at 25 C/60 mmHg to a total volume of -40 mL. The residue was diluted with 60 ml_ of methyl te/t-butyl ether. This solution was then directly used for the next step. |
|
With thionyl chloride; for 4h;Heating / reflux;Product distribution / selectivity; |
p-Fluorophenylacetic acid (31.4 g, 0.203 mol) is treated with SOCl2 (45 mL, 0.62 mol) and the resulting solution is heated at reflux for 4 hours. The reaction mixture is diluted with toluene and concentrated by distillation to remove remaining SOCl2. When the temperature of the distillate reaches 114C, the distillation is discontinued and the reaction mixture cooled to ambient temperature. |
|
With thionyl chloride; for 2h;Reflux; |
4-Fluorophenylacetic acid (900 mg) was dissolved in thionyl chloride (5 mL), and the solution was refluxed under heating for 2 hours. The reaction system was concentrated under reduced pressure and azeotroped with toluene, to thereby yield 4-fluorophenylacetyl chloride as a crude product. This acid chloride was dissolved in acetonitrile (20 mL), and potassium thioisocyanate (851 mg) was added to the solution, followed by stirring at 70 C. for 5 hours. The reaction mixture was cooled to room temperature and concentrated under reduced pressure. Subsequently, the product was separated with saturated aqueous solution of sodium hydrogencarbonate (100 mL) and ethyl acetate (50 mL). The organic layer was washed with saturated brine (100 mL) and dried over sodium sulfate, followed by concentration under reduced pressure, to thereby yield 4-fluorophenylacetyl thioisocyanate. This thioisocyanate was not subjected to further purification and dissolved in tetrahydrofuran (20 mL). A solution (20 mL) of compound 1c (374 mg) in tetrahydrofuran was added to the thioisocyanate solution and the mixture was stirred at room temperature for 12 hours. The reaction mixture was concentrated under reduced pressure, and the formed solid was filtrated, to thereby yield compound 19a (452 mg, yield: 79%).1H-NMR(CDCl3)δ: 12.47(1H,s), 11.82(1H,s), 8.73(1H,s), 8.65(1H,d,J=4.4 Hz), 7.95(1H, dd,J=11.2 Hz, 2.8 Hz), 7.49(1H,s), 7.43-7.40(1H,m), 7.31-7.25(3H,m), 7.15(2H,m), 6.42(1H,dd,J=5.2 Hz, 1.2 Hz), 4.03(3H,s), 3.74(2H,s), 1.64(9H,s); ESI-MS m/z 580(MH+). |
|
With thionyl chloride; In benzene; at 80℃; for 3h; |
The chemicals were purchased from the commercial venders and were used without purification. The reactions were monitored and the purity of the product was checked by thin layer chromatography (TLC). Silica gel 60 F254 chromatoplates were used for TLC. The solvent systems were chloroform/methanol (15:1). Thionyl chloride (1.5 ml) and 4-fluorophenyl acetic acid (0.5 mmol) were refluxed in benzene (5 ml) at 80 for 3 h, and then excess thionyl chloride was removed in vacuo [27]. The residue was dissolved in ether (10 ml) and the solution added during 1 h to a stirred, ice-cold mixture of 2-amino-5-nitrophenol (0.5 mmol), sodiumbicarbonate (0.5 mmol), diethyl ether (10 ml), and water (10 ml). The mixture was stirred overnight at room temperature and filtered. After the precipitate was washed with water, 2 N HCl and water, respectively, and finally with ether, compound was obtained. The crude product was purified by recrystallization from ethanol. |
|
With thionyl chloride; at 80℃; for 1h;Product distribution / selectivity; |
Synthesis of (4-Fluoro-phenyl) -acetic acid hydrazide; (4-Fluoro-phenyl)-acetyl chloride was readily prepared from thecorresponding acid by refluxing the acid in thionyl chloride for 1 h. However, the acid chloride was too reactive. Addition of hydrazine hydrate to a solution of the acid chloride gave only the dimer. Reverse addition of acid chloride to hydrazine hydrate at 0 C gave the desired hydrazide with some dimer. The acid chloride was then transformed to the corresponding ethyl ester. Reaction of the ethyl ester with 2 equiv. of hydrazine hydrate in refluxing EtOH gave cleanly the (4-Fluoro-phenyl)-acetic acid hydrazide in 76% yield. The results are summarized below. |
|
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; at 20℃;Inert atmosphere; |
General procedure: Acid (1 equiv) was placed in a round bottom flask and flushed with Ar (2x). Anhydrous DCM (7 mL) was then added followed by the dropwise addition of oxalylchloride (2 M in DCM, 3.1 equiv). After a few minutes, a few drops of anhydrous DMF were added to the reaction mixture and once the fizzing stopped, the reaction was allowed to stir overnight at R.T. Solvent was then evaporated using reduced pressure and the crude residue was used right away without any further purification. |
|
With thionyl chloride; at 80℃; for 3h; |
General procedure: For the synthesis of compounds 4a-4h and 5a-5h was depicted in Scheme 2. Firstly, 2-phenylacetic acid (1 mmol) and SOCl2 (4-6 mL) were refluxed at 80 C for 3 h. The reaction liquid was cooled to room temperature and then evaporated to give reactive acyl chloride. The product was obtained as an oil matter, which would be dissolved in acetone (5-6 mL) in the next step. Treatment of 3a (5-phenylthiazol-2-amine) with 2-phenylacetyl chloride in acetone for 5 h at ice-bath afforded the aimed amine. Meanwhile, K2CO3 (0.8 g) was added to the mixture. Then the mixture was evaporated under reduced pressure and the resulting solid was washed with diluted NaOH liquid. The aimed amide was extracted from the NaOH liquid with ethyl acetate for column chromatography. Column chromatography was performed using silica gel (200-300 mesh) eluting with ethyl acetate and petroleum ether to give the aimed amine (4a). Compounds of 4b-4h and 5a-5h could begot with corresponding acids by the procedures above. |
|
With thionyl chloride; for 3h;Reflux; |
A solution of 2-(4-fluorophenyl)acetic acid (5.0 g, 32.5 mmol, 1.00 equiv) in SOCl2 (30 mL) was heated for 3 hr at reflux in a 50-mL round-bottom flask. Most of SOCl2 was removed under vacuum, and the residue was added dropwise into a solution of AlCl3 (12.8 g, 96.2 mmol, 3.00 equiv) and fluorobenzene (6.1 g, 63.54 mmol, 2.00 equiv) in DCM (20 mL) at 0 C. The resulting solution was allowed to react, with stirring, for 2 hrs at room temperature. |
|
With thionyl chloride; In dichloromethane; at 0℃;Reflux; |
Example 2 Preparation of Intermediate Compound 2 Step A - Synthesis of Intermediate Compound 2b To a solution of compound 2a (100 g, 0.65 mol) in anhydrous DCM (1 L) was added SOCI2 (200 mL) dropwise at 0 C under a drying tube charged with CaCl2. After the addition, the mixture was heated to reflux and stirred overnight. The reaction was done in 2 batches, which were combined and concentrated in vacuo to provide crude compound 2b as an oil. |
|
With thionyl chloride; In N,N-dimethyl-formamide; toluene; at 70℃; |
The method of Kuethe et al. is depicted in FIG. 1. Briefly, commercially available 4-fluorophenylacetic acid g (Sigma-Aldrich Co. LLC, St. Louis, Mo.) is reacted with thionyl chloride in DMF/toluene to yield acid chloride (2). The acid chloride (6) is then reacted with the hydrochloride salt of the Weinreb amine (CH3NHOCH3.HCl) in the presence of sodium hydroxide to give 2-(4-fluorophenyl)-N-methoxy-N-methylacetamide (4). A vinyl Grignard reaction converts (4) to 1-(4-fluorophenyl)but-3-en-2-one (5). TES dienyl ether (6) is produced from the reaction of (5) with chlorotriethylsilane (TESCl) in the presence of iPr2NEt2. |
|
With trichlorophosphate; In 1,2-dichloro-ethane; for 3h;Reflux; |
General procedure: Aralkanoic acid chlorides 2a-g were synthesized by the reaction of aralkanoic acid 1a-g (1 mmol) in the presence of 1,2-dichloroethane (12 mL) solvent and phosphorous oxychloride(0.4 mL) chlorinating agent under reflux for 3hours. Then, the resulting solution was cooled to room temperature, and the solvent was removed under reduced pressureto afford aralkanoic acid chloride 2a-g, which was directly used in the next step without further purification. Acid chloride 2a-g was dissolved in acetonitrile (80 mL), addeddropwise to a solution containing hydrazine hydrate(1 mmol), TEA (0.5 mL) and acetonitrile (20 mL) and allowed to reflux for 3 hours with monitoring by TLC. After consumption of the starting material, the reaction mixture was cooled to room temperature. Evaporation of the solvent under reduced pressure yielded crude acid hydrazide 3a-g as a white solid on cooling, which was purified by column chromatography and crystallized in methanol [46]. |
|
With thionyl chloride; In dichloromethane; at 0℃;Reflux; |
To a solution of compound 2a (100 g, 0.65 mol) in anhydrous dichloromethane (1 L) was added SOCI2 (200 mL) dropwise at 0 C under a drying tube charged with CaCl2. After the addition, the mixture was heated to reflux and stirred overnight. The reaction was done in 2 batches, which were combined and concentrated in vacuo to provide crude compound 2b as an oil that was used without further purification. |
|
With thionyl chloride; In dichloromethane; at 0℃;Reflux; |
To a solution of compound 2a (100 g, 0.65 mol) in anhydrous dichloromethane (1 L) was added SOC12 (200 mL) dropwise at 0 C under a drying tube charged with CaC12. Afterthe addition, the mixture was heated to reflux and stirred overnight. The reaction was done in 2 batches, which were combined and concentrated in vacuo to provide crude compound 2b as an oil that was used without flirther purification. |
|
With thionyl chloride; In N,N-dimethyl-formamide; at 0℃; for 3h; |
General procedure: To the carboxylic acid (10mmol) was added thionyl chloride (20 mmol) followed by a catalytic amount ofDMF (1 drop). The mixture was stirred at 0 C until completion (ca. 3 h). Thesolvent was then removed under reduced pressure to afford the crude acidchloride. Alkoxyamine hydrochloride (11 mmol) was added to a mixture of K2CO3(20 mmol) in a 2:1 mixture of EA: H2O (0.2 M). The resultingsolution was cooled to 0 C followed by dropwise addition of the crude acidchloride dissolved in EtOAc (50 mL). The reaction was allowed to stir for 4 hwhile reaching room temperature. The two layers were separated, and the aqueousphase was extracted with EA (50 mL × 2). The combined organic phase was driedover anhydrous Na2SO4,filtered, and evaporated under reduced pressure. The residue was purified by flash columnchromatography on silica gel to give the desired products. |
|
With thionyl chloride; at 80℃; for 4h;Inert atmosphere; |
General procedure: A dry flask containing the corresponding carboxylic acid (1 mmol) and SOCl2 (0.6 mL) was heated at 80 C for 4 h under a nitrogen atmosphere. After the reaction period, the reaction mixture was concentrated under reduced pressure to remove the volatiles and then, the resultant crude reaction mixture was diluted with anhydrous DCM (2 mL). The DCM solution of corresponding acid chloride was slowly added to another RB flask containing the corresponding amine (1 mmol), Et3N (111 mg, 1.1mmol) and DCM (4 mL) under a nitrogen atmosphere. The resulting mixture was stirred at rt for 12 h. After this period, the reaction mixture was diluted with dichloromethane and washed with water and saturated aqueous NaHCO3 solution (twice). The combined organic layers were dried over anhydrous Na2SO4 and then, the solvent was evaporated in vacuo to afford a crude reaction mixture. Purification of the crude reaction mixture by column chromatography (neutral alumina (EtOAc/hexanes=25:75) furnished the corresponding products 1f-j. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In tetrahydrofuran; at 0℃; for 0.5h;Inert atmosphere; |
General procedure: A carboxylic acid (0.75 mmol) was added into a dry double-neckflask, and 5 mL of dry THF was added to the flask. N,N-dimethylformamide(1.50 mmol, 0.15 mL) was added into the solutionunder nitrogen at 0 C. When the solids were completely dissolved,oxalyl chloride (3.75 mmol) was added slowly. After 30 min, thesolvent and excess oxalyl chloride were removed under reducedpressure to obtain an acyl chloride which was kept dry. 100 mg ofcompound 2-4 (0.53 mmol) was added into a dry double-neckflask, and 5 mL of dry THF was added to the flask. When thesewere completely dissolved, dry pyridine (1.06 mmol, 0.01 mL) wasadded under nitrogen at 0 C. The THF solution of an acyl chloride(0.793 mmol) was added into the mixture slowly. After 30 min,20 mL of saturated ammonium chloride solution was slowly addedinto the mixture, and the mixture was extracted with EtOAc(3 x 20 mL). The organic layer was dried over anhydrous Na2SO4,filtered and concentrated to dryness, then purified through silicagel column (EA: PE 2: 1) to give B-1~B-15. |
|
With trichlorophosphate; In 1,2-dichloro-ethane; for 3h;Reflux; |
General procedure: Aralkanoic acid chlorides 2 were synthesized by the reaction ofaralkanoic acids 1 (1 mmol) in the presence of 1,2edichloroethane(12 mL) solvent and phosphorous oxychloride (0.4 mL) as chlorinatingagent under reflux for 3 h. Then, the resulting solution wascooled to room temperature, and the solvent was removed underreduced pressure to afford aralkanoic acid chlorides 2, which wasdirectly used in the next step without further purification. Aralkanoicacid chlorides 2 was dissolved in acetonitrile (80 mL), addeddrop wise to a solution containing hydrazine hydrate (1 mmol), TEA(0.5 mL) and acetonitrile (20 mL) and allowed to reflux for 3 h withmonitoring by TLC. After consumption of the starting material, thereaction mixture was cooled to room temperature. Evaporation ofthe solvent under reduced pressure yielded crude substituted aromaticacid hydrazides 3 as a white solid on cooling, which waspurified by column chromatography and crystallized on methanol. |
|
With thionyl chloride; In benzene; at 20℃; for 18h; |
General procedure: To a stirred solution of carboxylic acid 7a-h or 9c, i (0.55 mmol)in CH2Cl2 for 7a-h or benzene for 9c, i (3 mL) was added SOCl2(0.05 mL, 0.66 mmol), and the resulting mixture was stirred atroom temperature for 18 h. The solvent was removed, and thento the residue were added CH2Cl2 (3 mL), NH4SCN (63 mg,0.82 mmol), and PEG-400 (1 drop), and the resulting mixture wasstirred at room temperature for 2 h. The solvent was removed,and the residue was chromatographed on SiO2 (Hexane:Acetone= 50:1 40:1) to give corresponding isothiocyanate 8a-hand 10c, i which were used in the next step immediately. |
|
With thionyl chloride; In N,N-dimethyl-formamide; toluene; for 6h;Reflux; |
15.4 g (0.1 mol) of p-fluorophenylacetic acid, 24.0 g (0.2 mol) of thionyl chloride, 60 ml of toluene and 4 drops of DMF were placed in a reaction flask,The reaction was refluxed for 6 hours, evaporated to dryness under reduced pressure to give a light red liquid, diluted with 20ml of acetone, spare. |
|
With thionyl chloride; at 65 - 70℃; for 12h; |
General procedure: The substituted phenylacetic acid (3 mmol) and thionyl chloride (10 ml) were placed in a dry round-bottomed flask, and the mixture was heated to reflux at 65-70 C for 12 h, which was detected by TLC. After the completion of reaction, the solvent was removed in vacuo and the residue was washed with CH2Cl2. The above procedure was repeated twice to obtain the crude intermediates 4, which were used for the next reaction without further purification. |
|
With thionyl chloride; for 2h;Heating / reflux; |
Step A: Preparation of Ethyl 2-(4-fluorophenyl)acetate; [0282] A mixture of 2-(4-fluorophenyl)acetic acid (23.5 g, 152 mmol) in thionyl chloride 56 mL, 762 mmol) was refluxed for 2 hours and then concentrated in vacuo. The residue was diluted with 200 mL DCM, and stirred at O0C. The mixture was treated with EtOH (9.8 mL, 168 mmol) and TEA (26 mL) dropwise. The mixture was then stirred for 2 hours. The mixture was quenched with 20 mL H2O and extracted with DCM (3 x 50 mL). The combined organics were washed with H2O (3 x 50 mL) and brine 20 mL, dried over anhydrous Na2SO4, and concentrated in vacuo to give 24.7 g of a pale yellow oil. |
|
With thionyl chloride; |
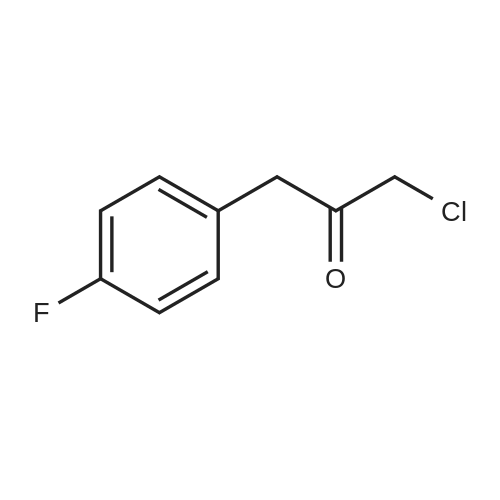
Example 12.7: Preparation of l-Amino-3-(4-fluoro-phenyl)-propan-2-one hyrdrochlorideCNCH2CO2Et 1BuOKThe proecdures as described in Tetrahedron. 1994, 50 (21), 6287-6298 and Chem. Pharm. Bull 1984, 32 (7), 2536-2543 were adapted to afford l-amino-3-(4-fluoro- phenyl)-propan-2-one hydrochloride. |
|
With thionyl chloride; In toluene; at 105℃; for 16h; |
4-Fluorophenylacetic acid (28Og, 1.82mol) was suspended in 1.9L toluene followed by the careful addition of 185mL thionyl chloride (303g, 2.545mol). The reaction was heated to 1050C for 16hr (overnight). The reaction was allowed to cool to room temperature and the volatiles were removed in vacuo. The crude acid chloride was dissolved in 1.9L THF, cooled to 00C, and 0.72L piperidine (618g, 7.27mol) was added. The reaction vessel was allowed to warm to ambient temperature for 18hr. The mixture was quenched with a saturated solution of aq. NaHCO3 and extracted several times withEtOAc. The combined organic extracts were washed with brine, dried over Na2SO4, and filtered on a fritted funnel. The volatiles were removed in vacuo and the crude residue was purified on silica gel and eluted with a mixture of EtO Ac/heptanes (0-75% gradient elution). This furnished the title compound. 1H-NMR (CDCI3): δ 1.32-1.42 (m, 2H), 1.48-1.64 (m, 4H), 3.34-3.42 (m, 2H)5 3.54-3.60 (m, 2H), 3.69(s, 2H), 6.90-7.05 (m, 2H), 7.18-7.24 (m, 2H) ppm. |
|
With thionyl chloride; In dichloromethane; at 50℃; for 2h; |
Dissolve 2- (4-fluorophenyl) acetic acid (100.0 g, 648 mmol, 1.0 eq) in dichloromethane (200 mL), add sulfoxide (193.0 g, 1622 mmol, 2.5 eq), and stir the reaction at 50 C. 2 The reaction was monitored for completion by TLC and concentrated under reduced pressure to give the product (116.0 g of crude product). |
|
With thionyl chloride; In dichloromethane; at 60℃; for 3h;Inert atmosphere; |
Add 2- (4-fluorophenyl) acetic acid (50.0 g, 324.380 mmol, 1.0 eq)And dichlorosulfoxide (77.18g, 648.761mmol, 2.0eq)Dissolved in dichloromethane (250.0mL),The temperature was raised to 60 C under N2 protection and refluxed for 3h. TLC showed that the reaction was complete.The reaction solution was concentrated under reduced pressure, and an appropriate amount of dichloromethane was added and concentrated.This was repeated twice to obtain a yellow oily product. |
|
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; for 2h;Cooling with ice; |
Add 473mg of 4-fluorophenylacetic acid and 25ml of DCM into a 100ml single-necked flask, After stirring for a while, add (2eq) 0.513ml of oxalyl chloride under ice bath, Two drops of DMF are used as a catalyst, and the reaction is very violent, releasing a lot of gas. Stir in an ice bath for 2 hours, evaporate the solvent to obtain 4-fluorophenylacetyl chloride, which is prepared for immediate use. |
|
With oxalyl dichloride; N,N-dimethyl-formamide; In N,N-dimethyl-formamide; at 0 - 20℃; for 6h;Inert atmosphere; |
General procedure: To a stirred solution of substituted or unsubstituted phenylaceticacid (14ae14f) (5.0 mmol, 1.0 eq) in anhydrous dichloromethane(20 mL)was successively added oxalyl chloride (6.0 mmol,1.2 eq) and DMF (0.5 mmol, 0.1 eq) at 0 C under argon atmosphere.The resulting mixture was subsequently warmed up to roomtemperature and stirred for 6 h. The reaction mixture wasconcentrated in vacuum to afford the corresponding phenylacetylchloride. The solution of the corresponding phenylacetyl chloridein anhydrous dichloromethane (10 mL) was slowly added to astirred solution of 2,2-dimethyl-1,3-dioxane-4,6-dione (5.0 mmol,1.0 eq), pyridine (10.0 mmol, 2.0 eq) in anhydrous dichloromethane(10 mL) at 0 C under argon atmosphere. The resulting mixture wassubsequently warmed up to room temperature and stirred for 5 h.The reaction mixture was concentrated and the resulting residuewas dissolved in anhydrous ethanol (20 mL), and the mixture washeated to 80 C for additional 4 h. After cooling, the mixture wasconcentrated to dryness and distributed inwater and ethyl acetate.The organic layer was separated and washed with water and brinesuccessively, dried over anhydrous sodium sulfate and concentratedin vacuum. The resulting residue was purified by silica gelchromatography (petroleum ether/ethyl acetate, v/v, 99:1 to 90:10)to give the desired product.5.1.1.1. Ethyl 4-(4-fluorophenyl)-3-oxobutanoate (16a). The titlecompound was prepared from 15a following general procedure A.Yield: 86%. 1H NMR (300 MHz, DMSO-d6) δ7.26e7.09 (m, 4H), 4.08(q, J 7.2 Hz, 2H), 3.88 (s, 2H), 3.66 (s, 2H), 1.17 (t, J 7.2 Hz, 3H). |
|
With thionyl chloride; at 0 - 80℃; for 7h; |
General procedure: The starting material of phenylacetic acid analogues (20mmol) was added into SOCl2 (25mL) at 0 , then the mixture was warmed to 80 and stirred for 7h. Then, the mixture was cooled to rt, the solvent was concentrated in vacuo to afford the crude product 1a-1k which was directly for next reaction without further purification. |
|
With thionyl chloride; N,N-dimethyl-formamide; In dichloromethane; at 20℃; for 2h; |
General procedure: Corresponding acid (1.0 equiv, 10 mmol) was dissolved in 25 mL CH2Cl2 in a 100 mL round-bottom flask with a stir bar, one drop of DMF was added, then SOCl2(2.0 equiv, 20 mmol) was slowly added into the mixture at room temperature, the obtained solution was stirred for 2 h at room temperature. The title acyl chloride was obtained after evaporating the solvent and used without further purification. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping