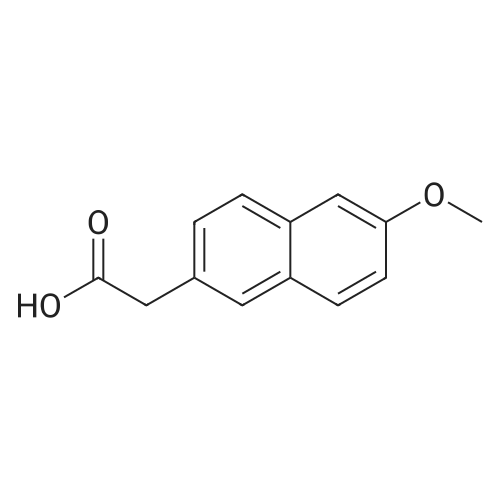
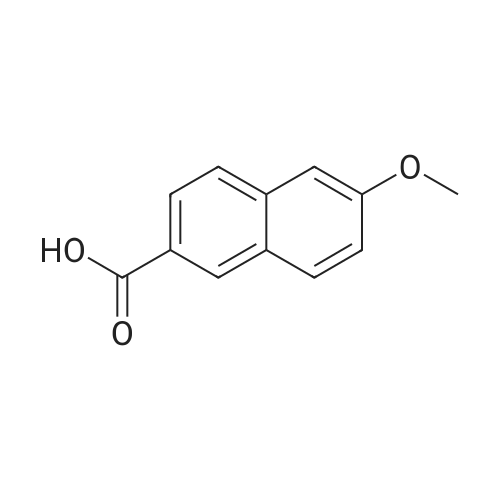
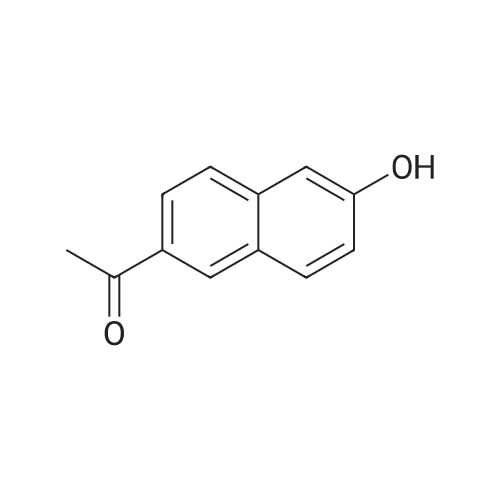
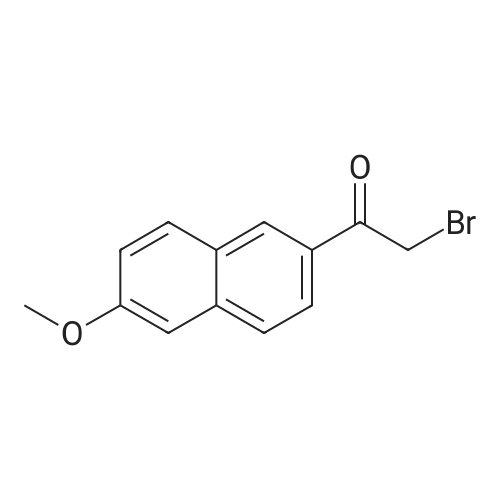
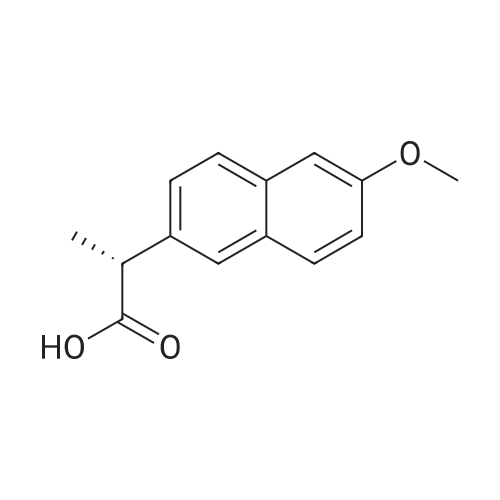
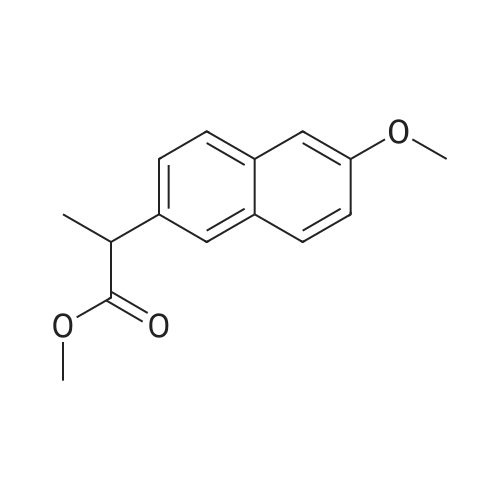
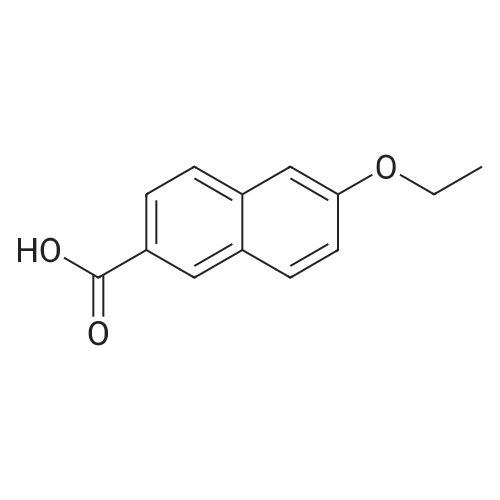
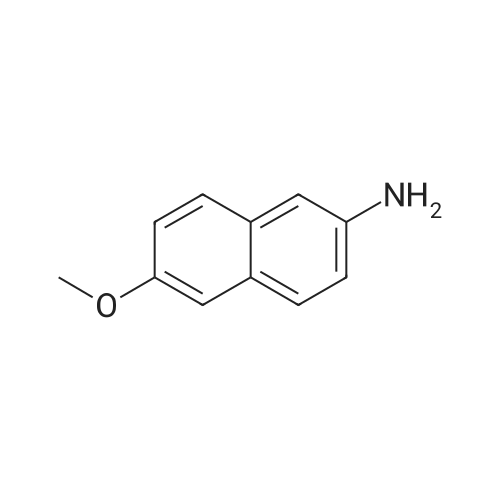
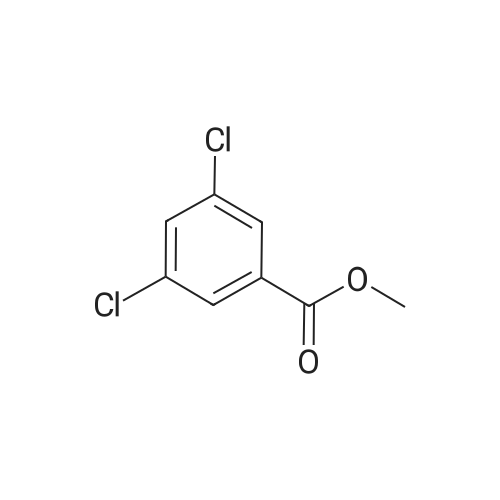
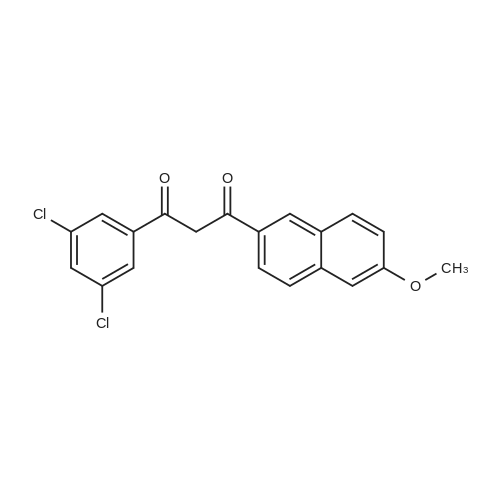
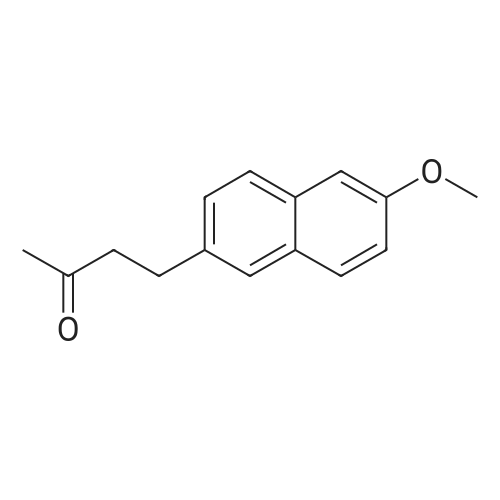
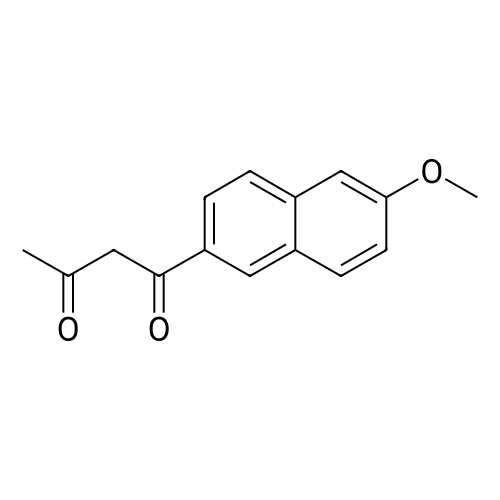
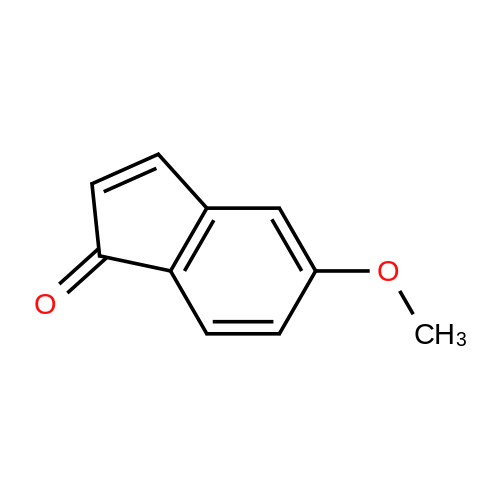
| 90.2% |
With hydrogenchloride; triethylamine In dichloromethane; water at 85℃; for 4h; |
A-1 Synthesis of Compound 1
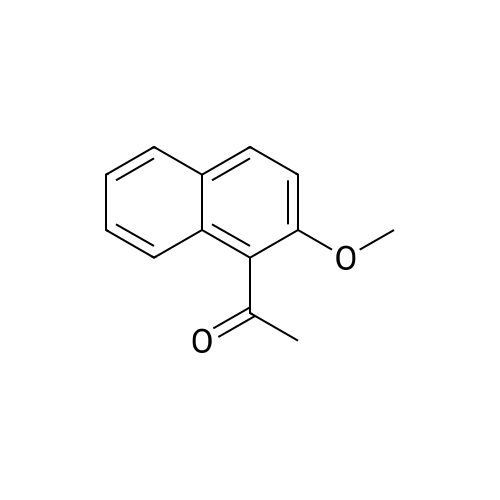
After 6-acetyl-2-methoxynaphthalene (1.00 g, 5 mmol) was dissolved in 4 mL of dichloromethane,A solution of 36% hydrochloric acid (80 mL, 0.93 mol) was added dropwise,While stirring side edge,Triethylamine (0.75 mL, 5.4 mmol) was added dropwise dropwise,Then stirred at 85 4h;After the reaction is complete,Excessive acid is neutralized with sodium hydroxide,Extracted with ethyl acetate,And washed with saturated sodium bicarbonate and brine,The organic phase was dried over anhydrous sodium sulfate,The solvent was distilled off under reduced pressure,And purified by silica gel column chromatography,Petroleum ether and ethyl acetate elution separation,The eluent ratio is ethyl acetate: petroleum ether = 1: 5,The yield was 90.2%. |
| 89% |
With hydrogenchloride In dichloromethane for 2h; Heating; |
|
| 89% |
With hydrogenchloride; water In dichloromethane for 2h; Heating / reflux; |
1.b Preparation of 2-(1-{6[ethyl-(2-{8-[4-(4-fluorophenyl)-4-oxobutyl]-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl}ethyl)-amino]-2-naphthyl}ethylidene)malononitrile
Example 1(b) Preparation of 2-(1-{6[ethyl-(2-{8-[4-(4-fluorophenyl)-4-oxobutyl]-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl}ethyl)-amino]-2-naphthyl}ethylidene)malononitrile In a 3 L two-neck round bottom flask, equipped with a reflux condenser and a dropping funnel, 2 L of hydrochloric acid (d=1.16) were stirred and heated to boiling. A solution of 6.06 g (30.3 mmol) of 1-(6-methoxy-2-naphthyl)-1-ethanone (prepared as described in Arsenijevic et al., Org. Synth. Coll. 6:34 (1988), the disclosure of which is incorporated herein by reference) in a minimum amount of dichloromethane was added, and the mixture was stirred and heated at reflux for 2 hours. The hot solution was filtered through a mineral wool plug to remove oily residue. The solid that separated after cooling was filtered on a glass frit and dissolved in 130 mL of ethyl acetate. The solution was washed with brine, dried with anhydrous magnesium sulfate and evaporated to give 5 g (89%) of 1-(6-hydroxy-2-naphthyl)1-ethanone. |
| 84% |
With hydrogenchloride In dichloromethane; water for 2h; Reflux; |
|
| 83% |
With boron tribromide In dichloromethane at -78 - 20℃; for 2.16667h; |
6-acetyl-2-hydroxy- 1 -naphthaldehyde.
To 1 -(6-metho xynaphthalen-2 - yl)ethan-l -one (0.200g, 0.99mmol) in anh. DCM (1 OmL) was added boron iribromide at - 78 °C. Mixture was stirred at -78 for 10 min, then at r.L for 2 hr. Water was added, followed by extraction with DCM. Purification by column chromatography to yield intermediate 1 -(6-hydroxynaphtlen-2-yl)ethan- 1 -one (0.155g, 83%). Formation of final product closely followed general procedure B. Purification by column chromatography yielded desired product 6-acetyl-2 -hydroxy- 1 -naphthaldehyde (0.047g, 29%). IH NMR (600 MHz, Chloroform-d) 8 13.32 (s, lH), 10.83 (s, IH), 8.44 - 8.40 (m, 2H), 8.19 (d, J= 9.5 Hz, IH), 8.10 (d, J === 9.2 Hz, IH), 7.23 (d, J === 9.2 Hz, 1H), 2.72 (s, 3H) ppm. l3C NMR (150 MHz, CDClj) 8 197.33, 193.35, 166.66, 140.27, 135.85, 133.31, 131.17, 127.68, 127.02, 120.47, 1 19.17, 1 11.44, 26.67 ppm. HRMS (ES ) calculated for [CuHtOtf 213.0557, found 213.0556. |
| 81% |
With hydrogenchloride; triethylamine In dichloromethane; water at 110℃; for 2h; Reflux; |
|
| 81% |
With hydrogenchloride; triethylamine In dichloromethane at 110℃; for 2h; |
1.1 (1) Synthesis of Compound 1
10 g of 1-(6-methoxy-2-naphthyl)-1-ethanone, 80 mL of concentrated hydrochloric acid,2 mL of dichloromethane and 15 drops (about 0.75 mL) of triethylamine were added to 100 mLIn a three-necked round bottom flask, stir and heat to 110 ° C for 2 h.After cooling, the reaction mixture was filtered to give a white solid crystals.The solvent was then removed under reduced pressure to give a crude material which was dissolved in 1 mol/L.In the NaOH solution, a 1 mol/L hydrochloric acid solution was added at 0 ° C until the yellow precipitate formed completely, and the pure compound 1 was filtered off, and dried to obtain 8.09 g, and the yield was 81%. |
| 75% |
With hydrogenchloride Reflux; |
|
| 74% |
With hydrogen bromide In acetic acid at 100℃; for 12h; |
|
| 74% |
With hydrogen bromide; acetic acid In water at 100℃; for 12h; |
1.1
Preparative Example 1.16-acetyl-2-hydroxynaphthalene (Formula 8) To a solution containing 6-acetyl-2-methoxynaphthalene (10.4 g, 52 mmol) in glacial acetic acid (100 mL), 48% HBr (43.0 g, 0.53 mol) was added. The mixture was stirred at 100° C. for 12 hr. Excess acetic acid was removed in vacuo, and the residue was taken up in ethyl acetate and washed with dilute NaHCO3 and brine. The organic layer was dried with MgSO4 and the solvent was removed in vacuo. The product was purified by column chromatography using ethyl acetate/hexane (1:1) as the eluent.Yield 7.2 g (74%); m.p. 173° C.; IR (KBr): 3,362, 1,664 cm-1; 1H NMR (300 MHz, CDCl3): δ 8.41 (d, 1H, J=2 Hz), 7.99 (dd, 1H, J=9, J=2 Hz), 7.87 (d, 1H, J=9 Hz), 7.70 (d, 1H, J=9 Hz), 7.20 (d, 1H, J=2 Hz), 7.18 (dd, 1H, J=9, J=2 Hz), 5.70 (br s, 1H), 2.71 (s, 3H). Anal. Calcd. for C12H10O2: C, 77.40; H, 5.41. Found: C, 77.52; H, 5.46. |
| 74% |
With hydrogen bromide In acetic acid at 100℃; for 122h; |
|
| 72% |
With potassium carbonate; thiophenol In 1-methyl-pyrrolidin-2-one at 194℃; for 0.75h; Inert atmosphere; |
|
| 60% |
With pyridine hydrochloride at 200℃; for 2h; |
|
| 60% |
With hydrogen bromide In acetic acid at 100℃; for 12h; |
|
| 50% |
With hydrogenchloride; triethylamine In dichloromethane; water at 85℃; for 4h; |
|
| 43% |
With hydrogen bromide In acetic acid at 100℃; for 12h; |
1.1 Example 1
(1) The synthesis (reference is carried out) of 6-acetyl-2-naphthol (abbreviated as compound 2):In a three-necked flask, 20 grams of 6-methoxy-2-acetylnaphthalene (abbreviated as compound) are added sequentially 1) Stir with 200 ml of glacial acetic acid, then add 86 g of hydrobromic acid, and stir and reflux at 100°C for 12 hours. The black reaction solution is heated at 60°C to remove the solvent under reduced pressure, and 10% sodium bicarbonate solution is added to neutralize it. After extraction with ethyl acetate, the organic phase was dried with anhydrous MgSO4, filtered, and the solvent was removed under reduced pressure. After column separation, 8.03g of 43.17mmol yellow powder was obtained as compound 2, with a yield of 43%; wherein the eluent used for column separation was V (petroleum ether): V (ethyl acetate) = 2:1; the structure and literature of the compound 2 (HMKim, C. Jung, BRKim, et al., Angew. Chem. Int. Ed., 2007 ,46:3460-3463) The report is consistent |
| 33% |
With hydrogen bromide In dichloromethane at 90℃; for 2h; |
Synthesis of 1-(6-hydroxynaphthalen-2-yl)ethanone (2)
To a stirring hot solution of concd. HBr (100ml), 1-(6-methoxynaphthalen-2-yl)ethanone (2.0 g, 10.00 mmol) in dry CH2Cl2 (5 mL) was added dropwise. The reaction mixture was heated at 90°C for 2 h, and treated with 6 N aqueous NaOH to make it basic. The black precipitate was ltered to give a kelly solution then added 2 N aqueous HCl, the pale white precipitate which formed was ltered and washed with water to yield 2 (614 mg, 33.0 % yield). IR: 3364, 1662, 1627, 1570, 1486, 1435, 1370, 1352, 1289, 1207, 1159, 972, 928, 901, 877, 816. 1H NMR (CDCl3, 400MHz): δ 2.65 (s, 3H), 5.60 (s, 1H), 7.12 (d, J = 7.4 Hz, 1H), 7.13 (s, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.81 (d, J = 9.2 Hz, 1H), 7.93 (d, J = 8.6 Hz, 1H), 8.34 (s, 1H). m.p. 156.3-160.0°C. |
| 28% |
With boron tribromide In dichloromethane at -40 - 20℃; for 1h; Inert atmosphere; |
|
|
With hydrogen bromide; acetic acid |
|
|
With pyridine hydrochloride |
|
|
With aluminium trichloride; xylene |
|
|
With pyridine hydrochloride at 210 - 225℃; |
|
|
With hydrogenchloride Heating; |
|
|
With hydrogenchloride; triethylamine In dichloromethane for 2h; Heating; |
|
|
With hydrogen bromide; acetic acid for 0.166667h; microwave irradiation; |
|
| 5 g (89%) |
With hydrogenchloride In dichloromethane; ethyl acetate |
1 Example 1(b)-Preparation of 2-(1-{6[ethyl-(2-{8-[4-(4-fluorophenyl)-4-oxobutyl]-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl}ethyl)-amino]-2-naphthyl}ethylidene)malononitrile
Example 1(b)-Preparation of 2-(1-{6[ethyl-(2-{8-[4-(4-fluorophenyl)-4-oxobutyl]-4-oxo-1-phenyl-1,3,8-triazaspiro[4.5]dec-3-yl}ethyl)-amino]-2-naphthyl}ethylidene)malononitrile In a 3 L two-neck round bottom flask, equipped with a reflux condenser and a dropping funnel, 2 L of hydrochloric acid (d=1.16) were stirred and heated to boiling. A solution of 6.06 g (30.3 mmol) of 1-(6-methoxy-2-naphthyl)-1-ethanone (prepared as described in Arsenijevic et al., Org. Synth. Coll. 6:34 (1988), the disclosure of which is incorporated herein by reference) in a minimum amount of dichloromethane was added, and the mixture was stirred and heated at reflux for 2 hours. The hot solution was filtered through a mineral wool plug to remove oily residue. The solid that separated after cooling was filtered on a glass frit and dissolved in 130 mL of ethyl acetate. The solution was washed with brine, dried with anhydrous magnesium sulfate and evaporated to give 5 g (89%) of 1-(6-hydroxy-2-naphthyl)-1-ethanone. |
| 7.2 g (74%) |
With hydrogen bromide In hexane; acetic acid; ethyl acetate |
6-acetyl-2-hydroxynaphthalene (Formula 8)
6-acetyl-2-hydroxynaphthalene (Formula 8) To a solution containing 6-acetyl-2-methoxynaphthalene (10.4 g, 52 mmol) in glacial acetic acid (100 mL), 48% HBr (43.0 g, 0.53 mol) was added. The mixture was stirred at 100° C. for 12 hr. Excess acetic acid was removed in vacuo, and the residue was taken up in ethyl acetate and washed with dilute NaHCO3 and brine. The organic layer was dried with MgSO4 and the solvent was removed in vacuo. The product was purified by column chromatography using ethyl acetate/hexane (1:1) as the eluent. Yield 7.2 g (74%); m.p. 173° C.; IR (KBr): 3,362, 1,664 cm-1; 1H NMR (300 MHz, CDCl3): δ 8.41 (d, 1H, J=2 Hz), 7.99 (dd, 1H, J=9, J=2 Hz), 7.87 (d, 1H, J=9 Hz), 7.70 (d, 1H, J=9 Hz), 7.20 (d, 1H, J=2 Hz), 7.18 (dd, 1H, J=9, J=2 Hz), 5.70 (br s, 1H), 2.71 (s, 3H). Anal. Calcd. for C12H10O2: C, 77.40; H, 5.41. Found: C, 77.52; H, 5.46. |
|
With hydrogen bromide; acetic acid |
|
|
With hydrogen bromide; acetic acid In water at 100℃; for 12h; |
|
|
With hydrogenchloride In water for 2h; Reflux; |
|
|
With hydrogenchloride In water for 48h; Reflux; |
|
|
With hydrogenchloride; triethylamine |
|
|
With hydrogen bromide |
|
|
With hydrogen bromide; acetic acid at 120℃; for 12h; |
1.1 (1) Synthesis of 2-acetyl-6-hydroxynaphthalene
5 g of 2-acetyl-6-methoxynaphthalene was dissolved in 50 mL of glacial acetic acid, to which was added 15 mL of HBr,120 reflux 12h. After completion of the reaction, the aqueous solution of saturated NaHCO3 was used to neutralize the acid in the solvent to pH7, then extracted with ethyl acetate 3 times, combined with organic phase, at room temperature over silica gel column, eluent is pureCH2Cl2, and the resulting eluent was dried to give the product as a white solid. |
|
With hydrogen bromide |
|
|
With hydrogenchloride; triethylamine In dichloromethane; water at 85℃; for 4h; |
|
|
With hydrogen bromide; acetic acid at 100℃; for 12h; |
|
|
With hydrogenchloride at 100℃; |
a
One equivalent of 2-acetyl-6-methoxynaphthalene (referred as a first compound i hereinafier) as a starting material is added in sixty equivalents of a i 2N hydrochloric acid (HC1) solution to be placed and mixed in a round- bottom flask to obtain a first mixed solution. The first mixed solution is heated with stirring by using magnet to be refluxed overnight until solution color is turned from yellow to black with precipitation. After the first mixed solution is filtered and cooled down, the following steps are processed:dichloromethane (CH2C12) is added for extraction; an aqueous sodium bicarbonate solution and a salt water are used for washing; a layer of organics is collected; dehydration is processed in an organic phase with sodium sulfate; and a second compound 2 is obtained after processing filtration and concentration. |
|
With hydrogenchloride In water at 100℃; |
A A preferred embodiment of the [F-18]FEONM precursor synthesis process of the present invention is as follows:
(A) A round bottom bottle is charged with 1 equivalent (eq) of 2-ethenyl-6-methoxynaphthalene.(2-Acetyl-6-methoxynaphthalene) as a starting material(hereinafter referred to as the first compound 1),Place it in 60 equivalents of 12N hydrochloric acid (HCl).Heating and refluxing with a magnetic stirring overnight, the solution is changed from yellow clarification to black, and the black precipitate is stopped, and the filtrate obtained by filtration is cooled.Add dichloromethane (CH2Cl2) for extraction,The organic layer was washed with an aqueous solution of sodium hydrogencarbonate and brine, and the organic layer was collected.And adding sodium sulfate to remove residual water of the organic phase, filtering and concentrating the solution,Obtaining a second compound 2; |
|
With hydrogenchloride In ethanol; water for 3h; Reflux; |
|
|
With hydrogenchloride Reflux; |
|
|
With hydrogenchloride In water at 90℃; |
|
|
With hydrogen bromide; acetic acid |
|
|
With hydrogenchloride In dichloromethane for 5h; Reflux; |
|






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping