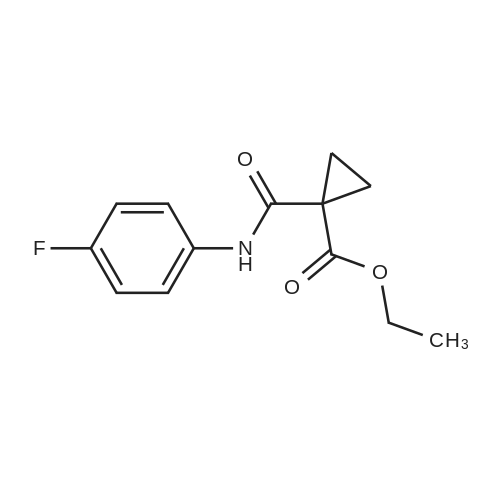
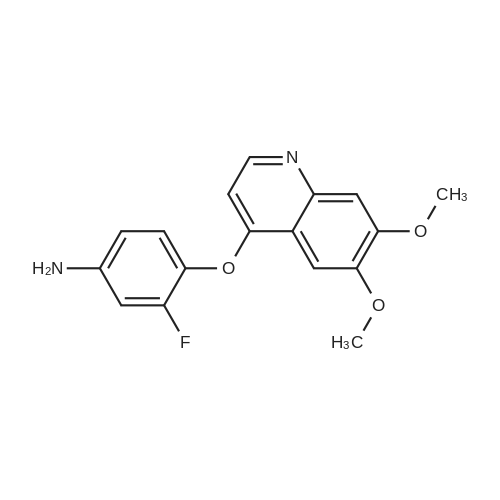
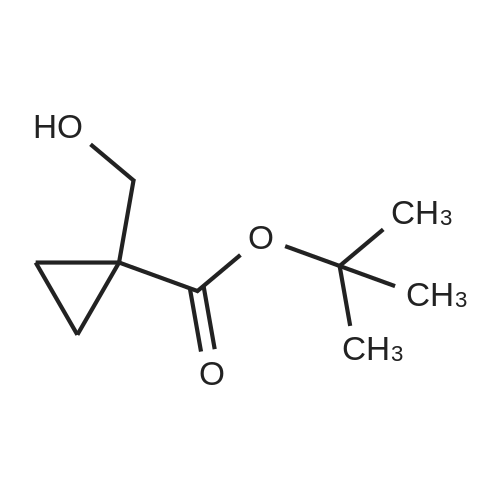
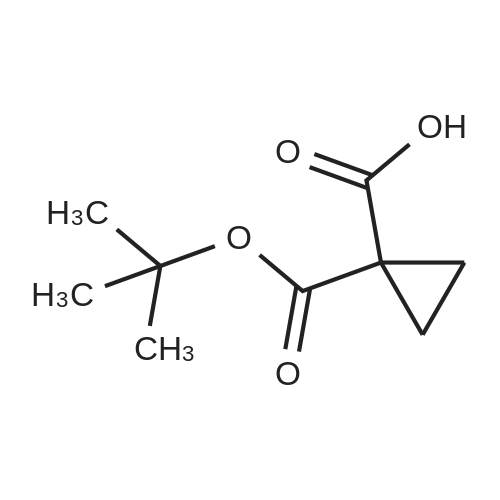
| 88.4% |
With potassium hydroxide; In ethanol; water; |
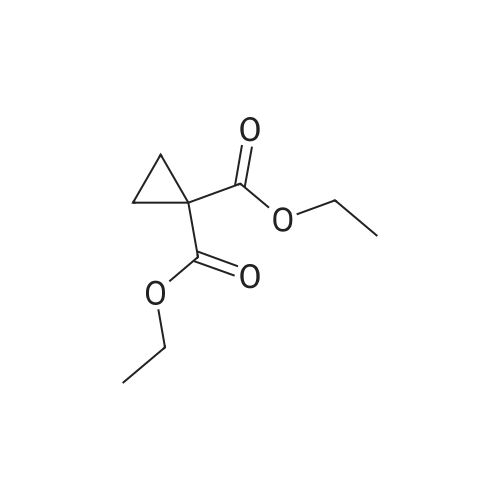
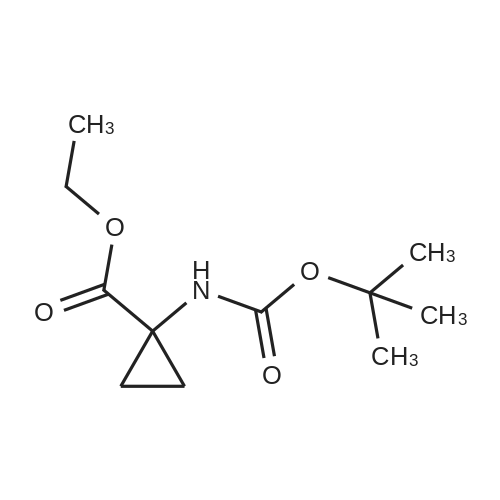
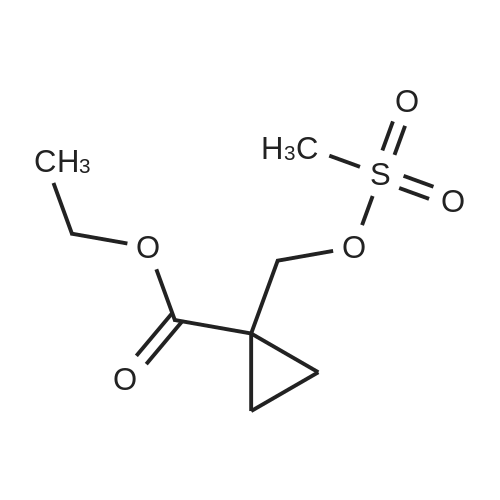
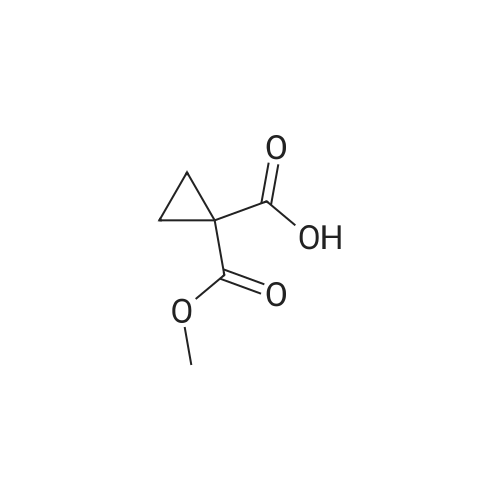
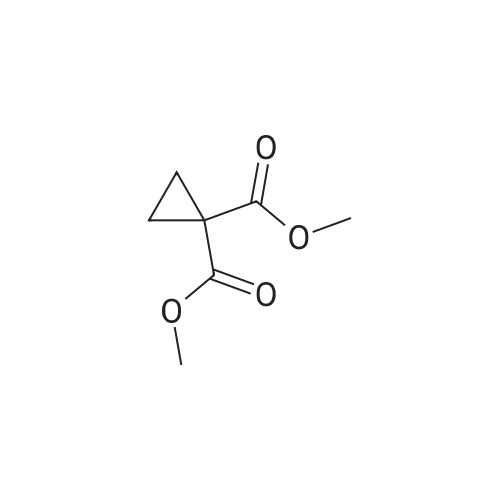
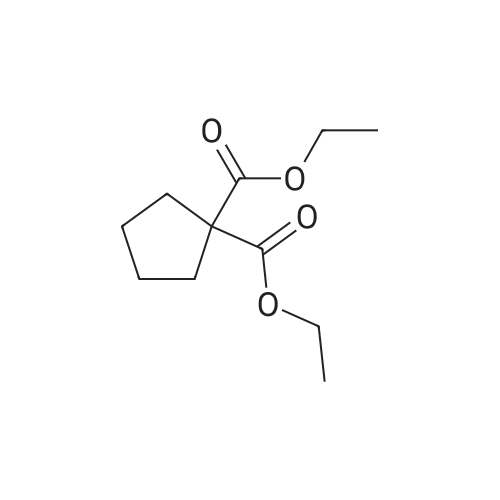
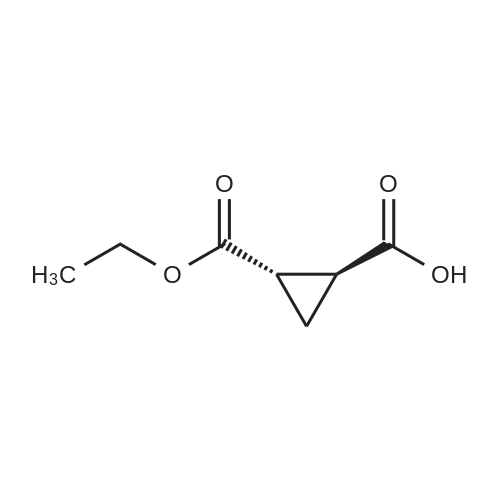
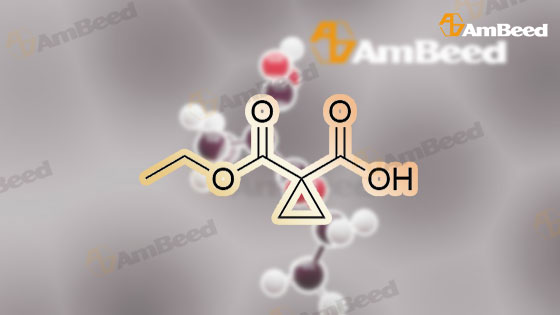
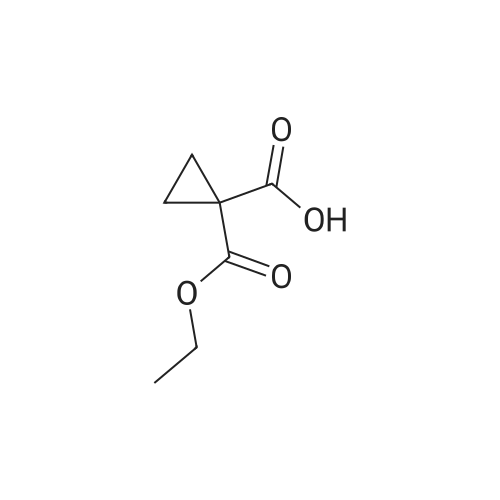
Step 2) 1-(Ethoxycarbonyl)cyclopropanecarboxylic acid To a solution of diethyl cyclopropane-1,1-dicarboxylate (4.77 g, 25.6 mmol) in ethanol (40 mL) was added KOH (1.43 g, 25.6 mmol) in H2O (8 mL), and the reaction mixture was stirred at room temperature overnight. The ethanol was removed under reduced pressure. The residue was neutralized with HCl (6 mL, 5 mol/L), then extracted with EtOAc (100 mL*3). The combined organic phases were dried over Na2SO4, filtered and the filtrate was concentrated in vacuo to give the title compound as a white solid (3.58 g, 88.4%). 1H NMR (400 MHz, CDCl3): delta 1.27 (t, J=6.7 Hz, 3H), 1.83 (m, 2H), 1.86 (m, 2H), 4.25 (m, 2H). |
| 88.4% |
With water; potassium hydroxide; at 20℃; |
To a solution of diethyl cyclopropane-1,1-dicarboxylate (4.77 g, 25.6 mmol) in ethanol (40 mL) was added KOH (1.43 g, 25.6 mmol) in H2O (8 mL), and the reaction mixture was stirred at room temperature overnight. The ethanol was removed under reduced pressure. The residue was neutralized with HCl (6 mL, 5 mol/L), then extracted with EtOAc (100 mL*3). The combined organic phases were dried over Na2SO4, filtered and the filtrate was concentrated in vacuo to give the title compound as a white solid (3.58 g, 88.4 %). 1H NMR (400MHz, CDCl3): delta 1.27 (t, J=6.7Hz, 3H), 1.83 (m, 2H), 1.86 (m, 2H), 4.25 (m, 2H). |
| 88.4% |
|
To a solution of diethyl cyclopropane- 1,1 -dicarboxylate (4.77 g, 25.6 mmol) in ethanol (40 mL) was added KOH (1.43 g, 25.6 mmol) in H2O (8 mL), and the reaction mixture was stirred at room temperature overnight. The ethanol was removed under reduced pressure. The residue was neutralized with HCl (6 mL, 5 mol/L), then extracted with EtOAc (100 mLx3). The combined organic phases were dried over Na2SO4, filtered and the filtrate was concentrated in vacuo to give the title compound as a white solid (3.58 g, 88.4 %).1H NMR (400MHz, CDCl3): delta 1.27 (t, J=6.7Hz, 3H), 1.83 (m, 2H), 1.86 (m, 2H), 4.25 (m, 2H). |
| 83% |
With potassium hydroxide; In ethanol; water; |
Monoethyl cyclopropane-1,1-dicarboxylate STR19 A solution of 53 g (285 mmol) of diethyl cyclopropane-1,1-dicarboxylate and 16.0 g (286 mmol) of potassium hydroxide in 300 ml of ethanol was stirred for 3 hours at 20 C. and then evaporated to dryness. The residue was dissolved in 100 ml of water, after which byproducts were extracted with 200 ml of methylene chloride. After the aqueous phase had been acidified to pH 2 with dilute hydrochloric acid, the product was extracted with 200 ml of methyl tert -butyl ether and isolated in a conventional manner. Purification was carried out by distillation. Yield: 83%; bp.: 99-102 C. at 2.5 mbar; colorless oil. |
| 79% |
With potassium hydroxide; In ethanol; at 20℃; |
KOH (2.8 g, 50 mmol), was added to a solution of IV-1 (9.3 g, 50 mmol) in 200 mL EtOH. The reaction mixture was stirred at rt overnight. After concentrated under reduced pressure, the residue was re-dissolved in 50 mL of NaHCO3 solution (w/w=5%) and extracted with DCM. The aqueous layer was separated, and adjusted pH to 2 with 1N HCl, and extracted with EtOAc. The combined organic layer was dried and concentrated to afford IV-2 (6.0 g, yield 79%), which was used to next step directly. |
| 78.5% |
With sodium hydroxide; In ethanol; at 20℃; for 3h; |
500ml three-necked flask to 200ml of ethanol and 1,1-diethyl-cyclopropylamino burn 30g (161mmol), coldBut to l C, added dropwise with stirring a solution of sodium hydroxide (NaOH 6. 4g, 161mmol; water 32ml). Dropping was completed, the reaction at room temperature for 3 hours When the end of the reaction, the solvent was evaporated under reduced pressure to give a white solid. The solid was added water and ethyl acetate 200ml 100ml, stirring pointsLayer, the organic phase was discarded. Aqueous layer was washed with 2mol / L hydrochloric acid, adjusted to pH rhoEta3~4, ethyl acetate (100ml X 2) and extracted with ethyl acetateLayer was dried over anhydrous sodium sulfate, the solvent was evaporated under reduced pressure to give a colorless oil translucent 20. 0g (78. 5%). |
| 78.5% |
With sodium hydroxide; In methanol; water; at 20℃; for 3h; |
To 500 ml of a three-necked flask was added 200 ml of methanol and 30 g of 1,1-cyclopropanedicarboxylic acid diethyl ester, cooled to 10 C, stir A solution of sodium hydroxide (NaOH 6.4 g, water 32 ml) was added dropwise, The reaction was carried out at room temperature for 3 hours, the reaction was terminated, the solvent was distilled off under reduced pressure, A white solid. To the solid was added 200 ml of water and 150 ml of ethyl acetate, ] Stir and layer, discard the organic phase. The aqueous layer was adjusted to pH 3 to 4 with 2 mol / L hydrochloric acid, Ethyl acetate extraction (200 ml x 2), The organic phase was dried over anhydrous sodium sulfate, filtered, The solvent was distilled off under reduced pressure, To give a colorless translucent oil |
| 64.6% |
With sodium hydroxide; In tetrahydrofuran; methanol; at 25℃; for 16h;Inert atmosphere; |
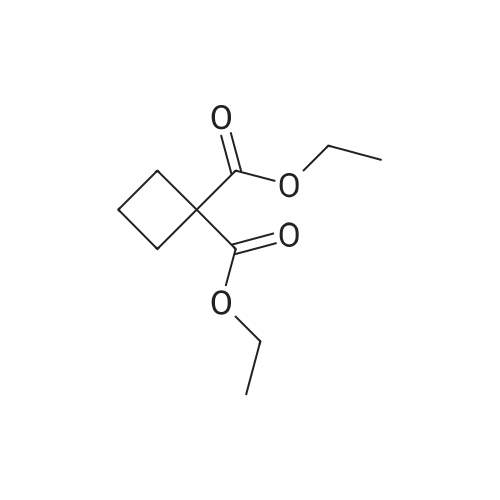
Ethyl 1,1-cyclopropyl dicarbonate (1.0 g, 5.37 mmol) was added to tetrahydrofuran (3 ml) at 25 C under the protection of nitrogen, then a mixed solution of sodium hydroxide/methanol (1 mol/L, 5.37 ml) was added thereto, and the mixture was stirred under the protection of nitrogen for 16 hours at 25 C. The solution was concentrated under reduced pressure at 30 C and then added to water (20 ml). The aqueous phase was washed with ethyl acetate (20 ml * 2), the pH was adjusted to 2 with hydrochloric acid (2 mol/L) (20 ml * twice) and extracted with ethyl acetate (20 ml * 2). The organic phase was dried over anhydrous sodium sulfate, filtered and concentrated to give compound 168A (creamy white solid, 500 mg, the yield was 64.6%). 1H NMR (400 MHz, CHLOROFORM-d) 3.79 (s, 3H), 1.80-1.88 (m, 2H), 1.72-1.79 (m, 2H) |
|
With sodium hydroxide; In ethanol; water; |
(a) 1-Ethoxycarbonylcyclopropanecarboxylic acid (1a) A solution of sodium hydroxide (2.15 g, 54 mmol) in water (15 ml) was added dropwise to a stirred solution of diethyl 1,1-cyclopropanedicarboxylate (10 g, 54 mmol) in ethanol (30 ml) while the temperature was maintained below 25 C. After the completion of the addition, the resulting mixture was stirred at room temperature for 60 hours. Most of ethanol was removed by evaporation in vacuo and the residue was diluted with brine, acidified with 6N hydrochloric acid and extracted with diethyl ether. The extract was dried over magnesium sulfate, filtered and concentrated. The residue was purified by distillation to give the title compound (bp 66-9 C./0.15 mm) as a viscous oil. |
|
|
(Example 23-1) To an ethanol solution (93 ml) of diethyl cyclopropane-1,1-dicarboxylate (9.31 g) was added 1 N aqueous sodium hydroxide solution (50 ml), followed by stirring at room temperature for 9 hours. Ethanol was distilled off under reduced pressure, and the remaining aqueous solution was washed with dichloromethane. Under ice cooling, 5 N hydrochloric acid was added to the aqueous layer, and extracted with dichloromethane. The organic layer was washed with water and saturated sodium chloride solution, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to afford 1-(ethoxycarbonyl)cyclopropanecarboxylic acid (6.38 g). 1H NMR (400 MHz, CDCl3) delta: 1.27 (t, 3H, J = 7.0 Hz), 1.72-1.77 (m, 2H), 1.81-1.86 (m, 2H), 4.24 (q, 2H, J = 7.0 Hz). |
|
With water; sodium hydroxide; In ethanol; at 20℃; |
To a stirring solution of compound 70 (55.8 g , 300 mmol) in 95% EtOH (200 ml), was added aqueous NaOH (12 g, 300 mmol) (50 ml) dropwise. The reaction mixture was stirred overnight at room temperature, decompression to remove EtOH, water (300 ml) was added and the stirred. After the aqueous phase was washed with EtOAc (50 ml x 2), the aqueous phase was acidified to pH 1.5 with aqueous 10% HCI and the aqueous phase was extracted with EtOAc (100 ml x 3). The combined organic phase was dried over Na2S04, filtered, and concentrated to give crude compound 71 (35 g, yield 73%) as a colorless oil, used directly in the next step without further purification. |
|
With water; sodium hydroxide; In ethanol; at 20℃; |
[0370] To a stirring solution of compound 70 (55.8 g, 300mmol) in 95% EtOH (200 ml), was added aqueous NaOH (12g, 300 mmol) (50 ml) dropwise. The reaction mixture wasstirred overnight at room temperature, decompression toremove EtOH, water (300 ml) was added and the stirred. Afterthe aqueous phase was washed with EtOAc (50 mlx2), theaqueous phase was acidified to pH 1.5 with aqueous 10% HCland the aqueous phase was extracted with EtOAc (1 00 mlx3). The combined organic phase was dried over N a2 SO 4 , filtered,and concentrated to give crude compound 71 (35 g, yield73%) as a colorless o |
|
With potassium hydroxide; In ethanol; at 0℃; for 5h; |
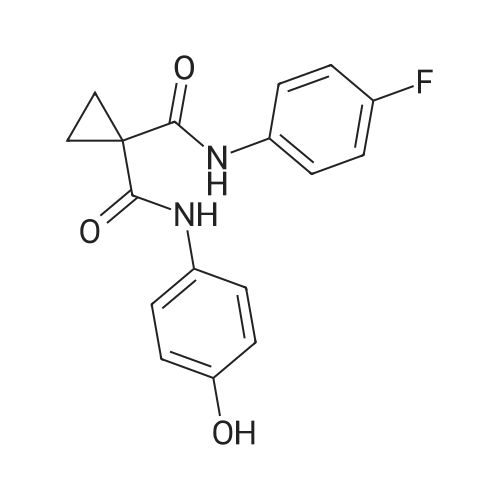
General procedure: Diethyl cyclopropane-1,1-dicarboxylatederivatives (1.0 eq.) were dissolved in EtOH. KOH (1.0 eq.)in ethanol solution was added and stirred for 5 h. After the completion of the reaction, the intermediatewas dissolved in THF solution at 15 C. 4-Methylmorpholine (1.2 eq.), isobutyl chloroformate(1.0 eq.) and 4-amino-2-fluorophenol (1.0 eq.) were added. The crude product was purified by columnchromatography, and the above process was repeated. The final product compounds 12 were obtained. |
|
With water; lithium hydroxide; In ethanol; at 10 - 35℃; for 48h; |
To 5L reaction flask was charged 372.4g (2.0mol) 1,1- diethyl-cyclopropyl, was added 1400mL 62.4g of ethanol was slowly dropping, the system was cooled ice-salt bath to 10 C after (2.6mol) of lithium hydroxide and 480mL Water to form a solution, drops complete, insulation 35 C for 48h the reaction the reaction was stopped. The solvent was distilled off under reduced pressure, to the system Was added 1400mL of water, after cooling to 10 C with 5% -10% of dilute hydrochloric acid to adjust the pH to about 4, with a system 3X1000mL extracted with ethyl acetate, the combined organic layers, the organic layer was dried, filtered, and evaporated to give 311g Pale yellow oil. The product was introduced directly into a 5L reaction flask was added 1500mL of tetrahydrofuran, body Cooled to 0 C line to the system after dropping 300g (2.4mol) of oxalyl chloride and 5gDMF, drops Bi, 15 C to Reaction 3-5h after TLC the reaction was complete testing of raw materials. System cooling again to 0 C, solution of 194.3g (1.92mol) Triethylamine, dropping was completed, the batch was added 222g (2mol) with fluorine aniline and 400mLTHF Into the solution, plus complete within 1h. After 0.5h the reaction system at room temperature, filtered, washed with 200mLTHF consider pie, The combined filtrate, the filtrate evaporated under reduced pressure to give the crude Intermediate 4 to about 510g, the crude product with ethyl acetate / n-hexane Alkyl recrystallized pure intermediate 4 about 465.6g, HPLC: 98.9%, yield: 92.7%. |
| 6 g |
With ethanol; sodium hydroxide; at 25℃; for 12h; |
A solution of 1,1-diethyl cyclopropane-1,1-dicarboxylate (10.00 g, 53.70 mmol, 1.00 equiv), sodium hydroxide (2.15 g, 53.75 mmol, 1.00 equiv) in ethanol (50 mL) was stirred for 12 h at 25 oC. After completion, the solids were collected by filtration and re-crystallized from ethanol. This resulted in 6 g of the title compound as a white solid. |
|
With water; sodium hydroxide; In ethanol; at 25℃; for 16h; |
To a mixture of diethyl 1 ,1-cyclopropanedicarboxylate (10.0 g, 53.7 mmol) in ethanol (70 mL) and water (35 mL) was added sodium hydroxide (2.1 g, 53.7 mmol). The reaction was stirred at 25 C for 16 h and diluted with ethyl acetate (60 mL). The organic layer was discarded. The aqueous phase was adjusted to pH = 3 by addition of aqueous hydrochloric acid (4 M). The mixture was extracted with ethyl acetate (3 x 80 mL). The combined organic layers were washed with brine (30 mL), dried over sodium sulfate and concentrated under reduced pressure to give crude 1- ethoxycarbonylcyclopropanecarboxylic acid (6.6 g, 78%) as a colorless oil. NMR (400MHz, CDCl3) delta 12.86 (br s, 1H), 4.31 - 4.17 (m, 2H), 1.87 - 1.81 (m, 2H), 1.77 - 1.69 (m, 2H), 1.32 - 1.24 |
|
With potassium hydroxide; In ethanol; at 20℃; |
A flask was charged with diethyl cyclopropane- 1, 1-dicarboxylate (10.0 g, 53.7 mmol, 1.00 equiv), KOH (3.00 g, 53.7 mmol, 1.00 equiv) and EtOH (100 mL). The resulting mixture was stirred overnight at room temperature and concentrated under reduced pressure. The residue was dissolved in water (100 mL) and extracted with diethyl ether (2 x 50 mL). The pH value of aqueous layer was adjusted to 4 with aqueous HC1 (1 M) and extracted with EtOAc (3 x 100 mL). The organic layers were combined, washed with water (50 mL) and brine (50 mL), dried over anhydrous Na2S04, filtered and concentrated under reduced pressure to provide 6.60 g (crude) of l-(ethoxycarbonyl)cyclopropane-l-carboxylic acid. LCMS (ESI, m/z): 159 [M+H]+. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping