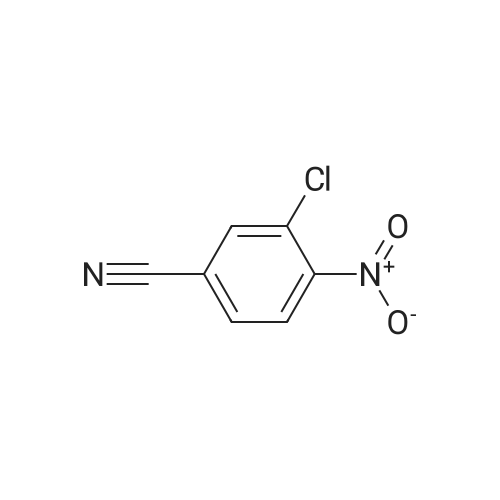
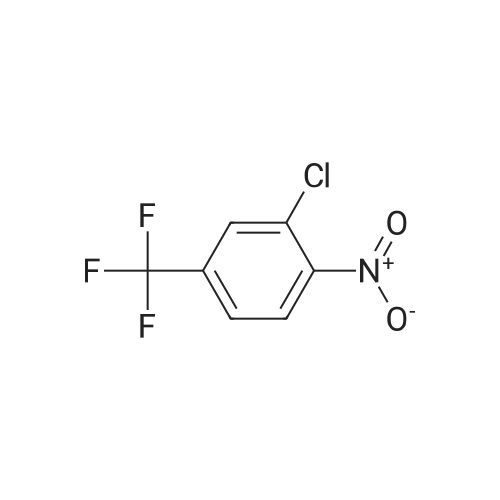
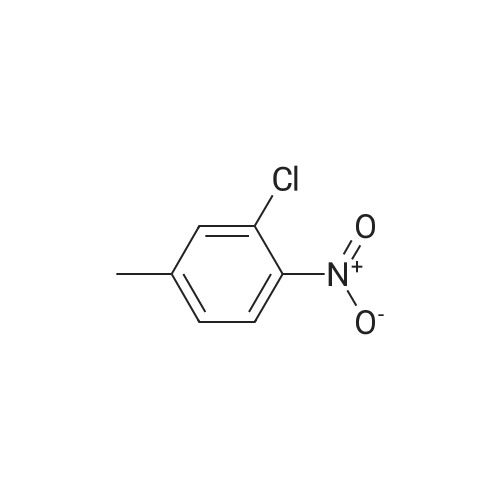
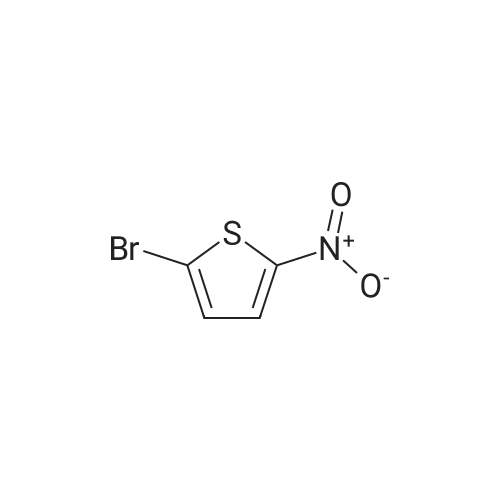
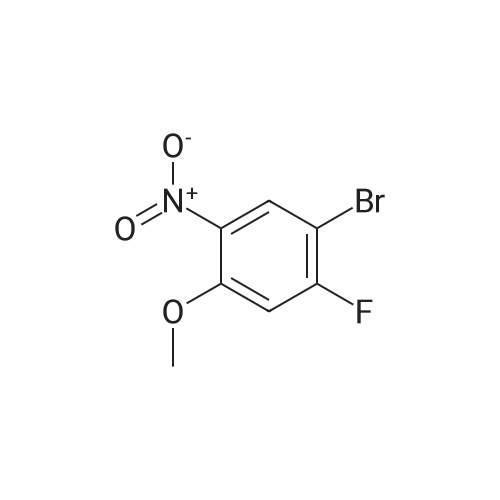
There will be a HazMat fee per item when shipping a dangerous goods. The HazMat fee will be charged to your UPS/DHL/FedEx collect account or added to the invoice unless the package is shipped via Ground service. Ship by air in Excepted Quantity (each bottle), which is up to 1g/1mL for class 6.1 packing group I or II, and up to 25g/25ml for all other HazMat items.
Type
HazMat fee for 500 gram (Estimated)
Excepted Quantity
USD 0.00
Limited Quantity
USD 15-60
Inaccessible (Haz class 6.1), Domestic
USD 80+
Inaccessible (Haz class 6.1), International
USD 150+
Accessible (Haz class 3, 4, 5 or 8), Domestic
USD 100+
Accessible (Haz class 3, 4, 5 or 8), International
USD 200+
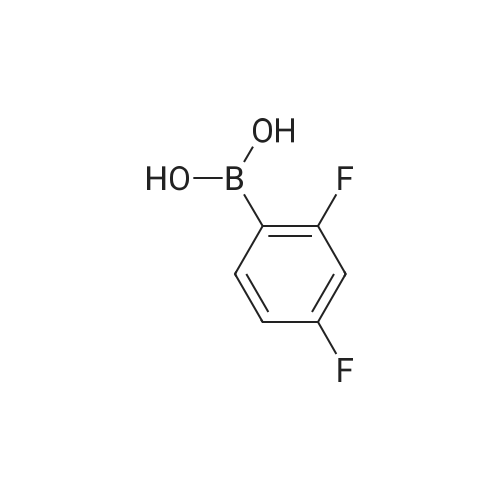
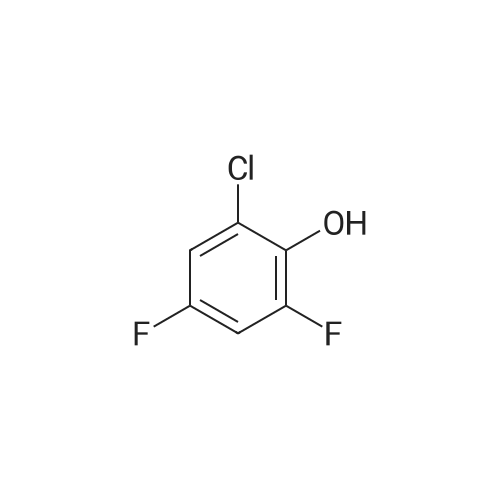
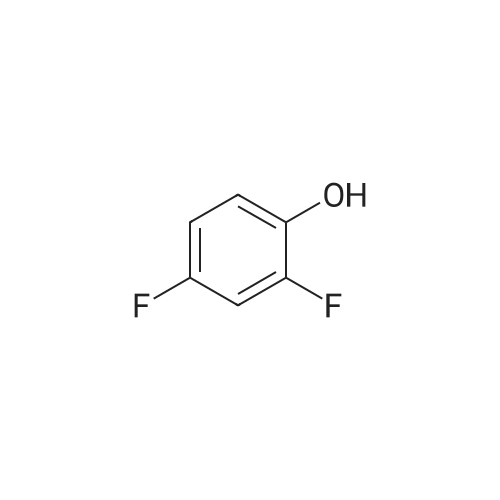
Structure of 367-27-1 * Storage: {[proInfo.prStorage]}
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
General procedure: In a 50 mL round-bottomed flask, a mixture of arylboronic acid(1 mmol), H2O2 (30percent aq, 0.2 mL), ZnO nanocatalyst (5 molpercent; sampleZnO-1) and 2 mL of water were stirred at room temperature under aerobic condition. The progress of the reaction was monitored by thin layer chromatography (TLC). After completion of the reaction, the reaction mixture was diluted with 20 mL of water and extracted with (3×20) mL of diethyl ether. The combined organic layer was washed with brine and dried over Na2SO4. The solvent was removed in a rotary evaporator under reduced pressure. The crude product was purified by column chromatography (hexane/ ethylacetate, 9:1) on silica (100–200mesh) to get the desired product. The products were identified by 1HNMR and 13C NMR.
91%
With dihydrogen peroxide In water at 20℃; for 0.166667 h; Green chemistry
General procedure: In a 50mL round-bottomed flask, a mixture of arylboronic acid (1mmol), H2O2 (30percent aq, 0.2mL), bio-silica (5mg) and 2mL of water was added and stirred at room temperature in aerobic condition. The reaction was monitored by TLC. After completion of the reaction the reaction mixture was diluted with 20mL of water and extracted with (3×20) mL of diethylether and the combined organic layer was washed with brine and dried over by Na2SO4 and evaporated in a rotary evaporator under reduced pressure. The crude was purified by column chromatography (hexane/ethylacetate, 9:1) on mesh silica (100–200) to get the desired product. The products were confirmed by 1H NMR, 13C NMR, FT-IR spectroscopy and mass spectrometry.
Reference:
[1] Applied Catalysis A: General, 2018, vol. 562, p. 58 - 66
[2] Tetrahedron Letters, 2015, vol. 56, # 14, p. 1780 - 1783
[3] Journal of Organic Chemistry, 2001, vol. 66, # 2, p. 633 - 634
With Selectfluor; 1-(n-butyl)-3-methylimidazolium triflate In methanol at 80℃; for 5 h; Inert atmosphere
General procedure: A mixture of phenol (20 mg), F‐TEDA‐BF4 (1.1 equivalents), IL(0‐15 equivalents) and the organic solvent (5 mL) was stirred for 5 h at various temperatures under an argo atmosphere (Tables 1‐5). The mixture was evaporated at a reduced pressure and analysed by 1H, 19F NMR assolution in CDCl3 or CDCl3‐DMSO‐d6. Cl2CHCHCl2 and PhCF3 were used as internal standards for peakintegration.
Reference:
[1] Journal of the American Chemical Society, 1990, vol. 112, # 23, p. 8563 - 8575
[2] Tetrahedron Letters, 1986, vol. 27, # 37, p. 4465 - 4468
[3] Tetrahedron Letters, 1986, vol. 27, # 37, p. 4465 - 4468
[4] Journal of Organic Chemistry, 1998, vol. 63, # 10, p. 3379 - 3385
[5] Tetrahedron Letters, 1986, vol. 27, # 37, p. 4465 - 4468
[6] Journal of Organic Chemistry, 1998, vol. 63, # 10, p. 3379 - 3385
[7] Journal of Organic Chemistry, 1998, vol. 63, # 10, p. 3379 - 3385
[8] Russian Journal of Organic Chemistry, 2009, vol. 45, # 10, p. 1468 - 1473
[9] Arkivoc, 2017, vol. 2018, # 2, p. 60 - 71
4
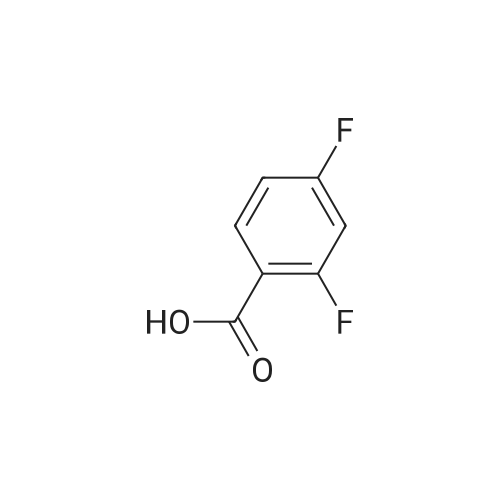
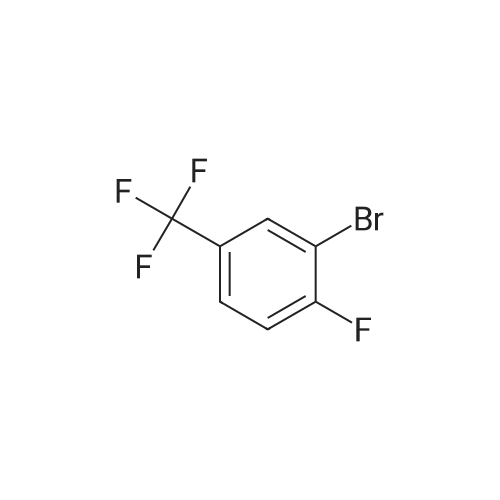
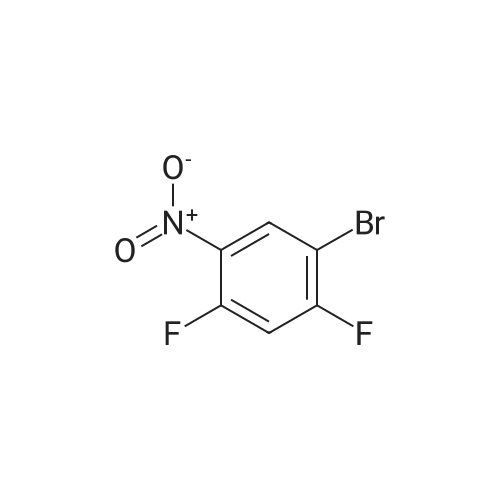
[ 540-36-3 ]
[ 367-27-1 ]
[ 2713-31-7 ]
Reference:
[1] Journal of the American Chemical Society, 2014, vol. 136, # 32, p. 11321 - 11330
5
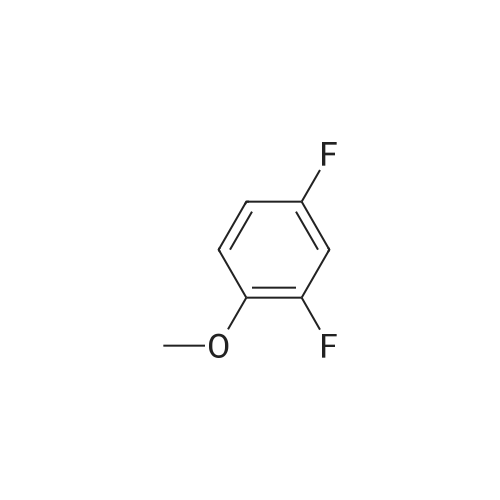
[ 108-95-2 ]
[ 371-41-5 ]
[ 367-12-4 ]
[ 367-27-1 ]
[ 28177-48-2 ]
Reference:
[1] Patent: CN105753655, 2016, A, . Location in patent: Paragraph 0028; 0030; 0038; 0054-0058
6
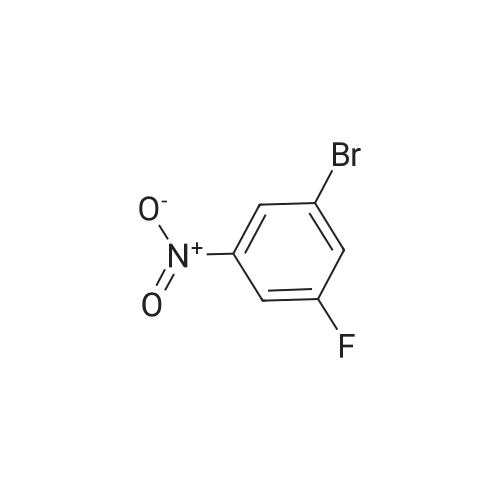
[ 367-25-9 ]
[ 367-27-1 ]
Reference:
[1] Chemische Berichte, 1937, vol. 70, p. 2396,2400
[2] Journal of the American Chemical Society, 1959, vol. 81, p. 94,95, 97
[3] Journal of Organic Chemistry USSR (English Translation), 1974, vol. 10, p. 1237 - 1238[4] Zhurnal Organicheskoi Khimii, 1974, vol. 10, # 6, p. 1228 - 1230
7
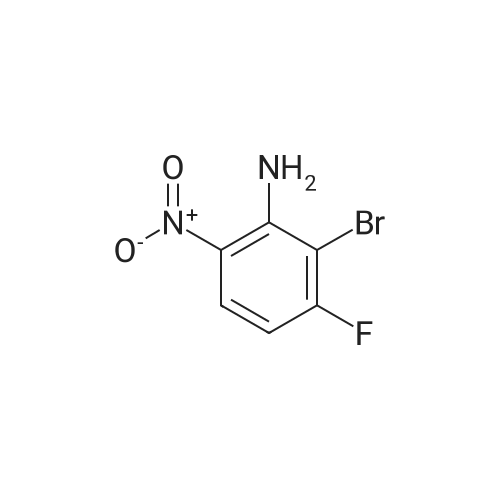
[ 364-83-0 ]
[ 367-27-1 ]
[ 36914-77-9 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2005, vol. 347, # 7-8, p. 1027 - 1034
8
[ 1550-35-2 ]
[ 367-27-1 ]
[ 1583-58-0 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2005, vol. 347, # 7-8, p. 1027 - 1034
Reference:
[1] Chemische Berichte, 1937, vol. 70, p. 2396,2400
11
[ 399-44-0 ]
[ 10034-85-2 ]
[ 367-27-1 ]
Reference:
[1] Journal of the American Chemical Society, 1958, vol. 80, p. 5960,5963
[2] Journal of the American Chemical Society, 1958, vol. 80, p. 5960,5963
12
[ 446-35-5 ]
[ 367-27-1 ]
Reference:
[1] Chemische Berichte, 1937, vol. 70, p. 2396,2400
13
[ 540-36-3 ]
[ 371-41-5 ]
[ 367-27-1 ]
[ 2713-31-7 ]
[ 7664-39-3 ]
Reference:
[1] Journal of the American Chemical Society, 2014, vol. 136, # 32, p. 11321 - 11330
14
[ 108-95-2 ]
[ 371-41-5 ]
[ 367-12-4 ]
[ 367-27-1 ]
[ 28177-48-2 ]
Reference:
[1] Patent: CN105753655, 2016, A, . Location in patent: Paragraph 0028; 0030; 0038; 0054-0058
15
[ 540-36-3 ]
[ 367-27-1 ]
[ 2713-31-7 ]
Reference:
[1] Journal of the American Chemical Society, 2014, vol. 136, # 32, p. 11321 - 11330
16
[ 540-36-3 ]
[ 371-41-5 ]
[ 367-27-1 ]
[ 2713-31-7 ]
[ 7664-39-3 ]
Reference:
[1] Journal of the American Chemical Society, 2014, vol. 136, # 32, p. 11321 - 11330
17
[ 367-27-1 ]
[ 5317-89-5 ]
Reference:
[1] Organic Process Research and Development, 1998, vol. 2, # 2, p. 71 - 77
18
[ 367-27-1 ]
[ 2267-99-4 ]
Reference:
[1] Journal of the American Chemical Society, 1959, vol. 81, p. 94,95, 97
Stage #1: at 0℃; for 0.5 h; Inert atmosphere Stage #2: at 20℃; for 5 h;
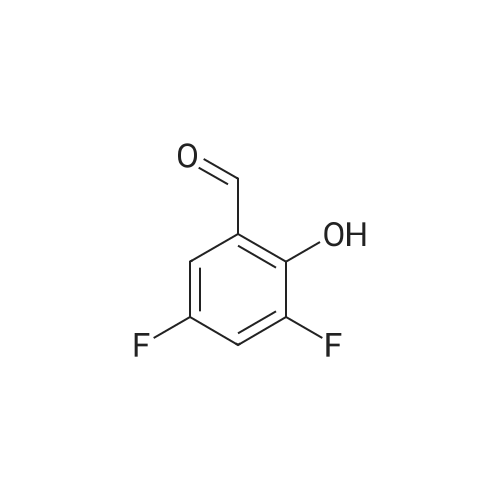
Phosphorus oxychloride (12 g, 77 mmol) was added dropwise to anhydrous dimethyl formamide (8.4 g, 0.12 mol) at 0 °C under nitrogen with stirring. The mixture was stirred at this temperature for 30 minutes, and 2,4-difluorophenol (5.0 g, 39 mmol) in N N-dimethyl formamide (50mL) was added dropwise. The reaction mixture was stirred at room temperature for 5 hours. On completion, the reaction mixture was quenched slowly with water and extracted with ethyl acetate (3 x 150 mL). The combined organic layers were washed with water and brine, dried over anhydrous sodium sulfate and concentrated in vacuo to give compound B-163 (5.5 g, 90percent yield) as a yellow oil.
Stage #1: at 78℃; for 0.5 h; Stage #2: at 78℃; for 1 h;
Step 1. A solution of HMTA (21.55 g, 153.74 mmol, 28.73 mL, 1.00 eq.) in TFA (350.00 mL) was stirred at 78 °C for 0.5 hour, then A-5-1 (20.00 g, 153.74 mmol, 1.00 eq.) in TFA (150.00 mL) was added drop-wise at 78 °C. The resulting mixture was stirred at 78 °C for 1 hour. Then the reaction mixture was concentrated under reduced pressure to remove TFA. The residue was poured into ice-water (500 mL) and stirred overnight. Then the mixture was filtered and filter cake was concentrated to give A-5-2 (8.00 g, 32.91percent yield) as a white solid. 1H NMR (400MHz, CDC13) δ 10.71 (br s, 1H), 9.89 (d, 7=2.0 Hz, 1H), 7.21 - 7.16 (m, 1H), 7.15 - 7.10 (m, 1H).
Step 1. A solution of HMTA (21.55 g, 153.74 mmol, 28.73 mL, 1.00 eq.) in TFA (350.00 mL) was stirred at 78 C for 0.5 hour, then A-5-1 (20.00 g, 153.74 mmol, 1.00 eq.) in TFA (150.00 mL) was added drop-wise at 78 C. The resulting mixture was stirred at 78 C for 1 hour. Then the reaction mixture was concentrated under reduced pressure to remove TFA. The residue was poured into ice-water (500 mL) and stirred overnight. Then the mixture was filtered and filter cake was concentrated to give A-5-2 (8.00 g, 32.91% yield) as a white solid. 1H NMR (400MHz, CDC13) delta 10.71 (br s, 1H), 9.89 (d, 7=2.0 Hz, 1H), 7.21 - 7.16 (m, 1H), 7.15 - 7.10 (m, 1H).
To a 100 L reactor was charged trifluoroacetic acid (11.25 L, 17.28 kg) and 2,4- difluorophenol (IVA, 2.25 kg); the resulting solution was adjusted to 20C. With good stirring hexamethylentetramine (2.70 kg) was charged over ~30 minutes; the reaction temperature was allowed to attain 40C. The reaction mixture was adjusted to 80C and held at 80C for at least 1.0 hour before heating to 115 C. The reaction mixture was held at 115 C for 18.0 to 20.0 hours whereupon the reaction was cooled to 30C and the reactor charged with water (76.5 L) over at least 30 minutes. The reaction was then <n="26"/>adjusted to 2C and held at 2C for at least 4.0 hours. The resulting suspension was then filtered and the filter cake washed twice with water (18.0 L and 13.5 L) and then pulled dry for at least 30 minutes.Two lots of the water wet aldehyde (HIA) were then employed in the following:To a 100 L reactor was charged the water wet aldehyde (IIIA), acrylonitrile (7.9 kg), dimethyl formamide (13.5 kg) and water (18.5 L). With good stirring DABCO (0.88 kg) was added to affording a clear yellow solution, he reaction mixture was then adjusted to 70C and the reaction mixture was held at 70C for 18.0 to 20.0 hours, whereupon the reaction mixture was cooled to 20C. Water (18.4 L) was then charged and the reaction mixture adjusted to 2C and held at 2C for 3 hours. The product was then filtered, washed with aqueous methanol (7.3 L) (5: 1, MeOtLFhO) and dried under vacuum at 45C to afford 6,8-difluoro-2H-chromene-3-carbonitrile (HA, 2.90 kg, 43.5 %) as a pale yellow crystalline solid.; Example IA: 6,8-difluoro-2H-chromene-3-carbonitrile - compound IVA to compound IIIA to compound HA; To a 100 L reactor was charged trifluoroacetic acid (20 L, 30.72 kg) and 2,4- difluorophenol (IVA, 4.0 kg); the resulting solution was adjusted to 20C. With good stirring hexamethylentetramine (4.80 kg) was charged over ~ 30 minutes; the reaction temperature was allowed to attain 40C. The reaction mixture was adjusted to 80C and held at 80C for at least 1.0 hour before heating to 115C. The reaction mixture was held at 115C for 18.0 to 20.0 hours whereupon the reaction was cooled to 90C and the reactor charged with water (8 L). The reaction mixture was maintained at 90C for 60 min. , then further water (52 L) was added at such a rate as to maintain a solution and the <n="27"/>resulting solution was held at 80C for 30 min. and then slowly cooled to 20C over at least 90 min. The resulting slurry was then aged at 20C for 30 min. The resulting slurry was then cooled to 2C and aged at this temperature for at least 3.0 h. The suspension was then filtered and subsequently washed with additional water. (32 L and 24 L) and then pulled dry for at least 30 minutes.To a 100 L reactor was charged the water wet aldehyde (IIIA), acrylonitrile (10.4 L)), dimethyl formamide (10.4 L)) and water (8 L). With good stirring DABCO (0.96 kg) was added to affording a clear yellow solution. The reaction mixture was then adjusted to 70C and the reaction mixture was held at 70C for 18.0 to 20.0 hours, whereupon the reaction mixture was cooled to 20C. Water (20 L) was then charged over 20 min and the reaction mixture adjusted to 2C and held at 2C for 3 hours. The product was then filtered, washed with aqueous methanol (10 L) (2:1, MeOHiH?O) and dried under vacuum at 45 C to afford 6,8-difluoro-2H-chromene-3-carbonitrile (IIA, 3.64 kg, 61.3 %) as a pale yellow crystalline solid.
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 160℃; for 2h;
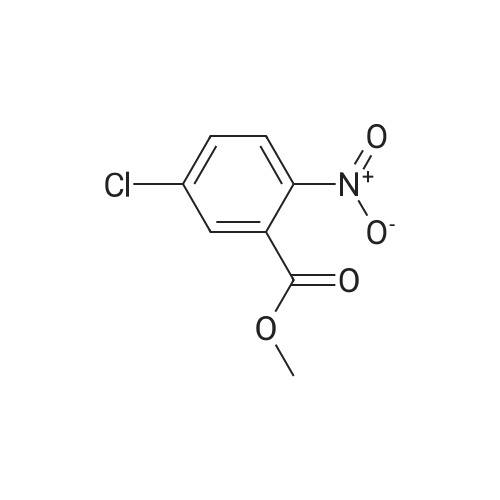
[0142] Methyl 5-chloro-2-nitrobenzoate (73 g, 0.34 mol), 2,4-difluorophenol (51.1 g, 0.39 mol) and potassium carbonate (73 g, 0.53 mol) in NMP (200 mL) were stirred at 160 C. for 2 h. The reaction mixture was cooled to room temperature poured into water (250 mL) and extracted into EtOAc (2*100 mL). The organic layer was washed several times with water, dried over sodium sulfate, concentrated in vacuo, and the residue was purified by flash chromatography eluting with hexane: EtOAc (95:5) to yield 66.8 g of 5-(2,4-difluoro-phenoxy)-2-nitro-benzoic acid methyl ester (1A).
With di-isopropyl azodicarboxylate; triphenylphosphine In tetrahydrofuran at 0 - 20℃; for 16h;
1.1
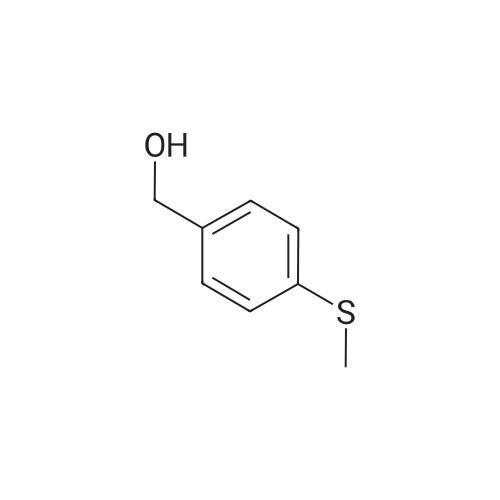
Intermediate 1 Sodium 4-[(2,4-difluorophcnoxy)mcthyl]bcnzcncsulfinatc EPO Method 1Step 1: 2,4-Difluoro-l-{r4-(metfayltnio)benzyl1oxy}benzene; To a solution of 2,4-difluorophenol (13 g, 100 mmol), 4-hydroxymethyl thioanisole (15.4 g, 100 mmol) and triphenylphosphine (28.93 g, 110 mmol) in THF (300 mL) at 0 0C was added diisopropylazodicarboxylate (22.22 g, 110 mmol). The reaction was allowed to warm to room temperature and stirred for 16 h. The solvent was removed in vacuo. The residue was purified by flash column chromatography on silica, eluting with 12% ethyl acetate/isohexane, followed by crystallisation from isohexane to give the title compound as a solid (13.5 g, 51%). 1H NMR (500 MHz, CDCl3): δ 7.33 (2 H, d, J 8.2 Hz), 7.26 (2 H, d, J 8.3 Hz), 6.94-6.84 (2 H, m), 6.76-6.72 (1 H, m), 5.05 (2 H, s), 2.49 (3 H, s).
2,4-Difluoro-6-aminophenol Hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; H2; nitrogen;palladium; In methanol;
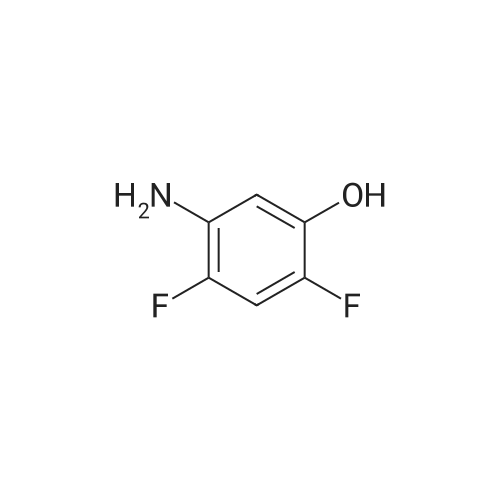
2,4-Difluoro-6-aminophenol Hydrochloride (715) A mixture of 2,4-Difluoro-6-nitrophenol (28.4 g, 0.162 mol; prepared by a similar method as 711 except replacing 2-chloro-4-fluorophenol with 2,4-difluorophenol) and 10% palladium on carbon (3.5 g) in absolute MeOH (120 mL) was placed under 1 atm of H2 and stirred until complete reduction had occurred. The H2 was replaced with nitrogen and the reaction was filtered through Celite. Gaseous HCl was bubbled through the filtrate and the resulting solution concentrated. The residue was taken up into H2 O, washed with Et2 O (2*), neutralized with solid NaHCO3 and the product extracted with Et2 O. The extracts were combined dried over MgSO4 and filtered. The filtrate was treated with gaseous HCl and resulting precipitate collected and dried under vacuum to provide 12.9 g of compound 715 as a beige solid. STR94
1-bromo-2-(2,4-difluorophenoxy)-4-methyl-5-nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With potassium carbonate; at 100℃; for 45.0h;
Step B: 1-Bromo-2-(2,4-difluorophenoxy)-4-methyl-5-nitrobenzene A mixture of 1,2-dibromo-4-methyl-5-nitrobenzene (84.3 g, 286 mmol), 2,4-difluorophenol (37.2 g, 286 mmol), and K2CO3 (43.5 g, 315 mmol) were heated to 100 C. for 45 hours. The reaction mixture was cooled to room temperature and then stored in a 5 C. refrigerator overnight. The reaction mixture was poured into 1200 mL of ice water. The resulting damp solid was collected, partially ground up, and stirred in 900 mL H2O for 45 minutes. The solid was collected by filtration and rinsed with 700 mL of water portion-wise. The resulting solid was dried under high vacuum overnight to yield 93.5 g of the desired product as a brown solid (95% yield). 1H NMR (400 MHz, CDCl3) delta 8.38 (s, 1H), 7.18 (m, 1H), 7.03 (m, 1H), 6.97 (m, 1H), 6.52 (s, 1H), 2.50 (s, 3H).
95%
With potassium carbonate; at 100℃; for 45.0h;
[00252] Step B: l-Bromo-2-(2,4-difluorophenoxyV4-methyl-5-m*trobenzene:; A mixture of l,2-dibromo-4-methy 1-5 -nitrobenzene (84.3 g, 286 mmol), 2,4-difluorophenol (37.2 g, 286 mmol), and K2CO3 (43.5 g, 315 mmol) were heated to 100 0C for 45 hours. The reaction mixture was cooled to room temperature and then stored in a 5 0C refrigerator overnight. The reaction mixture was poured into 1200 mL of ice water. The resulting damp solid was collected, partially ground up, and stirred in 900 mL H2O for 45 minutes. The solid was collected by filtration and rinsed with 700 mL of water portion- wise. The resulting solid was dried under high vacuum overnight to yield 93.5 g of the desired product as a brown solid (95 % yield). 1H NMR (400 MHz, CDCl3) delta 8.38 (s, IH), 7.18 (m, IH), 7.03 (m, IH), 6.97 (m, IH), 6.52 (s, IH), 2.50 (s, 3H).
With potassium carbonate; In (2S)-N-methyl-1-phenylpropan-2-amine hydrate; water;
Step B: 1-Bromo-2-(2,4-difluorophenoxy)-4-methyl-5-nitrobenzene A mixture of 1,2-dibromo-4-methyl-5-nitrobenzene (84.3 g, 286 mmol), 2,4-difluorophenol (37.2 g, 286 mmol), and K2CO3 (43.5 g, 315 mmol) were heated to 100 C. for 45 hours. The reaction mixture was cooled to room temperature and then stored in a 5 C. refrigerator overnight. The reaction mixture was poured into 1200 mL of ice water all at once. The resulting damp solid was collected, partially ground up, and stirred in 900 mL H2O for 45 minutes. The solid was collected by filtration and rinsed with 700 mL of water portion-wise. The resulting solid was dried under high vacuum overnight to yield 93.5 g of a brown solid (95%yield). 1H NMR (400 mHz, CDCl3) delta 8.38 (s, 1H), 7.18 (m, 1H), 7.03 (m, 1H), 6.97 (m, 1H), 6.52 (s, 1H), 2.50 (s, 3H).
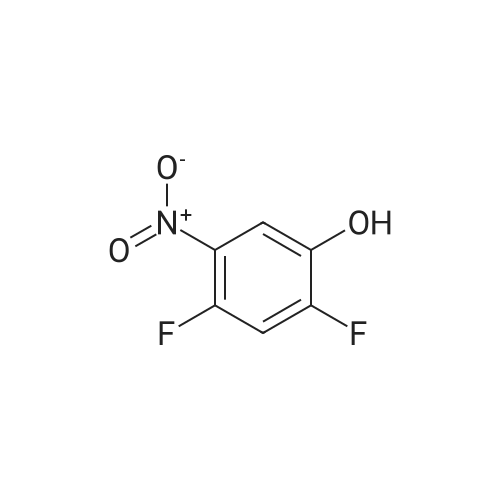
With nitric acid; acetic acid; at 10.0℃; for 1.5h;
Step 1: To a solution of 2A (100.0 g, 0.769 mol) in AcOH (800 mL) was added dropwise HNO3/AcOH (200 mL, v/v=1/1). The mixture was stirred at 10 C. for 90 min and then poured into ice-water (4 L). The solid was collected by filtration and washed by small amount of water to afford 2B (125 g, 93%), which was used in the next step without further purification.
With nitric acid; In acetic acid;
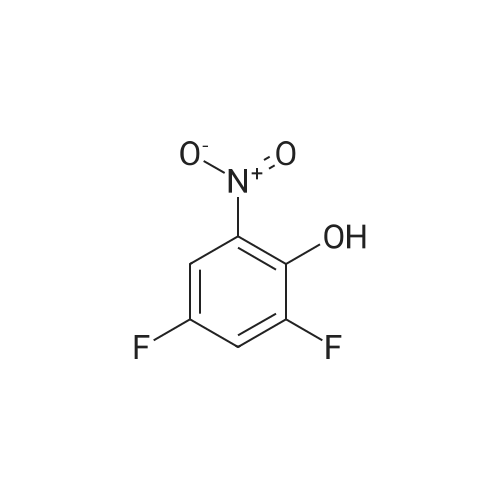
C.1 2,4-Difluoro-6-nitrophenol To a mixture of nitric acid (100 ml) and acetic acid (100 ml) was added a solution of 2,4-difluorophenol (13 g, 100 mmoles) in acetic acid (100 ml) dropwise at -5 under stirring. Stirring continued for 2 hours. The reaction mixture was poured into ice. The precipitate was collected, washed with water and dried to leave 11.26 g of a yellow solid. The material was used in the next step without further purification. 1H NMR: 11.15 (br.s., 1H), 7.68-7.79 (m, 2H).
With potassium carbonate; In N,N-dimethyl-formamide; at 120℃; for 18h;
<strong>[502496-33-5]1-Bromo-5-fluoro-2-methyl-3-nitro-benzene</strong> (9.40 g, 41.45 mmol) was dissolved in 90 mL dry DMF, and 2,4-difluorophenol (5.93 g, 45.58 mmol) and potassium carbonate (6.70 g, 45.58 mmol) were added. The reaction mixture was placed under nitronge and heated to 120 C with stirring for 18 hours. The reaction mixture was cooled and concentrated under reduced pressure, and the residue was partitioned between water and EtOAc. The organic phase was separated, washed with IN aqueous NaOH and brine, dried (MgSO4), filtered and concentrated under reduced pressure. The residue was chromatographed through silica (0%-6% EtOAc in hexanes) to give 8.484 g of 1-bromo-5-(2,4-difluoro-phenoxy)-2-methyl-3-nitro-benzene.
With citric acid;tris-(dibenzylideneacetone)dipalladium(0); palladium diacetate; In ethyl acetate; toluene;
Example 557 0.018 mL of 2,4-difluorophenol was added to 1 mL of toluene suspension containing 10 mg of 60percent sodium hydride at room temperature, and the resulting mixture was heated to reflux under nitrogen atmosphere for 15 minutes. After the reaction mixture was cooled to room temperature, 0.5 mL of toluene solution containing 4.7 mg of 2-(di-tert-butylphosphino)-2',4',6'-triisopropylbiphenyl, 1.7 mg of palladium acetate and 70 mg of tert-butyl 2-(benzamido)-4-bromobenzoate was added and the resulting mixture was heated to reflux under nitrogen atmosphere for 4 hours. After the reaction mixture was cooled to room temperature, 4.5 mg of 60percent sodium hydride, 4.7 mg of 2-(di-tert-butylphosphino)-2',4',6'-triisopropylbiphenyl and 1.7 mg of palladium acetate were added and the resulting mixture was heated to reflux under nitrogen atmosphere for 2 hours. After the reaction mixture was cooled to room temperature, 0.018 mL of 2,4-difluorophenol, 7.4 mg of 60percent sodium hydride, 4.7 mg of 2-(di-tert-butylphosphino)-2',4',6'-triisopropylbiphenyl and 1.7 mg of palladium acetate were added and the resulting mixture was heated to reflux under nitrogen atmosphere for 2 hours. After the reaction mixture was cooled to room temperature, 1.6 mg of tri-tert-butylphosphine tetrafluoroborate and 5.1 mg of tris(dibenzylideneacetone)dipalladium(0) were added, and the resulting mixture was heated to reflux under nitrogen atmosphere for 1 hour and 30 minutes. 10percent citric acid aqueous solution and ethyl acetate were added after the reaction mixture was cooled to room temperature. The organic layer was separated and dried over anhydrous magnesium sulfate after washed with a saturated sodium chloride aqueous solution, and the solvent was evaporated under reduced pressure. The obtained residue was purified with silica gel column chromatography [Flash Tube 2008 manufactured by Trikonex Company, eluent; hexane: ethyl acetate = 4:1] to obtain tert-butyl 2-(benzamido)-4-(2,4-difluorophenoxy)benzoate.
Step 2 r2-(2,4-Difluoro-phenoxy)-4-hydrazino-pyrimidine-5-carboxylic acid ethyl ester To a suspension of <strong>[98490-76-7]4-chloro-2-methanesulfonyl-pyrimidine-5-carboxylic acid ethyl ester</strong> (12.8 g) and 2,4-difluorophenol (6.42 g) in MTBE (100 mL) was added a solution of NaOEt (3.87 g) in water (64 mL) at 0 C. The reaction was warmed to room temperature and stirred for one hour. The mixture was then added into a solution of hydrazine hydrate (4.8 g) in MTBE (25 mL) at 0 C. The reaction mixture was slowly warmed to room temperature and stirred for four hours, then quenched by addition of water and extracted with methylene chloride. The combined organic layers were dried with MgSO4, filtered and concentrated under reduced pressure to give 2-(2,4-difluoro-phenoxy)-4-hydrazino-pyrimidine-5-carboxylic acid ethyl ester (8.0 g).
With di-isopropyl azodicarboxylate; triphenylphosphine; In toluene; at 25 - 30℃; for 7.5h;
The contents of the vessel were then cooled to below 30C using cooling water and the reactor was vented with nitrogen. 2,4-Difluorophenol (21.3kg) was charged to the vessel followed by triphenylphoshine (42.4 kg). The mixture was stirred out for 30 min. Maintaining the reaction temperature in the range 25-30C, the diisopropyl azodicarboxylate (DIAD, 40.9 kg) was charged over 3 hours and 30 min. The reaction mixture was stirred for a further 4 hours before sampling. The reaction was analysed by HPLC and showed no starting material.
With potassium carbonate; In N,N-dimethyl-formamide; at 60℃; for 3h;
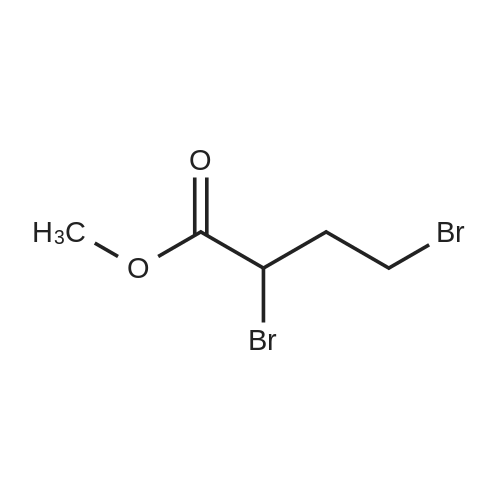
4-Bromo-2-(2,4-difluoro-phenoxy)-butyric acid methyl ester (Ll-I):To a solution of 2,4-difluro phenol ( 0.35 g, 2.73 mmol) in DMF (5 ml), added potassium carbonate ( 0.38 g, 2.73 mmol) and 2,4-Dibromo-butyric acid methyl ester (0.71 g, 2.74 mmol) and heated at 60 0C for 3 h. Reaction mixture was cooled to r.t. It was then extracted in EtO Ac( 30 ml) and washed with brine( 2X10 ml), dried over Na2SO4 and concentrated under reduced pressure to obtain crude product, purification on silica gel to yield 4-Bromo-2-(2,4-difluoro-phenoxy)-butyric acid methyl ester (0.52 g, 61 %). 1H NMR <n="70"/>(400 MHz, CDCl3): delta 12.42-2.56 (m, 2H), 3.53-3.71 (m, 2H), 3.77 (s, 3H), 4.81 (dd, J=3.6 and 8.8 Hz, IH), 6.72-6.80 (m,lH), 6.85-6.90 (m, IH), 6.95-7.02 (m, IH).
step 2-Sodium hydride (1.53 g, 38.27 mmol) was suspended in dry THF (70 mL) under a N2 atmosphere, cooled to 0 C. and 2,4-difluorophenol (3.31 mL, 34.94 mmol) was added dropwise, via syringe. After the addition was complete the mixture was stirred for 15 min, then the cooling bath was removed for 30 min and finally the solution was again cooled to 0 C. A solution of 28b (6.89 g, 33.28 mmol) in dry THF (20 mL) was added through a cannula. The resulting mixture was stirred at RT overnight and then heated to 50 C. for 3 h. The reaction was cooled to RT and saturated NH4Cl (40 mL) was added followed by water (60 mL). The mixture was thrice extracted with EtOAc, dried (MgSO4), filtered and evaporated. The crude product was purified by SiO2 chromatography eluting with an EtOAc/hexane gradient (10 to 20% EtOAc) to afford 8.15 g (82%) of 28c as a light yellow oil.
82%
step 2-Sodium hydride (1.53 g, 38.27 mmol) was suspended in dry THF (70 mL) under a N2 atmosphere, cooled to 0 C. and 2,4-difluorophenol (3.31 mL, 34.94 mmol) was added dropwise, via syringe. After the addition was complete the mixture was stirred for 15 min, then the cooling bath was removed for 30 min and finally the solution was again cooled to 0 C. A solution of R-74b (6.89 g, 33.28 mmol) in dry THF (20 mL) was added through a cannula. The resulting mixture was stirred at RT overnight and then heated to 50 C. for 3 h. The reaction was cooled to RT and saturated NH4Cl (40 mL) was added followed by water (60 mL). The mixture was thrice extracted with EtOAc, dried (MgSO4), filtered and evaporated. The crude product was purified by SiO2 chromatography eluting with an EtOAc/hexane gradient (10 to 20% EtOAc) to afford 8.15 g (82%) of R-74c as a light yellow oil.
1B. 2-(2,4-difluoro-phenoxy)-5-nitro-isonicotinic acid methyl ester To 2,4 difluorophenol (Aldrich Chemical Co.) (2.68 ML, 1.1 eq) in THF (60 ML) at 0 C. was added sodium hydride (60% in oil) (1.123 g, 1.1 eq), and the resulting mixture was stirred from 0 C. to room temperature over 1 hour.This solution was then added to compound (1A) (5.79 g, 26.7 mmol), and the resulting mixture was heated at reflux for 2 hours. TLC analysis confirmed that the reaction was complete, and the mixture was cooled to room temperature. EtOAc (300 ML) and water (100 ML) were added, the resulting mixture was partitioned, and the layers were separated.The organic layer was washed with water (3*100 ML) and brine (1*100 ML).Then the EtOAc layer was dried over magnesium sulfate, filtered, concentrated, and pumped to give the titled compound (1B) as an off-white powder (7.99 g, M+=310, Mp=90.2-91.4 C.)
With potassium carbonate; In 1-methyl-pyrrolidin-2-one; at 120℃; for 16h;
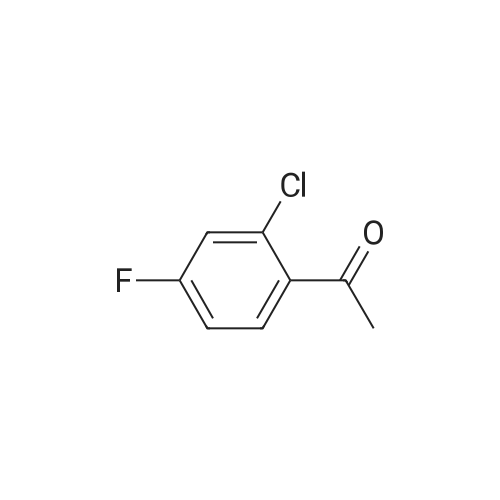
A solution of 50 g of 4-Fluoro-2-Chloro-acetophenone was dissolved in NMP (250 ml_). K2CO3 (52.1 g) and 2,4-difluorophenol (41 .5 g) was added and the reaction mixture was heated to 120C for 16h. MTBE (500ml_) was added and the organic phase was extract 3 times with sat-aq. NH4CI- solution. The organic phase was separated and the aqueous phase was three times extracted with MTBE (300ml_). The combined organic layers were washed with sat NaHC03-solution, sat aq. NaCI and dried over Na2S04. 1 -[2- chloro-4-(2,4-difluorophenoxy)phenyl]ethanone (100 g) was obtained as a brown oil and used in the next step without further purification (HPLC-MS, Rt=1.23 min, masse=283).
3-bromo-4-(2,4-difluorophenoxy)benzaldehyde[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With caesium carbonate; In dimethyl sulfoxide; at 100℃; for 1h;
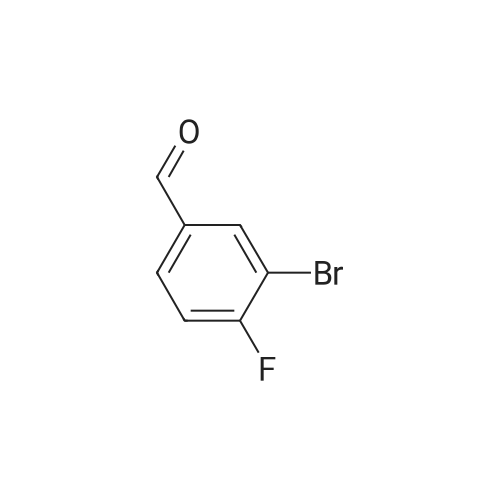
Example 52A 3-bromo-4-(2,4-difluorophenoxy)benzaldehyde [0967] A mixture of <strong>[77771-02-9]3-bromo-4-fluorobenzaldehyde</strong> (4.06 g, 20 mmol), 2,4-difluorophenol (2.60 g, 20 mmol) and cesium carbonate (7.17 g, 22 mmol) in dimethyl sulfoxide (20 mL) was heated at 100 C. for 1 hour. The reaction mixture was partitioned with ethyl acetate and water. The organic layer was washed with saturated aqueous sodium chloride twice, dried with anhydrous sodium sulfate, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, 20% ethyl acetate in heptanes) to provide the title compound (5.94 g, 95%).
95%
With caesium carbonate; In dimethyl sulfoxide; at 100℃; for 1h;
Example 68a 3-bromo-4-(2,4-difluorophenoxy)benzaldehyde A mixture of <strong>[77771-02-9]3-bromo-4-fluorobenzaldehyde</strong> (4.06 g, 20.0 mmol), 2,4-difluorophenol (2.60 g, 20.0 mmol) and cesium carbonate (7.17 g, 22.0 mmol) in dimethyl sulfoxide (20 mL) was heated at 100 C. for 1 hour. The reaction mixture was partitioned with ethyl acetate and water. The organic layer was washed with saturated aqueous sodium chloride twice, dried over anhydrous sodium sulfate, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, 20% ethyl acetate in heptanes) to provide the title compound (5.94 g, 95%).
47%
With caesium carbonate; In dimethyl sulfoxide; at 100℃; for 14h;
In a 100 mL three-neck flask, 2,4-difluorophenol (1.47 g, 11.33 mmol), <strong>[77771-02-9]3-bromo-4-fluorobenzaldehyde</strong> (2.30 g, 11.33 mmol), cesium carbonate (4.05 g, 12.46 mmol), and dimethyl Sulfoxide (10 mL) were sequentially added. The oil bath was heated to 100 C. and reacted for 1 hour and then cooled to room temperature. The reaction solution was diluted with ethyl acetate (30 mL) and washed with saturated brine (10 mL × 3). The organic phase was dried over anhydrous sodium sulfate, filtered, and spin-dried. The crude product was separated by column chromatography (petroleum ether / ethyl acetate: 5/1) and purified to obtain 3-bromo-4- (2,4-difluorophenoxy) benzaldehyde (2.60 g, pale yellow oil, Rate: 47%).
2.9 g
With caesium carbonate; In dimethyl sulfoxide; at 100℃; for 2h;Inert atmosphere;
<strong>[77771-02-9]3-bromo-4-fluorobenzaldehyde</strong> (2.1 g, 10 mmol, CAS: 77771-02-9),2,4-Difluorophenol (1.43 g, 11 mmol), cesium carbonate (3.6 g, 11 mmol) were sequentially added to 50 mL of dimethyl sulfoxide, and the mixture was heated to 100 C under argon atmosphere for 2 hours.500 mL of water was added to the reaction mixture, and the mixture was extracted with ethyl acetate 150 mL×3, and the organic phase was combined, washed three times with saturated brine, dried over anhydrous sodium sulfate and evaporated.Purification by silica gel column chromatography gave Compound E56-1 (2.9 g).
2-bromo-1-(2,4-difluorophenoxy)-4-nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1h;
Example 163a 2-bromo- 1 -(2,4-difluorophenoxy)-4-nitrobenzene 2-Bromo-l-fluoro-4-nitrobenzene (15 g, 68.2 mmol), 2,4-difluorophenol (7.82 mL, 82 mmol), and cesium carbonate (26.7 g, 82 mmol) were combined in DMSO (75 mL) then heated to 110C for 1 hour. After cooling, the reaction mixture was partitioned between water and ethyl acetate. The aqueous layer was extracted with additional ethyl acetate twice. The combined organic layers were washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate, filtered, and concentrated to give the title compound (22.51 g, 68.2 mmol, 100 %).
100%
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1h;
A mixture of 2-bromo-l-fluoro-4-nitrobenzene (15 g, 68 mmol), 2,4- difluorophenol (7.82 mL, 82 mmol), and cesium carbonate (26.7 g, 82 mmol) in dimethylsulfoxide (75 mL) was heated at 110 C for 1 hour. The reaction mixture was cooled to ambient temperature and water (1000 mL) and saturated aqueous sodium chloride (1000 mL) were added. The mixture was extracted with ethyl acetate (3x200 mL). The combined organics were washed with saturated aqueous sodium chloride, dried (anhydrous magnesium sulfate), filtered, and concentrated under reduced pressure to provide 2-bromo-l-(2,4-difluorophenoxy)-4-nitrobenzene (22.5 g, quantitative).
84%
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 2h;
2-Bromo-1-fluoro-4-nitrobenzene (14 g, 107.7 mmol), 2,4-difluorophenol (19.6 g, 89.7 mmol) was dissolved in DMSO (100 mL) and cesium carbonate was added at room temperature ( 35 g, 17.7 mmol), then stirred at 110 C for 2 hours. The reaction mixture was added water (150 mL), extracted with ethyl acetate (200mL * 1), the organic phase washed with brine (100mL * 1), dried over anhydrous sodium sulfate, and concentrated to give2-Bromo-1-(2,4-difluorophenoxy)-4-nitrobenzene (32 g, 86.16 mmol, molar yield 84%).
84%
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 2h;
<strong>[701-45-1]2-bromo-1-fluoro-4-nitrobenzene</strong> (14 g, 107.7 mmol),2,4-difluorophenol (19.6 g, 89.7 mmol) was dissolved in DMSO (100 mL).Cesium carbonate (35 g, 17.7 mmol) was added at room temperature and then stirred at 110 C for 2 hours.Water (150 mL) was added to the reaction mixture, followed by ethyl acetate (200 mL*1).The organic phase was washed with saturated brine (100 mL*1).Dry over anhydrous sodium sulfate and concentrate to give 2-bromo-1-(2,4-difluorophenoxy)-4-nitrobenzene (32 g, yield 84%).
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1h;
2-Bromo-1-fluoro-4-nitrobenzene (15 g, 68.2 mmol), 2,4-difluorophenol (7.82 ml, 82 mmol), and cesium carbonate (26.7 g, 82 mmol) were combined in DMSO (75 mL) then heated to 110C for 1 hour. After cooling, to the reaction mixture was added water (1000 mL) and brine (1000 mL), and the mixture was extracted with ethyl acetate (3 x 200 mL). The combined organics were washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure to give a crude solid which was used in the next step without additional purification.
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1h;
Example 5B 2-bromo-1-(2,4-difluorophenoxy)-4-nitrobenzene [0729] 2-Bromo-1-fluoro-4-nitrobenzene (15 g, 68.2 mmol), 2,4-difluorophenol (7.82 ml, 82 mmol), and cesium carbonate (26.7 g, 82 mmol) were combined in DMSO (75 mL) then heated to 110 C. for 1 hour. After cooling, to the reaction mixture was added water (1000 mL) and brine (1000 mL), and the mixture was extracted with ethyl acetate (3×200 mL). The combined organics were washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure to give a crude solid which was used in the next step without additional purification.
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1h;
[0363] 2-Bromo-1-fluoro-4-nitrobenzene (15 g, 68.2mmol), 2,4-difluorophenol (7 .82 mL, 82 mmol), and cesiumcarbonate (26.7 g, 82 mmol) were combined in DMSO (75mL), heated at 110 C. for I hour, and then cooled. To thecooled reaction mixture was added water (1000 mL) andsaturated aqueous sodium chloride (1000 mL). The mixturewas extracted with ethyl acetate (3x200 mL). The combinedorganics were washed with saturated aqueous sodium chloride,dried (anhydrous magnesium sulfate), filtered, and concentratedunder reduced pressure to give a crude solid whichwas used in the next step without additional purification.
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1h;
2-Bromo-1-fluoro-4-nitrobenzene (15 g, 68.2 mmol), 2,4-difluorophenol (7.82 mL, 82 mmol), and cesium carbonate (26.7 g, 82 mmol) were combined in DMSO (75 mL), heated at 110 C for 1 hour, and then cooled. To the cooled reaction mixture was added water (1000 mL) and saturated aqueous sodium chloride (1000 mL). The mixture was extracted with ethyl acetate (3 x 200 mL). The combined organics were washed with saturated aqueoussodium chloride, dried (anhydrous magnesium sulfate), filtered, and concentrated under reduced pressure to give a crude solid which was used in the next step without additional purification.
With caesium carbonate; In dimethyl sulfoxide; at 100℃;
3-Bromo-4-fluoronitrobenzene (5.0 g, 22.7 mmol, CAS: 701-45-1), 2,4-difluorophenol (3.0 g, 23.1 mmol), cesium carbonate (9.5 g, 29.2 mmol) The suspension was suspended in 6 mL of dimethyl sulfoxide and heated to 100 C overnight. After cooling to room temperature, water and ethyl acetate were added and the organic phase was washed with saturated brine, dried over anhydrous sodium Intermediate M-3-1.
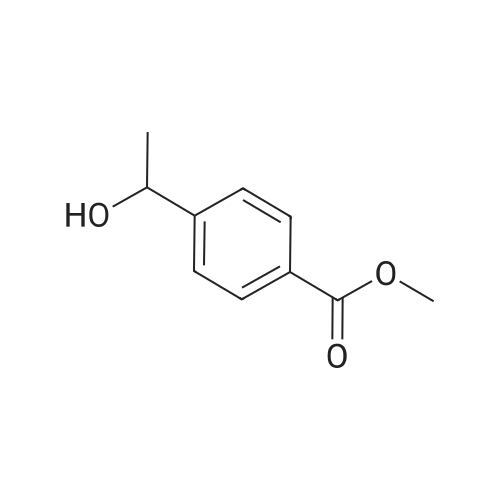
methyl 4-(1-(2,4-difluorophenoxy)ethyl)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
33%
Methyl 4-(l-(2,4-dtfluorophenoxy)ethyl)benzoate. To the solution of 4-(l- hydroxyethyl)benzoate (300 mg.1.67 mmol) in tetrahydrofuran (80 mL) was added triphenylphosphine (570 mg, 2.2 mmol) and 2,4-difluorophenol (159 mg,1.67 mmol) , the mixture was stirred for 30 minutes at room temperature, then diisopropylazodicarboxylate (568.3 mg, 2.2 mmol) was added dropwise to the solution at 0 C. The mixture was stirred at room temperature for 12 hours. Then the mixture was concentrated and the residue was purified by column chromatography (silica gel, Petroleum ether / ethyl acetate = 20: 1) to give methyl 4-(l-(2,4-difhiorophenoxy)ethyl)benzoate (151 mg, 33%) as oil. 1H-NMR (300 MHz, CD3OD) delta 8.03 (d, J = 8.1 Hz, 2H,), 7.460 (d, J = 8.1 Hz, 2H), 6.86-6.72 (m, 3H), 5.29 (q, 1H), 3.91(s, 3H), 1.67 (d, J = 6.3 Hz, 2H).
33%
To the solution of 4-(1-hydroxyethyl)benzoate (300 mg. 1.67 mmol) in tetrahydrofuran (80 mL) was added triphenylphosphine (570 mg, 2.2 mmol) and 2,4-difluorophenol (159 mg, 1.67 mmol), the mixture was stirred for 30 minutes at room temperature, then diisopropylazodicarboxylate (568.3 mg, 2.2 mmol) was added dropwise to the solution at 0 C. The mixture was stirred at room temperature for 12 hours. Then the mixture was concentrated and the residue was purified by column chromatography (silica gel, Petroleum ether/ethyl acetate=20:1) to give methyl 4-(1-(2,4-difluorophenoxy)ethyl)benzoate (151 mg, 33%) as oil. 1H-NMR (300 MHz, CD3OD) delta 8.03 (d, J=8.1 Hz, 2H), 7.460 (d, J=8.1 Hz, 2H), 6.86-6.72 (m, 3H), 5.29 (q, 1H), 3.91 (s, 3H), 1.67 (d, J=6.3 Hz, 2H).
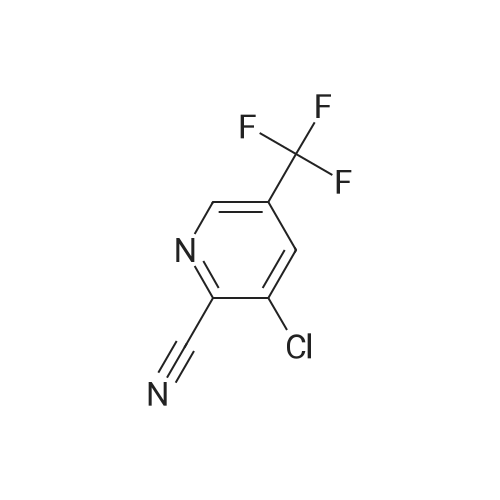
3-(2,4-difluorophenoxy)-5-trifluoromethylpyridine-2-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
71%
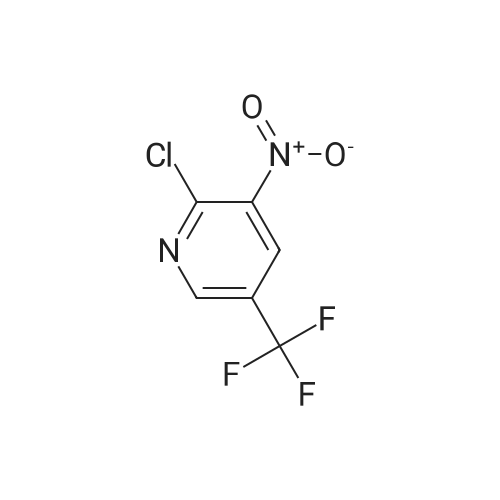
To an N,N-dimethylformamide solution (6 mL) of 2,4-difluorophenol (0.57 mL, 6.0 mmol), sodium hydride (content 63%) (0.23 g, 6.0 mmol) was added under a nitrogen stream at 0C, followed by stirring for 10 minutes. Subsequently, to the reaction solution, <strong>[80194-70-3]3-chloro-5-trifluoromethylpyridine-2-carbonitrile</strong> (2.8 g, 12.3 mmol) was added, followed by stirring for 1 hour. The reaction solution was poured into a saturated aqueous ammonium chloride solution, extraction was carried out with ethyl acetate, and the extract was washed with water and brine sequentially. The resulting organic layer was dried over anhydrous sodium sulfate and subsequently concentrated under reduced pressure to give a residue. The resulting residue was purified using silica gel column chromatography (hexane-ethyl acetate) to afford the desired title compound (1.3 g, 71 %). 1H-NMR (CDCl3, 400 MHz) delta: 8.68-8.67 (1H, m), 7.32-7.27 (2H, m), 7.11-7.02 (2H, m).
With potassium carbonate In dimethyl sulfoxide at 120℃; for 2h;
252a Example 252a 1-(2,4-difluorophenoxy)-4-(methylsulfonyl)-2-nitrobenzene
A mixture of 1-fluoro-4-(methylsulfonyl)-2-nitrobenzene (20 g, 91 mmol), 2,4-difluorophenol (11.87 g, 91 mmol) and potassium carbonate (12.6 g, 91 mmol) in DMSO (90 mL) was heated at 120° C. for 2 hours. The reaction mixture was quenched with water and extracted with ethyl acetate. The combined organic layers were washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, 1:1 ethyl acetate/hexanes) to provide the title compound (28 g, 89% yield).
89%
With potassium carbonate In dimethyl sulfoxide at 120℃; for 2h;
155.155a Example 155a l-(2,4-difluorophenoxy)-4-(methylsulfonyl)-2 -nitrobenzene
Example 155a l-(2,4-difluorophenoxy)-4-(methylsulfonyl)-2 -nitrobenzene A mixture of l-fluoro-4-(methylsulfonyl)-2-nitrobenzene (20 g, 91 mmol), 2,4- difluorophenol (11.87 g, 91 mmol) and potassium carbonate (12.6 g, 91 mmol) in DMSO (90 mL) was heated at 120 °C for 2 hours. The reaction mixture was quenched with water and extracted with ethyl acetate. The combined organic layers were washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate, filtered, and concentrated. The residue was purified by flash chromatography (silica gel, 1 : 1 ethyl acetate/hexanes) to provide the title compound (28 g, 89% yield).
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃; for 16h;
A mixture of <strong>[68322-84-9]3-bromo-4-fluorobenzotrifluoride</strong> (0.5 mL, 3.52 mmol), 2,4-difluorophenol (0.337 mL, 3.52 mmol), and potassium carbonate (0.486 g, 3.52 mmol) in dimethylformamide (7 mL) was heated at 80 C. for 16 hours. The reaction mixture was cooled to ambient temperature and partitioned between ethyl acetate and water. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organics were washed with water and saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, filtered, and concentrated. The crude material was purified by flash chromatography (silica gel, 0-10% ethyl acetate/hexanes gradient) to provide the title compound (1.0 g, 80% yield).
80%
With potassium carbonate; In N,N-dimethyl-formamide; at 80℃;
Example 34a 2-bromo-1-(2,4-difluorophenoxy)-4-(trifluoromethyl)benzene A mixture of <strong>[68322-84-9]3-bromo-4-fluorobenzotrifluoride</strong> (0.5 mL, 3.52 mmol), 2,4-difluorophenol (0.337 mL, 3.52 mmol), and potassium carbonate (0.486 g, 3.52 mmol) in dimethylformamide (7 mL) was heated at 80 C. overnight. The reaction mixture was cooled to ambient temperature and partitioned between ethyl acetate and water. The layers were separated, and the aqueous layer was extracted with ethyl acetate. The combined organics were washed with water and saturated aqueous sodium chloride, dried over anhydrous sodium sulfate, filtered, and concentrated by rotary evaporation. The crude material was purified by flash chromatography (ethyl acetate/hexanes) to provide the title compound (1.0 g, 80% yield).
With caesium carbonate; In dimethyl sulfoxide; at 110℃; for 1.0h;
Example 2a 4-bromo-5-(2,4-difluorophenoxy)-2-nitrobenzoic acid A mixture of <strong>[1020717-99-0]4-bromo-5-fluoro-2-nitrobenzoic acid</strong> (3.5 g, 13.3 mmol), 2,4- difluorophenol (1.5 mL, 15.70 mmol), cesium carbonate (9.5 g, 29.2 mmol) and dimethylsulfoxide (28 mL) was heated at 110 C for 1 hour. After cooling to ambient temperature, water (100 mL) was added. The solution was acidified with IN HC1 (60 mL) and was extracted with ethyl acetate. The ethyl acetate layer was washed with water, saturated aqueous sodium chloride, dried (anhydrous magnesium sulfate), filtered, and concentrated to provide the title compound (4.73 g, 12.64 mmol, 95 % yield)
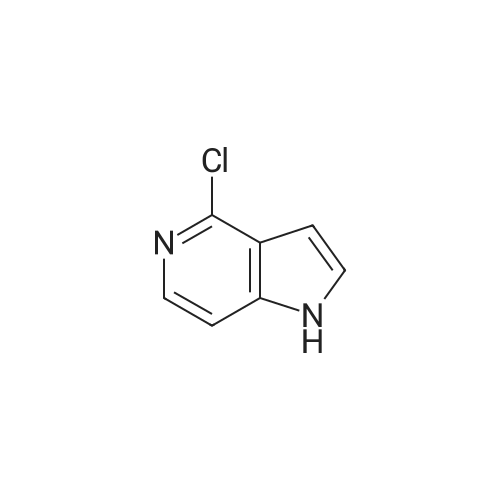
With sodium hydride; In N,N-dimethyl acetamide; mineral oil; at 250℃; for 0.05h;Inert atmosphere; Microwave irradiation;
A solution of <strong>[60290-21-3]4-chloro-1H-pyrrolo[3,2-c]pyridine</strong>(100 mg, 0.66 mmol) and 2,4-difluorophenol (213 mg, 1.64mmol) in DMA (2 mL) was stirred for 5 minutes under nitrogen. Sodium hydride (26.2 mg, 0.66 mmol; 60% inmineral oil) was heated with stirring at 250C for 3 minutesunder microwave irradiation. The mixture was diluted with THF (10 mL) and quenched with water and ice (2 mL). The solution was filtered through a VarianChemElute cartridge and concentrated to dryness. The material was partially purified by flash chromatography with gradient elution of heptane and ethyl acetate (1:0 to 0:1) to give a gum. The material was further purified by reverse phase preparative HPLC to provide the desired product (49.0 mg, 60.7 %) as asolid. HPLC purity: >99% (215 nM),>99% (254 nM), >99% (280 nM). 1H NMR (400 MHz, DMSO-d6) d ppm 6.63 (dd, J=3.3, 1.0 Hz, 1 H) 7.08 - 7.19 (m, 2 H) 7.35 - 7.46(m, 3 H) 7.58 (d, J=5.9 Hz, 1 H). HRMS m/z calcd for C13H8F2N2O[M+H]+ 247.0677, found 247.0678.
60.7%
A solution of <strong>[60290-21-3]4-chloro-1H-pyrrolo[3,2-c]pyridine</strong>(100 mg, 0.66 mmol) and 2,4-difluorophenol (213 mg, 1.64 mmol) in DMA (2 mL)was stirred for 5 minutes under nitrogen.Sodium hydride (26.2 mg, 0.66 mmol; 60% in mineral oil) was heated withstirring at 250 C for 3 minutes under microwave irradiation. The mixture was dilutedwith THF (10 mL) and quenched with water and ice (2 mL). The solution wasfiltered through a Varian ChemElute cartridge and concentrated to dryness. The materialwas partially purified by flash chromatography with gradient elution of heptaneand ethyl acetate (1:0 to 0:1) to give a gum. The material was further purifiedby reverse phase preparative HPLC to provide the desired product (49.0 mg, 60.7%) as a solid. HPLC purity: >99% (215nM), >99% (254 nM), >99% (280 nM). 1H NMR (400 MHz, DMSO-d6) d ppm 6.63 (dd, J=3.3, 1.0 Hz, 1 H) 7.08 - 7.19 (m, 2 H) 7.35 - 7.46(m, 3 H) 7.58 (d, J=5.9 Hz, 1 H). HRMS m/z calcd for C13H8F2N2O[M+H]+ 247.0677, found 247.0678.
3-bromo-2-(2,4-difluorophenoxy)-5-nitropyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
94%
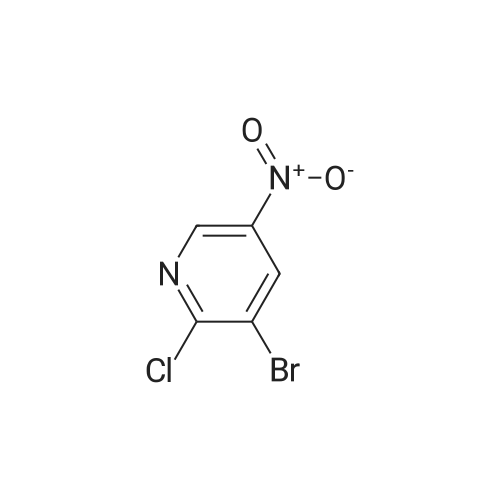
With caesium carbonate; In dimethyl sulfoxide; at 80℃; for 0.5h;Microwave irradiation;
Example 184A 3-bromo-2-(2,4-difluorophenoxy)-5-nitropyridine [1238] A mixture of <strong>[5470-17-7]3-bromo-2-chloro-5-nitropyridine</strong> (0.237 g, 1 mmol), 2,4-difluorophenol (0.13 g, 1 mmol) and cesium carbonate (0.326 g, 1 mmol) in dimethyl sulfoxide (2 mL) was reacted in a Biotage microwave reactor at 80 C. for 30 minutes. The reaction mixture was partitioned between ethyl acetate and water, washed with saturated aqueous sodium chloride, dried over anhydrous magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (silica gel, 0-20% ethyl acetate in heptanes) to provide 0.312 g (94%) of the title compound.
59%
With caesium carbonate; In 1-methyl-pyrrolidin-2-one; at 60℃; for 12h;
A solution of <strong>[5470-17-7]3-bromo-2-chloro-5-nitropyridine</strong> (2.4 g, 10 mmol) and 2,4-difluorophenol (1 mL, 11 mmol) in NMP (20 ml) was treated with cesium carbonate (3.9 g, 12 mmol). The resulting mixture was heated to 60 C. for 12 h. The mixture was treated with water (100 ml) and extracted with EtOAc (3*50 ml); the combined organic extracts were washed with saturated bicarbonate solution (aq), dried over sodium sulfate, filtered and concentrated in vacuo to afford a yellow solid. The solid was purified by silica gel column chromatography (gradient of 5 to 30% EtOAc in hexanes) to afford the free base of the title compound (2 g, 59%) as a yellow solid. LCMS (M+H)+=332.
7 g
With caesium carbonate; In dimethyl sulfoxide; at 100℃; for 1h;
2-Chloro-3-bromo-5-nitropyridine (5.0 g, CAS: 5470-17-7), 2,4-difluorophenol (2.3 g), cesium carbonate (8 g) suspended in 100 mL of dimethylsulfoxide,The mixture was heated to 100 C for 1 hour, cooled to room temperature, and filtered to remove solids.After adding water and ethyl acetate, the organic phase was washed with saturated brine and dried over anhydrous sodium sulfate.Concentrate to intermediate E92-1 (7 g).
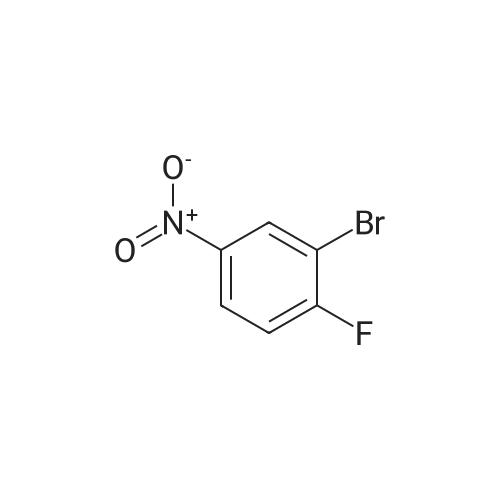
1-bromo-2-(2,4-difluorophenoxy)-4-fluoro-5-nitrobenzene[ No CAS ]
1-bromo-4-(2,4-difluorophenoxy)-2-fluoro-5-nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With caesium carbonate; In dimethyl sulfoxide; at 20℃; for 1h;
Example 11A 1-bromo-2-(2,4-difluorophenoxy)-4-fluoro-5-nitrobenzene [0864] A mixture of <strong>[345-24-4]1-bromo-2,4-difluoro-5-nitrobenzene</strong> (0.5 g, 2.10 mmol) and cesium carbonate (0.685 g, 2.10 mmol) in DMSO (10.50 mL) was treated dropwise with 2,4-difluorophenol (0.273 g, 2.10 mmol), stirred for 60 minutes at ambient temperature and partitioned between ethyl acetate and water, adjusting the pH to 6 with 1 M HCl. The organic layer was washed with water, saturated aqueous sodium chloride, dried (MgSO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-30% ethyl acetate in heptanes) afforded the title compound and 1-bromo-4-(2,4-difluorophenoxy)-2-fluoro-5-nitrobenzene as an inseparable mixture (0.59 g, 81%).
4,4'-(4-bromo-6-nitro-1,3-phenylene)bis(oxy)bis(1,3-difluorobenzene)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
100%
With caesium carbonate; In dimethyl sulfoxide; at 20℃; for 1h;
Example 13A 4,4?-(4-bromo-6-nitro-1,3-phenylene)bis(oxy)bis(1,3-difluorobenzene) [0872] To a solution of <strong>[345-24-4]1-bromo-2,4-difluoro-5-nitrobenzene</strong> (0.3 g, 1.26 mmol) and cesium carbonate (0.86 g, 2.65 mmol) in DMSO (6.3 mL) was added dropwise 2,4-difluorophenol (0.27 g, 2.10 mmol). The mix was stirred for 60 minutes at ambient temperature and partitioned between ethyl acetate and water, adjusting the pH to 6 with HCl. The organic layer was washed with water, saturated aqueous sodium chloride, dried (MgSO4), filtered, and concentrated to afford the title compound (0.578 g, 100%).
5-bromo-4-(2,4-difluorophenoxy)-2-fluoroaniline[ No CAS ]
5-bromo-2-(2,4-difluorophenoxy)-4-fluoroaniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
A mixture of <strong>[345-24-4]1-bromo-2,4-difluoro-5-nitrobenzene</strong> (0.5 g, 2.10 mmol) and cesium carbonate (0.685 g, 2.10 mmol) in DMSO (10.50 mL) was treated dropwise with 2,4-difluorophenol (0.273 g, 2.10 mmol), stirred for 60 minutes at ambient temperature and partitioned between ethyl acetate and water, adjusting the pH to 6 with 1 M HCl. The organic layer was washed with water, saturated aqueous sodium chloride, dried (MgSO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-30% ethyl acetate in heptanes) afforded the title compound and 1-bromo-4-(2,4-difluorophenoxy)-2-fluoro-5-nitrobenzene as an inseparable mixture (0.59 g, 81%). Example 11B 5-bromo-4-(2,4-difluorophenoxy)-2-fluoroaniline [0865] The product from Example 11A (0.59 g, 1.69 mmol), iron (0.47 g, 8.48 mmol) and ammonium chloride (0.136 g, 2.54 mmol) were combined in a solvent mixture of ethanol (9 mL), tetrahydrofuran (9 mL) and water (3 mL) and heated at 90 C. for 2 hours. The mixture was cooled, filtered through Celite and the Celite pad was washed repeatedly with methanol. The filtrate was concentrated and the residue was partitioned between ethyl acetate and water. The organic layer was washed with saturated aqueous sodium chloride, dried (anhydrous Na2SO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-60% ethyl acetate in heptanes) afforded the title compound and 5-bromo-2-(2,4-difluorophenoxy)-4-fluoroaniline as an inseparable mixture (0.43 g, 80%).
N-(5-bromo-4-(2,4-difluorophenoxy)-2-fluorophenyl)methanesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
A mixture of <strong>[345-24-4]1-bromo-2,4-difluoro-5-nitrobenzene</strong> (0.5 g, 2.10 mmol) and cesium carbonate (0.685 g, 2.10 mmol) in DMSO (10.50 mL) was treated dropwise with 2,4-difluorophenol (0.273 g, 2.10 mmol), stirred for 60 minutes at ambient temperature and partitioned between ethyl acetate and water, adjusting the pH to 6 with 1 M HCl. The organic layer was washed with water, saturated aqueous sodium chloride, dried (MgSO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-30% ethyl acetate in heptanes) afforded the title compound and 1-bromo-4-(2,4-difluorophenoxy)-2-fluoro-5-nitrobenzene as an inseparable mixture (0.59 g, 81%). Example 11B 5-bromo-4-(2,4-difluorophenoxy)-2-fluoroaniline [0865] The product from Example 11A (0.59 g, 1.69 mmol), iron (0.47 g, 8.48 mmol) and ammonium chloride (0.136 g, 2.54 mmol) were combined in a solvent mixture of ethanol (9 mL), tetrahydrofuran (9 mL) and water (3 mL) and heated at 90 C. for 2 hours. The mixture was cooled, filtered through Celite and the Celite pad was washed repeatedly with methanol. The filtrate was concentrated and the residue was partitioned between ethyl acetate and water. The organic layer was washed with saturated aqueous sodium chloride, dried (anhydrous Na2SO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-60% ethyl acetate in heptanes) afforded the title compound and 5-bromo-2-(2,4-difluorophenoxy)-4-fluoroaniline as an inseparable mixture (0.43 g, 80%).Example 11C N-(5-bromo-4-(2,4-difluorophenoxy)-2-fluorophenyl)methanesulfonamide [0866] A mixture of the product from Example 11B (0.428 g, 1.35 mmol) and triethylamine (0.469 mL, 3.36 mmol) in dichloromethane (13.5 mL) was treated dropwise with methanesulfonyl chloride (0.23 mL, 2.96 mmol), stirred for 2 hours and concentrated. The residue was dissolved in a mixture of dioxane (4 mL) and 1M sodium hydroxide (1 mL), heated for 1 hour at 90 C., cooled and diluted with ethyl acetate. The mixture was brought to pH 7 with 1 M HCl and the organic layer was separated, dried (anhydrous Na2SO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-60% ethyl acetate in heptanes) afforded the title compound as the second eluting regioisomer (0.346 g, 65%).
N-(5-bromo-2-(2,4-difluorophenoxy)-4-fluorophenyl)methanesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
11%
A mixture of <strong>[345-24-4]1-bromo-2,4-difluoro-5-nitrobenzene</strong> (0.5 g, 2.10 mmol) and cesium carbonate (0.685 g, 2.10 mmol) in DMSO (10.50 mL) was treated dropwise with 2,4-difluorophenol (0.273 g, 2.10 mmol), stirred for 60 minutes at ambient temperature and partitioned between ethyl acetate and water, adjusting the pH to 6 with 1 M HCl. The organic layer was washed with water, saturated aqueous sodium chloride, dried (MgSO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-30% ethyl acetate in heptanes) afforded the title compound and 1-bromo-4-(2,4-difluorophenoxy)-2-fluoro-5-nitrobenzene as an inseparable mixture (0.59 g, 81%). Example 11B 5-bromo-4-(2,4-difluorophenoxy)-2-fluoroaniline [0865] The product from Example 11A (0.59 g, 1.69 mmol), iron (0.47 g, 8.48 mmol) and ammonium chloride (0.136 g, 2.54 mmol) were combined in a solvent mixture of ethanol (9 mL), tetrahydrofuran (9 mL) and water (3 mL) and heated at 90 C. for 2 hours. The mixture was cooled, filtered through Celite and the Celite pad was washed repeatedly with methanol. The filtrate was concentrated and the residue was partitioned between ethyl acetate and water. The organic layer was washed with saturated aqueous sodium chloride, dried (anhydrous Na2SO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-60% ethyl acetate in heptanes) afforded the title compound and 5-bromo-2-(2,4-difluorophenoxy)-4-fluoroaniline as an inseparable mixture (0.43 g, 80%).Example 11D N-(5-bromo-2-(2,4-difluorophenoxy)-4-fluorophenyl)methanesulfonamide [0867] A mixture of the product from Example 11B (0.428 g, 1.35 mmol) and triethylamine (0.469 mL, 3.36 mmol) in dichloromethane (13.5 mL) was treated dropwise with methanesulfonyl chloride (0.23 mL, 2.96 mmol), stirred for 2 hours at ambient temperature and concentrated. The residue was dissolved in a mixture of dioxane (4 mL) and 1M sodium hydroxide (1 mL), heated for 1 hour at 90 C., cooled and diluted with ethyl acetate. The mixture was brought to pH 7 with 1 M HCl and the organic layer was separated, dried (anhydrous Na2SO4), filtered, and concentrated. Purification by chromatography (silica gel, 0-60% ethyl acetate in heptanes) afforded the title compound as the first eluting regioisomer (0.057 g, 11%).
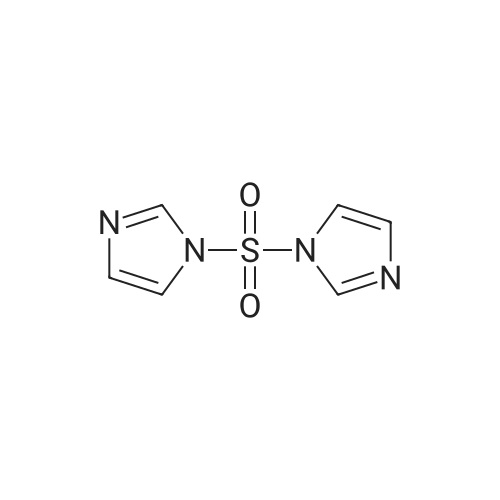
General procedure: A solution of 500 mg (4.46 mmol) of 2-fluorophenol 1.769 g (8.92 mmol) of 1,1?-sulfonyldiimidazole in 15 mL of tetrahydrofuran was treated with 727 mg (2.23 mmol) of cesium carbonate. The reaction was stirred for 12 h, concentrated, and the residue was partitioned between the ethyl acetate and water (10 mL each). The organic layer was washed sequentially with saturated ammonium chloride solution and brine. After drying over the anhydrous sodium sulfate, the organic layer was concentrated and chromatographed on silica with 1:5 ethyl acetate / hexane as the eluant to afford 880 mg (90%) of 11bas a clear oil:
1-(3-bromo-5-nitrophenoxy)-2,4-difluorobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
30%
With potassium carbonate; In N,N-dimethyl-formamide; at 100℃; for 5.0h;Microwave irradiation;
A mixture of 2,4-difluorophenol (286 mg, 2.2 mmol) and <strong>[7087-65-2]1-bromo-3-fluoro-5-nitrobenzene</strong> (440 mg, 2 mmol) in DMF (4.5 mL) was treated with K2CO3 (304 mg, 2.2 mmol). The mixture was heated to 100 C. by microwave irridation (normal) for 5 h. The resulting suspension was diluted with water and extracted with EtOAc (15 mL*3). The combined organic layers were washed with 1N NaOH (aq) (15 mL), water (15 mL), brine, dried over MgSO4, and concentrated in vacuo. The crude solid was purified by silica gel column chromatography using a gradient of EtOAc (5 to 25%) in hexanes to afford the title compound (200 mg, 30%) as a yellow solid. LCMS (M+H)+=339.
Phosphorus oxychloride (12 g, 77 mmol) was added dropwise to anhydrous dimethyl formamide (8.4 g, 0.12 mol) at 0 C under nitrogen with stirring. The mixture was stirred at this temperature for 30 minutes, and 2,4-difluorophenol (5.0 g, 39 mmol) in N N-dimethyl formamide (50mL) was added dropwise. The reaction mixture was stirred at room temperature for 5 hours. On completion, the reaction mixture was quenched slowly with water and extracted with ethyl acetate (3 x 150 mL). The combined organic layers were washed with water and brine, dried over anhydrous sodium sulfate and concentrated in vacuo to give compound B-163 (5.5 g, 90% yield) as a yellow oil.
2-bromo-3-(2,4-difluorophenoxy)-6-nitroaniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With caesium carbonate In dimethyl sulfoxide at 110℃; for 0.5h;
1.1 2-Bromo-3-(2,4-difluorophenoxy)-6-nitroaniline
A solution of 2-bromo-3-fluoro-6-nitroaniline (0.20 g, 0.851 mmol) [J&W PharmaLab, 20-0976] and cesium carbonate (0.333 g, 1.02 mmol) in dimethyl sulfoxide (0.94 mL) was treated with 2,4-difluorophenol (0.166 mL, 1.02 mmol) and stirred at 110° C. for 30 min. The reaction mixture was diluted with ethyl acetate and water. The organic layer was separated and washed with water and brine, dried with magnesium sulfate, filtered, and concentrated to give a crude oil. Purification by flash column chromatography (100% hexanes to 40% EtOAc/hexanes) gave the desired product (294 mg, 95%) as a yellow solid. LCMS calculated for C12HsBrF2N2O3 (M+H)+: m/z=345.0, 347.0. found: 345.0, 347.0.
4-bromo-5-(2,4-difluorophenoxy)-2-nitroaniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With caesium carbonate; In dimethyl sulfoxide; at 80℃; for 0.5h;
Step 1. 4-Bromo-5-(2,4-difluorophenoxy)-2-nitroaniline A solution of <strong>[153505-36-3]4-bromo-5-fluoro-2-nitroaniline</strong> (1.08 g, 4.60 mmol) [Combi-Blocks, AN-1501] and cesium carbonate (1.80 g, 5.52 mmol) in dimethyl sulfoxide (5.0 mL) was treated with 2,4-difluorophenol (0.90 mL, 5.53 mmol) [Acros Organics, 24320] and stirred at 80 C. for 30 min. The reaction mixture was diluted with ethyl acetate and water. The organic layer was separated and washed with water and brine, dried with magnesium sulfate, filtered, and concentrated to give the desired product (1.50 g, 95%) as a yellow solid that was used without further purification. LCMS calculated for C12H8BrF2N2O3 (M+H)+: m/z=345.0, 347.0. found: 345.0, 346.9.
4-bromo-3-(2,4-difluorophenoxy)-2-nitroaniline[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With caesium carbonate; In dimethyl sulfoxide; at 20 - 110℃; for 1h;
Step 1. 4-Bromo-3-(2,4-difluorophenoxy)-2-nitroaniline A solution of <strong>[886762-75-0]4-bromo-3-fluoro-2-nitroaniline</strong> (0.100 g, 0.426 mmol) [Combi-Blocks, AN-2785] and cesium carbonate (0.167 g, 0.511 mmol) in dimethyl sulfoxide (0.47 mL) was treated with 2,4-difluorophenol (0.083 mL, 0.512 mmol) and stirred at 20 C. for 30 min and heated at 110 C. for 30 min. The reaction mixture was diluted with ethyl acetate and water. The organic layer was separated and washed with water and brine, dried with magnesium sulfate, filtered, and concentrated to give a crude residue. Purification by flash column chromatography (100% hexanes to 40% EtOAc/hexanes) gave the desired product (140 mg, 95%) as an orange/brown oil. LCMS calculated for C12H8BrF2N2O3 (M+H)+: m/z=345.0, 347.0. found: 344.9, 346.9.
5-(2,6-dichloropyrimidin-4-yl)-1,3-dimethylpyridin-2-one[ No CAS ]
N-[4-(2,4-difluorophenoxy)-6-(1,5-dimethyl-6-oxopyridin-3-yl)pyrimidin-2-yl]ethanesulfonamide[ No CAS ]
N-[2-(2,4-difluorophenoxy)-6-(1,5-dimethyl-6-oxopyridin-3-yl)pyrimidin-4-yl]ethanesulfonamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
8%
A mixture of 2,4-difluorophenol (25 mg, 0.19 mmol) and 5-(2,6-dichloropyrimidin-4-yl)- 1 ,3-dimethylpy- ridin-2-one (50 mg, 0.19 mmol) in DMF (0.5 mE) and THF (0.5 mE) was treated with K2C03 (304 mg, 0.23 mmol). The mixture was stirred at rt for 3 h. The resulting suspension was diluted with water and extracted with EtOAc (10 mEx3). The combined organic layers were washed with iN NaOH (aq) (5 mE), water (15 mE), brine, dried over Mg504, and concentrated in vacuo. The crude solid was purified by silica gel column chromatography using a gradient of EtOAc (0 to 50percent) in DCM to afford an unseparated mixture of regioisomeric title compounds (66 mg, 96percent combined) as a white solid. ECMS (M+H)=364 for both regioisomers.11315] A solution of <strong>[1520-70-3]ethanesulfonamide</strong> (80 mg, 0.73 mmol) in DMF (2 mE) was treated with NaH (27 mg, 0.68 mmol, 60percent by weight). Afier 15 mm, the mixture was treated with a DMF (1 mE) solution of title compounds obtainedfrom Step 2. The reaction mixture was stirred at 500 C. for 14 h. The resulting suspension was diluted with water and extracted with EtOAc (10 mEx3). The combined organic layers were washed with brine, dried over Na2504, and concentrated in vacuo. Preparative HPEC isolated both regioisomers as Examples 346 and 347. The title compound (6 mg, 8percent) was obtained as a white solid. ?H NMR (400 MHz, DMSO-d5) oe ppm 1.08 (t, J=7.1 Hz, 3H) 2.07 (s, 3H) 3.11 (q, J=7.1 Hz, 2H) 3.54 (s, 3H) 6.81 (s, 1H) 7.17 (m, 1H) 7.47 (s, 2H) 7.79 (s, 1H) 8.37 (s, 1H) 11.37 (bs, 1H). ECMS (M+H7 =437.
1.30 g (10.00 mmol) 2,4-difluoro-phenol, 1.52 g (0.91 mL, 10 mmol) bromoacetic acid methyl ester, and 1.38 g (10 mmol) potassium carbonate were solved in 25 mL N-methylpyrrolidone, and the solution was stirred for 6 hours at room temperature. 0.92 g (5 mmol) 4-Methylamino-2-methylsulfanyl-pyrimidine-5-carbaldehyde and 1.38 g (10 mmol) potassium carbonate were added to the solution, and the mixture was stirred at 120 oC for 12 hours. The reaction mixture was cooled down to room temperature, and diluted with 75 ml water. After stirring a half an hour the solid product was filtered out, washed with water, and crystallized from acetonitrile.Yield: 0.9 g (55 %)MW calc: 335.054[M+H]+: 336.04[M-H]- : -Rt: 3.90 min1H NMR (300 MHz, DMSO-d6) delta ppm: 8.82 (s, 1H); 7.51 (ddd, J = 11.3, 8.8 and 2.9 Hz, 1H); 7.41 (s, 1H); 7.35 (ddd, J = 9.3, 9.0 and 5.6 Hz, 1H); 7.13 (dddd, J = 9.0, 8.3, 2.9 and 1.5 Hz, 1H); 3.67 (s, 3H); 2.60 (s, 3H)
(R)-methyl 3-(2,4-difluorophenoxy)-2-(tritylamino)propanoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
74%
With azodicarboxylic acid bis(2-methoxyethyl) ester; triphenylphosphine; In tetrahydrofuran; at 0 - 20℃; for 2h;
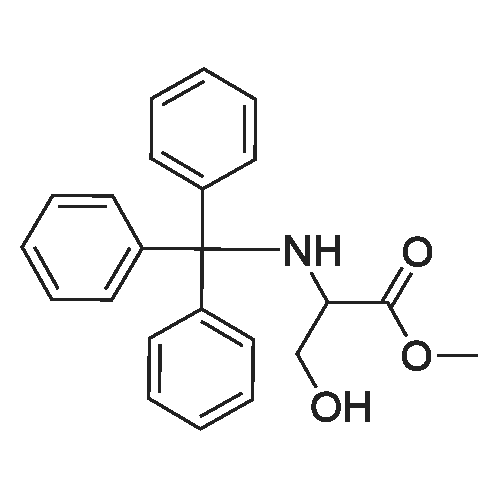
To a stirred solution of (R)-methyl3-hydroxy-2-(tritylamino)propanoate (800 mg, 2.2 mmol), 2,4-difluorophenol (370 mg, 2.9 mmol), and triphenylphosphine (760 mg, 2.9 mmol) in THF (15 mL) was added dropwise a solution of bis(2-methoxyethyl) azodicarboxylate (670 mg, 2.9 mmol) in THF (2 mL) at 0 C. The mixture was stirred at room temperature for 2 hrs. The mixture was concentrated, and the residue was diluted with EtOAc (50 mL). The diluted mixture was washed with a saturated aqueous NaHC03 solution and then brine, and was dried over Na2S04. After filtration, the filtrate was concentrated. The residue was purified by column chromatography (silica gel, eluted with 3% to 13% EtOAc in hexane) to give the title compound as a colorless gum (771 mg, 74% yield).‘H NMR (300 MHz, CDC13) delta 7.52(6H, d, J = 7.3 Hz), 7.30-7.16 (9H, m), 6.99-6.72 (3H, m), 4.29 (1H, d, J =9.5, 5.2 Hz), 4.02 (1H, dd, J = 9.5, 6.9 Hz), 3.78-3.71 (1H, m), 3.24 (3H, s),2.88 (1H, d, J = 10.3 Hz). MS (ESI)m/z: fragment signal of 243 (positive ion) was observed.
N-(4-cyano-3-(trifluoromethyl)phenyl)-3-(2,4-difluorophenoxy)-2-hydroxy-2-methylpropanamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
42%
General procedure: To a mixture of NaH (60% in mineral oil, 0.050 g, 1.23 mmol) in anhydrous THF (2 mL) at 0 C under Ar atmosphere was added a solution of the differently substituted phenol or thiophenol (1.11 mmol) in 1 mL of anhydrous THF. This mixture was stirred at r.t. for 20 min. A solution of the different intermediate 17-21 (0.74 mmol) in anhydrous THF (3 mL) was added slowly. The reaction mixture was stirred at r.t. o.n. The mixture was then diluted with ethyl acetate (30 mL), washed with brine (15 mL) and water (30 mL), dried over Na2SO4 and concentrated under vacuum. The crude residue was purified by flash column chromatography.
methyl 3-bromo-4-(2,4-difluorophenoxy)benzoate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
72%
With caesium carbonate; In water; dimethyl sulfoxide; at 80℃; for 12h;Inert atmosphere;
Cesium carbonate ((3.1 g, 9.6 mmol) was added to 2,4-difluorophenol (0.83 g, 6.4 mmol) and <strong>[82702-31-6]methyl 3-bromo-4-fluorobenzoate</strong> (1.57 g, 6.7 mmol)In a solution of dimethyl sulfoxide (10 mL). The reaction mixture was stirred at 80 C for 12 hr, then cooled to room temperature, then water (50mL), ethyl acetate (50mL*2). The organic phase was washed with brine (25 mL*4) and evaporated to dryness. The crude product was subjected to column chromatography (peel ether: ethyl acetate = 20:1) to give methyl 3-bromo-4-(2,4-difluorophenoxy)benzoate (1.58 g, 4. 72%).
1-bromo-2-(2,4-difluorophenoxy)-4-methyl-5-nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
99%
With potassium carbonate; In dimethyl sulfoxide; at 20℃;
<strong>[224185-19-7]1-bromo-2-fluoro-4-methyl-5-nitrobenzene</strong> (2.0 g, 8.55 mmol) was dissolved in dimethyl sulfoxide (20 mL) at room temperature, then 2,4-difluorophenol (1.67 g, 12.82 mmol) and potassium carbonate (2.36 g, 17.10 mmol) were added to the reaction mixture, and stirred at room temperature overnight. After completion of the reaction, the reaction mixture was diluted with ethyl acetate (40 mL). The organic phase is dried over anhydrous sodium sulfate. Filtration, spin-drying, and the crude product was separated by column chromatography (pure petroleum ether as mobile phase) to give 1-bromo-2-(2,4-difluorophenoxy)-4-methyl-5-nitrobenzene ( 2.90 g, white solid, yield: 99%).
1-bromo-2-(2,4-difluorophenoxy)-4-methoxy-5-nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
48%
With potassium carbonate; In dimethyl sulfoxide; at 20℃;
1-bromo-2-fluoro-4-methyl-5-nitrobenzene (2.0 g, 8.55 mmol) was dissolved in dimethyl sulfoxide (20 mL) at room temperature, then 2,4-difluorophenol (1.67 g, 12.82 mmol) and potassium carbonate (2.36 g, 17.10 mmol) were added to the reaction mixture, and stirred at room temperature overnight. After the reaction was completed, the reaction mixture was diluted with ethyl acetate (40 mL). The organic phase was washed with saturated brine (10 mL*2), dried over anhydrous sodium sulfate, filtered and evaporated. 2,4-Difluorophenoxy)-4-methoxy-5-nitrobenzene (2.90 g, white solid, yield: 99%).
With caesium carbonate; In dimethyl sulfoxide; at 95℃; for 3h;Inert atmosphere;
Methyl 5-bromo-6-chloronicotinate (10.0 g, 40.0 mmol) and 2,4-difluorophenol (7.8 g, 60.0 mmol) were dissolved in 100 mL of dimethyl sulfoxide (DMSO).Cesium carbonate (26.0 g, 80.0 mmol) was added and stirred at 95 C for 3 h under nitrogen.The TLC detects the reaction of the starting material completely. After cooling, pour into water, extract with EA, and wash with saturated brine.Dry over anhydrous sodium sulfate, concentrate and purify by column chromatography.A white solid 91-1 (4.9 g) was obtained.
2-(2,4-difluorophenoxy)-5-(trifluoromethyl)pyridine-3-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
With caesium carbonate; In acetonitrile; for 12h;Reflux;
<strong>[505084-59-3]2-chloro-5-(trifluoromethyl)pyridine-3-carboxylic acid</strong> (1.5 g, 6.7 mmol) was added to a round-bottom flask equipped with a magnetic stir bar, and acetonitrile (15 mL) was added. Cesium carbonate (4.3 g, 13.3 mmol) and 2,4-difluorophenol (952 mg, 7.3 mmol) were added, and the mixture was heated to reflux for 12 hours. The solvent was removed under reduced pressure, and the resulting residue was dissolved in water. pH was adjusted to about 3 by addition of 2N HC1, and the resulting precipitate was collected by filtration, rinsed with water, and sucked to dryness to afford the desired product as a white solid (1.9 g, 2.1 mmol, 91% yield). MS, ES+m/z 320[M+H]+.
1-(2,4-difluorophenoxy)-3-fluoro-2-nitrobenzene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
9.4 g
To a solution of 2,4-difluorophenol (5.00 g, 38.4 mmol) in DMF (50 mL) was added NaH (1.84 g, 46.1 mmol, 60% purity) at 0 C. The mixture was stirred at 25 C for 1 h. Then <strong>[19064-24-5]1,3-difluoro-2-nitrobenzene</strong> (6.11 g, 38.4 mmol) in DMF (10 mL) was added to the reaction mixture. The mixture was stirred at 25 C for 16 h. The resulting soluiton was diluted with water (100 mL), and extracted with ethyl acetate (50 mL × 3). The combined organic layers were washed with brine (50 mL × 3), dried over anhydrous Na2SO4, filtered and concentrated to give a residue. The residue was purified by column chromatography to give 1-(2,4- difluorophenoxy)-3-fluoro-2-nitrobenzene (I-178) (9.40 g) as a yellow oil.
With caesium carbonate; In N,N-dimethyl-formamide; at 80℃; for 3h;
5-Fluoro-2-formyl pyridine (0.25 g; 2.00 mmol) and Cs2CO3 (0.98 g; 3.00 mmol) were suspended in DMF (10 ml); 2,4-difluoro-phenol (0.31 g; 2.40 mmol) was added and the reaction mixture was stirred at 80 C. during 3 hours. Acetonitrile (20 ml) was added and the mixture was filtered before being purified via preparative HPLC. Obtained 0.25 g of the desired compound. (0273) Example 5k: HPLC-MS (Method):): Z011_S03;Rt [min]: 0.96 MS: 236 [M+H]+
tert-butyl 2-(2,4-difluorophenoxy)-7-azaspiro[3.5]nonane-7-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
511 mg
Stage #1: 2-hydroxy-7-azaspiro[3.5]nonane-7-carboxylic acid tert-butyl ester; 2,4-difluorophenol With triphenylphosphine In tetrahydrofuran at 20℃; Inert atmosphere;
Stage #2: With diethylazodicarboxylate In tetrahydrofuran at 0 - 20℃; Inert atmosphere;
Step a) tert-Butyl 2-(2,4-difluorophenoxy)-7-azaspiro[3.5]nonane-7-carboxylate
In a 25 mL four-necked sulphonation flask under argon, tert-butyl 2-hydroxy-7- azaspiro[3.5]nonane-7-carboxylate (401 mg, 1.66 mmol, CAS RN 240401-28-9) was dissolved in THF (6 mL), 2,4-difluorophenol (216 mg, 159 pL, 1.66 mmol) and triphenylphosphine (479 mg, 1.83 mmol) were added. The clear solution was stirred at RT for 5, then cooled to 0-2°C and DEAD (318 mg, 289 pL, 1.83 mmol) was added slowly within 10 min, stirring was continued for 1 hr at 2-4°C, then the cooling bath was removed and it was stirred over night at RT. 20 mL diethylether were added, the mixture was washed with water, 1M NaOH amd brine. The organic layer was dried over Na2S04 and concentrated in vacuo. The crude material was purified by flash chromatography (silica gel, 50 g, 0% to 40% EtOAc in heptane) to obtain 511 mg of product as a colorless oil. MS (ESI): m/z = 298.3 [M-56+H]+.
With potassium carbonate In N,N-dimethyl-formamide at 110℃; for 5h;
preparation of intermediate 11
General procedure: In a 250 cm3 three-neck bottle, added phenol derivative(22.0 mmol), 2-fluorobenzonitrile or 3-fluorobenzonitrilederivative (20.0 mmol), 4.56 g K2CO3(33 mmol), 80 cm3DMF. After the addition is completed, heated to 110 °Cfor 5 h. After the reaction was completed, 200 cm3EAwas added. Washed with NaCl solution (400 cm3× 3), theorganic phases were combined, dried over dry Na2SO4,andconcentrated to obtain the product 11.
With [4,4′-bis(1,1-dimethylethyl)-2,2′-bipyridine-N1,N1′]bis{3,5-difluoro-2-[5-(trifluoromethyl)-2-pyridinyl-κN]phenyl-κC}iridium(III) hexafluorophosphate; 4,4'-di-tert-butyl-2,2'-bipyridine nickel(II) bromide In ethyl acetate at 20℃; for 48h; Inert atmosphere; Glovebox; Irradiation;






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping