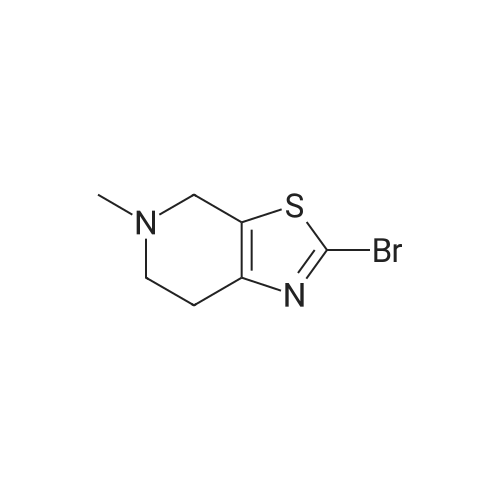
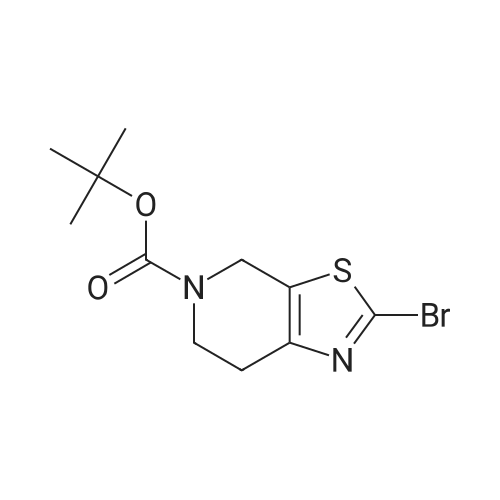
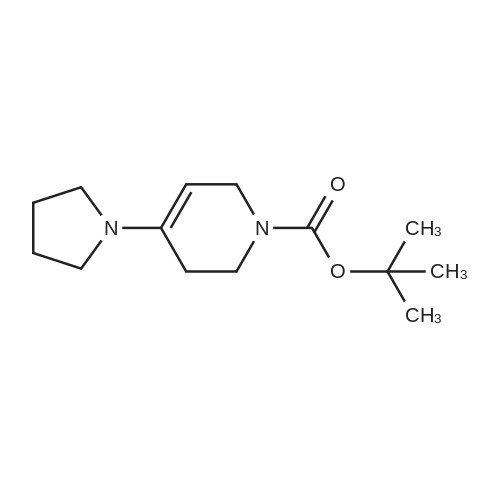
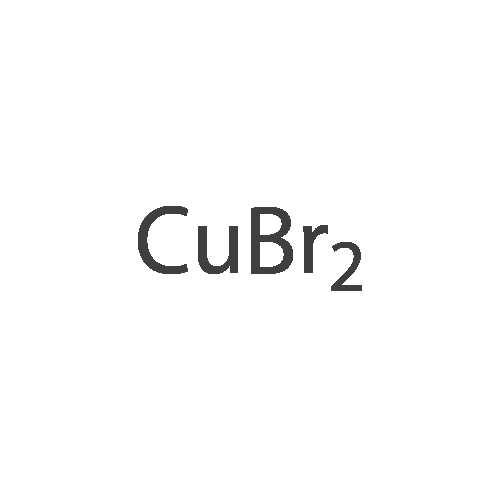* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With copper(ll) bromide; isopentyl nitrite In N,N-dimethyl-formamide at 0 - 40℃; for 0.5 h;
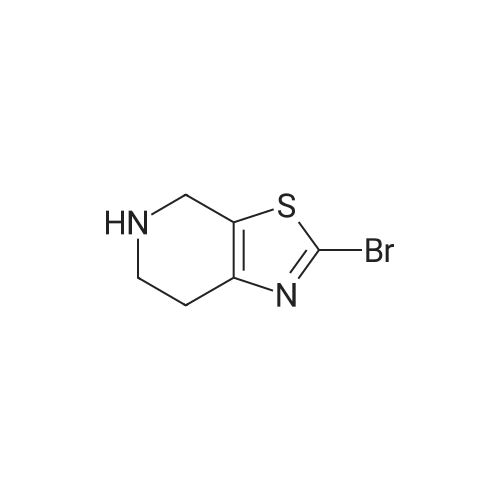
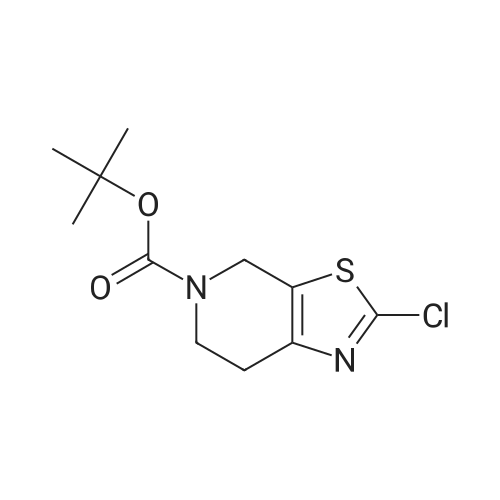
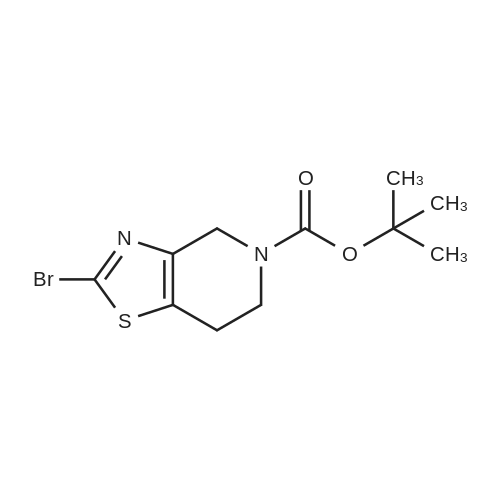
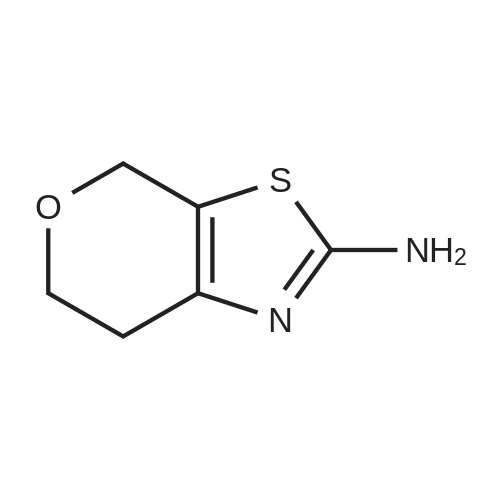
[0133] Method G-Step e: Tert-butyl 2-bromo-6,7-dihydrothiazolo[5,4-c]pyridine-5(4H)- carboxylate [0134] To a solution of isopentyl nitrite (8.8 mL, 62.8 mmol) and CuBr2 (10.7 g, 48 mmol) in 100 mL of DMF was added tert-butyl 2-amino-6,7-dihydrothiazolo[5,4-c]pyridine-5(4H)- carboxylate (10 g, 39.2 mmol) at 0 °C. The mixture was stirred at 40 °C for 30 min and evaporated. It was added to 50 mL of H20 and extracted with CH2C12 (100 mLx2). The combined organics were washed with brine (30 mLx2) and dried over Na2S04. Concentrated and purified by silica gel column chromatography (CH2C12) to give title compound (5.3g, 42.4percent) as a yellow solid.
41%
With tert.-butylnitrite; copper(I) bromide In N,N-dimethyl-formamide at 40℃; for 0.5 h; Cooling with ice
Reference Example 3 2-Bromo-5-tert-butoxycarbonyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine (Japanese Patent Laid-Open No. 2001/294572) Cupric bromide (1.05 g, 4.7 mmol) was suspended in N,N-dimethylformamide, and tert-butyl nitrite (696 mg, 6.5 mmol) was added to the suspension. 2-Amino-5-tert-butoxycarbonyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine (1.00 g, 5.9 mmol) was added thereto under ice cooling, and the reaction solution was then stirred under heating at 40° C. for 30 minutes. The reaction solution was concentrated under reduced pressure, and the residue was purified by silica gel column chromatography (ethyl acetate:hexane=1:5) to obtain the title compound (568 mg, 41percent) as a yellow solid. 1H NMR (CDCl3) 1.48 (9H, s), 2.85 (2H, br s), 3.72 (2H, t, J=5.6 Hz), 4.56 (2H, br s). MS (FAB) m/z 319 (M+H)+.
34%
With tert.-butylnitrite; copper(ll) bromide In acetonitrile at 20℃; for 1 h; Inert atmosphere
tert-Butyl nitrite (0.181 mL, 1.371 mmol) was added to copper (II) bromide (297mg, 1.332 mmol) in dry acetonitrile (3.0 mL) under argon. The reaction mixture wasstirred at room temperature for 10 mm. A suspension of tert-butyl 2-amino-6,7-dihydrothiazolo[5,4-cj pyridine-5 (4H)-carboxylate (200 mg, 0.783 mmol) in dry acetonitrile (4.0 mL) was added dropwise. The reaction mixture was stirred at room temperature for 1.0 h. Acetonitrile was removed under vacuum, the reaction mixture wasdiluted with EtOAc, quenched with 1.0 N HC1. The organic layer was collected, washed with 0.5 N HC1 (2X), saturated sodium bicarbonate, brine and dried over sodium sulfate. After evaporation of solvent, the crude product was dissolved in a small amount of chloroform and charged to a 12 g silica gel cartridge which was eluted with hexanes for 2 mm., then a 12 mm gradient from 0percent to 50percent. The desired fractions were combined,concentrated and lyophilized to give Intermediate 1 hA (85 mg, 0.266 mmol, 34.0 percent yield) as a white solid. ‘H NMR (500MHz, acetonitrile-d3) 4.56 (t, J1.8 Hz, 2H), 3.70 (t, J=5.8 Hz, 2H), 2.81-2.76 (m, 2H), 1.47 (s, 9H); LC-MS: method A, RT = 1.86 mm, MS (ESI) m/z: 319.0 and 321.0 (M+H)
8.78 g
With tert.-butylnitrite; copper(ll) bromide In N,N-dimethyl-formamide at 50℃; for 3 h; Cooling with ice
11.16 g (0. 08 mol) of cuprous chloride was dissolved in 100 mL of N, N-dimethylformamide, and 11.5 g (0.1 mol) of t-butyl nitrite was added dropwise to the reaction solution under ice-cooling , Then carefully batches of 18. 4g (0.072 mol) 2-amino-6,7-dihydrothiazole [5,4-c] pyridine-5 (4OH) -carboxylate was added to the reaction solution and heated to 50 ° C. After 3 hours, the TLC monitoring reaction was complete. Poured into water and extracted with 200 mL of ethyl acetate, washed with saturated aqueous sodium bicarbonate solution (200 mL) and saturated brine (200 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (ethyl acetate: Petroleum ether = 1: 5) to give a white solid 8. 78 g, m.p. 89. 1 ° C.2-amino-6,7-dihydrothiazole [5,4-c] pyridine-5 (4OH) -carboxylic acid tert-butyl ester was prepared according to the procedure described in Example 6, Step 3) Butyl ester was reacted with 21 g (0.094 mol) of cuprous bromide, 12.4 g (0.12 mol) of t-butyl nitrite to give 8. 78 g of a white solid, m.p. 100. 5 ° C.
8.78 g
With tert.-butylnitrite; copper(I) bromide In N,N-dimethyl-formamide at 50℃; for 3 h; Cooling with ice
Following the procedure described in Example 6, step 3)A mixture of 20 g (0.078 mol) of 2-amino-6,7-dihydrothiazole [5,4-c] pyridine-5 (4H) -carboxylate and 21 g (0.094 mol) of cuprous bromide, 12.4 g 0.12 mol) of t-butyl nitrite gave 8.78 g of a white solid The 11.6g (0.086mol) of cuprous chloride was dissolved in 100mL N, N- dimethyl formamide,The ice bath was 11.5g (0.11mol) t-butyl nitrite dropwise to the reaction mixture,Subsequently, 18.4 g (0.072 mol) of tert-butyl 2-amino-6,7-dihydrothiazole [5,4-c] pyridine-5 (4H) -carboxylate was added to the reaction solution,The temperature was raised to 50 ° C.After 3 hours,TLC monitoring reaction is complete. Poured into water and extracted with 200 mL of ethyl acetate, washed with saturated aqueous sodium bicarbonate (200 mL) and saturated brine (200 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (ethyl acetate: Petroleum ether = 1: 5)
With copper(ll) bromide In dichloromethane at 0℃; for 3 h;
To a stuffing solution of tert-butyl 2-amino-6,7-dihydrothiazolo[5,4-c]pyridine- 5(4H)-carboxylate (2.0 g, 7.84 mmol) in DCM (30 mL) was added tert-butyl nitrite (1.24 g, 12 mmol) and CuBr2 (1.78 g, 8 mmol). The solution was stirred at 0 °C for 3h. Once LCMS showed the reaction to be complete, solvents were then evaporated and the residue was purified with column separation to afford desired product as white solid (1.1 g, yield: 44percent);LCMS: 318.9/320.9 (M+1).
100mL flask A47-1 (10g, 50mmol), sulfur (3.22g, 100mmol), cyanamide (4.36g, 100mmol) and pyridine (40mL),The reaction was heated to 130 90 minutes. Cooling to room temperature, the solid was filtered off, washed with ethyl acetate, and dried to give a dark brown solid (9.7g, 76percent).
75%
Stage #1: With pyrrolidine In isopropyl alcohol at 55℃; for 2 h; Stage #2: With sulfur In isopropyl alcohol at 0 - 20℃; for 6 h;
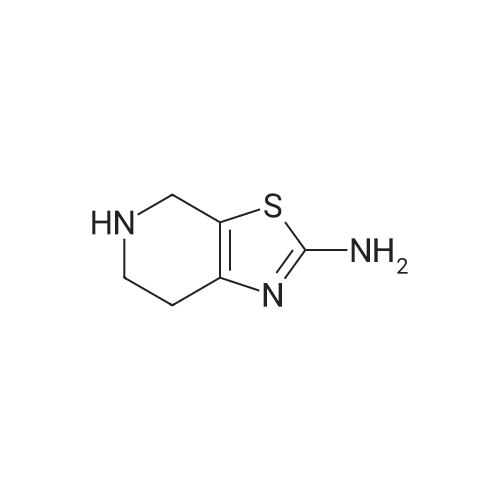
General procedure: Pyrrolidine (10.5 mmol, 1.05 equiv)was added to a solution of 4-piperidone (19am or 19or) (10 mmol, 1.0 equiv) in isopropanol(20 mL). The reaction mixture was stirred at 55 C for 2 h. The reactionmixture was cooled to room temperature. Then elementalsulfur (10 mmol, 1.0 equiv) was added in one portion, followed bydropwise addition of the solution of cyanamide (10.5 mmol, 1.05equiv) in isopropanol (10 mL) at 0 C. The reaction mixture wasstirred for 6 h at room temperature. The reaction mixture wasfiltered and solid was washed with ethyl acetate, then dried undervacuum to obtain the desired product.
74.2%
at 130℃; for 1.5 h;
[0124] Method F-Step a: Tert-butyl 2-amino-6,7-dihydrothiazolo[5,4-c]pyridine-5(4H)- carboxylate [0125] Tert-butyl 4-oxopiperidine-l-carboxylate (20 g, 101 mmol) was added to 100 mL of pyridine followed by addition of cyanamide (8.7 g, 201 mmol) and sublimed sulfur (6.4 g, 200 mmol). The mixture was stirred at 130 °C for 90 minutes and cooled down to room temperature. Filtered and washed with diethyl ether (100 mL) twice to give a pale yellow solid (19 g, 74.2percent). *H NMR (400 MHz, DMSO-i5) δ 6.83 (s, 2H), 4.29 (s, 2H), 3.56 (s, 2H), 2.43 (s, 2H), 1.41 (s, 9H).
47%
Stage #1: With pyrrolidine; toluene-4-sulfonic acid In cyclohexane for 2 h; Dean-Stark; Reflux Stage #2: With sulfur In methanol at 0 - 20℃; for 5 h;
Step i: tert -butyl 2-amino-6,7-dihydrothiazolor5,4-clpyridine-5(4H)-carboxylate To a 50 mL round bottom flask, were added tert-buty\ 4-oxopiperidine-l-carboxylate (5 g, 0.0251 mol), j?-toluenesulfonic acid monohydrate (0.023 g, 0.00012 mol), pyrrolidine (2.2 mL) and cyclohexane (10 mL). The round bottom flask was fitted with a Dean-Stark reflux condenser. The reaction mixture was stirred at reflux temperature for 2 h. The volatiles were evaporated under reduced pressure to get residue. The residue was dissolved in dry methanol (9 mL). To the same flask, sulfur powder (0.8 g, 0.0251 mol) was added. The reaction mixture was cooled to 0 °C. To the same flask, cyanamide (1.05 g, 0.0251 mol) in dry methanol (9 mL) was added. The resulting reaction mixture was stirred at RT for 5 h to get solid. The solid was collected by filtration to get the title compound [3 g, 47 percent]. NMR (600 MHz, CDCls): δ 6.82 (brs, 2H), 4.30 (s, 2H), 3.58 (t, 2H), 2.45 (t, 2H), 1.41 (s, 9H); LC-MS: 256.1 [M+H]+.
38 g
Stage #1: With pyrrolidine; toluene-4-sulfonic acid In cyclohexane for 5 h; Reflux Stage #2: With sulfur In methanol at 0 - 20℃; for 5 h;
In a Dean-Stark distiller, 50 g (0.25 mol) of 1-Boc-4-carbonylpiperidine was added to 300 mL of cyclohexane followed by the addition of 20 g of pyrrolidine (0.275mol) and p-toluenesulfonic acid monohydrate 0.5g (2.63mmol), the reaction was heated to reflux for 5h and cooled to room temperature, The reaction solution was filtered and the solvent cyclohexane in the filtrate was distilled off. The resulting distilled product was added to 500 mL of anhydrous methanol, 8 g (0.25 mol) of elemental sulfur was added thereto, The reaction temperature was set at 0 ° C, a solution of cyanamide dissolved in methanol (10.5 g, 0.25 mol) was slowly added dropwise and the temperature was allowed to warm to room temperature. After the reaction for 5h, the solvent was distilled off and the residue was purified by column chromatography (eluent: petroleum ether: ethyl acetate = 1: 2) to give 38 g of compound 3.
Reference:
[1] Patent: CN105254613, 2016, A, . Location in patent: Paragraph 0339; 0340; 0341
[2] European Journal of Medicinal Chemistry, 2017, vol. 139, p. 128 - 152
[3] Patent: WO2014/113191, 2014, A1, . Location in patent: Paragraph 0124; 0125
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 9, p. 3687 - 3706
[5] Heterocycles, 2004, vol. 63, # 7, p. 1555 - 1561
[6] Heterocyclic Communications, 2016, vol. 22, # 5, p. 291 - 294
[7] Patent: WO2015/101928, 2015, A1, . Location in patent: Page/Page column 90; 91
[8] Patent: EP1577302, 2005, A1, . Location in patent: Page/Page column 56-57
[9] Patent: WO2010/96389, 2010, A1, . Location in patent: Page/Page column 55-56
[10] Patent: CN104926839, 2017, B, . Location in patent: Paragraph 0012; 0013; 0014; 0015; 0016; 0017; 0018; 0019
6
[ 123-75-1 ]
[ 79099-07-3 ]
[ 365996-05-0 ]
Yield
Reaction Conditions
Operation in experiment
73.4%
Stage #1: With toluene-4-sulfonic acid In cyclohexane for 2.5 h; Reflux; Dean-Stark Stage #2: With sulfur In methanol at 20℃; for 0.166667 h; Stage #3: With CYANAMID In methanol at 0℃; for 5 h;
2-Amino-4,5,6,7-tetrahydrothia-zolo[5,4-c]pyridine was synthesized using a previously reported procedure[19]. 1-tert-Butoxycarbonyl-4-piperidone (1.0 g,5.0mM) was dissolved in 30ml of cyclohexane. To this solution,pyrrolidine (0.38 g, 5.3mM) and p-toluenesulphonicacid (catalytic quantity) were added and refluxed for 2.5 husingDean-Stark trap. Ten, the reaction was cooled to roomtemperature and filtered and the filtrate was concentrated todryness in vacuo.The formed residue was dissolved in 25mlof dry methanol; sulphur (S8, 0.16 g, 0.63mM) was addedat once and the reaction mixture was stirred for 10min atroom temperature. Then, the reaction mixture was cooledto 0°C, cyanamide (0.21 g, 5.0mM) in 5ml of dry methanolwas added slowly in dropwise manner, and the reaction wascontinued for 5 h at the same temperature. After completionof the reaction, the mixture was filtered and concentrated toget the crude TR-03a. TR-03a was further chromatographedover silica gel with dichloromethane/methanol (98 : 2)as eluent, yielding 73.4percent. The purity of the compoundwas confirmed by TLC using CHCl3/CH3OH [(95 : 5); Rf = 0.72], ESI-Q-TOF-MS: (m/z) 256.3 [M + H], and NMRspectra; 1H NMR (500MHz, DMSO-d6) δ = 1.41 (s, 9H),2.43 (t, 2H, J = 6.0Hz), 3.56 (t, 2H, J = 5.5Hz), 4.29 (s, 2H),6.80 (s, 2H, NH2).
Reference:
[1] Journal of Chemistry, 2017, vol. 2017,
7
[ 420-04-2 ]
[ 207691-65-4 ]
[ 365996-05-0 ]
Yield
Reaction Conditions
Operation in experiment
86 g
Stage #1: at 20℃; for 0.5 h; Stage #2: With sulfur In methanol at 20℃; for 2 h;
A solution of N-tert-butoxycarbonyl-4- (pyrrole-1-yl) -1,2,3,6-tetrahydropyridine 98. 8 g (0.39 mol) was dissolved in 100 mL of methanol, A mixture of 16. 5 g (0.9 mol) of monocarbicamide and 30 mL of methanol was added dropwise at room temperature with stirring, and the mixture was added dropwise over 30 minutes, Followed by the addition of 12. 5 g (0.9 mol) of sulfur, The mixture was stirred at room temperature for 2 hours to complete the TLC monitoring reaction. Filtered and washed with methanol to give 86 g of pale yellow solid, mp 208 ° C.
Reference:
[1] Patent: EP1405852, 2004, A1, . Location in patent: Page 65
[2] Patent: CN106467537, 2017, A, . Location in patent: Paragraph 0199; 0200; 0203; 0204
8
[ 17356-08-0 ]
[ 188869-05-8 ]
[ 365996-05-0 ]
Yield
Reaction Conditions
Operation in experiment
47%
at 120℃; for 3 h;
To the stuffing solution of tert-butyl 3-bromo-4-oxopiperidine-1-carboxylate (5.0 g,18 mmol) in DMF (50 mL) was added thiourea (1.37 g, 18 mmol), resulting solution wasthen heated at 120 °C for 3h. The solvents were evaporated and the residue purified bycolumn separation to afford desired product as pale yellow oil (2.2 g, yield:47percent). LCMS:256.1 (M+1).
With potassium carbonate In 1,4-dioxane; water at 0 - 20℃; for 3 h;
6.01.16.03 2-Amino-6,7-dihydro-4H-thiazolo(5,4-c)pyridine-5-carboxylic acid tert-butyl ester 15.8 g 4,5,6,7-tetrahydro-thiazolo(5,4-c)pyridin-2-ylamine and 100 mL dioxane was added to 15.2 g potassium carbonate in 158 mL water. 13.1 g di tert-butyl dicarbonate in 58 mL dioxane was added at 0° C. The reaction mixture was allowed to stir for 3 h at ambient temperature. The reaction mixture was diluted with water and the solid was filtered through silica gel, washed with water (2*50 mL) to afford the desired product. The filtrate was concentrated, diluted with waterand extracted with ethyl acetat. The organic layer was dried over magnesium sulfate and concentrated to afford 11.6 g desired product. 1H NMR (400 MHz, DMSO-d6): δ 1.41 (s, 9H), 2.43 (t, 2H), 3.56 (t, 2H), 4.28 (s, 2H), 6.80 (s, 2H)
11.6 g
With potassium carbonate In 1,4-dioxane; water at 0 - 20℃; for 3 h;
6.01.08.03 2-Amino-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester 15.8 g 4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridin-2-yl-amine and 100 mL dioxane was added to 15.2 g potassium carbonate in 158 mL water. 13.1 g di tert-butyl dicarbonate in 58 mL dioxane was added at 0° C. The reaction mixture was allowed to stir for 3 h at ambient temperature. The reaction mixture was diluted with water and the solid was filtered through silica gel, washed with water (2*50 mL) to afford the desired product. The filtrate was concentrated, diluted with water and extracted with ethyl acetat. The organic layer was dried over magnesium sulfate and concentrated to afford 11.6 g of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 1.41 (s, 9H), 2.43 (t, 2H), 3.56 (t, 2H), 4.28 (s, 2H), 6.80 (s, 2H); (M+H)+: 256
With potassium carbonate In 1,4-dioxane; water at 20℃; for 3 h; Enzymatic reaction
6.01.08.03 2-Amino-6, 7-dihydro-4H-thiazolo[5, 4-c]pyridine-5-carboxylic acid tert-butyl ester 15.8 g 4, 5, 6, 7-tetrahydro-thiazolo[5, 4-c]pyridin-2-yl-amine and 100 mL dioxane was added to 15.2 g potassium carbonate in 158 mL water. 13.1 g di tert-butyl dicarbonate in 58 mL dioxane was added at 0 °C. The reaction mixture was allowed to stir for 3 h at ambient temperature. The reaction mixture was diluted with water and the solid was filtered through silica gel, washed with water (2 x 50 mL) to afford the desired product. The filtrate was concentrated, diluted with water and extracted with ethyl acetat. The organic layer was dried over magnesium sulfate and concentrated to afford 11.6 g of the desired product. 1H NMR (400 MHz, DMSO-d6): δ 1.41 (s, 9H), 2.43 (t, 2H), 3,56 (t, 2H), 4.28 (s, 2H), 6.80 (s, 2H); (M+H)+: 256
The N- tert-butoxycarbonyl-4- (pyrrolidin-1-yl ) -1,2,3,6-tetrahydropyridine 98.8 g (0.39 mol) was dissolved in 100mL of methanol,Stirring at room temperature was added dropwise 16.5g (0.39mol) CyanamideAnd 30 mL of methanol,After 30 minutes drop finished,Followed by the addition of 12.5 g (0.39 mol) of sulfur,The mixture was stirred at room temperature for 2 hours to complete the TLC monitoring reaction.filter,Methanol washed with pale yellow solid 86g,
Reference:
[1] Patent: CN106467538, 2017, A, . Location in patent: Paragraph 0214; 0217; 0218
2-(3,5-Bis-trifluoromethyl-benzenesulfonyl-amino)-6,7-dihydro-4H-thiazolo [5,4-c]pyridine-5-carboxylic acid ter-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
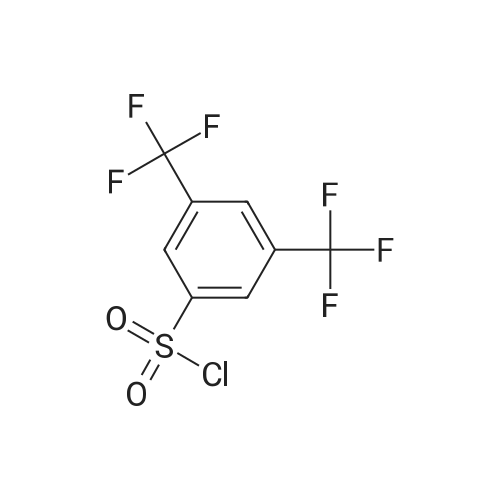
A mixture of 8.5 g of 2-amino-6, 7-dihydro-4H-thiazolo [5,4-c] pyridine-5-carboxylic acid tert.- butyl ester, 15.6 g of 3, 5-bis-trifluoromethyl-benzenesulfonyl chloride, and 8.1 g of DMAP in 100 ml of pyridine is stirred at 80 for 4 hours. From the mixture obtained solvent is evaporated, the evaporation residue obtained is treated with EtAc and the mixture obtained is extracted with aqueous NaHS04 solution and brine. The organic layer obtained is dried, from the solution obtained solvent is evaporated and the residue obtained is treated with a mixture of EtAc and c-Hex (+ 5% MeOH). 2- (3, 5-BIS-TRIFLUOROMETHYL-BENZENESULFONYL-AMINO)- 6, 7-dihydro-4H-thiazolo [5, 4-c] pyridine-5-carboxylic acid ter-butyl ester precipitates, is filtrated off and dried.
With toluene-4-sulfonic acid;sulfur; In methanol; at 0 - 20℃; for 5h;
[Referential Example 6] tert-Butyl 2-amino-6,7-dihydrothiazolo[5,4-c]pyridine-5[4H]-carboxylate: 1-tert-Butoxycarbonyl-4-piperidone (40.0 g) was dissolved in cyclohexane (80 ml), and to the solution p-toluenesulfonic acid monohydrate (191 mg) and pyrrolidine (17.6 ml) were added.. The mixture was heated under reflux for 2 hours while removing water using a Dean-Stark trap.. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in methanol (60 ml), and sulfur powder (6.42 g) was added.. A methanol solution (10 ml) of cyanamide (8.44 g) was slowly added dropwise to the solution with ice cooling, and the mixture was stirred at room temperature for 5 hours.. Precipitated solid materials were collected by filtration to obtain the title compound (31.0 g).1H-NMR (DMSO-d6) delta: 1.41(9H,s), 2.44(2H,t,J=5.6Hz), 3.57(2H,t,J=5.6Hz), 4.29(2H,s), 6.79(2H,s). MS (EI) m/z: 255(M+).
86 g
A solution of N-tert-butoxycarbonyl-4- (pyrrole-1-yl) -1,2,3,6-tetrahydropyridine 98. 8 g (0.39 mol) was dissolved in 100 mL of methanol, A mixture of 16. 5 g (0.9 mol) of monocarbicamide and 30 mL of methanol was added dropwise at room temperature with stirring, and the mixture was added dropwise over 30 minutes, Followed by the addition of 12. 5 g (0.9 mol) of sulfur, The mixture was stirred at room temperature for 2 hours to complete the TLC monitoring reaction. Filtered and washed with methanol to give 86 g of pale yellow solid, mp 208 C.
With sulfur; CYANAMID; In pyridine; for 1.66667h;Heating / reflux;
A mixture of 19.9 g of 1-BOC-piperidin-4-one, 8.4 g of cyanamide, and 6.4 g of sulfur in 100 ml of pyridine is REFLUXED under inert atmosphere for 100 minutes. From the mixture obtained solvent is evaporated and the evaporation residue obtained is subjected to flash chromatography over silica gel. 2-Amino-6, 7-dihydro-4H-thiazolo [5,4-c] PYRIDINE-5-CARBOXYLIC acid ter-butyl ester is obtained. 'H-NMR/CDCI3/D6-DMSO : 5.95 (bs, 2 H), 4.39 (s, 2 H), 3.67 (t, J = 5.2 Hz, 2 H), 3.08 (bs, 2 H), 1.43 (s, 9 H) ; 13C-NMR/CDCI3/Do-DMSO : 166. 17, 153.87, 79.19, 28.86, 27.69, 26.01
1-tert-Butoxycarbonyl-4-piperidone (40.0 g) was dissolved in cyclohexane (80 mL), and to the solution were added p-toluenesulfonic acid monohydrate (191 mg) and pyrrolidine (17.6 mL). The reaction mixture was heated under reflux for 2 hours while water was removed with Dean-Stark apparatus. The resultant mixture was concentrated under reduced pressure, and the residue was dissolved in methanol (60 mL). After sulfur powder (6.42 g) was added to the solution, a solution of cyanamide (8.44 g) in methanol (10 mL) was slowly added dropwise to the mixture under ice cooling, followed by stirring at room temperature for 5 hours. The resultant precipitated solid was collected by filtration, to thereby give the title compound (31.0 g).1H-NMR(DMSO-d6) delta:1.41(9H, s), 2.44 (2H, t, J=5. 6Hz), 3.57(2H, t, J=5.6Hz), 4.29(2H, s), 6.79(2H, s). MS(EI)m/z:255(M+).
[Reference Example 1] 2-Amino-6, 7-dihydrothiazolo[5,4-c]pyridine-5[4H]-carboxylic acid tert-butyl ester 1-Tert-butoxycarbonyl-4-piperidone (40.0 g) was dissolved in cyclohexane (80 ml), and p-toluenesulfonic acid monohydrate (191 mg) and pyrrolidine (17.6 ml) were added thereto. The mixture was heated to reflux for 2 hours, while dehydrating by means of a Dean-Stark apparatus. The reaction mixture was concentrated under reduced pressure, subsequently the residue was dissolved in methanol (60 ml), and powdered sulfur (6.42 g) was added thereto. Under ice cooling, a methanol solution (10 ml) of cyanamide (8.44 g) was gradually added dropwise, and the mixture was stirred for 5 hours at room temperature. The precipitated solid was collected by filtration, thus to obtain the title compound (31.0 g). 1H-NMR(DMSO-d6)delta:1.41(9H, s), 2.44(2H, t, J=5.6Hz), 3.57(2H, t, J=5.6Hz), 4.29(2H, s), 6.79(2H, s). MS (EI) m/z:255 (M+).
[Referential Example 9] 2-Amino-5-tert-butoxycarbonyl-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridine: 1-tert-Butoxycarbonyl-4-piperidone (40.0 g) was dissolved in cyclohexane (80 ml), and to the solution p-toluenesulfonic acid monohydrate (191 mg) and pyrrolidine (17.6 ml) were added. The mixture was heated under reflux for 2 hours while removing water using a Dean-Stark trap. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in methanol (60 ml), and sulfur powder (6.42 g) was added. A methanol solution (10 ml) of cyanamide (8.44 g) was slowly added dropwise with ice cooling, and the mixture was stirred at room temperature for 5 hours. Precipitated solid materials were collected by filtration to obtain the title compound (31.0 g) as a pale yellow solid. 1H-NMR (DMSO-d6) delta: 1.41(9H,s), 2.40-2.46(2H,m), 3.57(2H,t,J=5.6Hz), 4.29(2H,s), 6.79(2H,s). MS (EI) m/z: 255(M+).
REFERENTIAL EXAMPLE 6 tert-Butyl 2-amino-6,7-dihydrothiazolo[5,4-c]pyridine-5[4H]-carboxylate: 1-tert-Butoxycarbonyl-4-piperidone (40.0 g) was dissolved in cyclohexane (80 ml), and to the solution p-toluenesulfonic acid monohydrate (191 mg) and pyrrolidine (17.6 ml) were added. The mixture was heated under reflux for 2 hours while removing water using a Dean-Stark trap. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in methanol (60 ml), and sulfur powder (6.42 g) was added. A methanol solution (10 ml) of cyanamide (8.44 g) was slowly added dropwise to the solution with ice cooling, and the mixture was stirred at room temperature for 5 hours. Precipitated solid materials were collected by filtration to obtain the title compound (31.0 g). 1H-NMR (DMSO-d6) delta: 1.41(9H,s), 2.44(2H,t,J=5.6 Hz), 3.57(2H,t,J=5.6 Hz), 4.29(2H,s), 6.79(2H,s). MS (EI) m/z: 255(M+).
9
[ 7789-45-9 ]
[ 365996-05-0 ]
[ 365996-06-1 ]
Yield
Reaction Conditions
Operation in experiment
With tert.-butylnitrite; In N,N-dimethyl-formamide;
[Referential Example 10] 2-Bromo-5-tert-butoxycarbonyl-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridine: Copper(II) bromide (1.05 g) was suspended in N,N-dimethylformamide, and tert-butyl nitrite (0.696 ml) was added. After 2-amino-5-tert-butoxycarbonyl-4,5,6,7-tetrahydrothiazolo[5,4-c]pyridine (1.00 g) was added with ice cooling, the reaction mixture was heated and stirred at 40C for 30 minutes. The reaction mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel (ethyl acetate:hexane = 1:5) to obtain the title compound (568 mg) as a yellow solid. 1H-NMR (CDCl3) delta: 1.48(9H,s), 2.85(2H,br.s), 3.72(2H,t,J=5.6Hz), 4.56(2H,br.s). MS (FAB) m/z: 319(M+H)+.
In N,N-dimethyl-formamide;
REFERENTIAL EXAMPLE 7 tert-Butyl 2-bromo-6,7-dihydrothiazolo[5,4-c]pyridine-5[4H]-carboxylate: Copper(II) bromide (1.05 g) was suspended in N,N-dimethylformamide(20 ml), and tert-butyl nitrite (0.696 ml) and the compound (1.00 g) obtained in Referential Example 6 were added with ice cooling, the reaction mixture was heated and stirred at 40 C. for 30 minutes. The reaction mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel (ethyl acetate:hexane=1:5) to obtain the title compound (568 mg). 1H-NMR (CDCl3) delta: 1.48(9H,s), 2.85(2H,br.s), 3.72(2H,br.s), 4.56(2H,br.s). MS (FAB) m/z: 319(M+H)+.
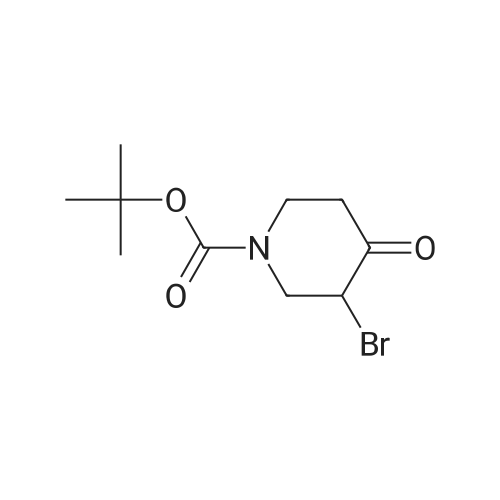
To a solution of i-butyl 3-bromo-4-oxopiperidine-1-carboxylate (1.0 g, 3.59 mmol) in isopropanol (10 mL), thiourea (0.33 g, 4.30 mmol) was added and the reaction mixture was refluxed at 90 °C for 1 h. Completion of the reaction was monitored by TLC. The reaction mixture was then concentrated under vacuum and the resulting crude material was washed with diethyl ether to afford the title compound. Yield: 99percent (0.9 g, white solid). 1H NMR (400 MHz, DMSO-d6): delta 9.05 (bs, 2H), 4.32 (m, 2H), 3.61-3.58 (m, 2H), 2.50 (s, 2H), 1.41 (s, 9H). LCMS: (Method A) 256.0 (M+H), Rt. 2.5 min, 64.8percent (Max).
47%
In N,N-dimethyl-formamide; at 120℃; for 3h;
To the stuffing solution of tert-butyl 3-bromo-4-oxopiperidine-1-carboxylate (5.0 g,18 mmol) in DMF (50 mL) was added thiourea (1.37 g, 18 mmol), resulting solution wasthen heated at 120 C for 3h. The solvents were evaporated and the residue purified bycolumn separation to afford desired product as pale yellow oil (2.2 g, yield:47%). LCMS:256.1 (M+1).
In isopropyl alcohol; for 1h;Reflux;
Step (ii): Preparation of 2-Amino-6,7-dihydro-4H-thiazolo[5,4-c|pyridine-5-carboxylic acid tert - butyl esterA suspension of 3-Bromo-4-oxo-piperidine- l-carboxylic acid tert-butyl ester (10 grams, 35 mmol, obtained in above step) and thiourea (3.28 grams, 42 mmol) in isopropanol ( 100 mL) was refluxed for 1 hour. After completion of reaction, reaction mass was concentrated and resulted crude was triturated with diethyl ether (50 mL), solids were filtered and dried under vacuum to obtain the title compound (10 grams). - NMR (delta ppm): 1 .39 (9H, s), 2.52 (2H, m), 3.56 - 3.59 (2H, t), 4.30 (2H, s), 7.10 (2H, bs); Mass (m/z): 256 (M+H)+.
In ethanol; for 2h;Reflux;
General procedure for the synthesis of Z2-a and Z2-b To a stirred solution of Zl-a or Zl-b (5.98 mmol) in EtOH (20 mL) was added thiourea (6.28 mmol) and the mixture was stirred at reflux temperature for 2 hours. After reaction completion, the reaction mixture was diluted with methylene chloride (20 mL) and washed with brine (20 mL). The organic layer was dried over anhydrous MgS04 and concentrated in vacuo to give Z2-a or Z2-b. Z2-a; ]H NMR (400 MHz, DMSO-< 6 6.62 (brs, 2H, N), 4.03 - 4.10 (m, 2H), 2.59 - 2.72 (m, 2H), 2.39 - 2.44 (m, 2H), 1.99 - 2.03 (m, 1H), 1.69 - 1.79 (m, 1H), 1.49 - 1.18 (m, 3H). Z2-b; FontWeight="Bold" FontSize="10" H NMR (400 MHz, CDC13) delta 4.77 (brs, 2H, NH2), 4.42 (s, 2H), 3.67 - 3.71 (m, 2H), 2.61 - 2.65 (m, 2H), 1.46 (s, 9H).
Intermediate 77 1.1-Dimethylethyl 2-amino-6.7-dihvdroH ,31thiazolof5.4-clpyridine-5(4/-/)-carboxylate; ourea (0.153g) was added to a solution of 1 ,1-dimethylethyl 3-bromo-4-oxo-1- piperidinecarboxylate (Ref. WO2004012684) (0.557g) in acetone (15ml) at ambient temperature and stirred overnight. Triethylamine (418ul) was then added and after 20min the reaction mixture was evaporated in vacuo. The residue was loaded onto a 2Og SCX cartridge preconditioned with MeOH and then eluted with 0%-20% 2M methanolic ammonia in MeOH. Appropriate f ractions were combined and evaporated in vacuo to provide a solid (0.383g) which was stirred in water (5ml) for 10min, filtered, then washed with water and dried in vacuo at 450C to give the title compound (0.319g) as a pale yellow solid.Mass spectrum: Found: MH+ 256 H.p.l.c. R,2.09min
Dissolved 4-Oxo-piperidine-l-carboxylic acid tert-butyl ester (15.Og, 75.28 mmol) in 75 rnL of cyclohexane. To this was added pyrrolidine (6.70 rnL, 79.00 mmol) and a catalytic amount of TsOH. The mixture was refluxed with a Dean-Stark trap until no water was collected (5h). The mixture was filtered and concentrated to an oily brown residue. The residue was dissolved in 25 mL of anhydrous MeOH. To this mixture was added S(O) (2.40 g). The mixture was cooled in and ice bath and acetamide (3.19 g, 76.00 mmol) was added in portions. After 2 h and warming to room temperature a thick tan ppt. appeared. Filtered the material and washed with cold MeOH to give 10.04g. A second ppt., 3.64g, was collected. 1H NMR indicated identical material and these were combined to give 2-amino- 6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester.[145] 3,4-Dimethoxy-Lambda/-[3-(4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridin-2-ylcarbamoyl)- benzylj-benzamide was prepared by the methods described in Example 23 and Example 24. MS, electrospray 453.3 (M+Eta).
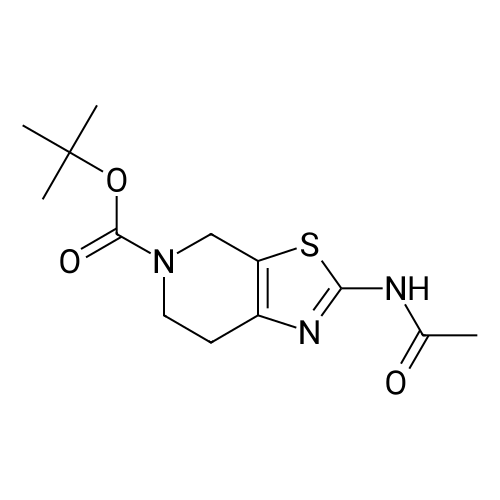
[00130] As shown in step 10-ii of Scheme 10, to Compound 1033 (10.0 g) in pyridine (80 mL) was added acetyl chloride (4.2 mL). After stirring at 5O0C for 30 min, water (200 mL) was added. The resulting solid was collected by filtration and washed with water (2x) gave tert-butyl 2-acetamido-6,7-dihydrothiazolo[5,4-c]pyridine-5(4H)-carboxylate (Compound1034, 10.873 g).
2-[(isoquinoline-3-carbonyl)-amino]-3H-benzoimidazole-4-carboxylic acid[ No CAS ]
[ 365996-05-0 ]
2-((2-[(isoquinoline-3-carbonyl)-amino]-3H-benzoimidazole-4-carbonyl)-amino)-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
55%
To a solution of 0.25 g (0.75 mmol) of 2-[(isoquinoline-3-carbonyl)-amino]-3H- benzoimidazole-4-carboxylic acid in DMF (4 mL) was added 0.4 g (1.05 mmol) of HBTU and DIEA (1 mL). The mixture was stirred for 5 min and then 0.18 g (0.7 mmol) of 2-amino- 6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester was added. The resulting reaction mixture was heated at 80 0C for 2h. After usual workup, the residue was purified by column chromatography to afford the 0.2 g (55%) of 2-({2-[(isoquinoline-3- carbonyl)-amino]-3H-benzoimidazole-4-carbonyl}-amino)-6,7-dihydro-4H-thiazolo[5,4- c]pyridine-5-carboxylic acid tert-butyl ester. LCMS: 570 (M+l)+. 1H NMR (DMSOd6, 400 MHz): delta 9.56 (s, IH), 8.84 (s, IH), 8.35 (m, 2H), 7.96 (m, 4H), 7.84 (d, IH), 7.35 (t, IH), 4.58 (bs, 2H), 3.70 (t, 2H), 2.73 (t, 2H), 1.44 (s, 9H) ppm
Example 79 Synthesis of 2-[6-(lH-indazol-6-ylcarbamoyl)-lH-benzimidazol-2-ylamino]-6,7-dihydro-4H- thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl esterTo a solution of l-Boc-4-piperidone (5 mmol) in dry THF (20 mL) was added solidBa2CC>3 (10 mmol). The resulting mixture was stirred vigorously. The reaction mixture was treated with pyrrolidone hydrotribromide (5.5 mmol) in portions at room temperature. After 3h, the contents were filtered and the solvent removed. The crude reaction mixture containing the product, 3-bromo-4-oxo-piperidine-l -carboxylic acid terf-butyl ester, was used for further transformation without further purification.To a solution of the bromo compound (5 mmol) , obtained as above, in acetone (20 mL) was added solid thiourea (6 mmol) and solid K2CO3 (10 mmol), and the reaction mixture was stirred at room temperature for 12h. To the reaction mixture was added BOC anhydride (5 mmol), and the reaction was stirred for 4h. The contents were then filtered, and the solvent was removed. The residue obtained was purified by silica gel chromatography using DCM/methanol as eluent. The product, 2-amino-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5- carboxylic acid tert-butyl ester, was obtained a light yellow solid. <n="66"/>The amine (0.5 mmol) from above was converted to corresponding isothiocyanate using general procedure A, which was then reacted with 334-diamino-N-(lH-indazol-6-yl)- benzamide (0.5 mmol; see Example 25) according to general procedure B to yield 2-[6-(1H- indazol-6-ylcarbamoyl)-lH-benzimidazol-2-ylamino]-6,7-dihydro-4H-thiazolo[5,4- cjpyridine-S-carboxylic acid tert-butyl ester. MS: m/z 531 (M+H)+.
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃; for 24h;
tert-Butyl 2-amino-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylate11 (179 mg, 0.700 mmol) and compound 28 (207 mg, 0.700 mmol) were dissolved in DMF (6 mL). To the solution were added HOBt (94.6 mg, 0.700 mmol) and EDC·HCl (268 mg, 1.40 mmol). After stirring at room temperature for 24 h, the solvent was distilled off in vacuo. To the residue were added CH2Cl2 and saturated NaHCO3 aqueous solution. After extraction with CH2Cl2, combined organics were dried over Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (CH2Cl2/MeOH = 24:1) to obtain the title compound (176 mg, 0.330 mmol, 47%) as a pale yellow amorphous solid. 1H NMR (CDCl3) delta: 1.50 (9H, s), 2.63-2.70 (2H, m), 3.69-3.77 (2H, m), 4.61 (2H, s), 4.67 (2H, d, J = 6.4 Hz), 6.86 (1 H, d, J = 3.9 Hz), 7.29 (1H, d, J = 3.9 Hz), 7.41-7.48 (3H, m), 7.56 (1H, td, J = 7.6, 1.2 Hz), 7.67 (1H, d, J = 7.3 Hz), 7.72 (1H, d, J = 7.3 Hz). ESI-MS m/z: 533 (M+H)+.
Step 1 Compound 21 was synthesized using a method described in Heterocycles, 63(7), 1555 (2004). To a suspension of Compound 21 (2.01 g, 7.87 mmol) in pyridine (10 mL), acetic anhydride (967 muL, 10.2 mmol) was added. The solution was then stirred under nitrogen atmosphere at room temperature overnight. To the reaction solution, acetic anhydride (372 muL, 3.94 mmol) was added. The reaction solution was then stirred at 40C for 3 hours. Water was added to the reaction solution, and then extracted with ethyl acetate. The extract was washed sequentially with water and saturated brine, and then dried with anhydrous magnesium sulfate. After concentrating in vacuo, purification by silica gel column chromatography (n-hexane:ethyl acetate = 1:1 ? 1:3) yielded Compound 23 (2.02 g, 86%). LC/MS (Method D): 1.43 min, [M+H]+ = 298 1H-NMR (CDCl3) delta: 10.81 (1H, br s), 4.57 (2H, s), 3.74 (2H, s), 2.73 (2H, s), 2.24 (3H, s), 1.49 (9H, s).
With triethylamine; at 0 - 20℃;
To a stirred solution of t-Butyl 2-amino-6,7-dihydrothiazolo[5,4-c]pyridine-5(4H)-carboxylate (0.9 g, 3.52 mmol) in TEA (5 mL), acetic anhydride (0.43 mL, 4.58 mmol) was added at 0 C and the reaction mixture was stirred at RT overnight. Completion of the reaction was monitored by TLC, the reaction mixture was concentrated under vacuum and the resulting mixture was dissolved in EtOAc (20 mL). The organic layer was washed with brine solution (5 mL), dried over anhydrous Na2S04 and concentrated under vacuum. The resulting crude material was used in the next step as such without any further purification. Yield: 82% (0.85 g, pale yellow liquid). 1H NMR (400 MHz, DMSO- d6): delta 1 1.9 (bs, 1 H), 4.46 (m, 2H), 3.63-3.44 (m, 2H), 2.62-2.49 (m, 2H), 2.21 (s, 3H), 1.41 (s, 9H). LCMS: (Method A) 298.0 (M+H), Rt. 3.4 min, 94.5% (Max).
With N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 80℃; for 2.5h;Inert atmosphere;
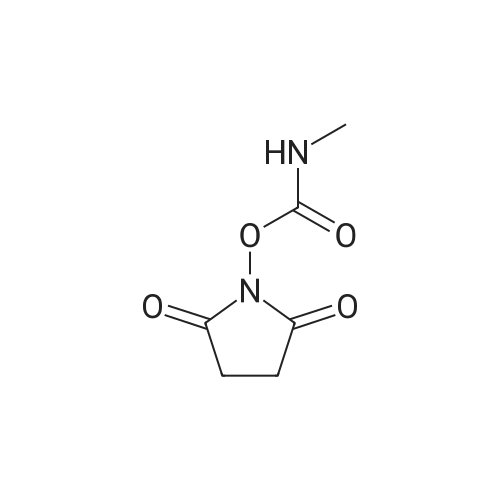
Step 1 Compound 21 was synthesized using a method described in Heterocycles, 63(7), 1555 (2004). To a solution of Compound 21 (97.0 mg, 0.380 mmol) in dimethylformamide (1.0 mL), diisopropylethylamine (199 muL, 1.140 mmol) and 2,5-dioxopyrrolidin-1-ylmethylcarbamate C (131.0mg, 0.706 mmol) were added. The reaction solution was then stirred under nitrogen atmosphere at 80C for 2 hours and a half. Aqueous saturated sodium bicarbonate solution was added to the reaction solution, and then extracted with ethyl acetate. The extract was washed sequentially with water (two times) and saturated brine, and then dried with anhydrous magnesium sulfate. After concentrating in vacuo, purification by aminosilica gel column chromatography (chloroform:methanol = 100:0 ? 95:5) yielded Compound I-66 (110.2 mg, 93%). LC/MS (Method D): 1.36 min, [M+H]+ = 313 1H-NMR (DMSO-d6) delta: 10.34 (1H, s), 6.41 (1H, br s), 4.42 (2H, s), 3.61 (2H, t, J = 5.3 Hz), 2.67 (3H, d, J = 4.5 Hz), 2.56 (2H, t, J = 5.3 Hz), 1.42 (9H, s).
With copper dichloride; isopentyl nitrite; In N,N-dimethyl-formamide; at 50℃; for 2h;
To a solution of copper chloride (II) (1.90 g, 14.10 mmol) in anhydrous dimethylformamide (5ml) were dropped under ice-cooling isoamyl nitrite (2.37 ml, 17.62 mmol) and a suspension of of Compound 10 (3 g, 11.75 mmol) in anhydrous dimethylformamide (10 ml), respectively. It was stirred at 50 C for 2 hours. To the reaction mixture were added saturation ammonium chloride solution (50ml) and ethyl acetate (50 ml). After extraction, the organic layer was washed with brine (40 ml) three times and dried over magnesium sulfate. The solvent was evaporated under reduced pressure. The residue was purified with silicagel chromatography (n-hexane : ethyl acetate = 5 : 1) to give Compound 11 (1.62 g, 50 %) as a white solid. 1H-NMR(CDCl3): delta(ppm) 1.49 (s, 9H), 2.82 (t, J=5.6 Hz, 2H), 3.73 (t, J=5.6 Hz, 2H), 4.55 (s, 2H).
8.78 g
With tert.-butylnitrite; copper(l) chloride; In N,N-dimethyl-formamide; at 50℃; for 3h;Cooling with ice;
11.16 g (0. 08 mol) of cuprous chloride was dissolved in 100 mL of N, N-dimethylformamide, and 11.5 g (0.1 mol) of t-butyl nitrite was added dropwise to the reaction solution under ice-cooling , Then carefully batches of 18. 4g (0.072 mol) 2-amino-6,7-dihydrothiazole [5,4-c] pyridine-5 (4OH) -carboxylate was added to the reaction solution and heated to 50 C. After 3 hours, the TLC monitoring reaction was complete. Poured into water and extracted with 200 mL of ethyl acetate, washed with saturated aqueous sodium bicarbonate solution (200 mL) and saturated brine (200 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (ethyl acetate: Petroleum ether = 1: 5) to give a white solid 8. 78 g, m.p. 89. 1 C
8.78 g
With tert.-butylnitrite; copper(l) chloride; In N,N-dimethyl-formamide; at 50℃; for 3h;Cooling with ice;
The 11.6g (0.086mol) of cuprous chloride was dissolved in 100mL N, N- dimethyl formamide,The ice bath was 11.5g (0.11mol) t-butyl nitrite dropwise to the reaction mixture,Subsequently, 18.4 g (0.072 mol) of tert-butyl 2-amino-6,7-dihydrothiazole [5,4-c] pyridine-5 (4H) -carboxylate was added to the reaction solution,The temperature was raised to 50 C.After 3 hours,TLC monitoring reaction is complete. Poured into water and extracted with 200 mL of ethyl acetate, washed with saturated aqueous sodium bicarbonate (200 mL) and saturated brine (200 mL), dried over anhydrous magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography (ethyl acetate: Petroleum ether = 1: 5) to give 8.78 g of a white solid,
With hydrogenchloride; In 1,4-dioxane; dichloromethane; at 20℃; for 1.5h;
Step 1; Compound 10 was synthesized in accordance with the reference (Heterocycles, 2004, 63(7), 1555-1561). To a solution of Compound 10 (5.00 g, 19.5 mmol) in methylene chloride (75 mL), hydrochloric acid (4 mol/L, a dioxane solution) (24.5 mL, 98.0 mmol) was added. The reaction mixture was then stirred at room temperature for 1 hour and a half. The precipitated solid was collected to yield mixture 11 (4.16 g). LC-MS (Method A): 0.18 min, [M+H]+ = 156 1H-NMR (DMSO-d6) delta: 9.86 (2H, s), 9.23 (2H, brs), 4.05 (2H, s), 3.34 (2H, s), 2.78 (2H, s).
With calcium carbonate; In tetrahydrofuran; for 15h;
6.01.16.04 2-Phenoxycarbonylamino-6,7-dihydro-4H-thiazolo(5,4-c)pyridine-5-carboxylic acid tert-butyl ester 39 g calcium carbonate and 36.8 g phenyl chloroformate in 250 mL THF was added to a stirred solution of 50 g 2-amino-6,7-dihydro-4H-thiazolo (5,4-c)pyridine-5-carboxylic acid tert-butyl ester in 1 L THF. The reaction mixture was allowed to stir for 15 h. The reaction mixture was filtered through silica gel and the filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate and the organic layer was dried over magnesium sulfate. The organic layer was concentrated under reduced pressure. The residue was washed with 20% ethyl acetate in hexane to give 60 g of the desired product. 1H NMR (400 MHz, CDCl3): delta 1.44 (s, 9H), 2.81 (s, 2H), 3.66 (s, 2H), 4.53 (s, 2H), 7.18 (d, 2H), 7.25-7.30 (m, 1H), 7.41 (t, 2H), 11.99 (br s, 1H)
60 g
With calcium carbonate; In tetrahydrofuran; at 20℃; for 15h;
6.01.08.04 2-Phenoxycarbonylamino-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester 39 g calcium carbonate and 36.8 g phenyl chloroformate in 250 mL THF was added to a stirred solution of 50 g <strong>[365996-05-0]2-amino-6,7-dihydro-4H-thiazolo[5,4-c]pyridine-5-carboxylic acid tert-butyl ester</strong> in 1 L THF. The reaction mixture was allowed to stir for 15 h at RT. The reaction mixture was filtered through silica gel and the filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate and the organic layer was dried over magnesium sulfate. The organic layer was concentrated under reduced pressure. The residue was washed with 20% ethyl acetate in hexane to give 60 g of the desired product. 1H NMR (400 MHz, CDCl3): delta 1.44 (s, 9H), 2.81 (s, 2H), 3.66 (s, 2H), 4.53 (s, 2H), 7.18 (d, 2H), 7.25-7.30 (m, 1H), 7.41 (t, 2H), 11.99 (br s, 1H); (M+H)+: 376
60 g
With calcium carbonate; In tetrahydrofuran; at 20℃; for 15h;
6.01.08.04 2-Phenoxycarbonylamino-6, 7-dihydro-4H-thiazolo[5, 4-c]pyridine-5-carboxylic acid tert-butyl ester 39 g calcium carbonate and 36.8 g phenyl chloro formate in 250 mL THF was added to a stirred solution of 50 g 2-amino-6, 7-dihydro-4H-thiazolo[5, 4-c]pyridine-5-carboxylic acid tert-butyl ester in 1L THF. The reaction mixture was allowed to stir for 15 h at RT. The reaction mixture was filtered through silica gel and the filtrate was concentrated under reduced pressure. The residue was diluted with water and extracted with ethyl acetate and the organic layer was dried over magnesium sulfate. The organic layer was concentrated under reduced pressure. The residue was washed with 20% ethyl acetate in hexane to give 60 g of the desired product. 1H NMR (400 MHz, CDC13): delta 1.44 (s, 9H), 2.81 (s, 2H), 3.66 (s, 2H), 4.53 (s, 2H), 7.18 (d, 2H), 7.25 - 7.30 (m, 1H), 7.41 (t, 2H), 11.99 (br s, 1H); (M+H)+: 376
4,5,6,7-tetrahydro-thiazolo(5,4-c)pyridin-2-ylamine dihydrobromide[ No CAS ]
[ 365996-05-0 ]
Yield
Reaction Conditions
Operation in experiment
11.6 g
With potassium carbonate; In 1,4-dioxane; water; at 20℃; for 3h;Enzymatic reaction;
6.01.08.03 2-Amino-6, 7-dihydro-4H-thiazolo[5, 4-c]pyridine-5-carboxylic acid tert-butyl ester 15.8 g 4, 5, 6, 7-tetrahydro-thiazolo[5, 4-c]pyridin-2-yl-amine and 100 mL dioxane was added to 15.2 g potassium carbonate in 158 mL water. 13.1 g di tert-butyl dicarbonate in 58 mL dioxane was added at 0 C. The reaction mixture was allowed to stir for 3 h at ambient temperature. The reaction mixture was diluted with water and the solid was filtered through silica gel, washed with water (2 x 50 mL) to afford the desired product. The filtrate was concentrated, diluted with water and extracted with ethyl acetat. The organic layer was dried over magnesium sulfate and concentrated to afford 11.6 g of the desired product. 1H NMR (400 MHz, DMSO-d6): delta 1.41 (s, 9H), 2.43 (t, 2H), 3,56 (t, 2H), 4.28 (s, 2H), 6.80 (s, 2H); (M+H)+: 256
General procedure: To a solution of 4-nitrophenyl chloroformate (2.01 g, 10 mmol) in CH2Cl2 (20 mL) was added a solution of 2-amino-4-methylthiazole (1.15 g, 10 mmol) and pyridine (0.97 mL, 12 mmol) in CH2Cl2 (5 mL) at 0 C. After 30 min, H2O (20 mL) was added and was stirred for 2 min. The resulting solid was filtered and washed with H2O (5 mL x 2) and Et2O (5 mL x 2). The product was dried in vacuo to give 1.45 g of intermediate C (ca. 60% purity). This product was used without further purification.
(2R,3R)-3-(2-(pyrimidin-5-yl)-6,7-dihydrothiazolo[5,4-c]pyridin-5(4H)-yl)-2-(2,4-difluorophenyl)-1-(1H-1,2,4-triazol-1-yl)butan-2-ol disulfate salt[ No CAS ]
With tris-(dibenzylideneacetone)dipalladium(0); potassium carbonate; 4,5-bis(diphenylphos4,5-bis(diphenylphosphino)-9,9-dimethylxanthenephino)-9,9-dimethylxanthene; In 1,4-dioxane; at 110℃; for 1h;Microwave irradiation;
General procedure: A microwave tube (10 mL) was charged with 24 (0.1 mmol),chloride 21b (0.12 mmol), Pd2(dba)3 (0.01 mmol), K2CO3 (0.15 mmol), Xantphos (0.02 mmol), and dioxane (2 mL). The mixture was heated to 110 C under 100 W for 1 h. The solution was concentrated in vacuum, washed with saturated NaHCO3, brine, dried over Na2SO4, and purified by chromatography (CHCl3/MeOH:15/1) to afford corresponding compounds 6a?, 6b-c. Compound 6a? (56 mg, 0.1 mmol) was dissolved in a mixed solvent system (CH2Cl2/TFA:1/1, 5 mL) and stirred at rt for 2 h. After removal of the solvent, the residue was taken up in water (5 mL), alkalified with NaHCO3 to pH >7, and then extracted with CHCl3. The combined organic layer was washed with brine, dried, filtered, and then evaporated. The residue was purified by chromatography (CHCl3/MeOH:10/1) to afford corresponding compounds 6a.
tert-butyl 2-(2-(4-(ethylsulfonyl)-2-fluorophenyl)acetamido)-6,7-dihydrothiazolo[5,4-c]pyridine-5(4H)-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
53%
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 20℃; for 12h;
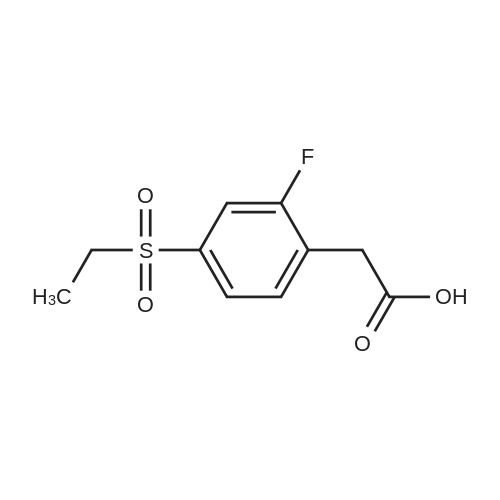
Step ii: tert-butyl 2-(2-(4-(ethylsulfonyl)-2-fluorophenyl)acetamido)-6,7-dihydrothiazolor5,4- clpyridine-5(4H)-carboxylate To a 50 mL round bottom flask, were added tert-butyl 2-amino-6,7-dihydrothiazolo[5,4- c]pyridine-5(4H)-carboxylate (0.3 g, 0.0012 mol), 2-(4-(ethylsulfonyl)-2-fluorophenyl)acetic acid (0.43 g, 0.0018 mol) and dichloromethane (15 mL). To the same flask, EDCI.HC1 (0.34 g, 0.0018 mol) and HOBt (0.24 g, 0.0018 mol) were added. The reaction mixture was stirred at RT for 12 h. The reaction mixture was diluted with dichloromethane. The diluted reaction mixture was washed with water. The organic layer was separated, washed with brine and dried over anhydrous sodium sulfate. The solvent was evaporated under reduced pressure to get crude product. The crude product was purified by column chromatography using 60-120 silica gel and 40 % ethyl acetate in hexane to get the title compound [0.3 g, 53 %]. NMR (300 MHz, CDCI3): delta 7.66-7.54 (m, 3H), 4.53 (s, 2H), 3.89 (s, 2H), 3.70-3.69 (m, 2H), 3.16 (q, 2H), 2.07- 2.62 (m, 2H), 1.46 (s, 9H), 1.29 (t, 3H); LC-MS: 482.0 [M-H]+.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping