* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
rhodamine and derivatives: ... rhodamine (Rhod) rhodamine B rhodamine 123 rhodamine X isothiocyanate sulforhodamine B sulforhodamine 101 N,N,N',N'-tetramethyl-6-carboxyrhodamine (TAMRA) tetramethyl rhodamine ...
rhodamine and derivatives: ... rhodamine (Rhod) rhodamine B rhodamine 123 rhodamine X isothiocyanate sulforhodamine B sulforhodamine 101 N,N,N',N'-tetramethyl-6-carboxyrhodamine (TAMRA) tetramethyl rhodamine
General procedure: (0090) Under nitrogen atmosphere, N-(3-amino-2,4,6-trimethylphenyl)-3,3-dimethylbutanamide (38 g, 0.153 mol) was added to an N-methyl-pyrrolidone solution (100 mL) of raw material A (15 g, 0.037 mol) having the following structure to cause a reaction at 150 C. for 6 hours. After completion of the reaction, the product was cooled to room temperature, to which 100 mL of 2 mol/L of hydrochloric acid was slowly added and stirred for 30 minutes. After filtration, the product was washed with 100 mL of water, and the obtained solid was dried. The dried solid was added to 30 g of fuming sulfuric acid ice-cooled at 5 C. or lower, and stirred at 30 to 32 C. for 24 hours. After completion of the reaction, the reaction liquid was slowly added onto 200 g of ice and stirred. After filtration, the product was washed with cold water, and the solid was suspended in 50 mL of water. A sodium hydroxide aqueous solution was used to adjust the pH at 7 to 8, and then acetone was used for crystallization to obtain 17.1 g of the compound (1).
2
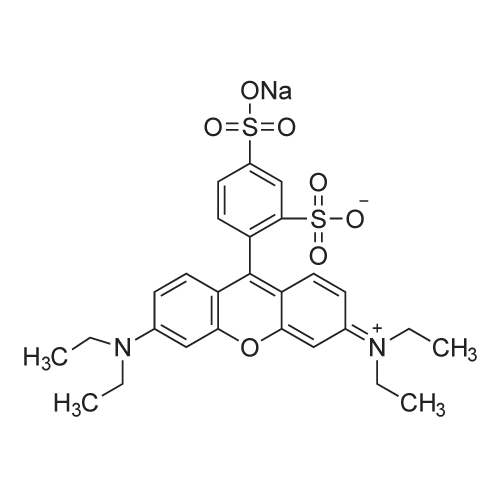
[ 3520-42-1 ]
[ 62796-29-6 ]
Yield
Reaction Conditions
Operation in experiment
With oxalyl dichloride; In dichloromethane; N,N-dimethyl-formamide; at 0 - 20℃; for 3.5h;
5.8 g (0.01 mol) of AR 52 was dispersed in 90 ml of dichloromethane, the reaction solution was maintained at 0 to 5 C,6.4 g (0.05 mol) of oxalyl chloride and 1.5 ml of dimethylformamide were added in this order, taking care not to raise the temperature of the solutionAfter stirring for 30 minutes, the reaction solution was slowly warmed to room temperature and stirred for 3 hours to synthesize Compound 2.Then, the reaction solution was cooled to 0 to 5 C and reacted with 24.0 g (0.04 mol) of SURFONAMINE B-60 {CH3-[OCH2CH2]x-[OCH2CH(CH3)]y-NH2, weight average molecular weight = 600, 9.1 g (0.09 mol) of triethylamine was added and the mixture was stirred for 1 hourAfter the reaction, the reaction solution was warmed to room temperature and stirred for 6 hours. When the reaction is complete, the solvent of the reaction product is distilled off under reduced pressureA product having a high viscosity was obtained. This was dispersed in 300 ml of a 5% hydrochloric acid aqueous solution and stirred at room temperature for 1 hourAfter filtration, it was washed several times with distilled water and dried to obtain a dark red 3,6-bis (diethylamino) -9- [4- [(polyetheralkylamino) sulfonyl] -2- sulfophenyl] (1) 7.0 g (61.4%).
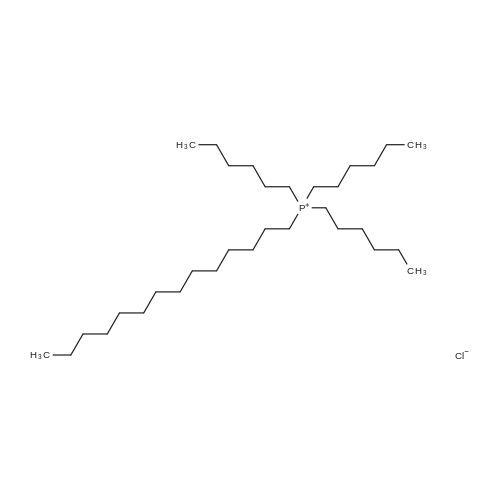
N-[6-(diethylamino)-9-(2,4-disulfophenyl)-3H-xanthen-3-ylidene]-N-ethyl-ethanaminium hydroxide inner salt trihexyltetradecylphosphonium salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In methanol; dichloromethane; for 24h;
4.0 grams (6.9 millimoles) of N-[6-(diethylamino)-9-(2,4-disulfophenyl)-3H-xanthen-3-ylidene]-N-ethyl-ethanaminium hydroxide, inner salt, trihexyltetradecylphosphonium salt, otherwise known in the art as SULFORHODAMINE B, or KITON RED 620, was dissolved in methanol (5 ml). Trihexyltetradecylphosphonium chloride (4.5 grams, 8.7 millimoles) was dissolved in 100 ml dichloromethane. The two solutions were combined into a round bottom flask and stirred for 24 hours. The mixture was transferred to a separatory funnel where the organic phase was separated from the aqueous phase. The organic phase was washed with 200 ml deionized water. The organic phase was separated and rotovapped. The ionic liquid product (i.e. N-[6-(diethylamino)-9-(2,4-disulfophenyl)-3H-xanthen-3-ylidene]-N-ethyl-ethanaminium, inner salt, trihexyltetradecylphosphonium salt) was collected.
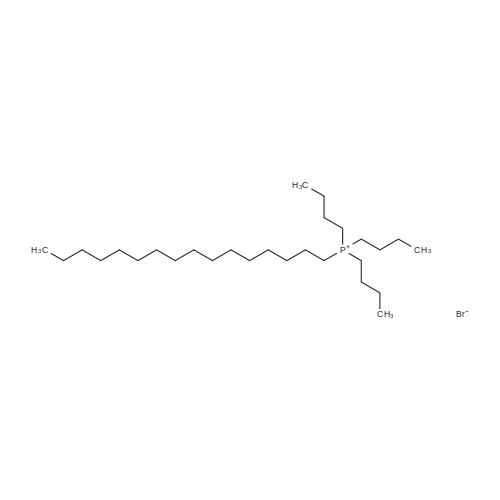
General procedure: Into a 200-mL eggplant-shaped flask containing a stirrer, 2.9 g (5.0 mmol) of acid red dye, acid red 52, 29 mL of ion-exchanged water was added and heated to 85C in an oil bath with stirring. Bath temperature. To this solution, a solution obtained by dissolving 5.2 g (10.26 mmol) of tributylhexadecylphosphonium bromide in 60 g of ion-exchanged water at room temperature was added little by little to the solution. At the time of complete addition, it was confirmed that a water-insoluble colored oily substance was formed. Then, it stirred at this temperature for 1 hour, and it cooled to the vicinity of room temperature using the ice bath. The supernatant was removed by decantation, and the residue was then washed with ion-exchanged water. After the residue was dissolved in methanol and recovered, it was concentrated under reduced pressure using a rotary evaporator. The obtained oily residue was dried under reduced pressure at 50C for 12 hours to obtain 6.1 g of a red-purple solid.
Into a 200-mL eggplant-shaped flask containing a stirrer, 2.9 g (5.0 mmol) of acid red dye, acid red 52, 29 mL of ion-exchanged water was added and heated to 85C in an oil bath with stirring. Bath temperature. To this solution, a solution obtained by dissolving 5.2 g (10.26 mmol) of tributylhexadecylphosphonium bromide in 60 g of ion-exchanged water at room temperature was added little by little to the solution. At the time of complete addition, it was confirmed that a water-insoluble colored oily substance was formed. Then, it stirred at this temperature for 1 hour, and it cooled to the vicinity of room temperature using the ice bath. The supernatant was removed by decantation, and the residue was then washed with ion-exchanged water. After the residue was dissolved in methanol and recovered, it was concentrated under reduced pressure using a rotary evaporator. The obtained oily residue was dried under reduced pressure at 50C for 12 hours to obtain 6.1 g of a red-purple solid.
Basic Red 1 (4.29 g) was completely dissolved in 50 mL of methanol. Then, 5 g of Acid Red 52 was dissolved completely in 50 mL of methanol, transferred to a buret, added dropwise to the above-prepared Basic Red 1 methanol solution, and stirred for 2 hours. After 2 hours, the methanol solvent was evaporated to remove all of the solvent, and the remaining product was dissolved in methylene chloride and extracted with water. At this time, unreacted materials and byproducts are dissolved out. After the extraction process is performed three times, Only the methylene chloride layer was separated, evaporated and dried to obtain 7.04 g (yield = 50%) of the complex of formula (1) having a molecular weight of 1,000.21 g / mol.
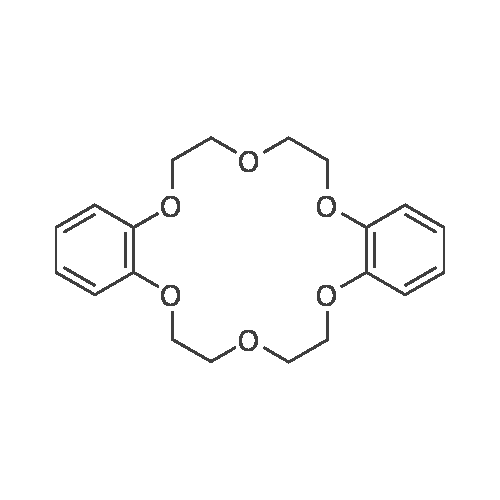
[(dibenzo-18-crown-6)(Na)(sulforhodamine B)][ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In dichloromethane;
[0157] A quantity of dry <strong>[3520-42-1]sulforhodamine B</strong> sodium (5.8 mg) is added to a flask. One molar equivalent of the cation ligand dibenzo-l8-crown-6 (3.6 mg) is measured out and added to the flask. A volume of dichloromethane (10 mL) is added to the dry mixture to yield a solution of desired concentration (e.g., 1 mM with respect to anionic dye). The solvent is then removed in vacuo and the resulting powder collected. A quantity (18.8 mg) of the dry SMILES powder [(dibenzo-l8-crown-6) (Na)(<strong>[3520-42-1]sulforhodamine B</strong>)] is added to a flask. The solid is then dissolved in a volatile organic solvent (dichloromethane) to a desired stock concentration (2 mM). A glass microscope slide is then affixed to a spin coater set to 2000 RPM. Spinning time is set for 30-45 seconds. The spinner is started and 20 pL of the dissolved stock solution of the SMILES [(dibenzo-l8-crown-6) (Na)(<strong>[3520-42-1]sulforhodamine B</strong>)] is then applied to the center of the slide. Once spinning is complete, the slide is removed and can be analyzed for optical properties without any additional processing.
With calcium chloride; In water; at 20 - 30℃; for 1h;
After dissolving 30.0 parts of C.I. Acid Red 52 in 300 parts of water in a reaction vessel, 5.7 parts of calcium chloride was added to the dissolving solution at 20 to 30 C and stirred at the same temperature for one hour.After filtering the precipitated crystals, the obtained wet cake was dried in a hot-air drier at 80 C., thereby obtaining 23.6 parts of a compound represented by No. 1.The maximum absorption wavelength lambdamax of this compound is 565nm (aqueous solution).
With barium(II) chloride; In water; at 20 - 30℃; for 1h;
After dissolving 50.0 parts of C.I. Acid Red 52 in 500 parts of water in a reaction vessel, 31.6 parts of barium chloride was added to the dissolving solution at 20 to 30 C, and stirred at the same temperature for one hour. After filtering the precipitated crystals, the obtained wet cake was dried in a hot-air drier at 80 C., thereby obtaining 46.4 parts of a compound represented by the aforementioned No. 5.The maximum absorption wavelength lambdamax of this compound was 565 nm (aqueous solution).
With hydrogenchloride; In water; at 20 - 30℃; for 1h;
After dissolving 50.0 parts of C.I. Acid Red 52 in 500 parts of water in a reaction vessel, 15.0 parts of concentrated hydrochloric acid was added to the solution at 20 to 30 C, and the mixture was stirred at the same temperature for one hour. After filtering the precipitated crystals, the obtained wet cake was dried in a hot-air dryer at 80 C., thereby obtaining 45.7 parts of a comparative compound represented by the following formula (x).The maximum absorption wavelength lambdamax of this compound was 565 nm (aqueous solution).
With acetic acid; In water; at 20 - 25℃; for 1.5h;
After dissolving 30.0 parts of C.I. Acid Red 52 in 300 parts of water in a reaction vessel,18-Amino-8,11,13-Abietin Triene23 parts dissolved in 3.3% acetic acid solution100 parts of water solutionThe solution was dripped into the dissolving solution at 20 to 25 C for 30 minutes, and stirred at the same temperature for one hour.After filtering the precipitated crystals,The obtained wet cake was dried in a hot-air dryer at 80 C., thereby obtaining 32.0 parts of a compound represented by the aforementioned No. 2.The maximum absorption wavelength lambdamax of this compound is 565nm (Aqueous solution).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping