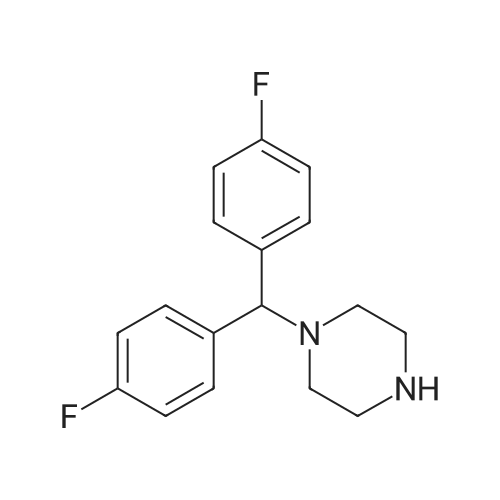
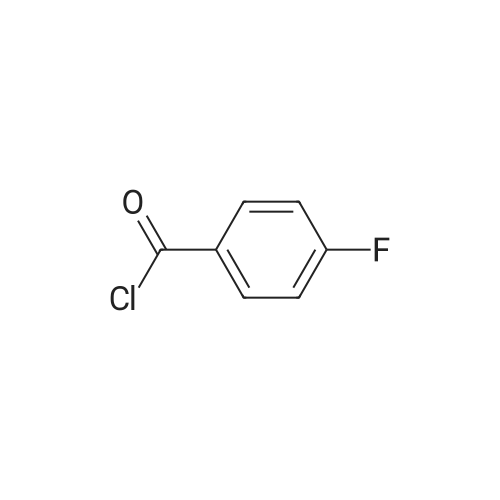
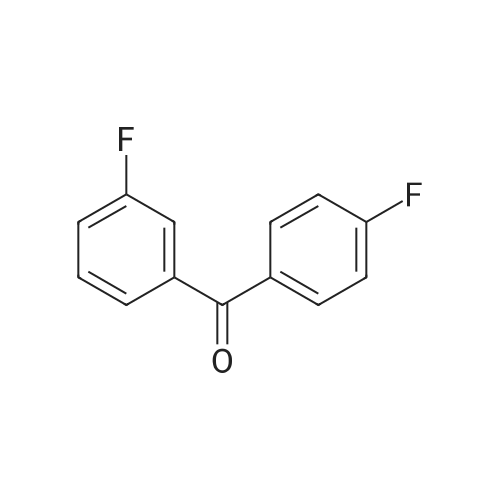
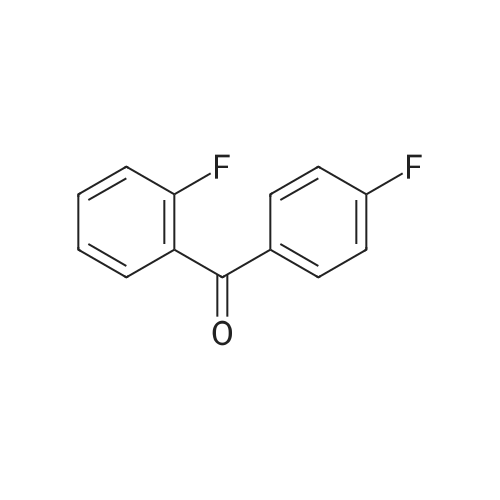
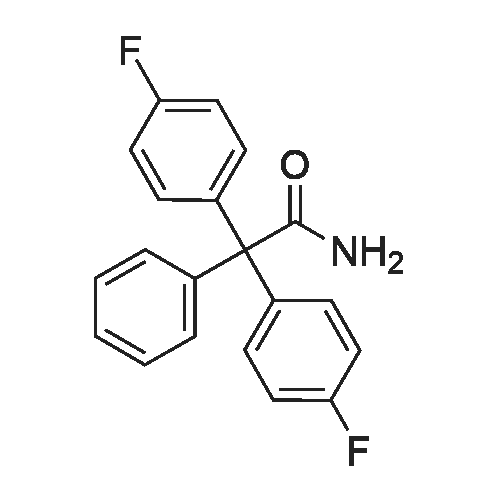
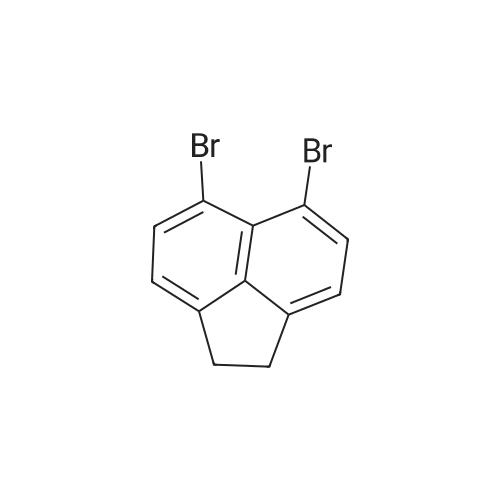
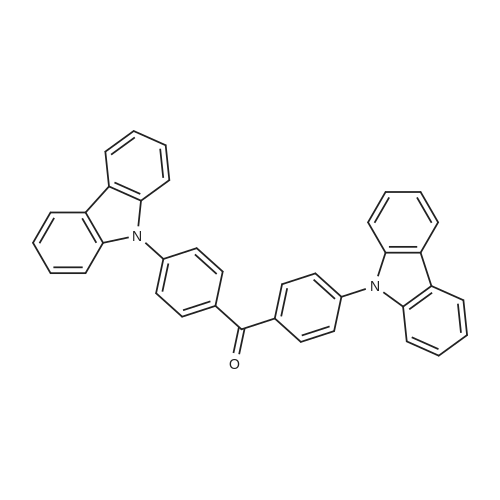
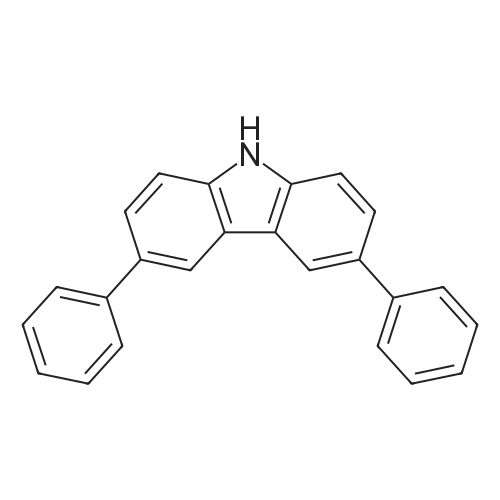
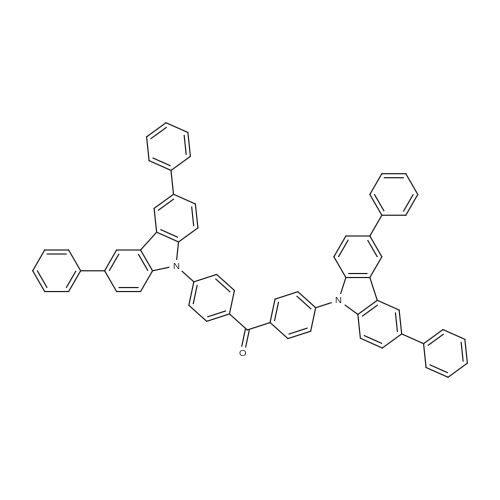
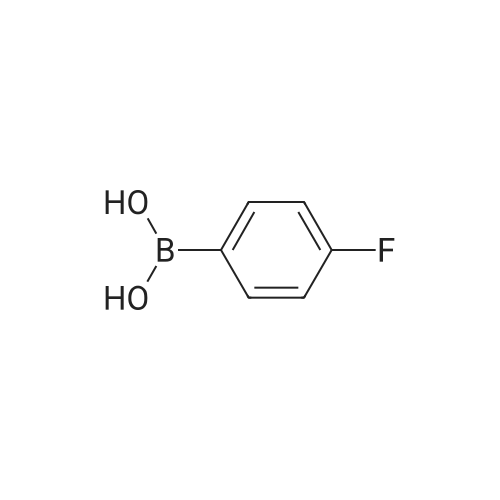
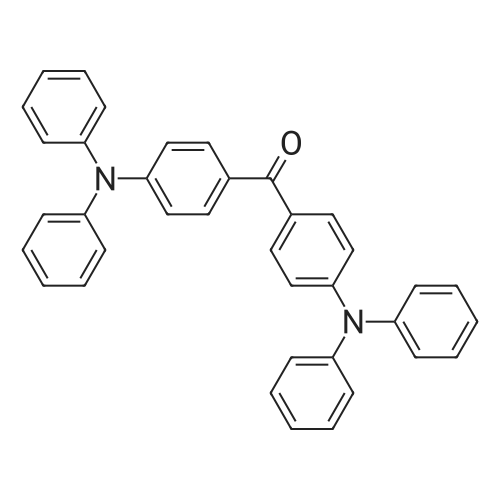
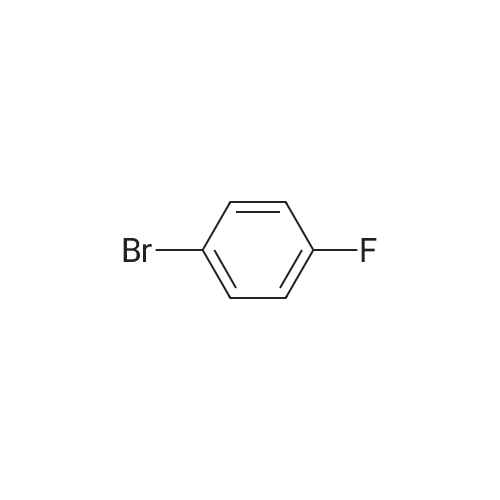
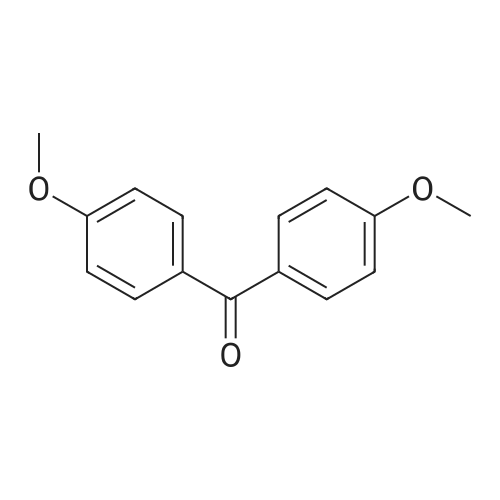
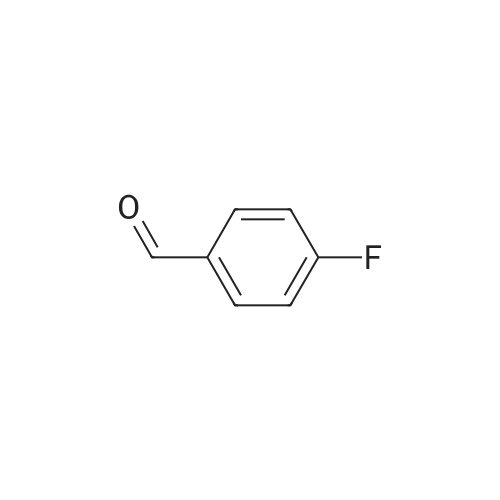
| 53% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1.5h; Inert atmosphere; Cooling with ice;
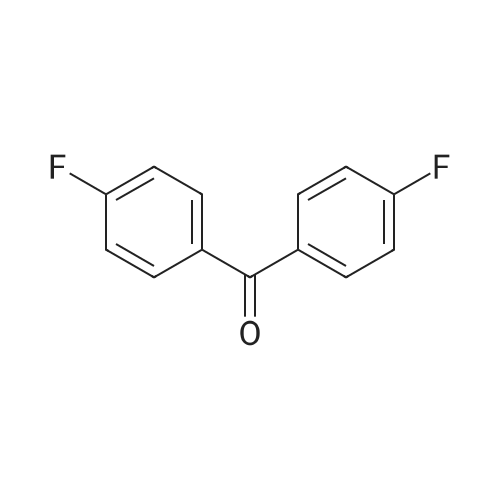
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; Inert atmosphere; |
1.3
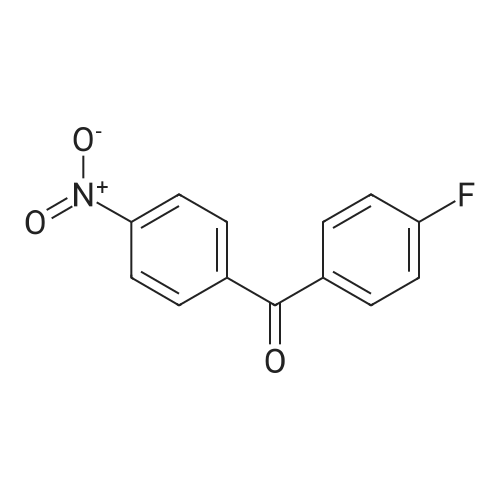
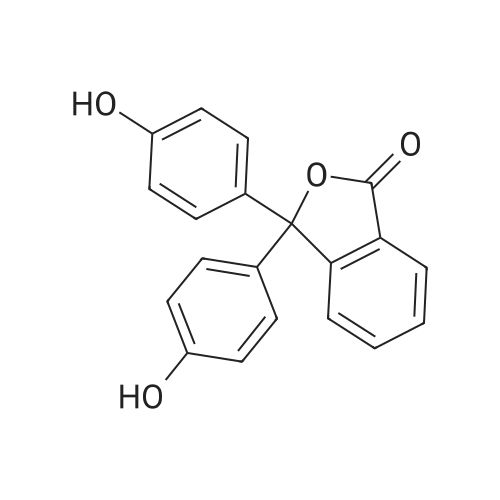
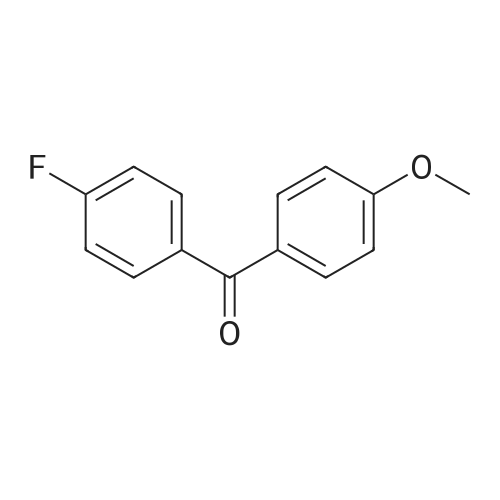
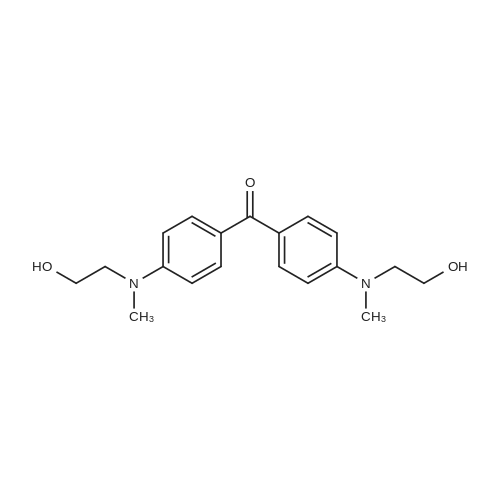
The following reaction, is carried out in a nitrogen atmosphere. A cooling pipe in a flask equipped with a stirrer and N-methylaniline (tokyo chemical industry (strain) of the company) 15.3 and N, N-dimethyl formamide 60 part of the part after charging, an ice cooled mixed solution. Under cold ice 60% sodium hydride (tokyo chemical industry (strain) of the company) 5.7 part after 30 minutes by adding little by little, 1 time while raising the temperature of the room. 4,4 '-difluorobenzophenone (tokyo chemical industry (strain) of the company) 10.4 part 24 at room temperature, the reaction solution little by little with time. The reaction liquid after 200 part slightly in addition to ice, to stand at room temperature for 15 hours, water is removed by decantation and viscous as solid residue is obtained. This viscous solid after adding methanol 60, stirred at room temperature for 15 hours. After the deposited solid is separated by filtration, purification column chromatography. Purified and dried under reduced pressure 60 °C corallite solid, a compound represented by obtd. eq. (C-I-1) 9.8 parts. Yield 53% |
| 53% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1.5h; Cooling with ice; Inert atmosphere;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide; mineral oil at 20℃; for 24h; Inert atmosphere; |
1
The following reaction was performed under a nitrogen atmosphere.N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) 15.3After 60 parts of N,N-dimethylformamide have been placed in a flask with a cooling tube and stirrer,The mixed solution was ice-cooling.It takes 30 minutes to add in a small amount under cold conditionsAfter 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) 5.7 parts,Stirred for 1 hour while warming to room temperature.4,4'-Difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) was added to the reaction solution in small amounts in an amount of 10.4 parts.And stirred at room temperature for 24 hours.After adding a small amount of the reaction solution to 200 parts of ice water,Let stand at room temperature for 15 hours,To collect water by decantation,Viscous solids are obtained in the form of residues.After adding 60 parts of methanol to the viscous solid,Stir at room temperature for 15 hours.The precipitated solid was filtered and purified by column chromatography.The purified pale yellow solid was dried under reduced pressure at 60C to obtain 9.8 parts of a compound represented by the formula (C-I-1).Yield 53% |
| 53% |
Stage #1: N-methylaniline With sodium hydride; N,N-dimethyl-formamide for 1.5h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone at 20℃; for 24h; |
1 Example-1:
The following reaction was carried out in a nitrogen atmosphere. 15.3 parts of N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) and 60 parts of N, N-dimethylformamide were fed into a flask equipped with a condenser and a stirrer, and then the mixed solution was ice-cooled. 5.7 parts of 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) was added little by little over 30 minutes under ice-cooling, and the mixture was stirred for 1 hour while raising the temperature to room temperature. And 10.4 parts of 4,4'-difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) were added little by little to the reaction solution, followed by stirring at room temperature for 24 hours. The reaction solution was added to ice water (200 parts) in small portions, and the mixture was allowed to stand at room temperature for 15 hours. When water was removed by decantation, a viscous solid was obtained as a residue. After adding 60 parts of methanol to the resulting solid, the mixture was stirred at room temperature for 15 hours. The precipitated solid was separated by filtration and then purified by column chromatography. The purified light yellow solid was dried at 60 under reduced pressure to obtain 9.8 parts of a compound represented by the formula (BP3). The yield was 53%. |
| 53% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1.5h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; Inert atmosphere; |
1 Synthesis example 1
The following reaction was performed under a nitrogen atmosphere.After 15.3 parts of N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) and 60 parts of N,N-dimethylformamide were put into a flask equipped with a condenser and a stirrer,The mixed solution was ice-cooled.Sodium hydride 60% (manufactured by Tokyo Chemical Industry Co., Ltd.) 5.7 parts was added little by little in 30 minutes while cooling in an ice bath.Then, warm up to room temperature and stir for 1 hour.40.4 parts of 4,4'-difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) were added to the reaction solution in small portions each time.Stir at room temperature for 24 hours.The reaction solution was added in small portions to 200 parts of ice water.Then let it sit at room temperature for 15 hours.If water is removed by decantation,A sticky solid is obtained as a residue.After adding 60 parts of methanol to the sticky solid,Stirring was performed at room temperature for 15 hours.After separating the precipitated solid by filtration,Purification by column chromatography.Purified pale yellow solid under reduced pressureDry at 60°C,In addition, 9.8 parts of a compound represented by formula (BP3) was obtained.The yield is 53%. |
| 53% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1.5h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; |
1
The following reaction was carried out under a nitrogen atmosphere.15.3 parts of N-methylaniline (manufactured by Tokyo Kasei Kogyo Co., Ltd.) and 60 parts of N, N-dimethylformamide were charged into a flask equipped with a condenser and a stirrer, and the mixed solution was cooled with ice.Under ice cooling, 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.)5.7 parts was gradually added over 30 minutes,The mixture was stirred for 1 hour while raising the temperature to room temperature.4,4'-Difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.)10.4 parts were gradually added to the reaction mixture and stirred at room temperature for 24 hours. The reaction mixture was added to 200 parts of ice water little by little,After standing at room temperature for 15 hours, water was removed by decantation to obtain a viscous solid as a residue. To this viscous solid was added 60 parts of methanol, followed by stirring at room temperature for 15 hours. PrecipitateThe solid was separated by filtration and purified by column chromatography. The purified pale yellow solid was dried under reduced pressure at 60 ° C. to obtain 9.8 parts of the compound represented by the formula (CI-2).The yield was 53%. |
| 53% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; |
2 Synthesis Example 2
The following reaction was carried out under a nitrogen atmosphere.After 15.3 parts of N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) and 60 parts of N,N-dimethylformamide were placed in a flask equipped with a condenser and a stirring device, the mixed solution was ice-cooled.After 5.7 parts of 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) was added little by little for 30 minutes under ice cooling, the mixture was stirred for 1 hour while warming to room temperature.10.4 parts of 4,4'-difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) was added little by little to the reaction mixture, and stirred at room temperature for 24 hours.The reaction mixture was added to 200 parts of ice water little by little, and then allowed to stand at room temperature for 15 hours, and water was removed by decantation to obtain a viscous solid as a residue.After 60 parts of methanol was added to the viscous solid, the mixture was stirred at room temperature for 15 hours.The precipitated solid was separated by filtration and purified by column chromatography.The purified pale yellow solid was dried under reduced pressure at 60 ° C to give 9.8 part of the compound of formula (CI-2).The yield was 53%. |
| 53% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1.5h; Inert atmosphere;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; |
2
The following reaction was carried out in a nitrogen atmosphere. After putting 15.3 parts of N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) and 60 parts of N, N-dimethylformamide into a flask equipped with a cooling tube and a stirrer, the mixed solution was ice-cooled. After adding 5.7 parts of 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) little by little over 30 minutes under ice-cooling, the mixture was stirred for 1 hour while raising the temperature to room temperature. Next, 10.4 parts of 4,4'-difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) was added little by little to the reaction mixture, and the mixture was stirred at room temperature for 24 hours. The reaction mixture was added little by little to 200 parts of ice water and then allowed to stand at room temperature for 15 hours, and the water was decanted to obtain a viscous solid as a residue. After adding 60 parts of methanol to this viscous solid, the mixture was stirred at room temperature for 15 hours. The precipitated solid was filtered off and then purified by column chromatography. The purified pale yellow solid was dried under reduced pressure at 60 ° C. to obtain 9.8 parts of the compound represented by the formula (C-II). The yield was 53%. |
| 5.3% |
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; Inert atmosphere; |
18 Synthesis Example 18
The following reaction was carried out in a nitrogen atmosphere.15.3 parts of N-methylaniline (manufactured by Tokyo Kasei Corporation) and 60 parts of N, N-dimethylformamide were added to a flask equipped with a condenser and a condenser, and the mixed solution was ice-cooled.Under ice-cooling, 60% sodium hydride (manufactured by Tokyo Kasei Co., Ltd.)5.7 parts were added in small portions over 30 minutes,The mixture was stirred for 1 hour while raising the temperature to room temperature.4,4'-difluorobenzophenone (manufactured by Tokyo Kasei Co., Ltd.)10.4 parts were added to the reaction solution in small amounts, and the mixture was stirred at room temperature for 24 hours.A small amount of the reaction solution was added to 200 parts of ice water, and the mixture was allowed to stand at room temperature for 15 hours.When the water was removed by decantation, a viscous solid was obtained as a residue.After adding 60 parts of methanol to the viscous solid, the mixture was stirred at room temperature for 15 hours.A precipitated solid was filtered and then purified by column chromatography.The purified light yellow solid was dried at 60°C under reduced pressure to obtain 9.8 parts of a compound represented by the formula (BP3). Yield 5.3% |
|
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1.5h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; Inert atmosphere; |
6
The following reaction was carried out under nitrogen atmosphere. In the apparatus equipped with a coolingtube and a stirring flask, into N- methylaniline (Tokyo Chemical Industry (Ltd.) Co., Ltd.) 15.3 parts and N, N-Dimethylformamide 60 parts, the mixed solution was cooled with ice liquid. Under ice-cooling, to take up to 30 minutes slowly into 60% sodium hydride (Tokyo Kasei Kogyo (Co., Ltd.) Inc. Shuzo) After 5.7 parts by raising the temperature to room temperature and stirred 1 hour. To the reaction mixture was slowly added 4,4-difluorobenzophenone (East Beijing Chemical Industry (Ltd.) Co., Ltd.) 10.4 parts, was stirred at room temperature for 24 hours. The reaction mixture was slowly added 200 parts of ice water After allowed to stand at roomtemperature for 15 hours, water was removed by decantation, the residue obtained as a sticky solid. In this sticky solid After addition of 60 parts of methanol was stirred at room temperature for 15 hours. The precipitated solid was filtered, purified by column chromatography. Light after purification Yellow solid under reduced pressure,60 ° C under dried, to give a compound of formula (C-I-18) 9.8 parts shown. |
|
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1.5h; Cooling with ice; Inert atmosphere;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; |
1
The following reaction was carried out under a nitrogen atmosphere.N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) was charged into a flask equipped with a cooling tube and a stirring device,15.3 parts and 60 parts of N, N-dimethylformamide, the mixed solution was ice-cooled.After 5.7 parts of 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) was added little by little over 30 minutes under ice,And the mixture was stirred at room temperature for 1 hour. 10.4 parts of 4,4'-difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) was added to the reaction solution little by little, and the mixture was stirred at room temperature for 24 hours.The reaction solution was added to 200 parts of ice water,The mixture was allowed to stand at room temperature for 15 hours, and the water was removed by decantation,As a residue to get a sticky solid. After 60 parts of methanol was added to the viscous solid, the mixture was stirred at room temperature for 15 hours. The precipitated solid was separated by filtration and purified by column chromatography. The purified pale yellow solid was dried under reduced pressure at 60 ° C to obtain 9.8 parts of the compound represented by the formula (C-I-18). |
|
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1h; Inert atmosphere; Cooling with ice;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; |
1
The following reaction is carried out under nitrogen. In the stirring device with cooling tube and put into a flask in N - methylaniline (Tokyo chemical industry Company) 15.3 parts and N, N - dimethyl formamide after 60 parts, but for cold mixed solution. In a cold but gradually added 30 minutes under 60% sodium hydride (Tokyo chemical industry Company) after 5.7 parts, raised to room temperature stirring 1 hour. At room temperature in 4, 4' - difluoro benzophenone (Tokyo chemical industry Company) 10.4 parts gradually a small amount of added into a reaction liquid stirring 24 hours. The reaction liquid after adding ice water 200 gradually small, 15 hours at room temperature, using decanting to remove water to obtain the as residue of sticky solid. The sticky solid in 60 parts after adding methanol, 15 hours stirring at room temperature. To precipitation of solids after filtration, chromatography method for the refining. In order to 60 °C under reduced pressure drying refined yellow solid, to obtain 9.8 parts of type (C CGI-I CGI-18) compound. |
|
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide at 20℃; for 1.5h; Cooling with ice; Inert atmosphere;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide at 20℃; for 24h; |
1 Production Example 1: Preparation of Lake Pigment (A1)
The following reaction was carried out under a nitrogen atmosphere. 15.3 parts of N-methyl aniline (manufactured by Tokyo Chemical Industry Co., Ltd.) and 60 parts of N, N-dimethyl formamide were placed in a flask equipped with a condenser and a stirrer, and the obtained mixed solution was cooled with ice . 5.7 parts of 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) was gradually added over 30 minutes under ice cooling and then stirred for 1 hour while raising the temperature to room temperature. 10.4 parts of 4,4'-difluoro benzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) was gradually added to the reaction solution and stirred at room temperature for 24 hours. The reaction solution was gradually added to 200 parts of ice water and then allowed to stand at room temperature for 15 hours. Water was removed by decantation to obtain a viscous solid as a residue. To this viscous solid was added 60 parts of methanol, and then mixture was stirred at room temperature for 15 hours. The precipitated solid was filtered off and then purified by column chromatography. The purified pale yellow solid was dried at 60 ° C under reduced pressure to obtain 9.8 parts of the compound represented by the formula (CI-18). |
|
Stage #1: N-methylaniline With sodium hydride In N,N-dimethyl-formamide; mineral oil at 20℃; for 1.5h; Cooling with ice; Inert atmosphere;
Stage #2: 4,4'-Difluorobenzophenone In N,N-dimethyl-formamide; mineral oil at 20℃; for 24h; |
1 Coloring agent (A-1)
The following reactions were carried out under a nitrogen atmosphere. Put 15.3 parts by weight of N-methylaniline (manufactured by Tokyo Chemical Industry Co., Ltd.) and 60 parts by weight of N,N-dimethylformamide into a flask equipped with a condenser and a stirring device, and then use the mixed solution Ice cooling. 5.7 parts by weight of 60% sodium hydride (manufactured by Tokyo Chemical Industry Co., Ltd.) was slowly added over 30 minutes under ice cooling, and then stirred for 1 hour while raising the temperature to room temperature. 10.4 parts by weight of 4,4'-difluorobenzophenone (manufactured by Tokyo Chemical Industry Co., Ltd.) was slowly added to the reaction liquid, and the mixture was stirred at room temperature for 24 hours. After slowly adding the reaction liquid to 200 parts by weight of ice water, it was allowed to stand at room temperature for 15 hours, and the water was removed by decantation to obtain a viscous solid as a residue. After adding 60 parts by weight of methanol to this viscous solid, it was stirred at room temperature for 15 hours. After separating the precipitated solid by filtration, it is purified by column chromatography. The purified light yellow solid was dried at 60° C. under reduced pressure to obtain 9.8 parts by weight of the compound of Chemical Formula 5. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping