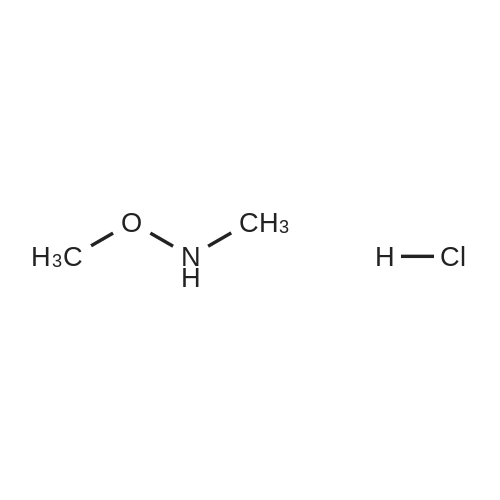
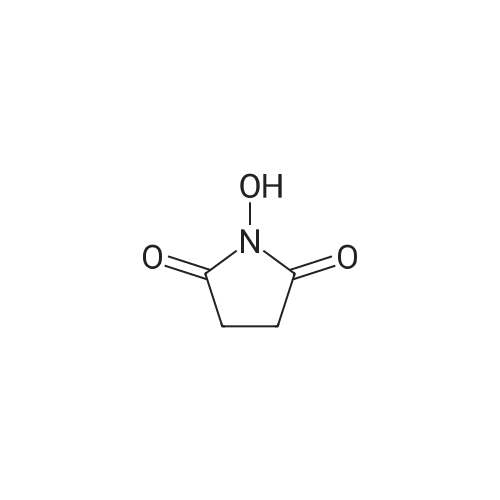
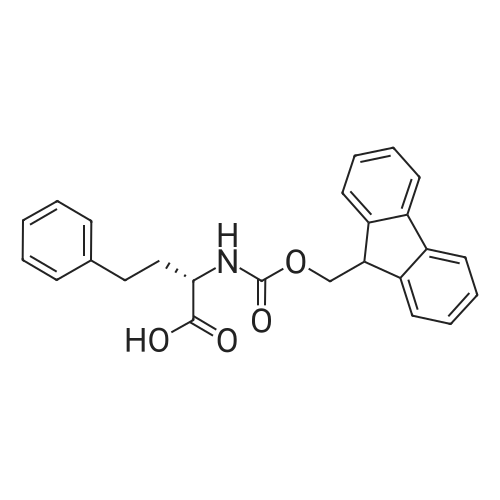
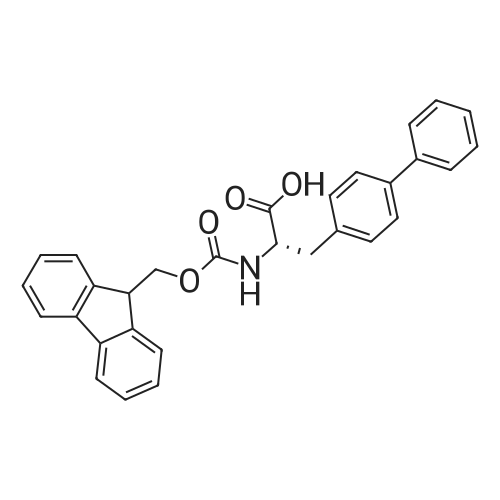
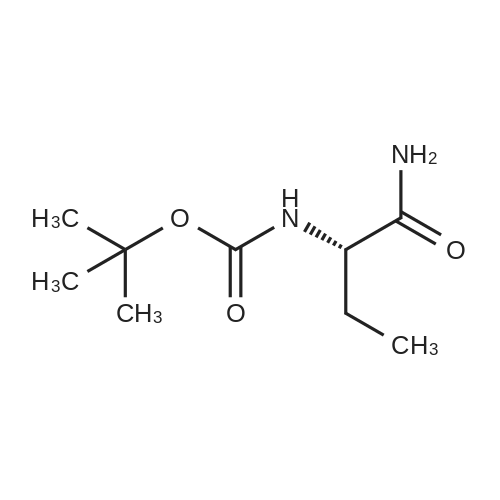
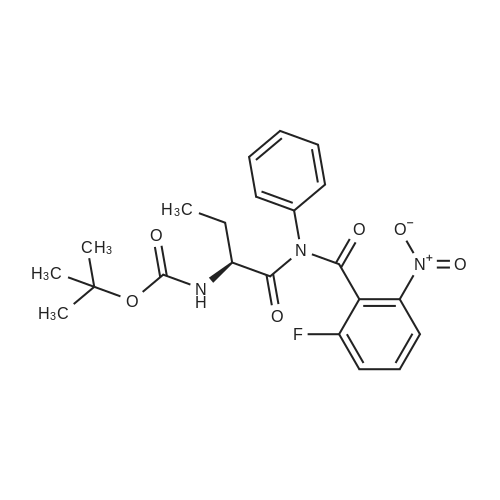
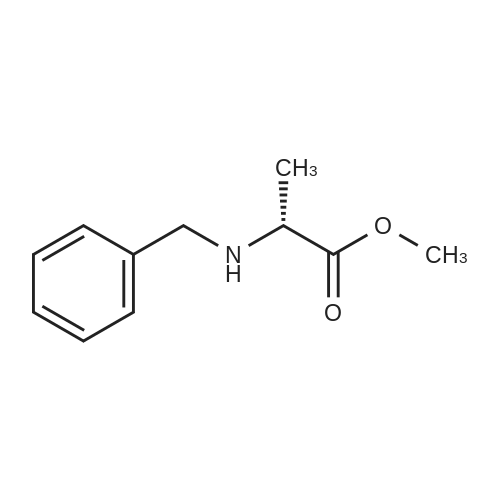
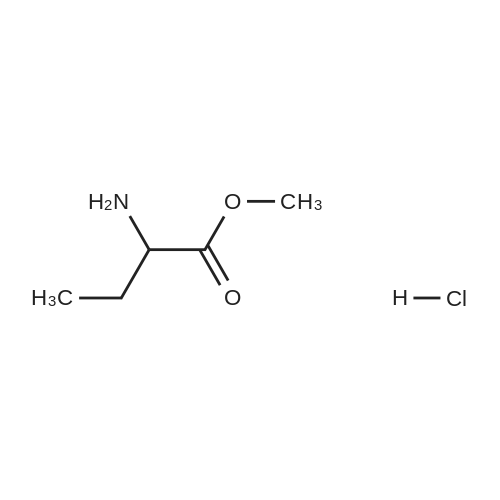
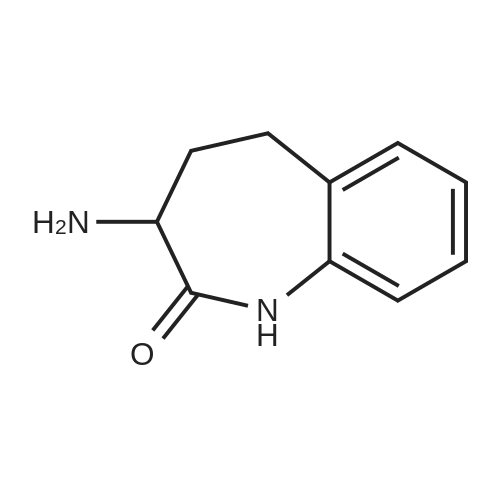
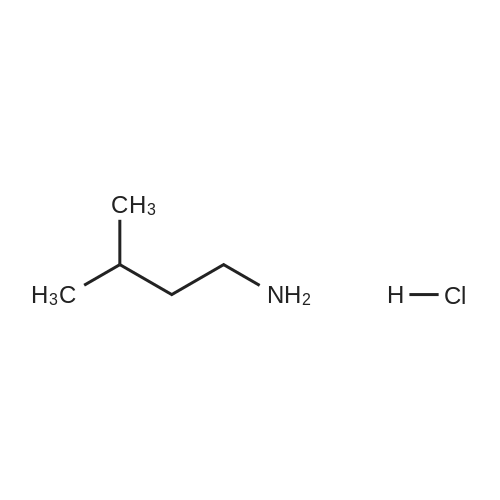
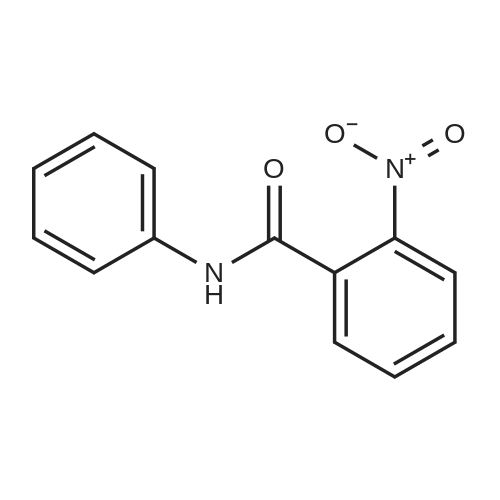
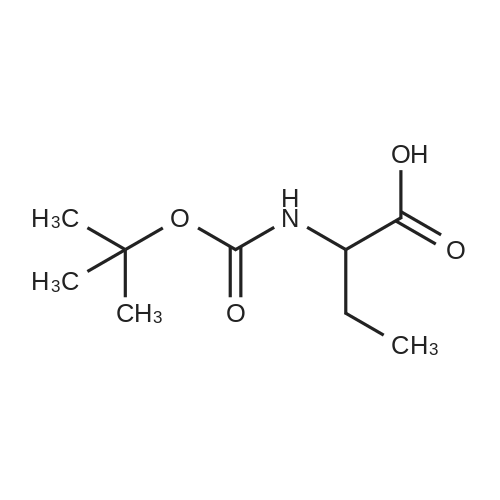
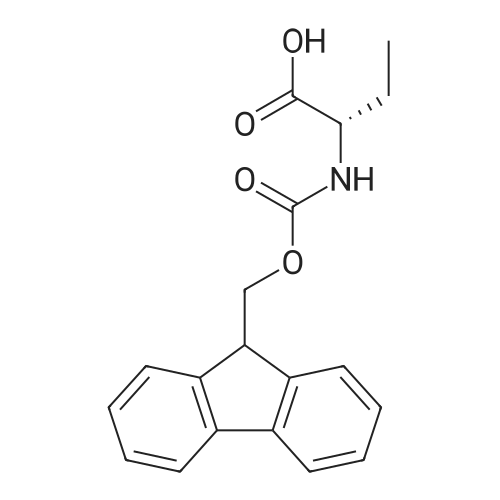
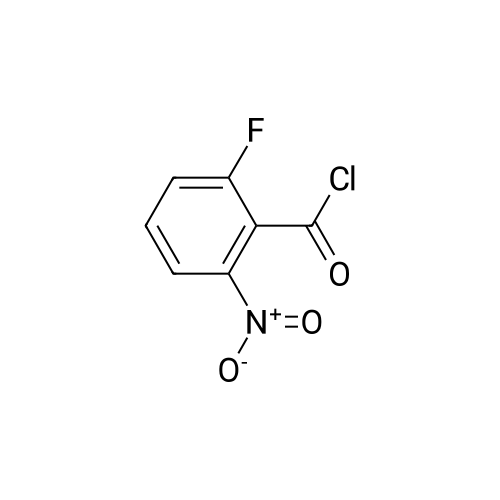
| 65% |
With 1-[(1-(cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino)]-uronium hexafluorophosphate; N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 0 - 20℃; for 16h; |
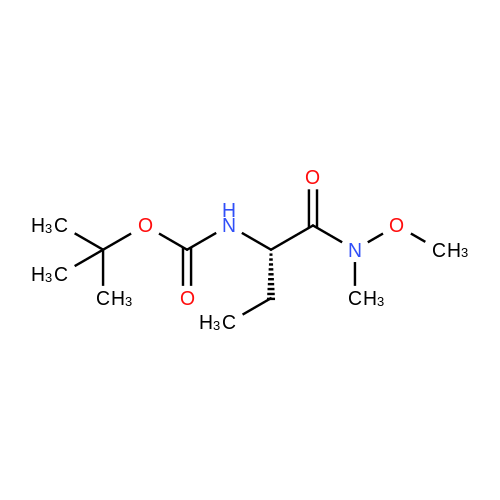
5 intermediate 15: 2-benz vi I -(tert-butyl) (2R, 4S)-4-((S)-2-((tert- butoxycarbon yi)amino)butanamido)-2-(4-(4 , 4,5,5-fetrameth vi- 1,3, 2-dioxaboroian-2- vi)butvi)piperidine-1, 2-dicarbox vi ate
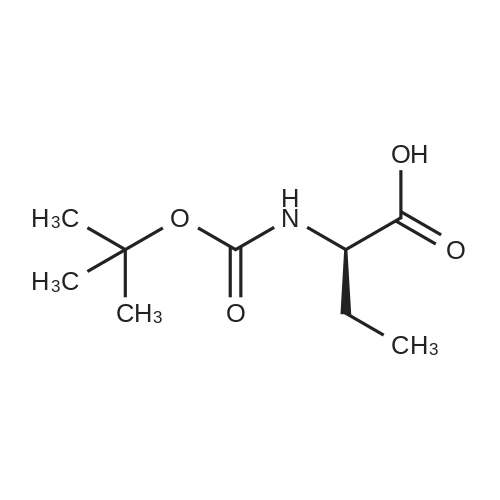
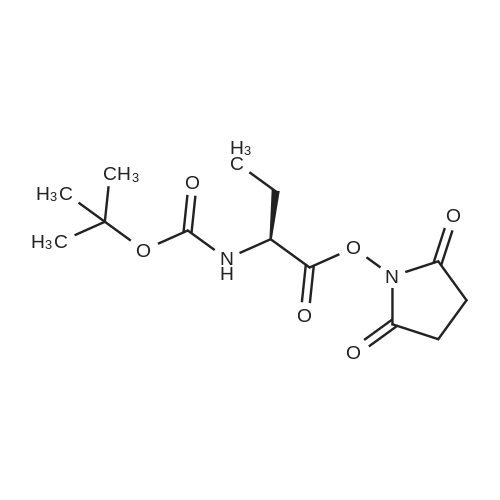
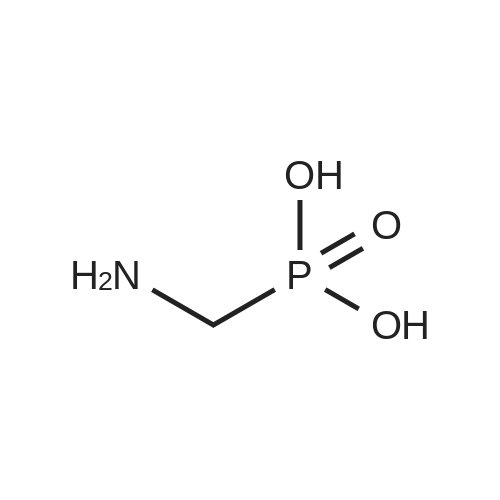
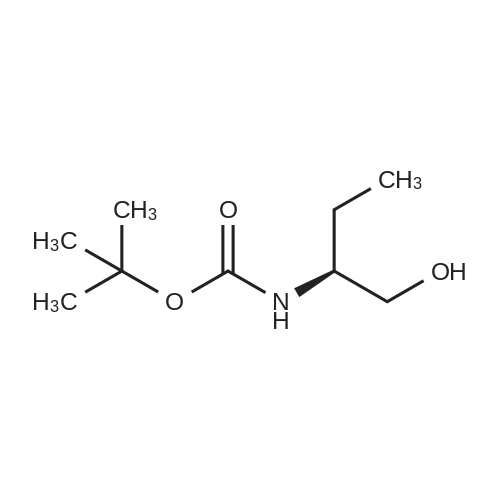
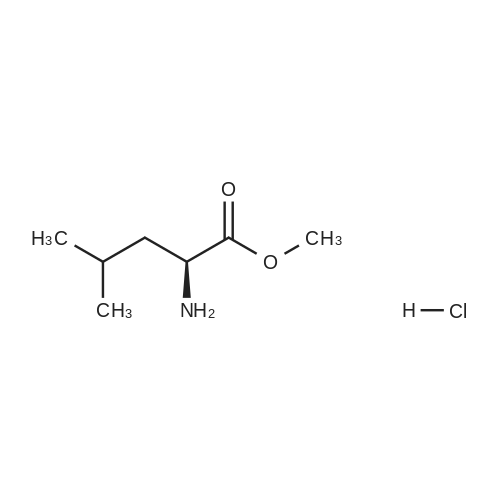
N,N-Diisopropylethylamine (0.165 mL, 0.94 mmcl) was added slowly to a stirred solutionof 2-benzyl I -(tert-butyl) (2R,4S)-4-amino-2-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)butyl)piperidine-1 ,2-dicarboxylate (Intermediate 8, 244 mg, 0.47 mmol), Boc-Abu-OH (96 mg,0.47 mmol) and COMU (206 mg, 0.48 mmol) in DMF (3 mL) at 0 °C. The reaction stirred for 16h while slowly warming to room temperature. The crude reaction mixture was diluted with water(30 mL) and extracted with EtOAc (3 x 10 mL). The combined organics were washedsequentially with saturated aqueous NaHCO3 (20 mL) and saturated aqueous NaCI (15 mL).The organic layer was dried over MgSO4, filtered and concentrated to dryness. The resulting residue was purified by flash silica chromatography (15 to 60% EtOAc in hexanes) to afford 2- benzyl I -(tert-butyl) (2R,4S)-4-((S)-2-((tert-butoxycarbonyl)amino)butanamido)-2-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2-yl)butyl)piperidine-1 ,2-dicarboxylate (Intermediate 15, 215mg, 65% yield) as clear gum and as a mixture of rotamers. 1H NMR (500MHz, CDCI3) 5 0.71 -0.79(2H, m), 0.87(3H, brt), 1.19(4H, brs), 1.21 (9H, s), 1.36(5H, brs), 1.38(8H, s), 1.39-1.41 (8H, m), 1.48 - 1.58 (2H, m), 1.68 (1 H, br dd), 1.72 - 1.81 (1 H, m), 1.84 - 1.98 (3H, m), 1.99- 2.02 (1 H, m), 2.88 - 3.04 (1 H, m), 3.89 (1 H, br d), 3.95 - 4.07 (2H, m), 5.00 (1 H, br d), 5.05 -5.22 (2H, m), 6.20 (1 H, br s), 7.27 - 7.36 (5H, m); mz: (ES) [M+H] = 703. |
| 215 mg |
With 1-[(1-(cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino)]-uronium hexafluorophosphate; N-ethyl-N,N-diisopropylamine In N,N-dimethyl-formamide at 0 - 25℃; for 16h; Inert atmosphere; |
5 Intermediate 15: 2-benzyl 1-(tert-butyl) (2R,4S)-4-((S)-2-((tert- butoxycarbonyl)amino)butanamido)-2-(4-(4,4.5,5-tetramethyl-1,3,2-dioxaborolan-2- yl)butyl)piperidine-1,2-dicarboxylate
N,N-Diisopropylethylamine (0.165 mL, 0.94 mmol) was added slowly to a stirred solution of 2-benzyl 1 -tert -butyl) (2R,4S)-4-amino-2-(4-(4,4,5,5-tetramethyl-1 ,3,2-dioxaborolan-2- yl)butyl)piperidine-1 ,2-dicarboxylate (Intermediate 8, 244 mg, 0.47 mmol), Boc-Abu-OH (96 mg, 0.47 mmol) and COMU (206 mg, 0.48 mmol) in DMF (3 mL) at 0 °C. The reaction stirred for 16 h while slowly warming to room temperature. The crude reaction mixture was diluted with water (30 mL) and extracted with EtOAc (3 x 10 mL). The combined organics were washed sequentially with saturated aqueous NaHCO3 (20 mL) and saturated aqueous NaCI (15 mL). The organic layer was dried over MgSO4, filtered and concentrated to dryness. The resulting residue was purified by flash silica chromatography (15 to 60% EtOAc in hexanes) to afford 2- benzyl 1 -tert -butyl) (2R,4S)-4-((S)-2-(tert -butoxycarbonyl)amino)butanamido)-2-(4-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)butyl)piperidine-1 ,2-dicarboxylate (Intermediate 15, 215 mg, 65% yield) as clear gum and as a mixture of rotamers. 1H NMR (500MHz, CDCI3) δ 0.71 - 0.79 (2H, m), 0.87 (3H, br t), 1.19 (4H, br s), 1.21 (9H, s), 1.36 (5H, br s), 1.38 (8H, s), 1.39 - 1.41 (8H, m), 1 .48 - 1 .58 (2H, m), 1 .68 (1 H, br dd), 1 .72 - 1.81 (1 H, m), 1.84 - 1 .98 (3H, m), 1.99 - 2.02 (1 H, m), 2.88 - 3.04 (1 H, m), 3.89 (1 H, br d), 3.95 - 4.07 (2H, m), 5.00 (1 H, br d), 5.05 - 5.22 (2H, m), 6.20 (1 H, br s), 7.27 - 7.36 (5H, m); m/z: (ES+) [M+H]+ = 703. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping