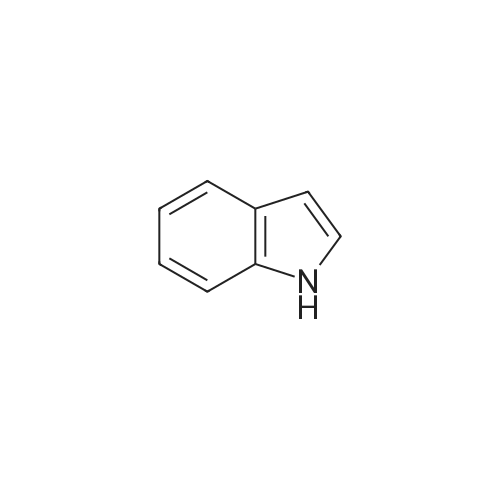
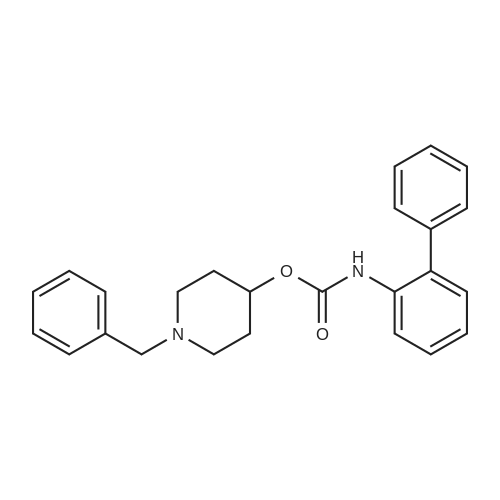
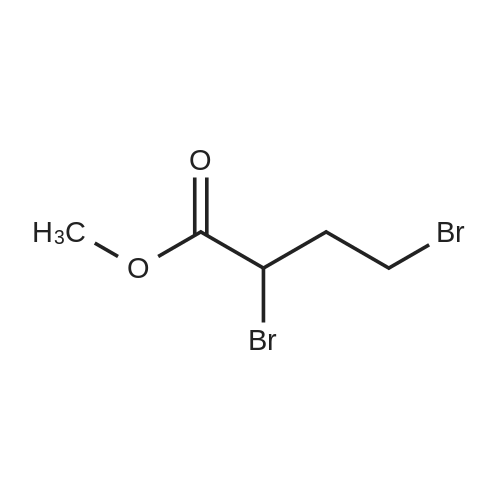
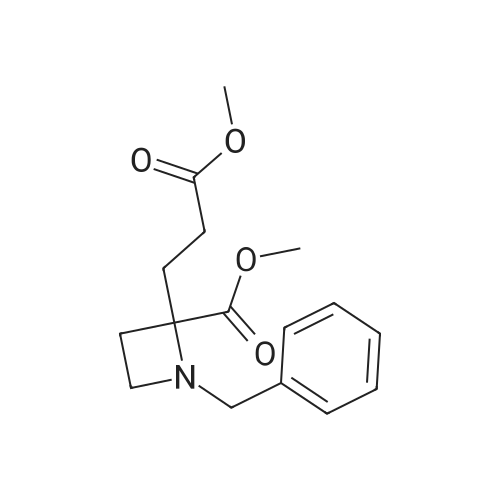
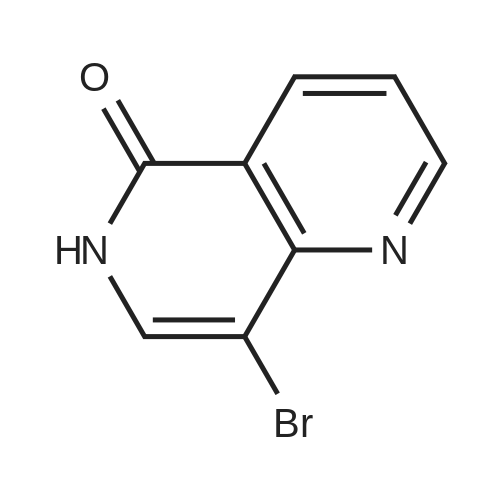
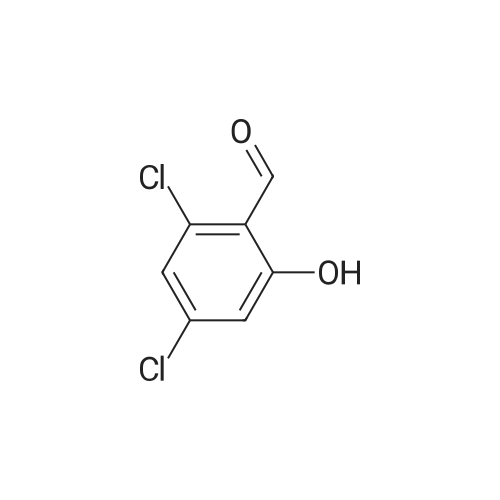
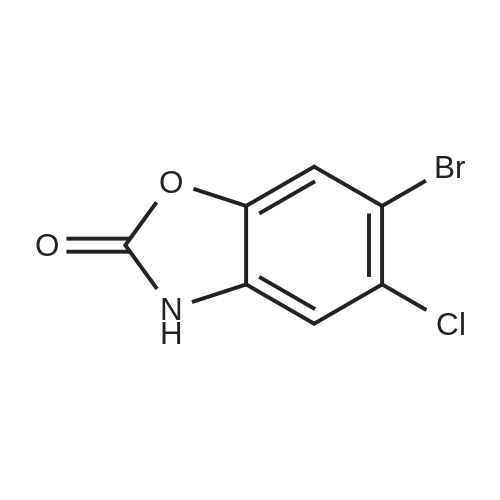
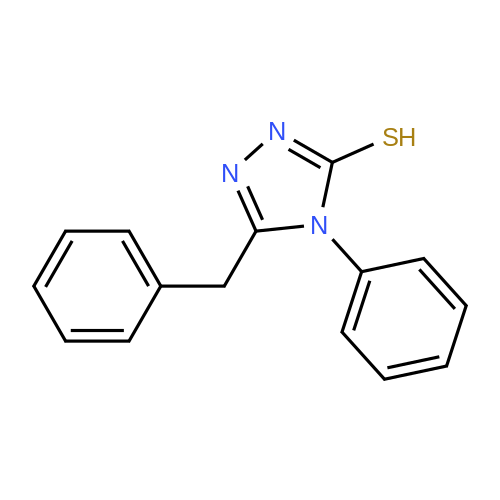
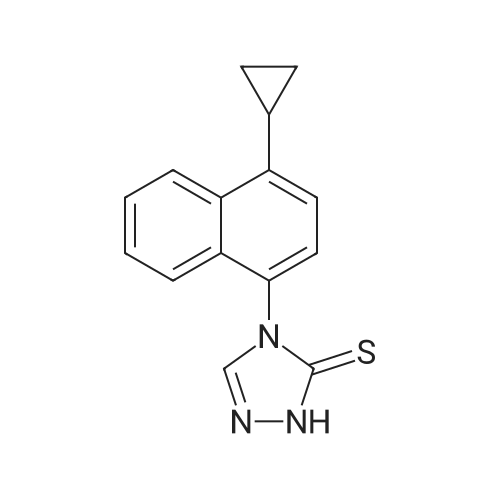
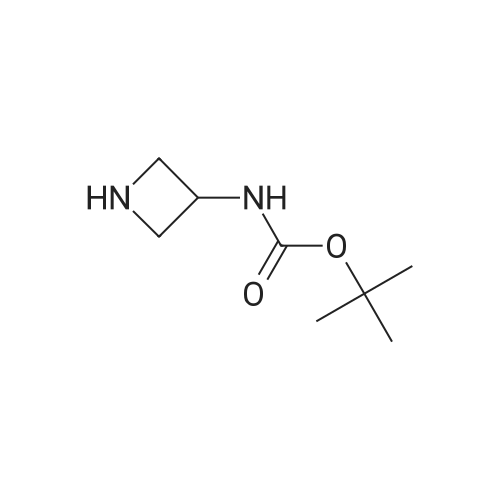
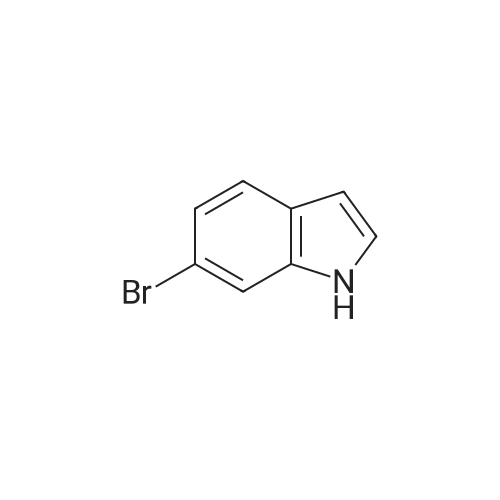
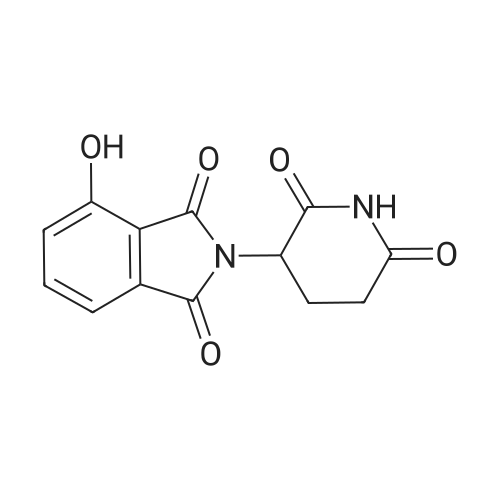
| 47% |
With triethylamine; In dichloromethane; at 30℃; |
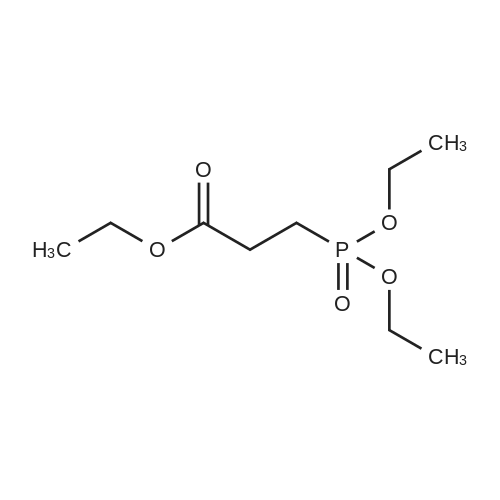
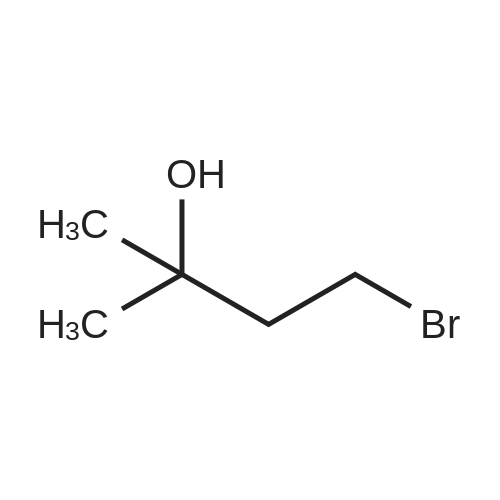
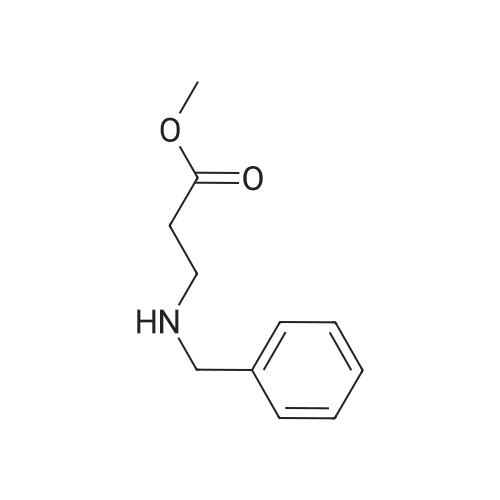
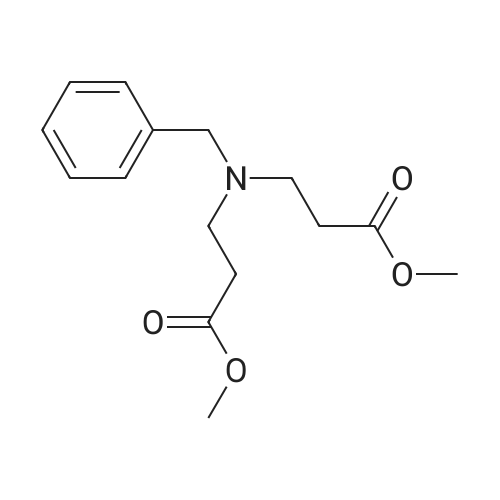
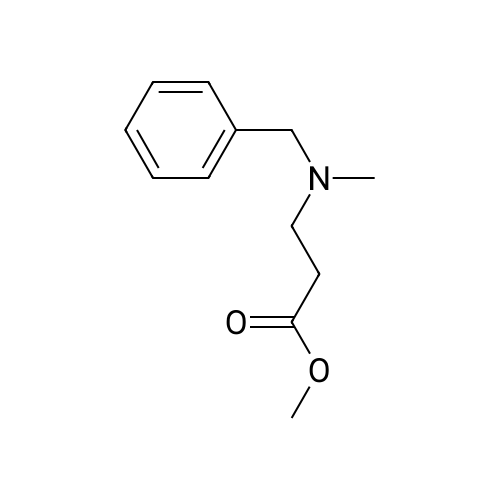
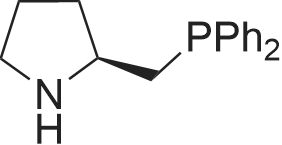
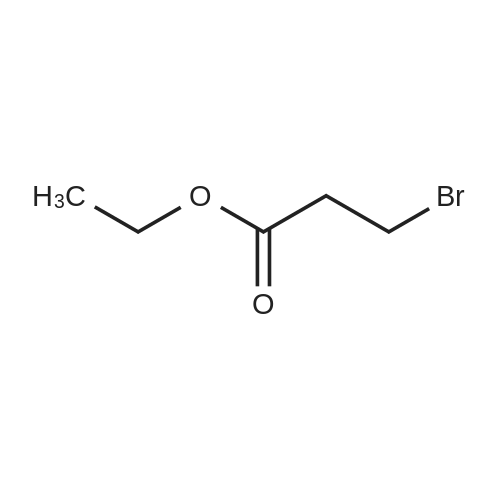
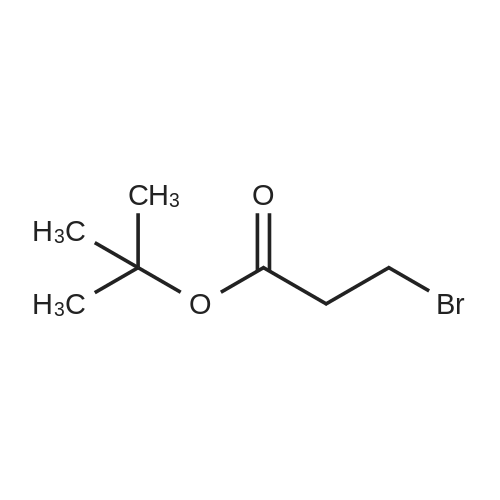
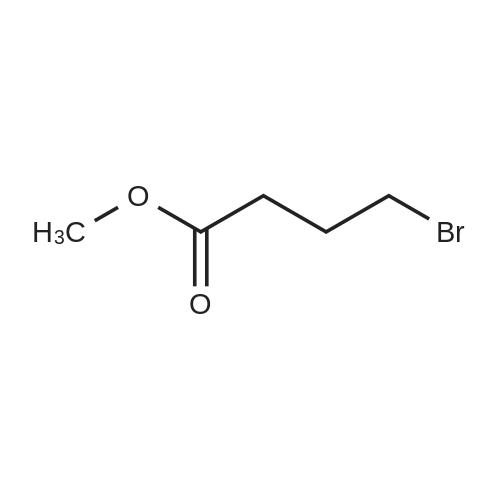
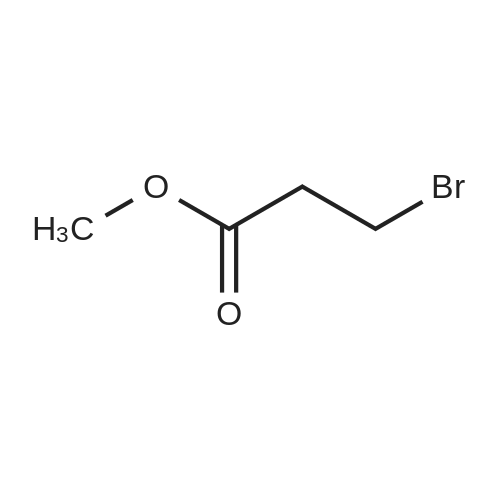
Synthesis Example 5a - methyl 3-{(2S)-2[(diphenylphosphino)methyl]pyrrolidin-1- yl}propanoate (13)(2S)-2-[(diphenylphosphino)methyl]pyrrolidine (shown below) was prepared following the procedure of Tomiaka Tetrahedron .Lett. 1996. 37. 7805 A solution of methyl-3-bromopropionate (1.55 g, 9.28 mmol, 1.01 ml) indichloromethane (8.0 ml) was added dropwise to a solution of the thethylamine (1.88 g, 18.57 mmol, 2.61ml) and (2S)-2-[(diphenylphosphino)methyl]pyrrolidine(2.50 g, 9.28 mmol) in dichloromethane (27 ml). The resulting solution was stirred at 30 C overnight. The reaction mixture was poured into water/dichloromethane (100 ml/100 ml). The crude residue was extracted with dichloromethane (100 ml), the organic phase was washed with water (100 ml), followed by brine (100 ml) and then dried over sodium sulfate and concentrated under reduced pressure.Purification by column chromatography ([5:95], methanol : dichloromethane) afforded the title compound as a yellow viscous oil (1.57 g) 47% yield.1H NMR (500 MHz, cdcl3) delta = 7.50 - 7.39 (m, 4H, CH-Ar), 7.37 - 7.28 (m, 6H, CH- Ar), 3.66 (s, 3H, CH3-9), 3. 9 - 3.03 (m, 2H, CH2-5,6), 2.54 (dt, J=3.3, 13.3, 1H, CH2-4), 2.49 - 2.29 (m, 4H, CH-2, CH2-6,7), 2.15 - 2.06 (m, 1H, CH2-5), 2.06 ~ 1.91 (m, 2H, CH2-1 ,3), 1.83 - 1.53 (m, 3H, CH2-3,4).13C NMR (126 MHz, cdcl3) delta = 172.85 (s, C-8), 139.28 (d, J=12.1 , Ar), 138.47 (d,J=13.3, Ar), 132.95 (d, J=19.3, Ar), 132.57 (d, J=18.7, Ar), 128.68 (s, Ar), 128.45 (s, Ar), 128.40 (s, Ar), 128.35 (s, Ar), 128.33 (s, Ar), 128.28 (s, Ar), 62.08 (d, J=19.3,C-2), 53.44 (d, J=0.8, C-5), 51.50 (s, C-9), 49.09 (s.C-6), 33.62 (d, J= 3.3, C- ),33.48 (s, C-7), 31.67 (d, J=7.8, C-3), 22.21 (d, J=0.6, C-4).IR (diamond, vWAx, cm'1) 2961 , 2802 (CH30 st), 1735 (C=0 st), 1433 (H-C-H st as), 1175 (C-0 st as).Acc. Mass (FAB): C2iH27 02PCalculated: 356.1774Found: 356.1778 error [ppm]: -1.28 |
| 47% |
With triethylamine; In dichloromethane; at 30℃; |
A solution of methyl-3-bromopropionate (1.55 g, 9.28 mmol, 1.01 ml) in dichloromethane (8.0 ml) was added dropwise to a solution of the triethylamine (1.88 g, 18.57 mmol, 2.61 ml) and (2S)-2-[(diphenylphosphino)methyl]pyrrolidine (2.50 g, 9.28 mmol) in dichloromethane (27 ml). The resulting solution was stirred at 30 C. overnight. The reaction mixture was poured into water/dichloromethane (100 ml/100 ml). The crude residue was extracted with dichloromethane (100 ml), the organic phase was washed with water (100 ml), followed by brine (100 ml) and then dried over sodium sulfate and concentrated under reduced pressure. Purification by column chromatography ([5:95], methanol:dichloromethane) afforded the title compound as a yellow viscous oil (1.57 g) 47% yield. [0466] 1H NMR (500 MHz, cdcl3) delta=7.50-7.39 (m, 4H, CH-Ar), 7.37-7.28 (m, 611, CH-Ar), 3.66 (s, 3H, CH3-9), 3.19-3.03 (m, 2H, CH2-5,6), 2.54 (dt, J=3.3, 13.3, 1H, CH2-4), 2.49-2.29 (m, 4H, CH-2, CH2-6,7), 2.15-2.06 (m, 1H, CH2-5), 2.06-1.91 (m, 21-1, CH2-1,3), 1.83-1.53 (m, 3H, CH2-3,4). [0467] 13C NMR (126 MHz, cdcl3) delta=172.85 (s, C-8), 139.28 (d, J=12.1, Ar), 138.47 (d, J=13.3, Ar), 132.95 (d, J=19.3, Ar), 132.57 (d, J=18.7, Ar), 128.68 (s, Ar), 128.45 (s, Ar), 128.40 (s, Ar), 128.35 (s, Ar), 128.33 (s, Ar), 128.28 (s, Ar), 62.08 (d, J=19.3, C-2), 53.44 (d, J=0.8, C-5), 51.50 (s, C-9), 49.09 (s, C-6), 33.62 (d, J=13.3, C-1), 33.48 (s, C-7), 31.67 (d, J=7.8, C-3), 22.21 (d, J=0.6, C-4). [0468] IR (diamond, VMAX, cm-1) 2961, 2802 (CH3O st), 1735 (C?O st), 1433 (H-C-H st as), 1175 (C-O st as). [0469] Acc. Mass (FAB): C21H27NO2P [0470] Calculated: 356.1774 [0471] Found: 356.1778 error [ppm]: -1.28 |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping