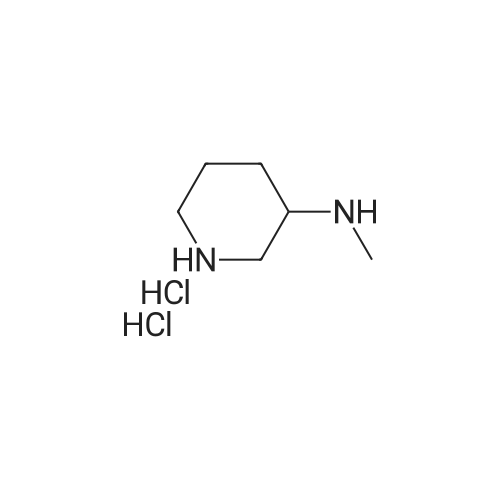
| 86% |
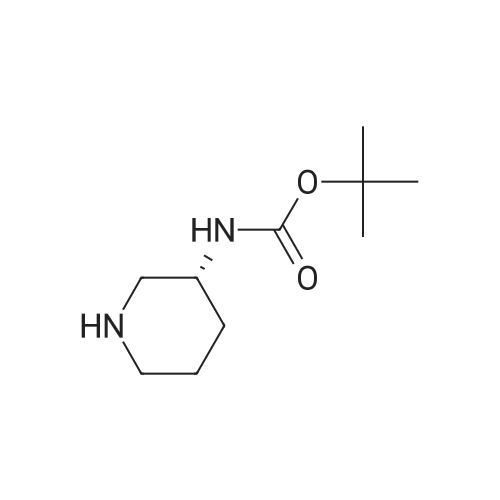
With 1,8-diazabicyclo[5.4.0]undec-7-ene In acetonitrile at 10 - 20℃; for 3h; |
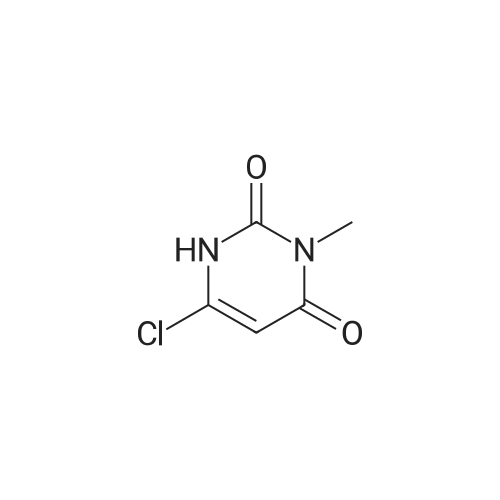
1 EXAMPLE 1 Preparation of Trelagliptin
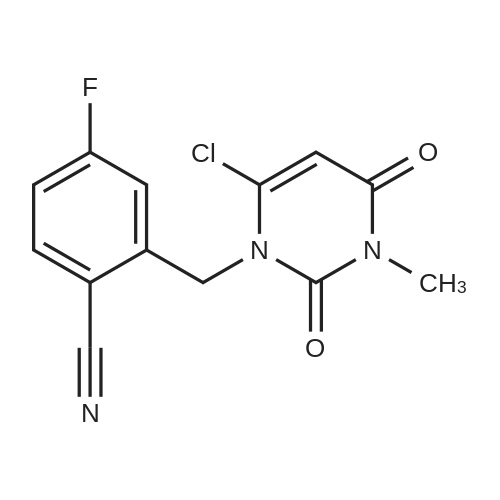
10L bottle was charged with three numbers I was 930.0g, RAPD603.0g, acetonitrile 3.72L, cooling to 10 , dropping DBU2122g. Plus finished warming to 10-20 , heat reaction 3 hours. The reaction solution was mixed with 15kg of ice water, then added 5.0L of methylene chloride, adjusted with 4M HCl pH = 1-4, separated, the aqueous phase was washed with 5.0L of dichloromethane; the combined aqueous phase was added dichloromethane 5.0L, saturated potassium carbonate solution adjusted to pH = 8-10, liquid separation, the aqueous phase was extracted with dichloromethane 5.0L, organic phases were combined, dried, filtered and concentrated to give a crude product No. II.10L reaction flask curved Ge Lieting crude isopropanol 5500ml, stirred and heated to reflux slowly dissolved. Qing retreat to dissolve the oil bath, cooled to an internal temperature of 0 ~ 10 , incubated for 1 hour, filtered, the filter cake washed with isopropanol. Set 45 vacuum oven drying to constant weight to give a white solid 973gYield 86%, HPLC purity 99.8% |
| 86% |
With potassium carbonate In isopropanol at 50 - 60℃; |
1.2 (2) INT2 synthesis
Add 1.00kg of INT1, 648g of R-3-aminopiperidine dihydrochloride, 8L of isopropanol, and 941g of potassium carbonate at room temperature. Slowly increase the temperature to 50-60 under stirring and react for 5-6h. The reaction liquid changes from a white turbid liquid. It is light yellow turbid liquid. TLC (developing reagent EA:PE=1:1, 254 color development) monitor the end of the reaction.After the reaction was completed, the isopropanol was concentrated and evaporated to dryness under reduced pressure to obtain a pale yellow solid, which was dissolved by adding 7L of dichloromethane and stirring. Filter to obtain a yellow organic phase, add 6L of water to the organic phase, add 2L of 2mol/L hydrochloric acid solution under stirring, precipitate solids, filter, and rinse the filter cake with 2L of dichloromethane. The filter cake was heated and dissolved in 3L of water, added 3L of dichloromethane, slowly added 2.1L of 2mol/L NaOH solution, adjusted PH=9-10, stirred and separated, the aqueous phase was extracted with dichloromethane once, and the organic phases were combined. Dry the organic phase with anhydrous sodium sulfate. After filtering to remove the desiccant, the filtrate was concentrated under reduced pressure to evaporate the solvent and recrystallized with methanol to obtain an off-white solid, namely INT2, with a yield of 86% and a product purity of 99.6%. |
| 86.74% |
With triethylamine In ethanol at 80℃; for 2h; |
3 Example 3: Preparation of Trelagliptin Free Base
9.0 g of 2-(6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethyl)-4-fluorobenzonitrile prepared by the above method, (R)-3-aminopiperidine dihydrochloride 8.5g, triethylamine 2g, 100ml of ethanol was added to the reaction flask, the stirring was started, and the temperature was raised to 80° C. to react for 2 hours. Cool to 0-5 and crystallize for 1 hour, filter, vacuum-drying at about 50°C for 6 hours to obtain 9.5 g of troxagliptin free base. Yield 86.74%. The purity determined by high performance liquid chromatography was: 99.927%. The liquid phase spectrum is shown in Figure 2. |
| 82% |
With Sodium hydrogenocarbonate In acetonitrile at 75 - 80℃; for 5h; Autoclave; Large scale; |
1 Example 1: Preparation of Tragostine M2
Acetonitrile (47.0 kg) was added to a 100 L autoclave, and M1 (6.0 kg), (R)-3-aminopiperidine dihydrochloride (4.3 kg), sodium hydrogen carbonate (11.0 kg) were added under stirring, and the temperature was gradually raised to 7580°C, reaction for 5 hours,Cool down to 25 ~ 35 °C, filtration, to obtain the filtrate, after adding the reaction tank to continue cooling 5 ~ 15 °C, slowly adding hydrochloric acid (3.4kg), stirring for 1 hour, filtering, filter cake dry.Add purified water (60.4kg) to the 100L reactor and the above filter cake, control the temperature at 5-15°C, add anhydrous sodium carbonate solid (4.0kg), and test the pH of the system with a wide pH test paper to be 8-9.Dichloromethane (5.0 kg×2 times) was added for extraction. The organic phase was separated and washed with purified water (12.4 kg). The organic phase was separated and filtered. The filtrate was obtained and transferred to a 50 L reaction vessel. The pressure was reduced at 40° C. Evaporate most of the solvent to the system volume of 12L,Suspension was added, and anhydrous ethanol (24L) was added under stirring. The volume was reduced to 24 L under reduced pressure at 30° C. The residue was then cooled to 10-15° C. with cooling water, and the mixture was stirred for 1 hour.Filter, drain, filter cake, blow drying at 50-55°C for 10 hours to obtain M2 boutique 6.0kg. Yield: 82.0%, purity 99.8%. |
| 79.8% |
With potassium carbonate In water monomer; isopropanol at 60 - 70℃; Large scale; |
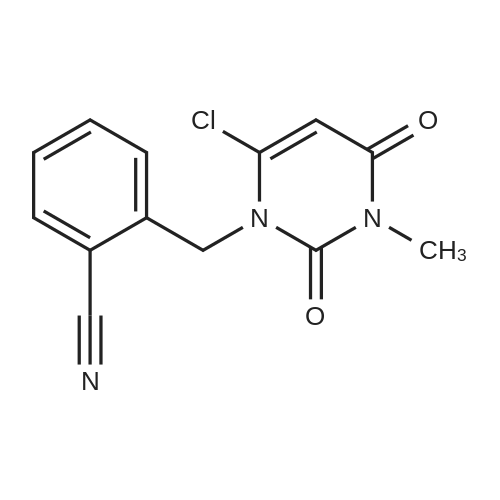
1.2; 2.2; 3.2 Step 2: Intermediates2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl] Methyl]-4-fluoro-benzonitrilePreparation:
7.25 kg of isopropanol, 2.95 kg of purified water, and 2.35 kg of 2-[(6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1) were sequentially added to a 20 L reactor. 2H)-pyrimidinyl)methyl]-4-fluorobenzonitrile, 1.35 kg of (R)-3-aminopiperidine dihydrochloride and 2.86 kg of potassium carbonate; the stirring and heating device was turned on, and the temperature was raised to 60-70 by heating. °C, the reaction is 8-9 hours;Reaction endpoint control method:Determination of 2-[(6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl)methyl]-4-fluoro by high performance liquid chromatography The benzonitrile content is less than 5%, which is regarded as the completion of the reaction;The temperature was lowered to 30-35 ° C, and isopropanol was distilled off under reduced pressure, and 7.0 kg of dichloromethane was entrained once; after the distillation was completed, it was transferred to a 50 L reactor, and the temperature was lowered to 20-25 ° C, and 10.75 kg of purified water was added. 22.2 kg of dichloromethane; vigorously stirred, and allowed to stand for liquid separation; washed with 10.75 kg of purified water for 3 times;To the dichloromethane phase, 14.5 kg of ethanol was added, and the mixture was distilled under normal pressure to a steam temperature of 75-77 ° C; the temperature was lowered to 20-25 ° C, stirred for 2-3 hours, and the filter cake obtained by filtration was washed with 0.55 kg of ethanol. And then drained to get a wet product;The wet product is added to 10.75 kg of ethanol, heated to reflux state, and beaten for 2-3 hours; the temperature is lowered to 20-25 ° C, stirred for 2-3 hours, and the filter cake obtained by filtration is washed twice with 1.3 kg of ethanol. Then drained; dried at 40-45 ° C under reduced pressure, dried to a loss on drying ≤ 0.5%, to obtain a white to off-white powder solid, yield 2.55 kg, yield 79.8%,The intermediate 2-[[6-[(3R)-3-amino-1-piperidinyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)- Pyrimidinyl]methyl]-4-fluoro-benzonitrile; |
| 78.72% |
With potassium carbonate In water monomer; isopropanol at 55℃; for 12h; |
7 Example 7
5.0 g of 2 - [(6-chloro-3,4-dihydro-3-methyl-2,4-dioxo-1 (2H) -pyrimidinyl) methyl]-4-fluorobenzonitrile(Compound III, R1 is F), 3.09 g of (R) -3-aminopiperidine dihydrochloride and 10.40 g of potassium carbonate were dissolved in a mixed solution of 50 ml of isopropanol and 10 ml of water, the temperature was raised to 55 ° C., and the mixture was stirred for 12 h . The reaction was cooled to room temperature and slowly mixed dropwise with 60ml of a mixed solution of acetonitrile and heptane. The mixture was stirred for 1 hour while dripping in an ice bath and filtered with suction to obtain 5.41g of crude compound V with a yield of 88.91% and a purity of 96.50%. The crude compound was recrystallized from ethanol, filtered, washed and dried to give 4.79 g of pure compound V. The purification yield was 88.54%. The total yield of compound V 78.72%, purity 99.80%. |
| 76.9% |
With anhydrous sodium carbonate; potassium iodide In water monomer; isopropanol at 65℃; for 20h; |
2.2
(2) Take another reaction bottle and add 940 ml of isopropanol in sequence.135.7 g of 2-[(6-chloro-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H))methyl]-4-fluoro prepared in the step (1) Benzonitrile,155.2 g of sodium carbonate, 86.7 g of (R)-3-aminopiperidine dihydrochloride,Mix 6.2g potassium iodide and 7.5ml pure water,Heating to 65 ° C for 20 h, stopping the reaction;The filter cake was obtained by hot filtration at 60 ° C.Then wash the filter cake 3 times with 80 ° C isopropanol,The amount of isopropanol used was 115 ml each time, and the mixture was steamed at 45 ° C after the completion of washing.Obtain a crude product; use ethanol,The crude solvent is recrystallized twice by a mixed solvent of petroleum ether and ethyl acetate.The amount of the mixed solvent used each time is 700 ml.After the crystallization is completed, it is cooled to room temperature for suction filtration, and after suction filtration, ethanol is used.Washing the mixed solvent of petroleum ether and ethyl acetate 3 times,The amount of the mixed solvent used per time is 120 ml, in a vacuum drying oven,Dry to constant weight at 55 ° C,2-({6-[(3R)-3-Amino-1-piperidin-1-yl]-3-methyl-2,4-dioxo-3,4-dihydro-1 (2H) )-yl}methyl)-4-fluorobenzonitrile 126.9g,The yield was 76.9%, and the purity was 98.7% as determined by liquid chromatography.The results are shown in Figures 5-1 and 5-2. |
| 72% |
Stage #1: (3R)-piperidin-3-amine dihydrochloride With potassium carbonate In water monomer; isopropanol at 20℃; for 0.5h;
Stage #2: 2-((6-chloro-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-4-fluorobenzonitrile at 65℃; for 8h;
Stage #3: In water monomer; isopropanol |
1.3 step 3:Synthesis of Troglitazone(2-[[6-[(3R)-3-amino-1-Piperidinyl]-3,4-dihydro-Methyl-2,4-dioxo-1(2H) -pyrimidinyl] methyl]-4-Fluorobenzonitrile)
R) -3-aminopiperidine dihydrochloride 17.3g(0.10mol) , potassium carbonate48.4g(0.35mol) and 7ml of Distilled water added into 150ml of Isopropanol.Stirred at room temperature for 30min, the intermediate -2(2-[(6-Chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-Pyrimidinyl)methyl]-4-Fluorobenzonitrile) 29.3g(0.10mol) added into thereaction solution , raised temperature to 65 ° C , keep reaction for 8h, TLC detection ofintermediate-2 when reaction iscompleted, hot Suction filtration, The filter cake was washed with hotisopropanol (60 ° C) (30 mL X 3), The filtrates were combined, washed , vacuumevaporated to obtain a red solid. Added 95% of ethanol 120ml and after heatedto dissolve added 1g of activated carbon, Reflux bleaching for 20min, Hotfiltration, at room temperature stirring crystallization for 1h, suctionfiltration, drying at 45 ° C for 6h to obtain the final product, 25.73g, 72% yield, purity99.6%. |
| 72.7% |
Stage #1: (3R)-piperidin-3-amine dihydrochloride With potassium carbonate In water monomer; isopropanol at 20℃; for 0.5h;
Stage #2: 2-((6-chloro-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-4-fluorobenzonitrile In water monomer; isopropanol at 45℃; for 6h; |
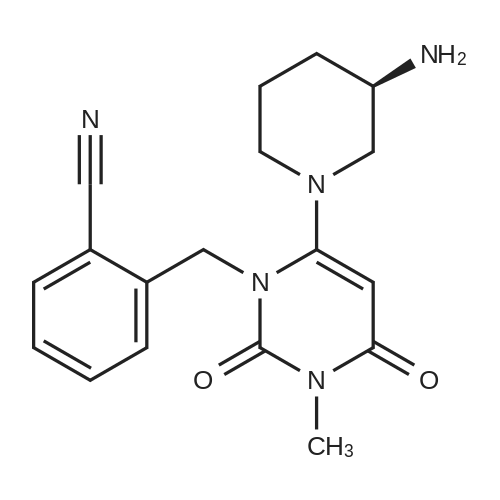
4 Preparation of (R)-2-((6-(3-aminopiperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydropyrimidine-1(2H)-yl)methyl)-4-fluorobenzonitrile
(R)-3-aminopiperidine dihydrochloride (27.03 g, 0.156 mol), potassium carbonate (24.9 g, 1.8 mol), 1.5 mL of water And isopropyl alcohol 360mL. Stir at room temperature for 0.5 h and add 2-((6-chloro-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidine-1-Methyl-4-fluorobenzonitrile (49.3g, 0.168mol), warmed to 45°C, reacted for 6h, decreased to room temperature, added acetonitrile 90mL, pumped After filtration, the filter cake was washed with acetonitrile (60 mL×3). The combined filtrates were evaporated to dryness under reduced pressure to give a crude product as a red solid, reconstituted with 150 mL of methanol. Crystalline, white solid, trodislip 26.52g, yield 72.7%, |
| 69.4% |
With tripotassium phosphate tribasic; tetra-n-propylammonium hydrogensulfate In ethanol at 50 - 60℃; |
4
trelagliptin Intermediate II 40 g was added to 631 g of ethanol,Further, 30.7 g of trelagliptin intermediate III,115 g of potassium phosphate and 200 mg of tetra-n-propylammonium hydrogen sulfate,And heated to 50 to 60 ° C for 2 hours to 3 hours.After concentration under reduced pressure, 200 mL of methylene chloride and 200 mL of water were added to separate the layers,The organic phase was washed three times.The aqueous phase was extracted once more with 100 mL of dichloromethane.The organic phases were combined,10 g of anhydrous magnesium sulfate was added to stand for 2 hours,filter,Concentration of reduced pressure after trelagliptin crude 42g.The whole batch of crude trelagliptin was added to 240 mL of methanol,Heating to 60 ° C to 65 ° C stirring until the clarification,Cooling to 20 DEG C to 25 DEG C and stirring for 1 hour to 2 hours,filter,(33.8 g) was obtained by washing twice with 25 mL of methanol cooled to 5 ° C to 10 ° C and vacuuming at a temperature of 45 ° C to 55 ° C (-0.01 MPa to -0.1 MPa) for 8 hours to 10 hours to obtain 33.8 g of the purified trelagliptin product,The yield was 69.4%HPLC purity 99.91%.The chiral isomer was not detected by chiral HPLC. |
| 66% |
With anhydrous sodium carbonate In water monomer; isopropanol at 70℃; for 18h; Large scale; |
2 Example 2: Preparation of Intermediate MII
Add 260.0L of isopropanol and 36.4L of purified water to the 800L reactor, turn on the stirring, and add intermediate MI in sequence26.0kg, (R)-3-aminopiperidine dihydrochloride 22.9kg and anhydrous sodium carbonate 32.8kg. The mixture is heated to70 ± 5°C, keep the temperature for about 18 hours, monitor the reaction by TLC (dichloromethane: methanol = 20:1), when the intermediate MI is basicallyComplete consumption is the end of the reaction.[0056] After the reaction is complete, add 260.0L of ethyl acetate to the reaction kettle, cool to 20±5°C, stir for 0.5~1h, separateHeart, collect the filtrate, and filter cake for later use. Put the filtrate into an 800L reactor, slowly add an appropriate amount of concentrated hydrochloric acid under stirring, and adjust the pHTo 3~5, a solid precipitated. Stir for 0.5h, centrifuge, and collect the solid. Add 260.0L of solid and purified water to the 800L reactorAfter stirring and dispersing evenly, add 130.0L of dichloromethane and the filter cake left for use in the previous step, and then use 10% NaOH solution toAdjust the pH to 8~10, stand still and separate into the organic phase, and then extract the aqueous phase with 78.0L of dichloromethane, separate the organic phase, and combine the two extractions.Take the organic phase.[0057] The combined organic phase was added to the 800L reactor, and an appropriate amount of concentrated hydrochloric acid was slowly added again under stirring, and the pHAdjust to 3~5, and precipitate a solid. Stir for 0.5h, centrifuge, collect the filter cake, add 260.0L of filter cake and purified water to the 800L reactorAdd 130.0L of dichloromethane after stirring and evenly dispersing. Use 10% NaOH solution to adjust the pH to 8~10, and let it separateThe organic phase and the aqueous phase were extracted with 78.0 L of dichloromethane, the organic phase was separated, and the organic phases extracted twice were combined.[0058] The filtrate was transferred to a 200L concentration reactor, concentrated under reduced pressure, and most of the dichloromethane was concentrated before adding isopropanol130.0L, a solid precipitated, and continued to concentrate until the dichloromethane was almost completely evaporated. Cool down to 20±5°C, and put the resulting suspension inBeat at this temperature for 2 hours, centrifuge, rinse the filter cake twice with isopropanol (26.0LX2), centrifuge, collect the filter cake, and drum at 50±5CDrying with wind, the compound of formula MII (20.94kg; yield: 66%, purity: 99.7%) is obtained |
| 31% |
With Sodium hydrogenocarbonate In methanol at 100℃; for 3h; Sealed tube; Molecular sieve; |
1.3 Step 3: Synthesis of (R)-2-[6-(3-aminopiperidin-1-yl)-3-methyl-2,4-oxo-3,4-dihydro-1(2H)-ylmethyl]-4-fluorophenylcyanide
To 100mL sealed tube was added successively 2- (3-methyl-6-chloro-2,4-dioxo-3,4-dihydro-pyrimidin -1 (2H) - yl methyl) fluorobenzamide carbonitrile (1.3 g, 4.426mM, 1.0equiv), (R) -3- aminopiperidine dihydrochloride (1.149g, 6.64mM, 1.5equiv), sodium bicarbonate (2g, 23.9mM, 5.4equiv), activated molecular sieves (1g,) and dry methanol (20mL), heated at 100 3h. The mixture was cooled to room temperature, filtered through celite, washed with methanol, the filtrate was concentrated. Column chromatography (dichloromethane: methanol = 20: 1) to give a white solid (490mg, 31% yield). |
|
With potassium carbonate In water monomer; isopropanol at 60℃; Inert atmosphere; |
Alternatively, the free base of 34 was prepared as follows. A mixture of 33 (1212 g), IPA (10.8 L), (R)-3-amino-piperidine dihydrochloride (785 g), purified water (78 mL) and potassium carbonate (2.5 kg, powder, 325 mesh) was heated at 60° C. until completion (e.g., for >20 hours) as determined, for example, by HPLC. Acetonitrile (3.6 L) was then added at 60° C. and the mixture was allowed to cool to <25° C. The resultant slurry was filtered under vacuum and the filter cake was washed with acetonitrile (2×3.6 L). The filtrate was concentrated at 45° C. under vacuum (for >3 hours) to afford 2.6 kg of the free base of 34. |
|
With potassium carbonate In water monomer; isopropanol at 60℃; for 5h; |
2 Second Embodiment
The formula (2) 29.4g (0.1mol) and (R) -3- amino - piperidine dihydrochloride 19.0g (0.11mol) was added to a 500mL three-neck flask, was added potassium carbonate 51.1g (0.37mol), 1.9mL of water and isopropanol 260mL, stirred and heated to 60 , the reaction at that temperature for 5 hours.Heating was stopped and acetonitrile 87mL, cooled to room temperature with stirring.Filter cake were washed twice with 87mL acetonitrile, the filtrate and washings were combined, 45 under reduced pressure by rotary evaporation to dryness, to give a pale yellow solid of formula (3) 43.2 g of a crude product; pale yellow solid of formula (3) crude recrystallization, follow these steps:D1: First 238ml ethanol was heated to reflux until a pale yellow solid (3) was added all crude dissolved active carbon 3.1g, heated at reflux, filtered hot;D2: The filtrate was allowed to stand slowly cooled to room temperature and allowed to stand for 2 hours, and then continue to cool to 2 ~ 5 , Paul was allowed to stand overnight to complete crystallization temperature;D3: filter cake was washed with ethanol and once with beating, filtered, drained, crystal at 45 vacuum dried to constant weight to give the formula (3) ethanol solvate as white crystals 38.8 g, total yield 89.7%, purity 99.84 %, . |
|
With triethylamine In 1,4-dioxane at 70℃; for 3h; |
2 Example 2
In the reaction flask by adding 2 - (6-chloro-3-methyl -2,4-dioxo -3,4-dihydropyrimidine -1 (2H)-methyl) - 4-fluorobenzonitrile (29.3g, 0 . 1mol), dioxane (200 ml), respectively added under stirring (R) - 3-amino-piperidine dihydrochloride (20.7g, 0 . 12mol), triethylamine (30.3g, 0 . 3mol). In the 70 degree Celcius stirring for 3 hours, LC-Ms detection reaction is complete, the reaction cooling to room temperature, poured into a 500 ml ice water, 5-10 degree Celcius stirring 2 hours, filtration, solid product is collected. |
| 502 g |
Stage #1: (3R)-piperidin-3-amine dihydrochloride; 2-((6-chloro-3-methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)methyl)-4-fluorobenzonitrile With potassium carbonate In acetonitrile
Stage #2: With hydrogenchloride In dichloromethane; water monomer at 20 - 30℃; for 1h; |
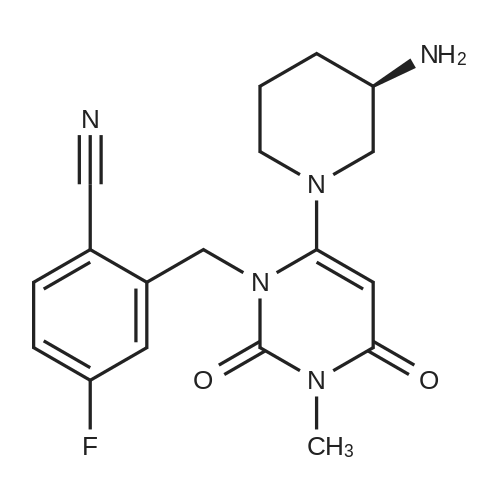
1.2 preparation of 2-[[6-[(3R)-3-amino-1-piperidyl]-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidyl]methyl]-4-fluoro-benzonitrile (I, Trelagliptin)
684 g of 2 - [(6-chloro-3,4-dihydro-3-methyl-2,4-dioxo- 1 (2H) - pyrimidyl) methyl] -4-fluoro-benzonitrile (IV)441.8 g of (R) -3-amino-piperidine dihydrochloride (III, obtained as a commercial product), 6150 ml of isopropanol,1420 g of potassium carbonate and 45 ml of water were mixed and reacted by stirring at 55 to 60 ° C .; after completion of the reaction was monitored by TLC,2000 ml of acetonitrile was added, cooled to room temperature,After stirring for about 3 hours, filtration, washing the filter cake with 2,000 ml of acetonitrile,It was combined with the filtrate; the filtrate was concentrated under reduced pressure at 45-50 ° C. The obtained concentrate was dissolved in 7500 ml of dichloromethane,A 2 mol / L hydrochloric acid aqueous solution was added dropwise at 20 to 30 ° C. so that the pH was about 3, after completion of dropwise addition of hydrochloric acid,Subsequently, the mixture was stirred for about 1 hour, then filtered, and the filter cake was washed successively with 4000 ml of dichloromethane and 2750 ml of tetrahydrofuran,And dried under reduced pressure at 45 to 50 ° C. The solid obtained by drying was added to a mixture consisting of 4500 ml of dichloromethane and 9000 ml of water,The aqueous phase was extracted with 3700 ml of dichloromethane and the organic phase was combined; the organic phase was washed successively with 3700 ml of water × 2, Dried over anhydrous sodium sulfate,Concentrated under reduced pressure to obtain a white solid; and the obtained solid was dried under reduced pressure at 45 to 50 ° C. to obtain 502 g of tolaglistin (I). |
|
With anhydrous sodium carbonate In water monomer; isopropanol at 70℃; for 16h; |
1.2
(2) adding isopropanol and purified water to the reaction vessel at a volume ratio of 10:1.4, and stirring was started.Then, the intermediate 1, (R)-3-aminopiperidine dihydrochloride and anhydrous sodium carbonate were successively added, and the mixture was heated to 70 ° C under stirring, and the reaction was kept for 16 h, and TLC (dichloromethane: methanol = 20: 1) Monitor the reaction. When the intermediate 1 is consumed as the end of the reaction, add ethyl acetate after the reaction is completed, cool to 20 ° C, stir for 0.5-1 h, and filter to obtain the filtrate and filter cake, and add to the filtrate.Concentrated hydrochloric acid was added to make the filtrate pH 3-4, stirred for 0.5 h, filtered, and the solid was collected. The solid and purified water were uniformly mixed, dichloromethane and filter cake were added, mixed again, and mixed with 10 wt% NaOH solution. The pH of the solution is 9-10, the organic phase is separated by standing, the aqueous phase is extracted again with dichloromethane, the organic phase is separated, and the extracted organic phase is combined; Add concentrated hydrochloric acid to the combined organic phase, adjust the pH of the solution to 3-4, stir for 0.5 h, filter, solid and pureThe water was mixed well, dichloromethane was added, and the pH of the mixed solution was adjusted to 9-10 with a 10 wt% NaOH solution.The organic phase, the aqueous phase is again extracted with dichloromethane, the organic phase is separated, the extracted organic phase is combined, and then the saturated salt is used.Wash the organic phase with water, let stand, separate the organic phase, dry with anhydrous Na2SO4, filter, and wash the filter cake with dichloromethane.The filtrate was concentrated under reduced pressure. Most of the dichloromethane was evaporated, then isopropyl alcohol was added and crystallised until the methylene chloride evaporated.After cooling to 20 ° C, the resulting suspension was beaten at this temperature for 2 h, filtered, and the filter cake was washed with isopropyl alcohol to collect the filter cake at 50Drying at ° C to give a white solid intermediate 2; wherein, intermediate 1, (R)-3-aminopiperidine dihydrochloride and anhydrous sodium carbonateThe molar ratio is 1:1.5:3.5; |
|
With potassium carbonate In water monomer; isopropanol at 66℃; for 10h; |
1.3.b; 1.3.c; 2.3.b; 2.3.c; 3.3.b; 3.3.c; 4.3.b; 4.3.c
(b) (R)-3-aminopiperidine dihydrochloride and potassium carbonate are added together in a molar ratio of 1:3.5 to a mixed solvent to obtain a mixed reaction solution; wherein the mixed solvent is made up of isopropyl alcohol and water in a volume ratio Mixed for 18 to 20:1;(c) 2-[(6-Chloro-3,4-dihydro-3-methyl-2,4-dioxo-1(2H)-pyrimidinyl)methyl]-4-fluorobenzonitrile and (R) molar ratio of 3-amino-piperidine dihydrochloride is 1:1.24 the step (a) to give 2 - [(6-chloro-3,4-dihydro-3-methyl-2, 4- dioxo -1 (2H) - pyrimidinyl) methyl] -4-fluorobenzonitrile was added in step (b) mixing the reaction mixture, followed by reaction at 66 °C for 10h, to give the final product |
|
In toluene at 40 - 50℃; for 2h; |
1.2 Step 2: Preparation of troxagliptin
Add 113.2 g of (R)-3-aminopiperidine dihydrochloride (1.05 equivalent) to the reaction flask in the first step, stir the reaction at 40-50° C., detect by TLC, and complete the reaction in 2 hours.Filter and evaporate the solvent of the filtrate to obtain troxagliptin. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping