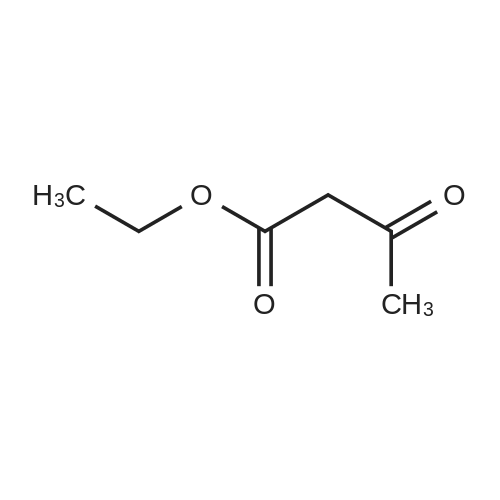
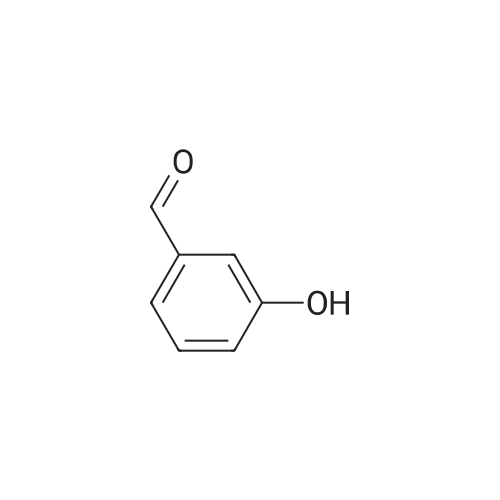
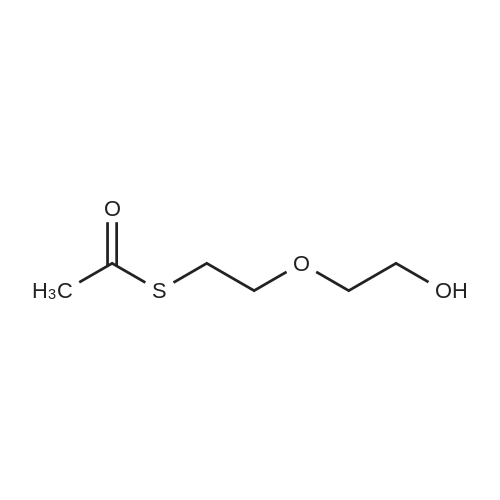
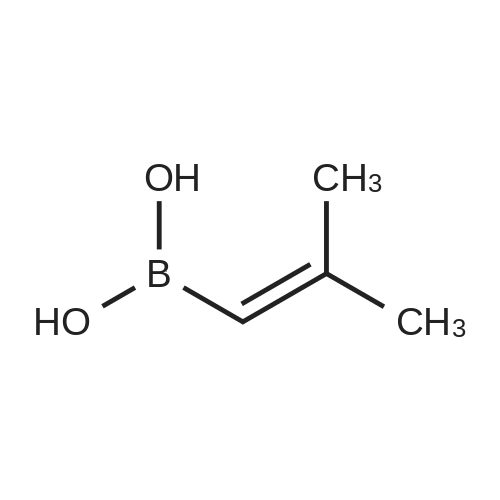
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With sodium hydrogensulfate monohydrate In hexane; acetonitrile for 0.5 h; Reflux; Sonication; Green chemistry
General procedure: To a solution of an aldehyde (1 mmol), ethyl/methyl acetoacetate or acetylacetone(1 mmol) and urea/thiourea (1.2 mmol) in n-hexane/CH3CN (2.5:0.5 mL), NaHSO4·H2O(3 mmol) was added. The reaction mixture was then heated to reflux and synchronously irradiated by ultrasound (240 W) via a micro-tip probe for an appropriate time. Progress of the reactions was monitored by TLC (n-hexane/EtOAc: 5/3). After completion of the reaction, the solution was cooled to room temperature and H2O (5 mL) was then added followed by stirring for 5 min. The mixture was extracted with EtOAc (3 × 8 mL) anddried over anhydrous Na2SO4. Evaporation of the solvent and recrystallization of the crude product in hot MeOH affords the pure DHPM (4–69) in 91–98percent yield (Tables 2 and 3).
96%
With alumina In neat (no solvent) at 100℃; for 0.15 h; Microwave irradiation; Green chemistry
General procedure: A mixture of the aldehyde 1 (2 mmol), the 1,3-dicarbonyl compound 2 (2 mmol), the corresponding urea derivative 3(3 mmol) and the 3D printed Al2O3 structure (0.350 g) was submitted to microwave irradiation (100 °C) in coated vial. After completion of the reaction, as indicated by TLC, the mixture was cooled and the desired compound solidified. For those 1,2,3,4-tetrahydropyrimidine-5-carboxylates that not solidified, the reaction mixture was poured onto crushed ice and stirred for 5-10 min. The solid obtained was filtered under suction, washed with ice-cold water (20 mL) and then purified by column chro-matography or recrystallization from the appropriate solvent.
94%
With Graphite In neat (no solvent) at 120℃; for 0.166667 h; Green chemistry
General procedure: A mixture of benzaldehyde (106mg, 1mmol), urea (60mg, 1mmol), ethylacetoacetate (130mg, 1mmol) and (10mg, 10percentw/w) graphite was heated at 70°C (120°C in case of thiourea). The heterogenous mixture slowly became clear and a solid product started to seperate out. After completion of the reaction (1h, TLC) the entire mass solidified. The solid mass was crushed, washed with 5mL of cold water to remove unreacted urea and filtered.The solid was then dissolved in hot ethanol, and the catalyst was separated by filtration. On cooling the filtrate pure crystals of the product (1a) was obtained, yield 97percent (237mg). In all the cases, the product obtained was characterized by comparing spectral data and melting points with literature data.
90%
With o-phthalimide-N-sulfonic acid In neat (no solvent) at 120℃; for 3 h;
General procedure: To a mixture of aryl aldehyde 4 (1 mmol), β-dicarbonyl 5 (1 mmol), and urea or thiourea 6 (1.2 mmol) was added 10 mol percent of phthalimide-N-sulfonic acid (PISA). The reaction mixture was heated to 120 °C on a heating mantle for the appropriate time. After completion of the reaction, as indicated by TLC analysis, the system was cooled to room temperature. Ethanol (5 mL) was added to the reaction mixture, and the mixture was heated until a homogeneous solution was obtained. Next, ethyl acetate was added to the resulting mixture and then cooled to RT, and the catalyst was recovered by filtration and washed thoroughly with ethyl acetate and then diethyl ether. The recovered catalyst was then reused under the same conditions as above for at least five reactions. After this, the organic phase was concentrated by evaporation, distilled water was added to the residue, and the solid thus obtained.The resulting solid product was filtered off, washed with cold water, and then thereaction mixture was subjected to isolation with preparative TLC using a mixture of ethyl acetate and n-hexane (3:10) as eluent.
90%
at 120℃; for 3 h; Green chemistry
General procedure: A mixture of aryl aldehyde 1 (1 mmol), b-dicarbonyl 2 (1 mmol), urea or thiourea 3 (1.2 mmol), and 10 mol percent of PPI was added and the reaction mixture was heated at 120 C for 1–3 h. During the heating, the progress of the reaction mixture was monitored by TLC analysis. After completion of the reaction, the reaction was cooled to room temperature. The solid product was dissolved in ethanol and water, filtered by simple filtration, and washed with water (5 mL), and the residue was recrystallized from ethanol to give the desired compounds in high yields. The filtrate was evaporated to remove water and leave the catalyst. The same catalyst was employed to synthesize further derivatives. The products are known and their identity was confirmed by comparison of their physical and spectroscopic data with those available in the literature.
88%
With tris(pentafluorophenyl)borate In ethanol for 3 h; Reflux; Green chemistry
General procedure: A mixture of benzaldehyde 1a (106 mg, 1 mmol), ethyl acetoacetate 2a (130 mg, 1 mmol) and urea 3a (90 mg,1.5 mmol) in EtOH (10 mL) was refluxed in the presence of B(C6F5)3 (18.1 mg, 1 molpercent). After completion of reaction, as indicated by TLC analysis, the solvent was evaporated. The resulting mass was treated with ice-cold water and the solid obtained was filtered, washed with cold water, dried and re-crystallized from ethanol to give pure product (4a).
87%
With molybdophosphoric acid supported on Y zeolite In acetonitrile for 7 h; Reflux
Method A. A mixture of 1 (122 mg, 1.0 mmol), ethylacetoacetate (130 mg, 1.0 mmol), and thiourea (190 mg,2.5 mmol) in MeCN (15 ml) was mixed with HPA supportedon Y zeolite (8 wtpercent NaY + 0.5 mM HPA) and refluxed for7 h. After cooling, the heteropoly acid (HPA) supported onHY filtered off and washed with hot water and ethanol toremove thiourea from the surface of the catalyst. Then, thecatalyst dried and was maintained for new runs. The filtratewas evaporated to dryness and the residue was recrystallized from EtOH to afford 2 (254 mg, 87 percent), m.p. 181–185 °C (Lit.[6, 7] 184–186 °C), Rf = 0.71. 1H NMR (DMSO-d6): δ 10.29(s, 1 H, NH), 9.60 (s, 1 H, NH), 9.44 (s, 1 H, OH), 7.10 (t,J = 7.8 Hz, H-5), 6.65 (d, 1 H, J2’,4′ = 2.4Hz, Harom.-2′), 6.64(m, 2 H, Harom.-4 + Harom.-6), 5.09 (d, 1 H, JNH,4 = 5.5 Hz,H-4), 4.02 (q, 2 H, J = 7.8Hz, CH2CH3), 2.28 (s, 3 H, C6-Me),1.12 (t, 3 H, J = 7.8 Hz, CH2CH3). 13C NMR (DMSO-d6): δ174.6 (C = S), 165.7 (CO2Et), 157.9 (C-6), 145.3 (C3’-OH + Carom.-1′), 130.0 (Carom.-5′), 117.7 (Carom.-2′ +Carom.-6′), 133.6 Carom.-4′), 101.2 (C-5), 60.1 (CH2CH3),54.5 (C-4), 17.7, 17.6 (C6-Me), 14.5 (CH2CH3). EI-MS:m/z (percent) = 292 [M]+. Anal. Calcd. for C14H16N2O3S(292.35): C, 57.52; H, 5.52; N, 9.58. Found: C, 57.32;H, 5.39; N, 9.32 percent. Method B. A mixture of 1 (244 mg, 2.0 mmol), ethylacetoacetate (300 mg, 2.30 mmol), and thiourea (380 mg,5.0 mmol) in EtOH (15 ml) in the presence conc. Hydrochloricacid (1 ml) was heated under reflux for 3 h. After cooling,the mixture was poured onto ice (20 g). The precipitate wasfiltered and dried and recrystallized from EtOH to give 2(292 mg, 50 percent), TheNMR spectra, m.p. and mixed m.p. werealmost similar for those of 2 prepared in method A.
86%
for 4 h; Reflux
General procedure: A mixture of 0.025 mol of urea or thiourea, 0.025 mol of an arylaldehyde, 0.025 mol of ethyl acetoacetate or acetylacetone, 10 mL ethanol and 3 drops of a liquid or 2-3 mg of solid phenol was heated under reflux for 6-24 h and the reaction mixture was then cooled to 0 °C and the product was filtered, washed with water, dried and recrystallized from a suitable solvent. The progress of the reaction was monitored by TLC using ethyl acetate and n-hexane (1:2) as eluent.
85%
With chloro-trimethyl-silane In acetonitrile for 24 h; Inert atmosphere; Reflux
General procedure: A 10 mmol mixture of the corresponding benzaldehyde (4),15 mmol of methyl or ethyl acetoacetate (5), 761 mg (10 mmol,1.0 equiv) of thiourea (6), 1.27 mL (10 mmol, 1.0 equiv) oftrimethylsilylchloride (TMSCl) and 7.0 mL of anhydrous acetonitrileunder nitrogen atmosphere was formulated in a 50 mL roundbottom flask, equipped with a magnetic stirring bar. The watercondenser was set at the neck of round bottom flask then the mixturewas stirred for 1 min at room temperature to allow homogenization,followed by reflux for 24 h at oil bath. After cooling thereaction mixture, solvent was evaporated under reduced pressurethrough rotary evaporator. Thereafter, 500 mL of ice water waspoured into reaction mixture and allowed to stir for 1 h in orderto obtain maximum precipitation. Filtration delivered the pureDHPMs. Where needed, the product was recrystallized or directlypurified by gradient dry flash chromatography using appropriatesolvents. The physical and spectroscopic data of DHPM 7–12 werefound in good agreement with the literature [11,13,20]. 4.2.1.1.
83%
With psychortria douarrei catalyst dispersed on montmorillonite K10 In neat (no solvent) at 80℃; for 1.2 h;
A mixture of ethyl acetoacetate (781 mg, 6.Ommol),3-hydrobenzaldehyde (488 mg, 4.0 mmol), thiourea (457 mg,6.0 mmol) and P douarrei crude catalyst (265 mg, amount corresponding to 1.0 mmol of nickel following previous dosing), supported on montmorillonite K10 (265 mg) was placed in a 10 mE sealed tube. The tube was heated to 80° C. in oil bath, under magnetic stirring for 1.2 h. The mixture was then extracted with hot ethanol (10 mE, 70° C.) and filtered in order to remove the catalyst, which was reactivated by heating (150°C.). The solutionwas poured into crushed ice (20 g) and stirred for 20 mm. The solid separated was filtered under suction, washed with cold water (30 mE) and recrystallized from hot ethanol, affording pure product, as colorless crystals (973 mg, 83percent). The same procedure was followed with G. pruinosa catalyst and commercial NiCl2. Mp 185-186° C. (184-186°C.);IR 3298,3181,3115,2982, 1663, 1617, 1573 cm’; ‘H NMR (DMSO-d5, 300 MHz) ö: 1.14 (t, J=7.4 Hz, 3H), 2.29 (s, 3H), 4.04 (q, J=7.4 Hz, 2H), 5.11 (d, J=3.5 Hz, 1H), 6.60-6.71 (m, 3H), 7.06-7.15 (m, 1H), 9.42 (brs, 1H),9.62 (brs, 1H), 10.29 (brs, 1H); ‘3C NMR (DMSO-d5, 75 MHz) ö: 14.0, 17.1, 54.2, 59.6, 100.8, 113.0, 114.4, 117.0, 129.3, 144.8, 144.9, 157.4, 165.4, 174.2. MS (EI+) calculated for C,4H,6N2035 [M]292.1. found 293.1 [M+H].
80%
With bis(p-sulfoanilino)triazine-functionalized silica-coated magnetite nanoparticles In neat (no solvent) at 100℃; for 1.16667 h;
General procedure: A mixture of aromatic aldehyde (1 mmol), b-keto ester or dimedone (1 mmol) and urea or thiourea (1.2 mmol) was stirred in presence MNPs-BSAT (20 mg) at 100 °C under solvent-free condition for the appropriate time (Scheme 1). After completion of the reaction as indicated by TLC (using n-hexane-ethyl acetate as eluent), the resulting mixture was diluted with hot ethanol (15 mL) and the catalyst separated by an external magnet and washed with hot ethanol (5 mL) two times. The filtrate was cooled to room temperature and the crude products which precipitated were collected and recrystallized from ethanol if necessary.
79%
With tin(II) chloride dihdyrate In acetonitrile at 70 - 75℃; Sonication
General procedure: A mixture of an aldehyde (10 mmol), a diamino compound (12 mmol), a dicarbonyl compound (10 mmol, mL), SnCl2·2H2O (10 molpercent) and acetonitrile (10 mL) was mixed in a pyrex tube. The mixture was then irradiated in ultrasonic bath at 70–75 °C. The reaction was monitored by TLC. After the completion of the reaction, the resulting precipitate was filtered and crude product was recrystallized from an appropriate solvent or purified through columnchromatography.
75%
With ytterbium(III) triflate In acetonitrile at 120℃; for 1 h; Microwave irradiation
General procedure: The appropriate β-keto ester (0.75–3.0mmol, 1.5 equiv.), the appropriate aldehyde (0.5–2.0mmol, 1 equiv.), the appropriate urea or thiourea (0.5–2.0mmol, 1 equiv.) and Yb(OTf)3 (0.05–0.20mmol, 0.10 equiv.) were placed in a microwave oven vial (0.5–2.0mL) with a magnetic stirring bar and dissolved in THF or MeCN (0.5–1.0mL). The resulting mixture is heated to 120°C for 30–60min using microwave irradiation. The resulting mixture is poured onto an ice-water mixture and left for precipitation. The resulting crude solids were purified by recrystallization, DCVC or flash chromatography.
74%
With copper dichloride In ethanol at 80℃; for 4 h;
General procedure: To a stirred mixture of thiourea (1.00 mmol), substituted benzaldehydes E–G (Scheme 1)(1.00 mmol), ethyl acetoacetate (130 mg, 1.00 mmol), and anhydrous cupric chloride(10 molpercent) were added. The mixture was heated at 80°C for 4 h under stirring. After thereaction was completed (checked by TLC), a mixture of H2O:EtOH 8:5 (13 mL) was added andthe resulting slurry was stirred at 80°C until total dissolution. After being cooled to roomtemperature, the reaction mixture was poured onto crushed ice (30 g) and stirred for5–10 min. The separated solid was filtered under suction (water aspirator), washed withice-cold water (50 mL), and then recrystallized from hot ethanol to afford the pureproduct.2.3. Ethyl-4-(3-hydroxyphenyl)-6-methyl-2-thioxo-pyrimidine-5-carboxylate(monastrol) L1))From 3-hydroxybenzaldehyde (E) (122 mg). Yield: 216 mg (74percent); mp 185–186°C (Lit. [23, 25]184–186°C).
72%
With 1-butyl-1,2,4-triazolium triflate In ethanol at 80℃; for 1 h; Green chemistry
General procedure: Catalyst 1a-c (10 molpercent) was added to a solution of aldehyde (1.0 mmol), β-ketoester (1.5 mmol) and urea or thiourea (2.0 mmol) in ethanol (0.5 mL). The reaction mixture was heated at 80 °C using oil bath for the specified time (0–5 h). The progress of the reaction was monitored by TLC. After completion, the reaction mixture was cooled to room temperature and subsequently quenched with a mixture of water:ethanol (5:0.5 mL). The solid product was filtered and washed with n-hexane (5mL ×2), which afforded pure 3,4-dihydropyrimidin-2(1H)-ones or 3,4-dihydropyrimidin-2(1H)-thiones in pure form.
71%
at 100℃; for 3 h;
General procedure: Aldehyde (3 mmol), ethyl acetoacetate (0.586 g, 4.5 mmol), urea (0.270 g, 4.5 mmol) or thiourea (0.343 g, 4.5 mmol), and β-cyclodextrin (17 mg, 0.5 molpercent) were mixed in a 25 mL round bottom flask for 3h at a temperature of 100 °C. The reaction mixture was cooled and the solid was solubilized in ethanol and the addition of few drops of cold water to precipitate the product. The precipitated solid was filtered on sintered funnel. The crude product was further purified by recrystallization from ethanol to afford pure 3,4-dihydropyrimidin-2(1H)-ones. After separation of the product by filtration, the filtrate was washed with ethyl acetate and the aqueous phase was concentrated on rotary evaporator obtaining the catalyst.
64%
With 1-ethyl-1,2,4-triazolium phenylsulfonate In ethanol at 70 - 80℃; for 2.5 h;
General procedure: Catalyst 1-ethyl-1,2,4-triazolium phenylsulfonate (TrHEtPS) (10 mol) was added to a solution of urea or thiourea (2.0 mmol), β-ketoester (1.5 mmol) and aldehyde (1.0 mmol) in ethanol (0.5 mL). The reaction mixture was heated at 70 to 80 °C for the specified time (0-5 h) (Scheme-II). The completion of the reaction was monitored by thin layer chromatography (ethyl acetate:hexane: :4:1) and then the reaction mixture was cooled to room temperature. Then, quenched with mixture of water: ethanol (5:0.5 mL). The obtained solid product was filtered, and washed with n-hexane (5mL) and afforded 3,4-dihydropyrimidin-2(1H)-ones or 3,4-dihydropyrimidin-2(1H)-thiones.
52%
With 25,26,27,28-terahydroxycalix[4]arene-5,11,7,23-tetrasulfonic acid In ethanol for 8 h; Reflux
General procedure: Aldehydes (3 mmol), ethyl acetoacetate (4.5 mmol) and (thio)urea (4.5 mmol) were dissolved in 3 mL of ethanol containing p-sulfonic acid calix[4]arene (0.5 mol percent). The mixture was heated under reflux and stirred for 8 h.20 All DHPMs were characterized by NMR (1H and 13C), infrared, melting point and elemental analysis. Characterization data for compounds BA9, BA11-BA14, BA16-BA21, BA24-BA28, BA30 and BA32 were recently reported by da Silva et al.20 Data for compounds BA2, BA10, BA15, BA22, BA29, BA31 and BA33 are listed as Supplementary data.
Reference:
[1] Synthetic Communications, 2007, vol. 37, # 22, p. 3907 - 3916
[2] ACS Catalysis, 2013, vol. 3, # 7, p. 1420 - 1430
[3] Molecules, 2006, vol. 11, # 8, p. 649 - 654
[4] Phosphorus, Sulfur and Silicon and the Related Elements, 2009, vol. 184, # 9, p. 2465 - 2471
[5] Synthetic Communications, 2013, vol. 43, # 11, p. 1477 - 1483
[6] Phosphorus, Sulfur and Silicon and the Related Elements, 2013, vol. 188, # 11, p. 1634 - 1642
[7] Applied Catalysis A: General, 2017, vol. 530, p. 203 - 210
[8] Tetrahedron Letters, 2002, vol. 43, # 34, p. 5913 - 5916
[9] Synlett, 2004, # 2, p. 279 - 282
[10] Phosphorus, Sulfur and Silicon and the Related Elements, 2012, vol. 187, # 4, p. 544 - 553
[11] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2005, vol. 44, # 4, p. 823 - 826
[12] Heterocyclic Communications, 2006, vol. 12, # 1, p. 61 - 66
[13] Synthetic Communications, 2009, vol. 39, # 5, p. 880 - 886
[14] Beilstein Journal of Organic Chemistry, 2009, vol. 5,
[15] Bioorganic and Medicinal Chemistry Letters, 2014, vol. 24, # 13, p. 2897 - 2899
[16] Phosphorus, Sulfur and Silicon and the Related Elements, 2009, vol. 184, # 7, p. 1722 - 1728
[17] Chemistry - A European Journal, 2013, vol. 19, # 13, p. 4156 - 4168
[18] Tetrahedron Letters, 2003, vol. 44, # 14, p. 2889 - 2891
[19] Journal of Heterocyclic Chemistry, 2007, vol. 44, # 4, p. 979 - 981
[20] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2007, vol. 46, # 11, p. 1886 - 1889
[21] Asian Journal of Chemistry, 2011, vol. 23, # 9, p. 3993 - 3995
[22] RSC Advances, 2015, vol. 5, # 60, p. 48506 - 48515
[23] RSC Advances, 2014, vol. 4, # 67, p. 35559 - 35567
[24] Heterocycles, 2003, vol. 60, # 11, p. 2435 - 2440
[25] Heterocycles, 2006, vol. 68, # 6, p. 1217 - 1224
[26] Journal of Heterocyclic Chemistry, 2007, vol. 44, # 1, p. 211 - 214
[27] Zeitschrift fur Naturforschung - Section B Journal of Chemical Sciences, 2008, vol. 63, # 2, p. 178 - 182
[28] Research on Chemical Intermediates, 2015, vol. 41, # 9, p. 6635 - 6648
[29] Research on Chemical Intermediates, 2015, vol. 41, # 8, p. 5177 - 5203
[30] Synthesis, 2005, # 11, p. 1748 - 1750
[31] Catalysis Communications, 2011, vol. 15, # 1, p. 123 - 126
[32] Synthetic Communications, 2013, vol. 43, # 1, p. 139 - 146
[33] New Journal of Chemistry, 2016, vol. 40, # 1, p. 838 - 843
[34] Journal of the Chinese Chemical Society, 2007, vol. 54, # 2, p. 263 - 266
[35] Bulletin of the Korean Chemical Society, 2010, vol. 31, # 2, p. 351 - 354
[36] Chinese Journal of Chemistry, 2010, vol. 28, # 3, p. 388 - 392
[37] Journal of the Brazilian Chemical Society, 2011, vol. 22, # 7, p. 1379 - 1388
[38] Journal of Chemical Sciences, 2015, vol. 127, # 6, p. 1047 - 1052
[39] Antiviral Research, 2012, vol. 95, # 2, p. 118 - 127
[40] Catalysis Letters, 2012, vol. 142, # 12, p. 1505 - 1511
[41] Journal of Fluorescence, 2016, vol. 26, # 1, p. 31 - 35
[42] Green Chemistry, 2011, vol. 13, # 4, p. 1009 - 1013
[43] Asian Journal of Chemistry, 2016, vol. 28, # 1, p. 23 - 26
[44] Synthesis, 2011, # 14, p. 2261 - 2267
[45] Asian Journal of Chemistry, 2012, vol. 24, # 5, p. 1906 - 1908
[46] Asian Journal of Chemistry, 2013, vol. 25, # 8, p. 4588 - 4590
[47] Bioorganic Chemistry, 2016, vol. 64, p. 85 - 96
[48] Bulletin of the Korean Chemical Society, 2011, vol. 32, # 2, p. 656 - 658
[49] RSC Advances, 2016, vol. 6, # 112, p. 110928 - 110934
[50] RSC Advances, 2013, vol. 3, # 44, p. 22340 - 22345
[51] ChemCatChem, 2014, vol. 6, # 12, p. 3455 - 3463
[52] Patent: US2015/376224, 2015, A1, . Location in patent: Paragraph 0643; 0660
[53] Tetrahedron, 2008, vol. 64, # 9, p. 2035 - 2041
[54] Tetrahedron, 2009, vol. 65, # 51, p. 10608 - 10611
[55] Journal of Heterocyclic Chemistry, 2010, vol. 47, # 1, p. 136 - 146
[56] Bioorganic Chemistry, 2006, vol. 34, # 4, p. 173 - 182
[57] Journal of Organic Chemistry, 2012, vol. 77, # 22, p. 10184 - 10193
[58] Research on Chemical Intermediates, 2018, vol. 44, # 7, p. 4083 - 4101
[59] Journal of Molecular Graphics and Modelling, 2013, vol. 43, p. 47 - 57
[60] Synthesis, 2004, # 13, p. 2091 - 2093
[61] European Journal of Medicinal Chemistry, 2017, vol. 138, p. 300 - 312
[62] Journal of Coordination Chemistry, 2017, vol. 70, # 12, p. 2061 - 2073
[63] Journal of Chemical Sciences, 2015, vol. 127, # 9, p. 1539 - 1545
[64] Bioorganic and Medicinal Chemistry Letters, 2006, vol. 16, # 9, p. 2463 - 2466
[65] Tetrahedron, 2013, vol. 69, # 38, p. 8245 - 8249
[66] Amino Acids, 2013, vol. 44, # 3, p. 1031 - 1037
[67] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 15, p. 4679 - 4682
[68] Asian Journal of Chemistry, 2018, vol. 30, # 9, p. 1999 - 2002
[69] Tetrahedron, 2000, vol. 56, # 13, p. 1859 - 1862
[70] Bioorganic and Medicinal Chemistry, 2012, vol. 20, # 8, p. 2645 - 2650
[71] Asian Journal of Chemistry, 2010, vol. 22, # 4, p. 2518 - 2528
[72] RSC Advances, 2016, vol. 6, # 107, p. 105087 - 105093
[73] ChemMedChem, 2010, vol. 5, # 4, p. 567 - 574
[74] Synthetic Communications, 2011, vol. 41, # 15, p. 2200 - 2208
[75] ChemMedChem, 2013, vol. 8, # 8, p. 1345 - 1352
[76] Tetrahedron, 2014, vol. 70, # 45, p. 8582 - 8587
[77] European Journal of Medicinal Chemistry, 2016, vol. 115, p. 230 - 244
[78] ChemMedChem, 2017, vol. 12, # 13, p. 1022 - 1032
[79] MedChemComm, 2018, vol. 9, # 8, p. 1282 - 1288
With sodium hydrogensulfate monohydrate; In hexane; acetonitrile; for 0.5h;Reflux; Sonication; Green chemistry;
General procedure: To a solution of an aldehyde (1 mmol), ethyl/methyl acetoacetate or acetylacetone(1 mmol) and urea/thiourea (1.2 mmol) in n-hexane/CH3CN (2.5:0.5 mL), NaHSO4·H2O(3 mmol) was added. The reaction mixture was then heated to reflux and synchronously irradiated by ultrasound (240 W) via a micro-tip probe for an appropriate time. Progress of the reactions was monitored by TLC (n-hexane/EtOAc: 5/3). After completion of the reaction, the solution was cooled to room temperature and H2O (5 mL) was then added followed by stirring for 5 min. The mixture was extracted with EtOAc (3 × 8 mL) anddried over anhydrous Na2SO4. Evaporation of the solvent and recrystallization of the crude product in hot MeOH affords the pure DHPM (4-69) in 91-98% yield (Tables 2 and 3).
96%
With alumina; In neat (no solvent); at 100℃; for 0.15h;Microwave irradiation; Green chemistry;
General procedure: A mixture of the aldehyde 1 (2 mmol), the 1,3-dicarbonyl compound 2 (2 mmol), the corresponding urea derivative 3(3 mmol) and the 3D printed Al2O3 structure (0.350 g) was submitted to microwave irradiation (100 C) in coated vial. After completion of the reaction, as indicated by TLC, the mixture was cooled and the desired compound solidified. For those 1,2,3,4-tetrahydropyrimidine-5-carboxylates that not solidified, the reaction mixture was poured onto crushed ice and stirred for 5-10 min. The solid obtained was filtered under suction, washed with ice-cold water (20 mL) and then purified by column chro-matography or recrystallization from the appropriate solvent.
94%
With Graphite; In neat (no solvent); at 120℃; for 0.166667h;Green chemistry;
General procedure: A mixture of benzaldehyde (106mg, 1mmol), urea (60mg, 1mmol), ethylacetoacetate (130mg, 1mmol) and (10mg, 10%w/w) graphite was heated at 70C (120C in case of thiourea). The heterogenous mixture slowly became clear and a solid product started to seperate out. After completion of the reaction (1h, TLC) the entire mass solidified. The solid mass was crushed, washed with 5mL of cold water to remove unreacted urea and filtered.The solid was then dissolved in hot ethanol, and the catalyst was separated by filtration. On cooling the filtrate pure crystals of the product (1a) was obtained, yield 97% (237mg). In all the cases, the product obtained was characterized by comparing spectral data and melting points with literature data.
90%
With o-phthalimide-N-sulfonic acid; In neat (no solvent); at 120℃; for 3h;
General procedure: To a mixture of aryl aldehyde 4 (1 mmol), beta-dicarbonyl 5 (1 mmol), and urea or thiourea 6 (1.2 mmol) was added 10 mol % of phthalimide-N-sulfonic acid (PISA). The reaction mixture was heated to 120 C on a heating mantle for the appropriate time. After completion of the reaction, as indicated by TLC analysis, the system was cooled to room temperature. Ethanol (5 mL) was added to the reaction mixture, and the mixture was heated until a homogeneous solution was obtained. Next, ethyl acetate was added to the resulting mixture and then cooled to RT, and the catalyst was recovered by filtration and washed thoroughly with ethyl acetate and then diethyl ether. The recovered catalyst was then reused under the same conditions as above for at least five reactions. After this, the organic phase was concentrated by evaporation, distilled water was added to the residue, and the solid thus obtained.The resulting solid product was filtered off, washed with cold water, and then thereaction mixture was subjected to isolation with preparative TLC using a mixture of ethyl acetate and n-hexane (3:10) as eluent.
90%
With potassium phtalimide; at 120℃; for 3h;Green chemistry;
General procedure: A mixture of aryl aldehyde 1 (1 mmol), b-dicarbonyl 2 (1 mmol), urea or thiourea 3 (1.2 mmol), and 10 mol % of PPI was added and the reaction mixture was heated at 120 C for 1-3 h. During the heating, the progress of the reaction mixture was monitored by TLC analysis. After completion of the reaction, the reaction was cooled to room temperature. The solid product was dissolved in ethanol and water, filtered by simple filtration, and washed with water (5 mL), and the residue was recrystallized from ethanol to give the desired compounds in high yields. The filtrate was evaporated to remove water and leave the catalyst. The same catalyst was employed to synthesize further derivatives. The products are known and their identity was confirmed by comparison of their physical and spectroscopic data with those available in the literature.
88%
With tris(pentafluorophenyl)borate; In ethanol; for 3h;Reflux; Green chemistry;
General procedure: A mixture of benzaldehyde 1a (106 mg, 1 mmol), ethyl acetoacetate 2a (130 mg, 1 mmol) and urea 3a (90 mg,1.5 mmol) in EtOH (10 mL) was refluxed in the presence of B(C6F5)3 (18.1 mg, 1 mol%). After completion of reaction, as indicated by TLC analysis, the solvent was evaporated. The resulting mass was treated with ice-cold water and the solid obtained was filtered, washed with cold water, dried and re-crystallized from ethanol to give pure product (4a).
87%
With molybdophosphoric acid supported on Y zeolite; In acetonitrile; for 7h;Reflux;
Method A. A mixture of 1 (122 mg, 1.0 mmol), ethylacetoacetate (130 mg, 1.0 mmol), and thiourea (190 mg,2.5 mmol) in MeCN (15 ml) was mixed with HPA supportedon Y zeolite (8 wt% NaY + 0.5 mM HPA) and refluxed for7 h. After cooling, the heteropoly acid (HPA) supported onHY filtered off and washed with hot water and ethanol toremove thiourea from the surface of the catalyst. Then, thecatalyst dried and was maintained for new runs. The filtratewas evaporated to dryness and the residue was recrystallized from EtOH to afford 2 (254 mg, 87 %), m.p. 181-185 C (Lit.[6, 7] 184-186 C), Rf = 0.71. 1H NMR (DMSO-d6): delta 10.29(s, 1 H, NH), 9.60 (s, 1 H, NH), 9.44 (s, 1 H, OH), 7.10 (t,J = 7.8 Hz, H-5), 6.65 (d, 1 H, J2?,4? = 2.4Hz, Harom.-2?), 6.64(m, 2 H, Harom.-4 + Harom.-6), 5.09 (d, 1 H, JNH,4 = 5.5 Hz,H-4), 4.02 (q, 2 H, J = 7.8Hz, CH2CH3), 2.28 (s, 3 H, C6-Me),1.12 (t, 3 H, J = 7.8 Hz, CH2CH3). 13C NMR (DMSO-d6): delta174.6 (C = S), 165.7 (CO2Et), 157.9 (C-6), 145.3 (C3?-OH + Carom.-1?), 130.0 (Carom.-5?), 117.7 (Carom.-2? +Carom.-6?), 133.6 Carom.-4?), 101.2 (C-5), 60.1 (CH2CH3),54.5 (C-4), 17.7, 17.6 (C6-Me), 14.5 (CH2CH3). EI-MS:m/z (%) = 292 [M]+. Anal. Calcd. for C14H16N2O3S(292.35): C, 57.52; H, 5.52; N, 9.58. Found: C, 57.32;H, 5.39; N, 9.32 %. Method B. A mixture of 1 (244 mg, 2.0 mmol), ethylacetoacetate (300 mg, 2.30 mmol), and thiourea (380 mg,5.0 mmol) in EtOH (15 ml) in the presence conc. Hydrochloricacid (1 ml) was heated under reflux for 3 h. After cooling,the mixture was poured onto ice (20 g). The precipitate wasfiltered and dried and recrystallized from EtOH to give 2(292 mg, 50 %), TheNMR spectra, m.p. and mixed m.p. werealmost similar for those of 2 prepared in method A.
86%
In ethanol; for 4h;Reflux;
General procedure: A mixture of 0.025 mol of urea or thiourea, 0.025 mol of an arylaldehyde, 0.025 mol of ethyl acetoacetate or acetylacetone, 10 mL ethanol and 3 drops of a liquid or 2-3 mg of solid phenol was heated under reflux for 6-24 h and the reaction mixture was then cooled to 0 C and the product was filtered, washed with water, dried and recrystallized from a suitable solvent. The progress of the reaction was monitored by TLC using ethyl acetate and n-hexane (1:2) as eluent.
85%
With chloro-trimethyl-silane; In acetonitrile; for 24h;Inert atmosphere; Reflux;
General procedure: A 10 mmol mixture of the corresponding benzaldehyde (4),15 mmol of methyl or ethyl acetoacetate (5), 761 mg (10 mmol,1.0 equiv) of thiourea (6), 1.27 mL (10 mmol, 1.0 equiv) oftrimethylsilylchloride (TMSCl) and 7.0 mL of anhydrous acetonitrileunder nitrogen atmosphere was formulated in a 50 mL roundbottom flask, equipped with a magnetic stirring bar. The watercondenser was set at the neck of round bottom flask then the mixturewas stirred for 1 min at room temperature to allow homogenization,followed by reflux for 24 h at oil bath. After cooling thereaction mixture, solvent was evaporated under reduced pressurethrough rotary evaporator. Thereafter, 500 mL of ice water waspoured into reaction mixture and allowed to stir for 1 h in orderto obtain maximum precipitation. Filtration delivered the pureDHPMs. Where needed, the product was recrystallized or directlypurified by gradient dry flash chromatography using appropriatesolvents. The physical and spectroscopic data of DHPM 7-12 werefound in good agreement with the literature [11,13,20]. 4.2.1.1.
83%
With psychortria douarrei catalyst dispersed on montmorillonite K10; In neat (no solvent); at 80℃; for 1.2h;
A mixture of ethyl acetoacetate (781 mg, 6.Ommol),3-hydrobenzaldehyde (488 mg, 4.0 mmol), thiourea (457 mg,6.0 mmol) and P douarrei crude catalyst (265 mg, amount corresponding to 1.0 mmol of nickel following previous dosing), supported on montmorillonite K10 (265 mg) was placed in a 10 mE sealed tube. The tube was heated to 80 C. in oil bath, under magnetic stirring for 1.2 h. The mixture was then extracted with hot ethanol (10 mE, 70 C.) and filtered in order to remove the catalyst, which was reactivated by heating (150C.). The solutionwas poured into crushed ice (20 g) and stirred for 20 mm. The solid separated was filtered under suction, washed with cold water (30 mE) and recrystallized from hot ethanol, affording pure product, as colorless crystals (973 mg, 83%). The same procedure was followed with G. pruinosa catalyst and commercial NiCl2. Mp 185-186 C. (184-186C.);IR 3298,3181,3115,2982, 1663, 1617, 1573 cm?; ?H NMR (DMSO-d5, 300 MHz) oe: 1.14 (t, J=7.4 Hz, 3H), 2.29 (s, 3H), 4.04 (q, J=7.4 Hz, 2H), 5.11 (d, J=3.5 Hz, 1H), 6.60-6.71 (m, 3H), 7.06-7.15 (m, 1H), 9.42 (brs, 1H),9.62 (brs, 1H), 10.29 (brs, 1H); ?3C NMR (DMSO-d5, 75 MHz) oe: 14.0, 17.1, 54.2, 59.6, 100.8, 113.0, 114.4, 117.0, 129.3, 144.8, 144.9, 157.4, 165.4, 174.2. MS (EI+) calculated for C,4H,6N2035 [M]292.1. found 293.1 [M+H].
80%
With bis(p-sulfoanilino)triazine-functionalized silica-coated magnetite nanoparticles; In neat (no solvent); at 100℃; for 1.16667h;
General procedure: A mixture of aromatic aldehyde (1 mmol), b-keto ester or dimedone (1 mmol) and urea or thiourea (1.2 mmol) was stirred in presence MNPs-BSAT (20 mg) at 100 C under solvent-free condition for the appropriate time (Scheme 1). After completion of the reaction as indicated by TLC (using n-hexane-ethyl acetate as eluent), the resulting mixture was diluted with hot ethanol (15 mL) and the catalyst separated by an external magnet and washed with hot ethanol (5 mL) two times. The filtrate was cooled to room temperature and the crude products which precipitated were collected and recrystallized from ethanol if necessary.
79%
With tin(II) chloride dihdyrate; In acetonitrile; at 70 - 75℃;Sonication;
General procedure: A mixture of an aldehyde (10 mmol), a diamino compound (12 mmol), a dicarbonyl compound (10 mmol, mL), SnCl2·2H2O (10 mol%) and acetonitrile (10 mL) was mixed in a pyrex tube. The mixture was then irradiated in ultrasonic bath at 70-75 C. The reaction was monitored by TLC. After the completion of the reaction, the resulting precipitate was filtered and crude product was recrystallized from an appropriate solvent or purified through columnchromatography.
75%
With ytterbium(III) triflate; In acetonitrile; at 120℃; for 1h;Microwave irradiation;
General procedure: The appropriate beta-keto ester (0.75-3.0mmol, 1.5 equiv.), the appropriate aldehyde (0.5-2.0mmol, 1 equiv.), the appropriate urea or thiourea (0.5-2.0mmol, 1 equiv.) and Yb(OTf)3 (0.05-0.20mmol, 0.10 equiv.) were placed in a microwave oven vial (0.5-2.0mL) with a magnetic stirring bar and dissolved in THF or MeCN (0.5-1.0mL). The resulting mixture is heated to 120C for 30-60min using microwave irradiation. The resulting mixture is poured onto an ice-water mixture and left for precipitation. The resulting crude solids were purified by recrystallization, DCVC or flash chromatography.
74%
With copper dichloride; In ethanol; at 80℃; for 4h;
General procedure: To a stirred mixture of thiourea (1.00 mmol), substituted benzaldehydes E-G (Scheme 1)(1.00 mmol), ethyl acetoacetate (130 mg, 1.00 mmol), and anhydrous cupric chloride(10 mol%) were added. The mixture was heated at 80C for 4 h under stirring. After thereaction was completed (checked by TLC), a mixture of H2O:EtOH 8:5 (13 mL) was added andthe resulting slurry was stirred at 80C until total dissolution. After being cooled to roomtemperature, the reaction mixture was poured onto crushed ice (30 g) and stirred for5-10 min. The separated solid was filtered under suction (water aspirator), washed withice-cold water (50 mL), and then recrystallized from hot ethanol to afford the pureproduct.2.3. Ethyl-4-(3-hydroxyphenyl)-6-methyl-2-thioxo-pyrimidine-5-carboxylate(monastrol) L1))From 3-hydroxybenzaldehyde (E) (122 mg). Yield: 216 mg (74%); mp 185-186C (Lit. [23, 25]184-186C).
72%
With 1-butyl-1,2,4-triazolium triflate; In ethanol; at 80℃; for 1h;Green chemistry;
General procedure: Catalyst 1a-c (10 mol%) was added to a solution of aldehyde (1.0 mmol), beta-ketoester (1.5 mmol) and urea or thiourea (2.0 mmol) in ethanol (0.5 mL). The reaction mixture was heated at 80 C using oil bath for the specified time (0-5 h). The progress of the reaction was monitored by TLC. After completion, the reaction mixture was cooled to room temperature and subsequently quenched with a mixture of water:ethanol (5:0.5 mL). The solid product was filtered and washed with n-hexane (5mL ×2), which afforded pure 3,4-dihydropyrimidin-2(1H)-ones or 3,4-dihydropyrimidin-2(1H)-thiones in pure form.
71%
With beta?cyclodextrin; at 100℃; for 3h;
General procedure: Aldehyde (3 mmol), ethyl acetoacetate (0.586 g, 4.5 mmol), urea (0.270 g, 4.5 mmol) or thiourea (0.343 g, 4.5 mmol), and beta-cyclodextrin (17 mg, 0.5 mol%) were mixed in a 25 mL round bottom flask for 3h at a temperature of 100 C. The reaction mixture was cooled and the solid was solubilized in ethanol and the addition of few drops of cold water to precipitate the product. The precipitated solid was filtered on sintered funnel. The crude product was further purified by recrystallization from ethanol to afford pure 3,4-dihydropyrimidin-2(1H)-ones. After separation of the product by filtration, the filtrate was washed with ethyl acetate and the aqueous phase was concentrated on rotary evaporator obtaining the catalyst.
70%
With hydrogenchloride; In neat (no solvent); at 100℃;
General procedure: The procedure described in the literature [51] was followed. To atwo-necked round-bottom flask, the appropriated aldehyde(10 mmol), ethyl acetoacetate or dimedone or cyclohexanedione(10 mmol), thiourea (20 mmol) and 5 drops of concentrated HCl were added. The reaction mixturewas stirred at 100 C for the time required for consumption of the starting materials, which was verified by TLC. After this time, the reaction mixture was pouredinto crushed ice and water. The precipitate was filtered and dried.The compounds were used in the next step without further purification.
64%
With 1-ethyl-1,2,4-triazolium phenylsulfonate; In ethanol; at 70 - 80℃; for 2.5h;
General procedure: Catalyst 1-ethyl-1,2,4-triazolium phenylsulfonate (TrHEtPS) (10 mol) was added to a solution of urea or thiourea (2.0 mmol), beta-ketoester (1.5 mmol) and aldehyde (1.0 mmol) in ethanol (0.5 mL). The reaction mixture was heated at 70 to 80 C for the specified time (0-5 h) (Scheme-II). The completion of the reaction was monitored by thin layer chromatography (ethyl acetate:hexane: :4:1) and then the reaction mixture was cooled to room temperature. Then, quenched with mixture of water: ethanol (5:0.5 mL). The obtained solid product was filtered, and washed with n-hexane (5mL) and afforded 3,4-dihydropyrimidin-2(1H)-ones or 3,4-dihydropyrimidin-2(1H)-thiones.
52%
With 25,26,27,28-terahydroxycalix[4]arene-5,11,7,23-tetrasulfonic acid; In ethanol; for 8h;Reflux;
General procedure: Aldehydes (3 mmol), ethyl acetoacetate (4.5 mmol) and (thio)urea (4.5 mmol) were dissolved in 3 mL of ethanol containing p-sulfonic acid calix[4]arene (0.5 mol %). The mixture was heated under reflux and stirred for 8 h.20 All DHPMs were characterized by NMR (1H and 13C), infrared, melting point and elemental analysis. Characterization data for compounds BA9, BA11-BA14, BA16-BA21, BA24-BA28, BA30 and BA32 were recently reported by da Silva et al.20 Data for compounds BA2, BA10, BA15, BA22, BA29, BA31 and BA33 are listed as Supplementary data.
With chloro-trimethyl-silane; ytterbium(III) triflate; In acetonitrile; at 120℃; for 0.666667h;Inert atmosphere; Sealed tube; Microwave irradiation;
General procedure: A mixture containing 10 mmol of the corresponding benzaldehyde(1a), 15 mmol of corresponding methyl 2a or ethyl acetoacetate2b, 761 mg (10 mmol, 1.0 equiv) of thiourea 3a, and 620 mg(1.0 mmol, 10 mol %) of Yb(OTf)3 or 1.27 mL (10 mmol, 1.0 equiv) ofTMSCl was suspended in 10 mL of anhydrous acetonitrile undera nitrogen atmosphere in a 20 mL microwave vial (Pyrex) equippedwith a magnetic stirring bar. The vial was sealed and stirred for2 min at room temperature to allow homogenization then themixture was heated for 40 min at 120 C by microwave irradiation.After cooling the reaction mixture was poured onto 500 mL of icewater and allowed to stir for 1 h in order to obtain maximumprecipitation. Filtration then delivers the pure DHPMs. Whereneeded the product was directly purified by gradient dry flashchromatography using appropriate solvents. The physical andspectroscopic data of DHPM (9aef) were found in agreement withthe literature.
With tin(II) chloride dihdyrate; In acetonitrile;Sonication; Heating;
General procedure: A mixture of an aldehyde (10mmol), a diamino compound (12mmol), a dicarbonyl compound (10mmol, mL), SnCl2.2H2O (10mol %) and acetonitrile (10mL) was mixed in a pyrex tube. The mixture was then irradiated in ultrasonic bath at 70-75C. The reaction was monitored by TLC. After the completion of the reaction, the resulting precipitate was filtered and crude product was recrystallized from an appropriate solvent or purified through column chromatography.
With 1H-imidazole; In N,N-dimethyl-formamide;Inert atmosphere;
<strong>[329689-23-8]Monastrol</strong> (1.46g, 5mmol), TBDMSCl (1.51g, 10mmol) and imidazole (1.02g, 15mmol) was dissolved in dry DMF (5mL) under N2 and stirred over night. The reaction mixture was diluted with MeOH (10mL) and stirred for 30min and thereafter concentrated under vacuum. The residue was dissolved in EtOAc (100mL) and the organic phase was washed with water (3×30mL), dried with MgSO4, filtered and evaporated. The crude product was purified by DCVC (heptane:EtOAc) to give the product as a white solid on 80% yield (1.63g). Purity by anal. HPLC (254nm)>97%, LC-MS(ESP): m/z=407 [M+H]+. 1H NMR (DMSO): delta 10.34 (d, J=0.8Hz, 1H), 9.63 (dd, J=3.5, 1.5Hz, 1H), 7.22 (t, J=7.8Hz, 1H), 6.82 (d, J=7.8Hz, 1H), 6.76-6.71 (m, 2H), 5.12 (d, J=3.8Hz, 1H), 4.02 (q, J=7.1Hz, 2H), 2.27 (s, 3H), 1.11 (t, J=7.1Hz, 3H), 0.93 (s, 9H), 0.17 (s, 6H). 13C NMR (DMSO): delta 174.4, 165.1, 155.2, 144.97, 144.94, 129.7, 119.2, 119.0, 117.6, 100.7, 59.6, 53.6, 17.9, 17.1, 14.0,-4.5.
4-[3-((2S,3R,4R,5R)-3,4-Bis-benzyloxy-5-benzyloxymethyl-tetrahydro-furan-2-carbonyloxy)-phenyl]-6-methyl-2-thioxo-1,2,3,4-tetrahydro-pyrimidine-5-carboxylic acid ethyl ester[ No CAS ]
4-(3-hydroxyphenyl)-6-methyl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
22%
With water; potassium hydroxide; at 20℃; for 72h;
KOH (0.10g, 1.8mmol) was dissolved in H2O (5mL). <strong>[329689-23-8]Monastrol</strong> (180mg, 0.62mmol) was added and the mixture was stirred for 72hat room temperature. The mixture was acidified with 1M HCl. The water was extracted with EtOAc. The organic phase was dried over MgSO4, filtered and evaporated to dryness. The crude white product was washed with Et2O to obtain the product in 22% yield (35mg). Purity by anal. HPLC (254nm)>97%, mp: 163-165C, LC-MS(ESP): m/z=264. The spectroscopic data is in agreement with data previously reported in literature [41].
(Z)-ethyl 2-((1-([1,1’-biphenyl]-4-yl-methyl)-1H-indol-3-yl)methylene)-5-(3-acetoxyphenyl)-7-methyl-3-oxo-3,5-dihydro-2H-thiazolo[3,2-a]pyrimidine-6-carboxylate[ No CAS ]
ii) Synthesis of ethyl 6-methyl-4-(3-hydroxyphenyl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylate (monastrol) A mixture of ethyl acetoacetate (781 mg, 6.0 mmol), 3-hydrobenzaldehyde (488 mg, 4.0 mmol), thiourea (457 mg, 6.0 mmol) and P. douarrei crude catalyst (265 mg, amount corresponding to 1.0 mmol of nickel following previous dosing), supported on montmorillonite K10 (265 mg) was placed in a 10 mL sealed tube. The tube was heated to 80C in oil bath, under magnetic stirring for 12 h. The mixture was then extracted with hot ethanol (10 mL, 70C) and filtered in order to remove the catalyst, which was reactivated by heating (150C). The solution was poured into crushed ice (20 g) and stirred for 20 min. The solid separated was filtered under suction, washed with cold water (30 mL) and recrystallized from hot ethanoL affording pure product, as colorless crystals (973 mg, 83%). The same procedure was followed with G. pruinosa catalyst and commercial NiCl2. Mp 185-186C (184-186C) ; IR 3298, 3181, 3115, 2982, 1663, 1617, 1573 cm1 ; 1H NMR (DMSO-d6, 300 MHz) delta: 1.14 (t, J=7.4 Hz, 3H), 2.29 (s, 3H), 4.04 (q, J=7.4 Hz, 2H), 5.11 (d, J=3.5 Hz, 1H), 6.60-6.71 (m, 3H), 7.06-7.15 (m, 1H), 9.42 (brs, 1H), 9.62 (brs, 1H), 10.29 (brs, 1H) ; 13C NMR (DMSO-d6, 75 MHz) delta: 14.0, 17.1, 54.2, 59.6, 100.8, 113.0, 114.4, 117.0, 129.3, 144.8, 144.9, 157.4, 165.4, 174.2. MS (EI+) calculated for C14H16N203S [M]+ 292.1, found 293.1 [M+H]+.
ethyl 6-(3-hydroxyphenyl)-1,4-dimethyl-2-(methylthio)-1,6-dihydropyrimidine-5-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
85%
With potassium carbonate; In acetonitrile; at 80℃; for 6h;Sealed tube; Inert atmosphere;
General procedure: A mixture of 0.25 mmol of the corresponding DHPMs 9aef,69.17 mg (0.50 mmol, 2.0 equiv) of potassium carbonate, and 78 mL(1.25 mmol, 5.0 equiv) of methyl iodide was suspended in 1.2 mL ofanhydrous acetonitrile in a 5 mL microwave vial equipped withmagnetic stirrer. The reaction vessel was sealed and flushed withnitrogen then heated for 6 h at 80 C using a conventional oil bath.Thereafter, the solvent was removed under reduced pressure. Theproduct was purified by automated flash chromatography (hexane/ethyl acetate) to provide the desired 2-substituted dihydropyrimidines11aef as yellow solids.
85%
With potassium carbonate; In acetonitrile; at 80℃; for 6h;Inert atmosphere; Sealed tube;
General procedure: A mixture containing 0.25 mmol of the corresponding DHPMs7-12, 69.17 mg (0.50 mmol, 2.0 equiv) of potassium carbonate,78 mL (1.25 mmol, 5.0 equiv) of methyl iodide and 1.2 mL of anhydrousacetonitrile in a 5 mL microwave vial, equipped with a magneticstirring bar. Thereafter, the reaction vessel was sealed andpurged with nitrogen following 6 h conventional oil bath heatingat 80 C. After completion of the reaction, the solvent was removedunder reduced pressure, and the product was purified byautomated flash chromatography system (hexane/ethyl acetate) to provide the desired dimethylated dihydropyrimidines (13-18)as yellow solids. The physical and spectroscopic properties ofdimethylated dihydropyrimidines (13-18) were found in goodagreement with the literature [14].
(E)-1-(4-aminophenyl)-3-(4-hydroxyphenyl)prop-2-en-1-one[ No CAS ]
[ 329689-23-8 ]
(E)-4-(3-hydroxyphenyl)-N-(4-(3-(4-hydroxyphenyl)acryloyl)phenyl)-6-methyl-2-thioxo-3,4-dihydropyrimidine-5-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
67%
In tetrahydrofuran;Sonication; Heating;
General procedure: In a pyrex tube, a mixture of 4?-aminochalcones (2mmol) and thioxo DHPM 12 (0.58 gm, 2mmol) and 13 (0.585 gm, 2mmol) in THF (5mL) was heated in ultrasonic bath at 40C. The reaction was monitored by TLC. The precipitate obtained was filtered, washed with sodium bicarbonate (2×10mL, 0.1M). The crude solid was recrystallized from a mixture of petroleum ether/ethanol (1:1).
(2E)-1-(4'-aminophenyl)-3-(3-nitrophenyl)prop-2-en-1-one[ No CAS ]
[ 329689-23-8 ]
(E)-4-(3-hydroxyphenyl)-6-methyl-N-(4-(3-(3-nitrophenyl)acryloyl)phenyl)-2-thioxo-3,4-dihydropyrimidine-5-carboxamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
63%
In tetrahydrofuran;Sonication; Heating;
General procedure: In a pyrex tube, a mixture of 4?-aminochalcones (2mmol) and thioxo DHPM 12 (0.58 gm, 2mmol) and 13 (0.585 gm, 2mmol) in THF (5mL) was heated in ultrasonic bath at 40C. The reaction was monitored by TLC. The precipitate obtained was filtered, washed with sodium bicarbonate (2×10mL, 0.1M). The crude solid was recrystallized from a mixture of petroleum ether/ethanol (1:1).
[{(η6-p-cymene)Ru(ethyl-4-(3-hydroxyphenyl)-6-methyl-2-thioxo-pyrimidine-5-carboxylate)}2](Cl)2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
68%
With potassium carbonate; In tetrahydrofuran; for 6h;Inert atmosphere; Reflux;
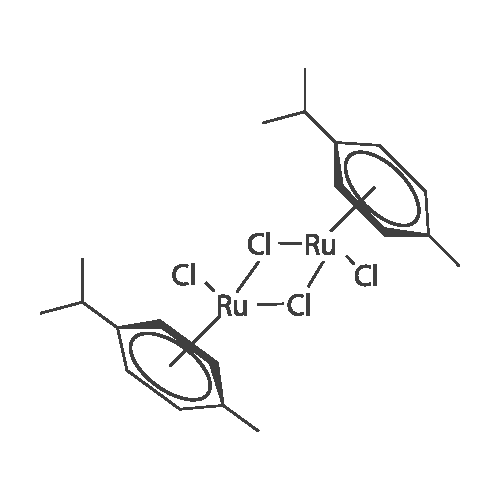
General procedure: To a stirred solution of pyrimidine derivatives (L1-L3) (0.20 mmol) in dry THF (10 mL), solid[(eta6-p-cymene)RuCl2]2 (H) (62 mg, 0.10 mmol) and potassium carbonate (0.20 mmol) wereadded. The solution was heated under reflux for 6 h under nitrogen. After cooling, the mixturewas evaporated to dryness and the residue was washed with diethylether and dried undervacuum to give the desired complex.
ethyl 4-(3-hydroxyphenyl)-6-methyl-2-(methylthio)-1,4-dihydropyrimidine-5-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
91%
In ethanol; for 1h;Reflux;
General procedure: The procedures described previously in the literature [75] were followed. In a two-necked round-bottom flask, the appropriate thioureas 1a-k, 6a-b or selenourea 3j (5 mmol) were solubilized in ethanol, followed by the addition of methyl iodide (6 mmol). Thereaction mixture was stirred at reflux temperature for the time required for consumption of the starting material, which was verified by TLC. After this time, the reaction mixture was treated with 20 mL of a NaHCO3 (Sat.) solution. The reaction mixture was extracted with ethyl acetate/water and the organic phase driedover MgSO4 and concentrated under vacuum. The resulting products presented high purity and were used in the next step without further purification.
[Ru(triphenylphosphine)2(ethyl 4-(3-hydroxyphenyl)-6-methyl-2-thioxo-pyrimidine-5-carboxylate)2]*2H3O+*2Cl-*H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
General procedure: A solution of hydrated RuCl3 (65 mg, 0.25 mmol) in MeOH(7 mL) was refluxed under nitrogen for 5 min, then allowedto cool to room temperature. To this solution, triphenylphosphine(40 mg, 0.15 mmol) was added and the brownsolution was refluxed under nitrogen for 2 h. After cooling,the ligands (L1-L3) (0.50 mmol) and K2CO3 (69 mg, 0.50mmol) were added. The mixture was heated at 60 C for 5h under nitrogen atmosphere. After cooling, the crude precipitatewas filtered and dried. Recrystallization from EtOHafforded the desired complexes.
dichlorotetrakis(dimethylsulfoxide)ruthenium(II)[ No CAS ]
[ 329689-23-8 ]
[RuCl(dmso)3(ethyl 4-(3-hydroxyphenyl)-6-methyl-2-thioxo-pyrimidine-5-carboxylate)][ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With potassium carbonate; In dichloromethane; at 50℃; for 6h;Inert atmosphere;
General procedure: A solution of the ligands L1-L3 (0.23 mmol) and K2CO3 (32mg, 0.23 mmol) in CH2Cl2 (15 mL) was added to a suspensionof dichlorotetrakis(dimethylsulfoxide) ruthenium[RuCl2(dmso)4] (111 mg, 0.23 mmol), in CH2Cl2 (10 mL)and the mixture was heated at 50 C under nitrogen atmospherefor 6 h. After cooling, the solvent was partiallyremoved under reduced pressure, and a brown solidappeared. It was filtered and washed with diethyl ether followedby drying to give the desired complexes.
[RuCl2(ethyl 4-(3-hydroxyphenyl)-6-methyl-2-thioxo-pyrimidine-5-carboxylate)]2[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
50%
With potassium carbonate; In tetrahydrofuran; methanol; for 5h;Reflux; Inert atmosphere;
General procedure: A mixture of L1-L3 (0.14 mmol), hydrated RuCl3 (36 mg,0.14 mmol), and K2CO3 (19 mg, 0.14 mmol) in THF-MeOH(1:1) (10 mL) was heated under reflux under nitrogen for 5h. After cooling, the solvent was evaporated to dryness andthe residue was washed with ether (3x10 mL) and dried togive the desired complexes as deep brown solid.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping