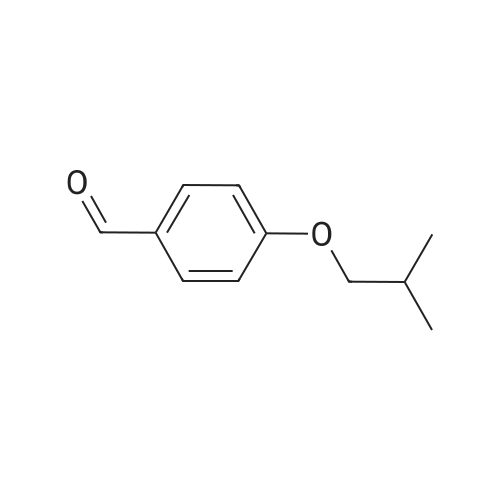
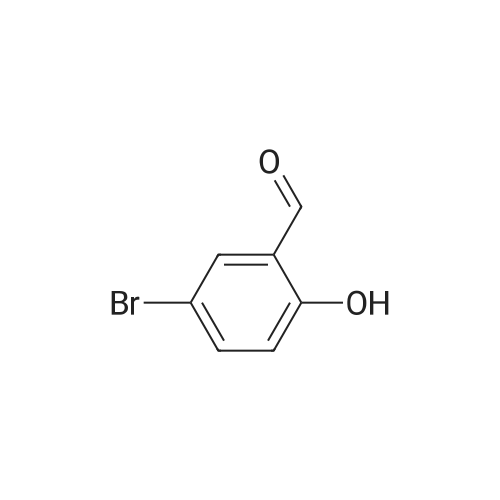
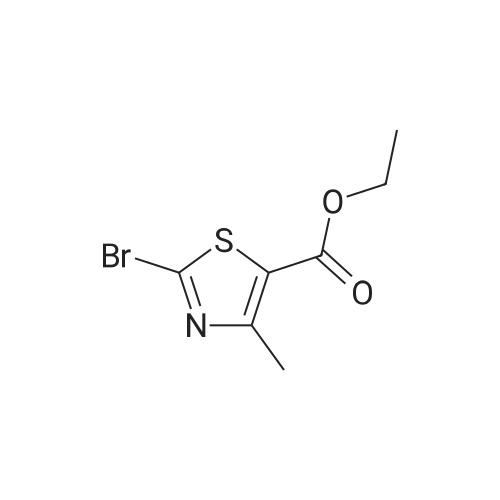
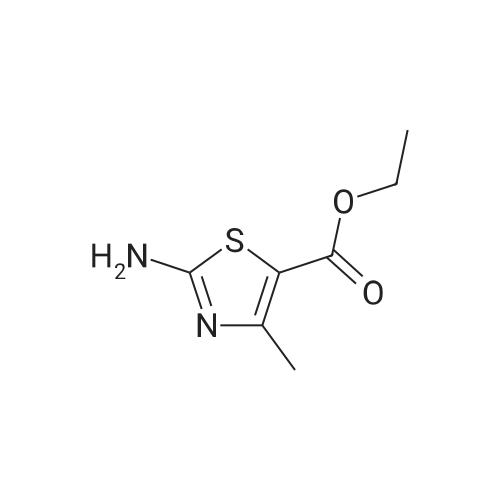
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Reference:
[1] Chemische Berichte, 1917, vol. 50, p. 1354
2
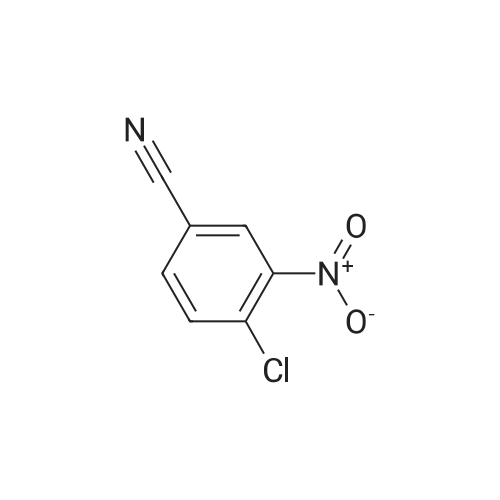
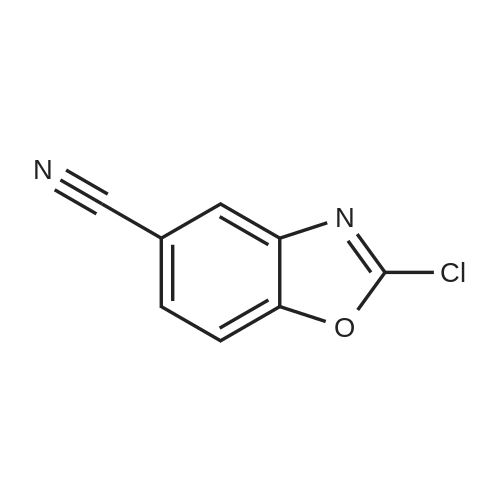
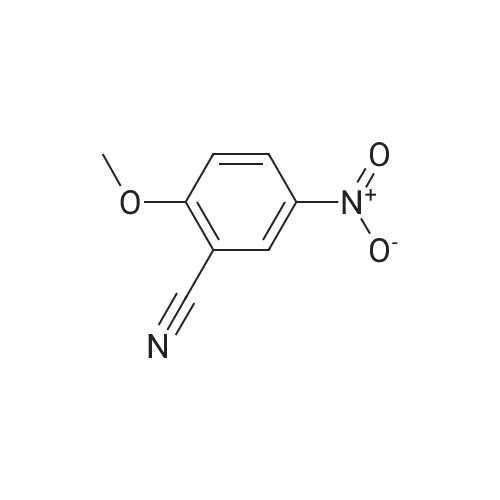
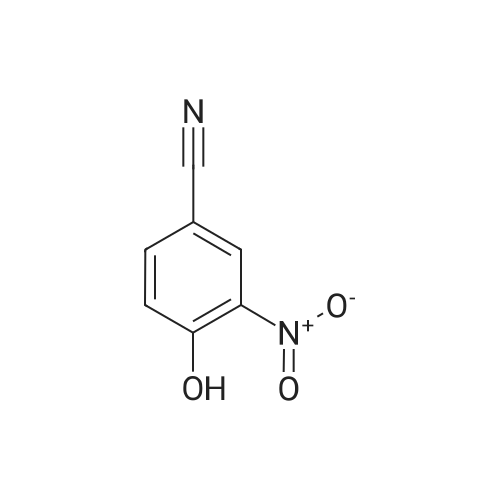
[ 3011-34-5 ]
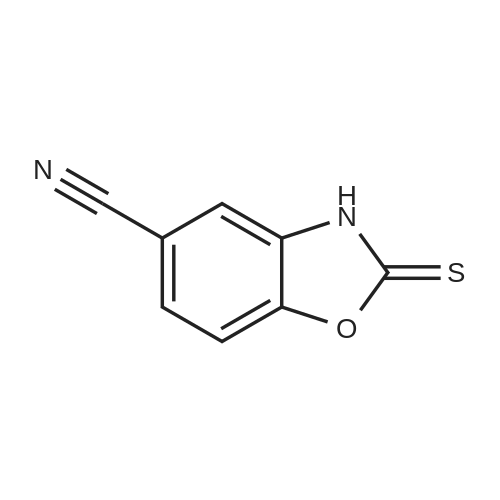
[ 3272-08-0 ]
Yield
Reaction Conditions
Operation in experiment
82%
With trifluorormethanesulfonic acid; trimethylsilylazide In acetonitrile at 20℃; for 0.75 h; Sealed tube; Inert atmosphere
General procedure: To a solution of an aromatic aldehyde 1 (0.500 mmol, 1.0 equiv) and TMSN3 (115 mg, 1.00 mmol,2.0 equiv) in a premixed HFIP/ACN mixture (2.0 mL, 1:1) in a nitrogen-flushed two dram vial wasadded triflic acid (TfOH; 17.7 L, 0.200 mmol, 0.40 equiv) (exotherm and brisk effervescence due tonitrogen gas evolution was immediately observed). The vial was capped and the reaction mixture wasallowed to stir at rt for 20–75 min. The reaction mixture was concentrated under nitrogen. The residueobtained was suspended in CH2Cl2/hexanes mixture and loaded on a silica gel in a 5 g samplecartridge. Purification using a normal phase silica flash column on a CombiFlash purification systemafforded a corresponding aromatic nitrile 2 upon concentration of appropriate fractions.
Reference:
[1] Journal of Organic Chemistry, 2012, vol. 77, # 12, p. 5364 - 5370
[2] Molecules, 2016, vol. 21, # 1,
[3] Patent: US5614520, 1997, A,
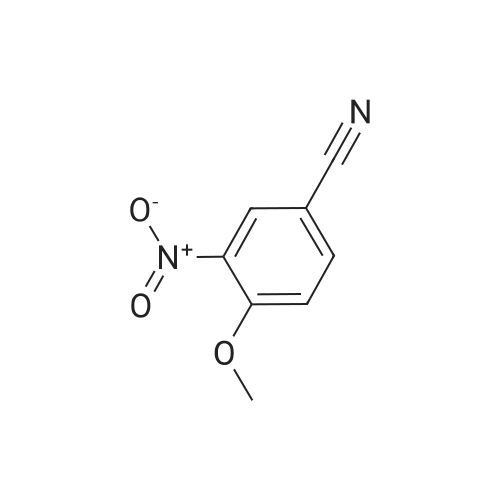
3
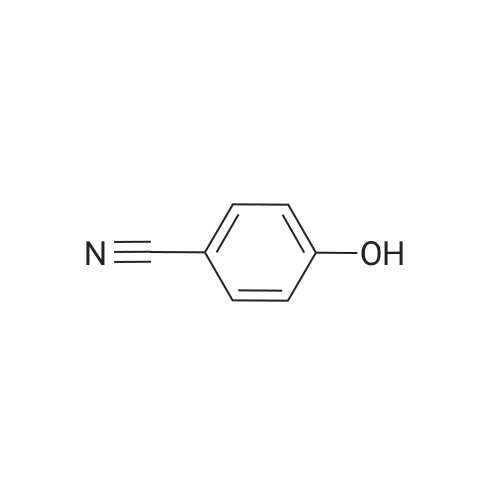
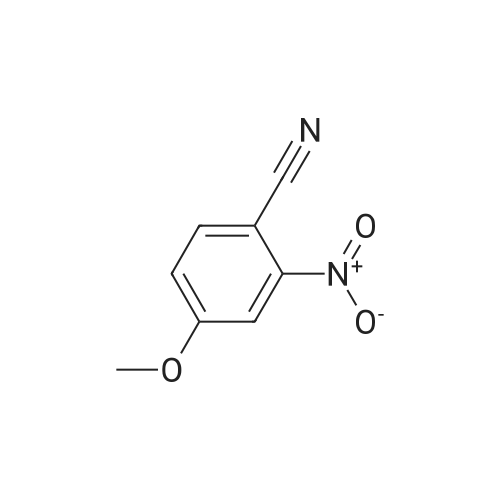
[ 767-00-0 ]
[ 3272-08-0 ]
Yield
Reaction Conditions
Operation in experiment
94.5%
at 45 - 50℃; for 1.5 h;
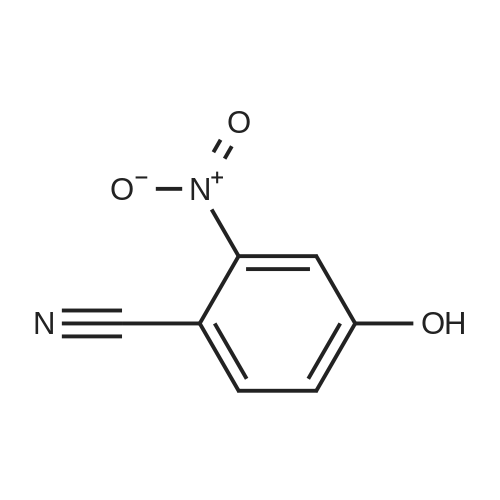
11.9 g of p-hydroxybenzonitrile (0.1 mol) and 50 mL glacial acetic acid were added to a 250 mL three-necked flask, added dropwise with the mixture of 8 mL concentrated nitric acid and 12 mL glacial acetic acid. After dripping, the solution was slowly heated to 45-50° C. to react 1.5 hours, tested by TLC, the starting materials were reacted completely; then cooled down, added with dichloromethane, standing, to separate the upper organic layer, and then washed by water, 5percent sodium bicarbonate solution and saturated brine, 50 mL each. Solvent was recovered under reduced pressure to get 15.5 g of light yellow solid 3-nitro-4-hydroxybenzonitrile (VII), with a yield of 94.5percent.
79%
at 40 - 60℃; for 0.333333 h;
Step A:4-Hydroxy-3-nitrobenzonitrile; <n="62"/>A mixture of nitric acid (1.16 g, 12 mmol) and glacial acetic acid (1 mL) was heated to 40 0C. To this mixture was added rapidly a solution of 4-hydroxybenzonitrile (1 g, 8.4 mmol) in glacial acetic acid (4 mL) until the flask temperature rose to 50 0C. Then the solution was added at a rate such that the temperature was maintained at 50-60 0C. When addition was complete the mixture was stirred for another 20 min at 55 0C, and then poured into ice-water (24 mL). The mixture was filtered and the solid was washed with water to give 4-hydroxy-3-nitro- benzonitrile 1.09 g (79 percent) as a solid.1H NMR (300 MHz, CDCI3): δ 10.91 (s, 1 H), 8.48 (d, 1 H), 7.82 (dd, 1 H), 7.28 (dd, 1 H).
67%
With copper(II) nitrate trihydrate In tetrahydrofuran for 3 h; Reflux
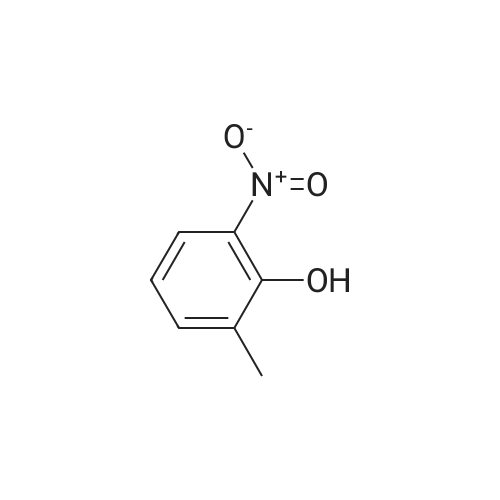
General procedure: A suspension of 2-methylphenol(18.5 mmol, 1.0 eq) and Cu(NO3)2.3H2O (27.7 mmol, 1.5 eq) in THF was stirred magnetically at 60°C or reflux for several hours. Then after the solvent was removed under vacuum, the mixture was extracted with EtOAc (3×30 mL). The combined organic layers were washed with brine (5mL), dried over anhydrous MgSO4 and concentrated under vacuum. The crude residue was purified by column chromatography to afford the product (67-90percent).
67%
at 40 - 55℃; for 0.333333 h;
To a mixture of HNO3 (2.7mL, 63.Ommol) and AcOH (5mL) was added 4- hydroxybenzonitrile (5g, 42mmol) in AcOH (5mL) dropwise at 40 00. The reaction mixture was heated at 55 00 for 20 mm. The TLC showed reaction to be complete. The reaction mixture was poured into ice-water (lOOmL). The precipitated solid was filtered, washed with water (200mL) and dried under vacuum to afford 4-hydroxy-3- nitrobenzonitrile as a yellow solid. Yield: 2.5g (67percent); 1H NMR (400 MHz, DMSO-d612.34 (bs, 1H), 8.43 (5, 1H), 7.94 (d, J= 10.5 Hz, 1H), 7.24 (d, J= 8.7 Hz, 1H); MS (ESl+) for CHNOS m/z 163.03 [M+H].
55%
With uronium nitrate In water; acetonitrile at 80℃; for 1 h; Microwave irradiation
General procedure: Phenol (10 mmol) and urea nitrate (10 mmol)were mixed together in acetonitrile–water (95:5, 5 ml) in a 25 ml round bottomed flask and placed in a Milestone’s Start SYNTH microwave reactor. The reaction mixture was heated at 80°C for 40–50 min. At the end of the reaction, the reaction mixture was allowed to cool at room temperature,treated with water, and extracted with dichloromethane. After removing the solvent under reduced pressure, the residue was purified by column chromatography on silica gel to give the corresponding nitrophenol. In allcases ortho-nitrophenols were obtained selectively without any evidence forthe formation of the para-substituted nitrophenols. All the compoundsobtained were characterized by 1H NMR, 13C NMR, mp (for solids), GC–MSand in comparison with authentic samples.
Reference:
[1] Chinese Chemical Letters, 2011, vol. 22, # 7, p. 827 - 830
[2] Phosphorus, Sulfur and Silicon and the Related Elements, 2003, vol. 178, # 9, p. 2019 - 2025
[3] Turkish Journal of Chemistry, 2010, vol. 34, # 5, p. 753 - 759
[4] Patent: US2016/83373, 2016, A1, . Location in patent: Paragraph 0043; 0044
[5] Journal of Organic Chemistry, 1996, vol. 61, # 10, p. 3289 - 3297
[6] Mendeleev Communications, 2006, vol. 16, # 1, p. 41 - 42
[7] Tetrahedron, 1989, vol. 45, # 5, p. 1415 - 1422
[8] Molecules, 2001, vol. 6, # 7, p. 614 - 620
[9] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2001, vol. 40, # 12, p. 1191 - 1195
[10] Synlett, 2003, # 2, p. 191 - 194
[11] South African Journal of Chemistry, 2007, vol. 60, p. 109 - 112
[12] Bulletin des Societes Chimiques Belges, 1984, vol. 93, # 11, p. 961 - 972
[13] Synthetic Communications, 2008, vol. 38, # 19, p. 3366 - 3374
[14] Molecules, 2002, vol. 7, # 10, p. 734 - 742
[15] Patent: WO2007/110364, 2007, A1, . Location in patent: Page/Page column 60-61
[16] Arkivoc, 2014, vol. 2014, # 5, p. 64 - 71
[17] Patent: WO2018/37223, 2018, A1, . Location in patent: Page/Page column 77
[18] Journal of Chemical Research - Part S, 2001, # 4, p. 140 - 142
[19] Tetrahedron Letters, 2014, vol. 55, # 7, p. 1320 - 1322
[20] Journal of the Chemical Society, 1949, p. 642,645
[21] Proceedings of the Royal Society of London, Series B: Biological Sciences, 1946, vol. 133, p. 20,30
[22] Bioorganic and Medicinal Chemistry Letters, 2011, vol. 21, # 22, p. 6842 - 6851
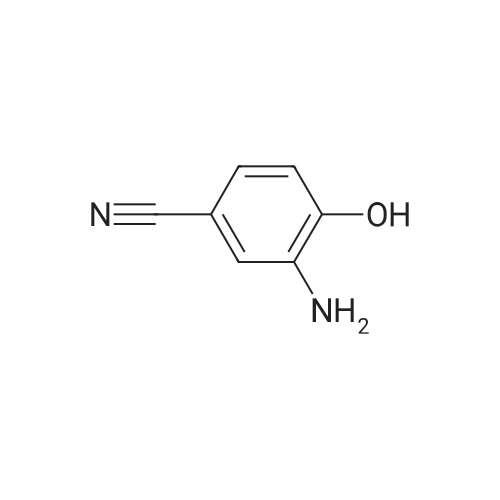
4
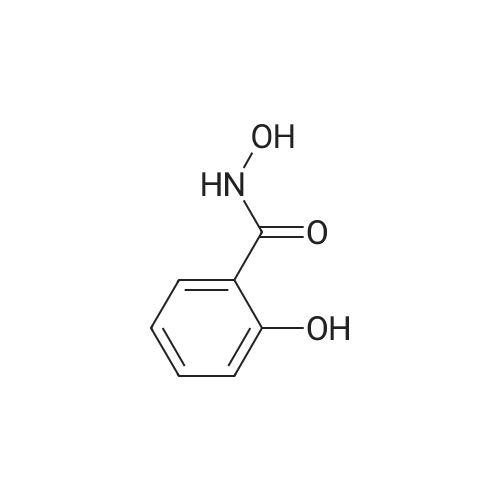
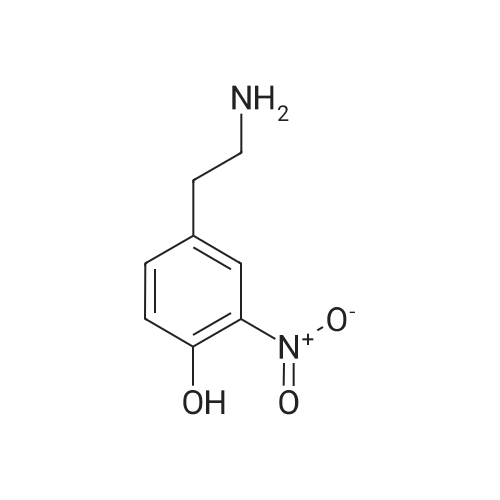
[ 89-73-6 ]
[ 939-80-0 ]
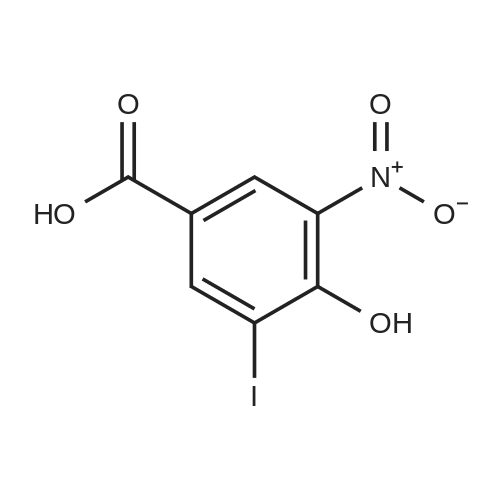
[ 59-49-4 ]
[ 3272-08-0 ]
Yield
Reaction Conditions
Operation in experiment
84%
With potassium carbonate In N,N-dimethyl-formamide at 20℃; for 18 h;
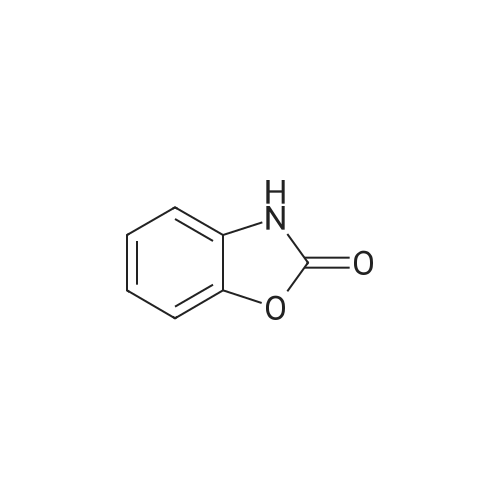
General procedure: To a solution of N-hydroxysalicylamide (0.5 g, 3.26 mmol) in anhydrous DMF (5 ml) was added 2a or 2b (3.26 mmol) and freshly calcinated K2CO3 (1.35 g, 9.78 mmol) and the mixture was kept, with stirring at the room temperature overnight. DMF was removed in vacuo and the residue was treated with water (10 ml) and then extracted with CH2Cl2 (5 ml). The organic layer was separated, washed with water, dried over anhydrous CaCl2, and purified by flesh chromatography on silica gel using CH2Cl2 as eluent. Dichloromethane was evaporated in vacuo. The resulting residue was benzo[d]oxazol-2(3H)-one 11. The water layer was acidify by hydrochloric acid, filtered and washed with water. The resulting residue was compound 12a(b).
Preparation of 3-amino-4-hydroxybenzonitrile. A solution of 4-hydroxy-3-nitrobenzonitrile (10 g, 61 mmol) in MeOH (200 mL) was treated with hydrogen gas through a balloon in the presence of 5percent Pd on active carbon (0.5 g) at room temperature for 24 hours. The mixture was filtered, and the filtrate was concentrated to give the product (8.2 g, 100percent).
77%
With hydrogenchloride; tin In ethanol; water for 0.75 h; Heating / reflux
3-AMINO4-HYDROXYBENZONITRILE : To a mixture of 5 G (30.5 MMOT) of 4-hydroxy-3-nitrobenzonitrile, 18.1 G of powder tin metal (152.5 mmol, 325 mesh), and 45 mL of ethanol was added with stirring a solution of 10 mL of concentrated HCI in 30 mL of H2O. The suspension was heated at reflux for 45 min and the resulting hot solution was poured into 100 ML of H20. Saturated aqueous NAHCO3 solution was slowly added to bring the pH to ca. 7. The suspension was filtered, and the residue was washed with MeOH giving the title compound (3.15 G, 77percent) as a white powder. Rf (CH2CI2/MEOH 9: 1) O. 47 ;1H NMR (CD30D) U 6. 96 (d, 1H, J=1. 3 Hz, H-2), 6.92 (d, 1H, J=8.1 Hz, H-6), 6.75 (d, 1H, J=8.1 Hz, H-5) ; 13C NMR (CD30D) 8 151.1 (C-4), 138.6 (C-3), 124.7 (C-6), 121. 6 (CN), 119. 1 (C- 2), 115.9 (C-5), 103.6 (C-1).
73%
With hydrogen In ethanol; ethyl acetate at 20℃; for 2 h;
Step B: 3-Amino-4-hydroxybenzonitrile; A mixture of 4-hydroxy-3-nitrobenzonitrile (100 mg, 0.61 mmol), palladium on charcoal (10 mg, 10 percent), EtOH (0.67 mL) and ethyl acetate (0.33 mL) was hydrogenated at rt for 2 h.Then the mixture was filtered and concentrated to give 3-amino-4-hydroxybenzonitrile 60 mg(73 percent) as a solid.1H NMR (300 MHz, CD3OD): δ 6.92 (dd, 1 H), 6.88 (d, 1 H), 6.74 (dd, 1 H).
67%
With 10% palladium on activated carbon; Degussa type; hydrogen In ethanol at 20℃; for 4 h;
To a solution of 4-hydroxy-3-nitrobenzonitrile (5g, 30.4mmol) in EtOH (lOOmL) was added 10percent Pd/C (4g). The reaction mixture was stirred at rt under H2 balloon atmosphere for 4 h. The TLC showed reaction to be complete. The reaction mixture was passed through a pad of celite. The celite was washed with EtOH (lOOmL). The filtrate was concentrated under reduced pressure to afford 3-amino-4- hydroxybenzonitrile as a black solid. Yield: 2.5g (67percent); 1H NMR (400 MHz, DMSOd 6): 9.02 (bs, 1H), 6.81-6.86 (m, 1H), 6.73 (d, J= 7.8 Hz, 1H), 6.49 (d, J= 7.8 Hz, 1H), 6.38 (bs, 1H), 6.17 (d, J = 6.5 Hz, 1H); MS (ESl+) for CHNOS m/z 165.10 [M+H].
49%
With hydrazine In methanol; ethanol; water at 20℃;
4-Hydroxy-3-nitrobenzonitrile (0.5 g, 3.05 mmol) was dissolved in ethanol (20 ml_) and methanol (10 ml_), and Raney-Nickel (1 ml_, 10percent aqueous solution) was added at rt followed by hydrazine mono hydrate (0.296 ml_, 6.09 mmol). The mixture was stirred at rt overnight. The mixture was then filtered through celite and concentrated by rotary evaporation. The residue was purified via chromatography (silica, n-hexane to ethyl acetate) to give 3-amino-4-hydroxybenzonitrile (D19, 200 mg, 49 percent) as an orange solid.M+H+ 135 1H NMR: δ (DMSO-d6, 400 MHz) 4.99 (2 H, br. s), 6.74 (1 H, d, J 7.9), 6.82-6.89 (2 H, m), 10.23 (1 H, br. s).
49%
With hydrazine hydrate In ethanol; water at 20℃;
Description 1. 3-Amino-4-hydroxybenzonitrile. (D1)4-Hydroxy-3-nitrobenzonitrile (0.5 g, 3.05 mmol) was dissolved in ethanol (20 ml.) and methanol (10 ml_), and Raney-Nickel (1 ml_, 10percent aqueous solution) was added at rt followed by hydrazine mono hydrate (0.296 ml_, 6.09 mmol). The mixture was stirred at rt overnight. The mixture was then filtered through celite and concentrated by rotary evaporation. The residue was purified via chromatography (silica, n-hexane to ethyl acetate) to give 3-amino-4-hydroxybenzonitrile (D1 , 200 mg, 49 percent) as an orange solid.M+H+ 135.
13 g
With 10% palladium on activated carbon; Degussa type; hydrogen In methanol at 20℃; for 8 h;
15 g of 2-nitro-4-cyanophenol and 1 g of 10percent palladium on carbon were added to 120 ml of methanol,Access to hydrogen,Stirred at room temperature for 8 hours,filter,Collecting filtrate,concentrate,To give 13 g of 3-amino-4-hydroxybenzonitrile.
Reference:
[1] Patent: US6207697, 2001, B1,
[2] Patent: US5886044, 1999, A,
[3] Patent: US5780483, 1998, A,
[4] Patent: US6262113, 2001, B1,
[5] Bioorganic and Medicinal Chemistry Letters, 2001, vol. 11, # 12, p. 1545 - 1548
[6] Journal of Medicinal Chemistry, 2010, vol. 53, # 1, p. 254 - 272
[7] European Journal of Medicinal Chemistry, 1998, vol. 33, # 12, p. 957 - 967
[8] Bioorganic and Medicinal Chemistry, 2002, vol. 10, # 8, p. 2663 - 2669
[9] Journal of Organic Chemistry, 1996, vol. 61, # 10, p. 3289 - 3297
[10] Patent: WO2005/12323, 2005, A2, . Location in patent: Page/Page column 55
[11] Patent: WO2007/110364, 2007, A1, . Location in patent: Page/Page column 61
[12] Journal of Medicinal Chemistry, 2006, vol. 49, # 17, p. 5080 - 5092
[13] Patent: WO2018/37223, 2018, A1, . Location in patent: Page/Page column 77
[14] Patent: WO2009/37294, 2009, A1, . Location in patent: Page/Page column 73
[15] Patent: WO2010/37723, 2010, A1, . Location in patent: Page/Page column 51
[16] Chemische Berichte, 1897, vol. 30, p. 991
[17] Proceedings of the Royal Society of London, Series B: Biological Sciences, 1946, vol. 133, p. 20,30
[18] Patent: WO2007/70173, 2007, A2, . Location in patent: Page/Page column 35; 42; 92
[19] Patent: US6921763, 2005, B2,
[20] Patent: EP2003132, 2008, A1, . Location in patent: Page/Page column 31
[21] Bioorganic and Medicinal Chemistry Letters, 2010, vol. 20, # 3, p. 1019 - 1022
[22] Patent: US2004/6083, 2004, A1, . Location in patent: Page/Page column 79
[23] Patent: CN106588808, 2017, A, . Location in patent: Paragraph 0021; 0022
21
[ 3272-08-0 ]
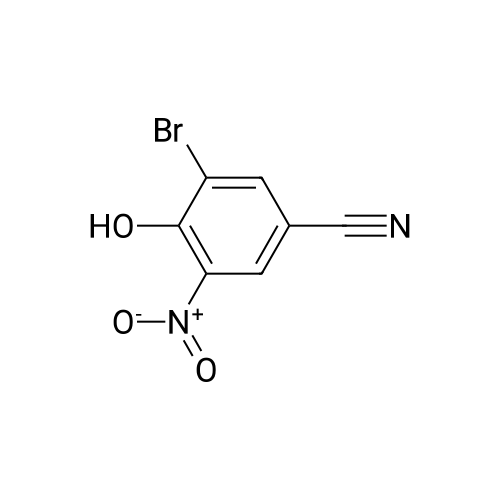
[ 14543-43-2 ]
Reference:
[1] Patent: US2002/156081, 2002, A1,
22
[ 3272-08-0 ]
[ 134997-74-3 ]
Reference:
[1] Bioorganic and Medicinal Chemistry, 2002, vol. 10, # 8, p. 2663 - 2669
[2] European Journal of Medicinal Chemistry, 1998, vol. 33, # 12, p. 957 - 967
Preparation of 3-amino-4-hydroxybenzonitrile. A solution of 4-hydroxy-3-nitrobenzonitrile (10 g, 61 mmol) in MeOH (200 mL) was treated with hydrogen gas through a balloon in the presence of 5% Pd on active carbon (0.5 g) at room temperature for 24 hours. The mixture was filtered, and the filtrate was concentrated to give the product (8.2 g, 100%).
97%
palladium-carbon; In methanol;
a) Preparation of 2-amino-4-cyanophenol To a solution of 2-nitro-4-cyanophenol(10 g, 61 mmol) in methanol(250 mL) was added 10% Pd/C (1 g). The mixture was flushed with argon, then hydrogen was bubbled through the solution for 10 min. and a hydrogen atmosphere was maintained at balloon pressure overnight The mixture was filtered through celite and the celite was washed with methanol. The solvent was evaporated and chromatography of the resulting solid on silica gel (5% MeOH/CH2 Cl2) gave the desired product(8.0 g, 97%). 1 H NMR (CD3 OD): delta 6.96 (d, 1H), 6.90 (dd, 1H), 6.77 (d, 1H).
97%
palladium-carbon; In methanol;
a)Preparation of 2-amino-4-cyanophenol To a solution of 2-nitro-4-cyanophenol(10 g, 61 mmol) in methanol(250 mL) was added 10% Pd/C (1 g). The mixture was flushed with argon, then hydrogen was bubbled through the solution for 10 min. and a hydrogen atmosphere was maintained at balloon pressure overnight. The mixture was filtered through celite and the celite was washed with methanol. The solvent was evaporated and chromatography of the resulting solid on silica gel (5%MeOH/CH2 Cl2) gave the desired product(8.0 g, 97%). 1 H NMR (CD3 OD): d 6.96 (d, 1H), 6.90 (dd, 1H), 6.77 (d, 1H).
97%
palladium-carbon; In methanol;
a) Preparation of 2-amino-4-cyanophenol To a solution of 2-nitro-4-cyanophenol (10 g, 6 mmol) in methanol (250 mL) was added 10% Pd/C (1 g). The mixture was flushed with argon, then hydrogen was bubbled through the solution for 10 min. and a hydrogen atmosphere was maintained at balloon pressure overnight. The mixture was filtered through celite and the celite was washed with methanol. The solvent was evaporated and chromatography of the resulting solid on silica gel (5%MeOH/CH2CH2) gave the desired product (8.0 g, 97%). 1H NMR (CD3OD): d 6.96 (d, 1H), 6.90 (dd, 1H), 6.77 (d, 1H).
92.3%
With palladium on activated charcoal; hydrogen; In methanol; for 6h;
Add 39.0g (0.238mol) to a 1000mL single-mouth bottle 3-nitro-4-hydroxybenzonitrile, 700 mL methanol, 3.9 g Pd/C under hydrogen Stir for 6h, the reaction is complete,After suction filtration, the filter cake was washed with a small amount of anhydrous methanol (5 mL×2).The filtrate was collected, and all the solvent was distilled off under reduced pressure.Concentrate to dryness and scrape off the residue.Obtained 29.5 g of a brown solid, yield: 92.3%,
92.3%
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 12h;
A mixture of the compound 2 (39.0 g, 0.238 mol) and 10% Pd/C(3.9 g) in methanol was stirred at room temperature for 12 h underhydrogen atmosphere. After the completion of the reaction, the Pd/C was filtered out and the filtrate was evaporated to give a brownsolid (29.5 g, 92.3%), which was used directly in the next step. Mp135.6 C-136.6 C. MS (ESI) m/z: 480.8 [2M Na]; 1H NMR(600 MHz, DMSO-d6) delta 9.79 (s, 1H), 8.26 (d, J 2.1 Hz, 1H), 8.07 (dd,J 8.8, 2.1 Hz, 1H), 7.58 (d, J 8.9 Hz, 1H), 4.92 (hept, J 6.0 Hz, 1H),1.27 (d, J 6.0 Hz, 6H).
84.5%
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 2h;
4-hydroxy-3-nitrobenzonitrile (100 g, 0.61 mol), Pd-C (15 g, 10%) and 500 mL of anhydrous methanol were added to a 1000 mL vial.The reaction was hydrogen-protected for 2 hours at room temperature. After the reaction was completed, Pd-C was removed by suction filtration, and the filtrate was concentrated to dryness under reduced pressure to give a dark brown solid powder. Yield: 84.5%. Mp 137.1 C -140.6 C.
77%
With hydrogenchloride; tin; In ethanol; water; for 0.75h;Heating / reflux;
3-AMINO4-HYDROXYBENZONITRILE : To a mixture of 5 G (30.5 MMOT) of 4-hydroxy-3-nitrobenzonitrile, 18.1 G of powder tin metal (152.5 mmol, 325 mesh), and 45 mL of ethanol was added with stirring a solution of 10 mL of concentrated HCI in 30 mL of H2O. The suspension was heated at reflux for 45 min and the resulting hot solution was poured into 100 ML of H20. Saturated aqueous NAHCO3 solution was slowly added to bring the pH to ca. 7. The suspension was filtered, and the residue was washed with MeOH giving the title compound (3.15 G, 77%) as a white powder. Rf (CH2CI2/MEOH 9: 1) O. 47 ;1H NMR (CD30D) U 6. 96 (d, 1H, J=1. 3 Hz, H-2), 6.92 (d, 1H, J=8.1 Hz, H-6), 6.75 (d, 1H, J=8.1 Hz, H-5) ; 13C NMR (CD30D) 8 151.1 (C-4), 138.6 (C-3), 124.7 (C-6), 121. 6 (CN), 119. 1 (C- 2), 115.9 (C-5), 103.6 (C-1).
73%
With hydrogen;palladium 10% on activated carbon; In ethanol; ethyl acetate; at 20℃; for 2h;
Step B: 3-Amino-4-hydroxybenzonitrile; A mixture of 4-hydroxy-3-nitrobenzonitrile (100 mg, 0.61 mmol), palladium on charcoal (10 mg, 10 %), EtOH (0.67 mL) and ethyl acetate (0.33 mL) was hydrogenated at rt for 2 h.Then the mixture was filtered and concentrated to give 3-amino-4-hydroxybenzonitrile 60 mg(73 %) as a solid.1H NMR (300 MHz, CD3OD): delta 6.92 (dd, 1 H), 6.88 (d, 1 H), 6.74 (dd, 1 H).
67%
With palladium 10% on activated carbon; hydrogen; In ethanol; at 20℃; for 4h;
To a solution of 4-hydroxy-3-nitrobenzonitrile (5g, 30.4mmol) in EtOH (lOOmL) was added 10% Pd/C (4g). The reaction mixture was stirred at rt under H2 balloon atmosphere for 4 h. The TLC showed reaction to be complete. The reaction mixture was passed through a pad of celite. The celite was washed with EtOH (lOOmL). The filtrate was concentrated under reduced pressure to afford 3-amino-4- hydroxybenzonitrile as a black solid. Yield: 2.5g (67%); 1H NMR (400 MHz, DMSOd 6): 9.02 (bs, 1H), 6.81-6.86 (m, 1H), 6.73 (d, J= 7.8 Hz, 1H), 6.49 (d, J= 7.8 Hz, 1H), 6.38 (bs, 1H), 6.17 (d, J = 6.5 Hz, 1H); MS (ESl+) for CHNOS m/z 165.10 [M+H].
49%
With hydrazine;nickel; In methanol; ethanol; water; at 20℃;
4-Hydroxy-3-nitrobenzonitrile (0.5 g, 3.05 mmol) was dissolved in ethanol (20 ml_) and methanol (10 ml_), and Raney-Nickel (1 ml_, 10% aqueous solution) was added at rt followed by hydrazine mono hydrate (0.296 ml_, 6.09 mmol). The mixture was stirred at rt overnight. The mixture was then filtered through celite and concentrated by rotary evaporation. The residue was purified via chromatography (silica, n-hexane to ethyl acetate) to give 3-amino-4-hydroxybenzonitrile (D19, 200 mg, 49 %) as an orange solid.M+H+ 135 1H NMR: delta (DMSO-d6, 400 MHz) 4.99 (2 H, br. s), 6.74 (1 H, d, J 7.9), 6.82-6.89 (2 H, m), 10.23 (1 H, br. s).
49%
With hydrazine hydrate;Raney nickel; In ethanol; water; at 20℃;
Description 1. 3-Amino-4-hydroxybenzonitrile. (D1)4-Hydroxy-3-nitrobenzonitrile (0.5 g, 3.05 mmol) was dissolved in ethanol (20 ml.) and methanol (10 ml_), and Raney-Nickel (1 ml_, 10% aqueous solution) was added at rt followed by hydrazine mono hydrate (0.296 ml_, 6.09 mmol). The mixture was stirred at rt overnight. The mixture was then filtered through celite and concentrated by rotary evaporation. The residue was purified via chromatography (silica, n-hexane to ethyl acetate) to give 3-amino-4-hydroxybenzonitrile (D1 , 200 mg, 49 %) as an orange solid.M+H+ 135.
With ammonium formate;palladium 10% on activated carbon; In methanol; at 20℃; for 1h;Heating / reflux;Product distribution / selectivity;
To a suspension of 10.0 g of 4-hydroxy-3-nitrobenzonitrile and 19.0 g of ammonium formate in 150 mL of MeOH was added portionwise 1.0 g of 10% palladium on carbon. The mixture spontaneously refluxed after addition of the catalyst was complete, and was maintained at reflux with the use of a heating bath for a total of 1 h. The mixture was cooled, filtered through a pad of Celite, washing liberally with MeOH, and concentrated. The residue was preadsorbed onto ca. 25 g of silica gel and purified by flash column chromatography on a Biotage Horizon, 65i column, eluting with 1 column volume of 5% EtOAc in CH2Cl2, followed by a linear gradient of EtOAc in CH2Cl2 from 5 to 100% over 10 column volumes to provide the title compound. Mass spectrum (ESI) 135.0 (M+l).; A mixture of 4-hydroxy-3-nitrobenzonitrile (328 mg, 2.00 mmol), NH4O2CH (631 mg, 10 mmol) and 10% Pd/C (55 mg) in MeOH (5 mL) was stirred at room temperature overnight. The mixture was concentrated under reduced pressure and diluted with EtOAc and brine. The aqueous layer was extracted with EtOAc (3 x) and the combined organic extracts were dried (MgSO4) and concentrated under reduced pressure to afford the crude product. This was purified by flash column chromatography (Si, 40 x 230 mm, 0-30% EtOAc in CH2Cl2 gradient) to afford 2-amino-4-bromo-6-methylphenol as a solid. LCMS calc. = 135.03; found = 135.06 (M+l)+. 1H NMR (500 MHz, CD3OD) delta 6.97 (d, J = 2.0 Hz, IH), 6.92 (dd, J = 8.0, 2.0 Hz, IH), 6.77 (d, J = 8.0 Hz, IH).
With sodium thiosulfate; In ethanol; water;
A. 3-amino-4-hydroxybenzonitrile To a mixture of 4-hydroxy-3-nitrobenzonitrile (4 g, 0.0244 mol) in ethanol (180 mL) and water (90 mL) was added sodium thiosulfate (17 g, 0.0976 mol) at room temperature. The heterogeneous mixture was stirred at 80 C. under an atmosphere of nitrogen for 1 hour. The reaction mixture was cooled to room temperature, and ethanol was removed under reduced pressure. The yellow solid was filtered, washed with water, and dried under reduced pressure to yield 3-amino-4-hydroxybenzonitrile (1.46 g, 0.011 mol). RP-HPLC (Delta Pak C18, 5 mum, 300 A, 15 cm; 5%-95% acetonitrile-0.1M ammonium acetate over 10 min, 1 mL/min) Rt 4.5 min. MS: MH: 133.
With iron; ammonium chloride; In tetrahydrofuran; ethanol; water; at 70℃; for 0.5h;Celite; Heating / reflux;
To a mixed solution of 4-hydroxy-3-nitrobenzonitrile (1 g) and NH4Cl (163 mg) in ethanol (20 ml), THF (10 ml), and water (10 ml) were added Celite (5 g) and reduced iron (1.7 g), followed by heating under reflux at 70C for 30 min. The reaction solution was cooled to room temperature, diluted with EtOAc (200 ml), and then filtered through celite. The solution was washed with saturated brine, the organic layer was dried over anhydrous MgSO4, and filtered, and the filtrate was concentrated under reduced pressure to obtain 3-amino-4-hydroxybenzonitrile (740 mg) as a pale brown solid.
With water; sodium thiosulfate; In ethanol; at 80℃; for 1h;
[0971] To a mixture of 4-hydroxy-3-nitrobenzonitrile (4 g, 0.0244 mol) in ethanol (180 mL) and water (90 mL) was added sodium thiosulfate (17 g, 0.0976 mol) at room temperature. The heterogeneous mixture was stirred at 80 C. under an atmosphere of nitrogen for 1 hour. The reaction mixture was cooled to room temperature, and ethanol was removed under reduced pressure. The yellow solid was filtered, washed with water, and dried under reduced pressure to yield 3-amino-4-hydroxybenzonitrile (1.46 g, 0.011 mol). [0972] RP-HPLC (Delta Pak C18, 5 mum, 300A, 15 cm; 5%-95% acetonitrile-0.1M ammonium acetate over 10 min, 1 mL/min) Rt 4.5 min. MS: MH-: 133
13 g
With palladium 10% on activated carbon; hydrogen; In methanol; at 20℃; for 8h;
15 g of 2-nitro-4-cyanophenol and 1 g of 10% palladium on carbon were added to 120 ml of methanol,Access to hydrogen,Stirred at room temperature for 8 hours,filter,Collecting filtrate,concentrate,To give 13 g of 3-amino-4-hydroxybenzonitrile.
With potassium carbonate; In DMF (N,N-dimethyl-formamide); acetonitrile; at 60℃; for 4h;
A. A solution of <strong>[3272-08-0]3-nitro-4-hydroxybenzonitrile</strong> (10 g, 61 mmol) in a mixture of 40 mL of dimethylformamide and 100 mL of acetonitrile was stirred as anhydrous potassium carbonate (10.1 g, 73.2 mmol) and benzyl bromide (10.4 g, 61 mmol) were added. The reaction was heated at 60 C. for about 4 h. The reaction was filtered and the solid was washed with ethyl acetate. The filtrate was washed with water. The organic layers were washed with water and brine, dried and concentrated. The solid was recrystallized from acetone/hexane to give 14 g (90%) of 3-nitro-4-(benzyloxy)benzonitrile, as a white solid.
With iron(III) chloride; bromine; acetic acid; at 50℃; for 2h;
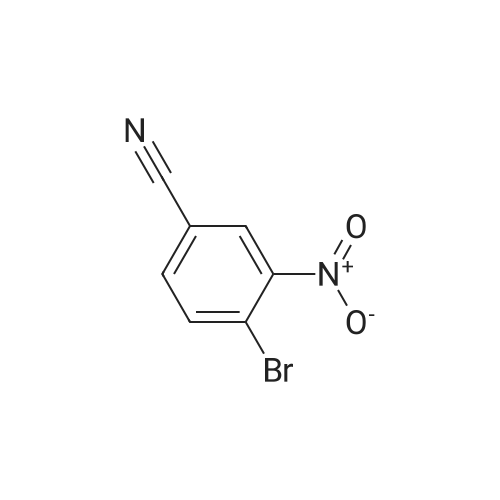
Step 1. 3-bromo-4-hydroxy-5-nitrobenzonitrile Bromine (500 mg, 3 mmol) was added to a mixture of <strong>[3272-08-0]4-hydroxy-3-nitrobenzonitrile</strong> (500 mg, 3 mmol) (Aldrich cat 344575), ferric chloride (100 mg, 0.9 mmol) and acetic acid (20 mL) at room temperature. The reaction mixture was heated to 50 C for 2 h, allowed to cool to room temperature, and water (100 mL) was added. A precipitate slowly formed, was collected, washed with water, and dried to obtain 3-bromo-4-hydroxy-5-nitrobenzonitrile (0.50 g, 70%) as a yellow solid. LCMS calculated for C7H4BrN203 (M+H)+: m/z = 242.9, 244.9; found = 242.9, 244.9.
57%
With potassium bromate; sulfuric acid; In water; at 20 - 35℃; for 22h;
3-Bromo-4-hydroxy-5-nitrobenzonitrile : To a mixture of 5 G (30.5 MMOL) of <strong>[3272-08-0]4-hydroxy-3-nitrobenzonitrile</strong> in H2SO4 solution (50 mL of concentrated H2SO4 + 50 mL of H2O) at 25 C, 7.9 G (47.3 MMOL) of potassium bromate were added in small portions cooling the flask with an ice-bath and maintaining the temperature between 25 and 35 C. After the addition was completed, the reaction was stirred at room temperature for 22 h and then filtered. The pale yellow solid was washed with water and dried to give 4.2 G (57%) of the title compound. M. p.: 162-164 C ; 1H NMR (CD30D) 8 8.54 (d, 1H, J=2. 0 Hz, H-6), 8.32 (d, 1H, J=2. 0 Hz, H-2) ; 13C NMR (DMSO-d6) 5 153.9 (C-4), 140.8 (C-2), 138.2 (C- 5), 130.1 (C-6), 117.0 (CN), 115.9 (C-3), 101.6 (C-1).
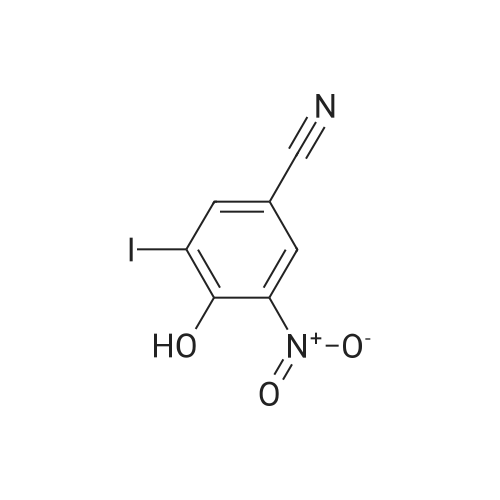
With potassium carbonate; In N,N-dimethyl-formamide; at 10 - 35℃; for 2h;
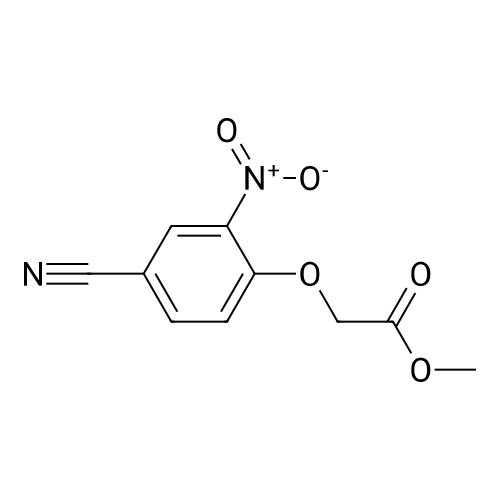
To a solution of <strong>[3272-08-0]4-cyano-2-nitrophenol</strong> (8.21 g, 50.0 mmol) and methyl bromoacetate (5.2 ml, 55 mmol) in N,N-dimethylformamide (50 ml) was added potassium carbonate (8.30 g, 60.1 mmol), and the mixture was stirred for 2 hours at room temperature. The reaction mixture was combined with water, and extracted twice with ethyl acetate. The combined organic layer was washed twice with water, 0.5 M hydrochloric acid and water, and concentrated under reduced pressure. The residue was washed with a mixed solution of ethyl acetate-diisopropyl ether to give the title compound (6.41 g, yield 54%). 1H NMR (CDCl3) delta 3.83 (3H, s), 4.89 (2H, s), 7.06 (1H, d, J = 8.9 Hz), 7.81 (1H, dd, J = 8.9, 2.1 Hz), 8.19 (1H, d, J = 2.1 Hz).
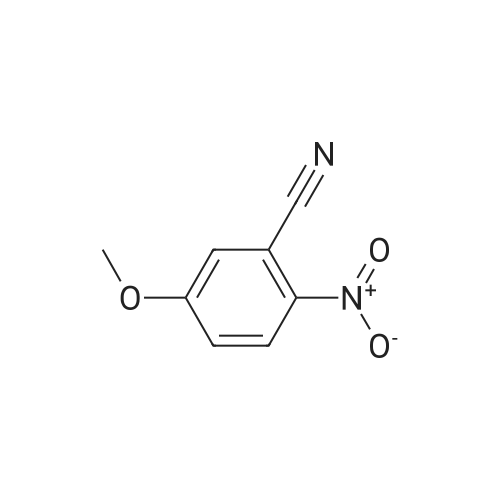
a) 5-Cyano-2-methoxynitrobenzene (Compound 18A) A suspension of 8.37 g of <strong>[3272-08-0]4-hydroxy-3-nitrobenzonitrile</strong>, 27.64 g of anhydrous potassium carbonate and 6 mL of dimethyl sulphate in 60 mL of acetonitrile was stirred at reflux for 2 hours. The solvent was removed by evaporation in vacuo. The residue was taken up in 100 mL of water and extracted with 3*80 mL of chloroform. The combined organic layers were dried over sodium sulphate and the solvent was evaporated off to afford 8.8 g (98.8%) of Compound 18A as an ivory solid.
8.8 g (98.8%)
With potassium carbonate; In water; acetonitrile;
a) 5-Cyano-2-methoxynitrobenzene (Compound 18A) A suspension of 8.37 g of <strong>[3272-08-0]4-hydroxy-3-nitrobenzonitrile</strong>, 27.64 g of anhydrous potassium carbonate and 6 ml of dimethyl sulphate in 60 ml of acetonitrile was stirred at reflux for 2 hours. The solvent was removed by evaporation in vacuo. The residue was taken up in 100 ml of water and extracted with 3 x 80 ml of chloroform. The combined organic layers were dried over sodium sulphate and the solvent was evaporated off to afford 8.8 g (98.8%) of Compound 18A as an ivory solid. 1H-NMR (200MHz, DMSO-d6, delta): 8.46, d, 1H, H6; 8.13, dd, 1H, H4; 7.54, d, 1H, H3; 4.00, s, 3H, CH3O.
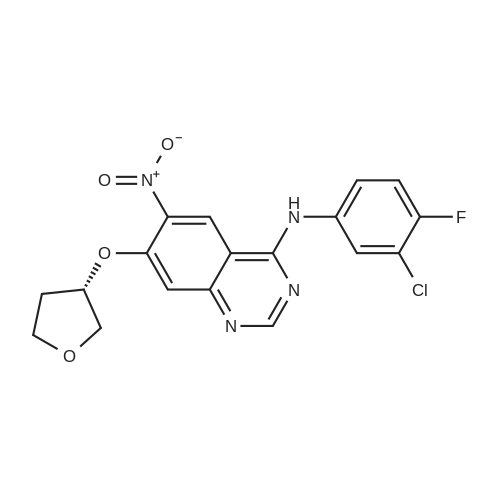
trans-N2-(4-{4-Amino-1-[4-(4-methylpiperazino)cyclohexyl]-1H-pyrazolo [3,4-d]pyrimidin-3-yl}-2-methoxyphenyl)benzo[b]furan-2-carboxamide P[ No CAS ]
[ 3272-08-0 ]
[ 14543-43-2 ]
Yield
Reaction Conditions
Operation in experiment
With sodium thiosulfate; In ethanol; water;
A. 3-amino-4-hydroxybenzonitrile To a mixture of 4-hydroxy-3-nitrobenzonitrile (4 g, 0.0244 mol) in ethanol (180 mL) and water (90 mL) was added sodium thiosulfate (17 g, 0.0976 mol) at room temperature. The heterogeneous mixture was stirred at 80 C. under an atmosphere of nitrogen for 1 hour. The reaction mixture was cooled to room temperature, and ethanol was removed under reduced pressure. The yellow solid was filtered, washed with water, and dried under reduced pressure to yield 3-amino-4-hydroxybenzonitrile (1.46 g, 0.011 mol). RP-HPLC (Delta Pak C18, 5 mum, 300 A, 15 cm; 5%-95% acetonitrile-0.1M ammonium acetate over 10 min, 1 mL/min) Rt 4.5 min. MS: MH-: 133
With trifluorormethanesulfonic acid; trimethylsilylazide; In acetonitrile; at 20℃; for 0.75h;Sealed tube; Inert atmosphere;
General procedure: To a solution of an aromatic aldehyde 1 (0.500 mmol, 1.0 equiv) and TMSN3 (115 mg, 1.00 mmol,2.0 equiv) in a premixed HFIP/ACN mixture (2.0 mL, 1:1) in a nitrogen-flushed two dram vial wasadded triflic acid (TfOH; 17.7 L, 0.200 mmol, 0.40 equiv) (exotherm and brisk effervescence due tonitrogen gas evolution was immediately observed). The vial was capped and the reaction mixture wasallowed to stir at rt for 20-75 min. The reaction mixture was concentrated under nitrogen. The residueobtained was suspended in CH2Cl2/hexanes mixture and loaded on a silica gel in a 5 g samplecartridge. Purification using a normal phase silica flash column on a CombiFlash purification systemafforded a corresponding aromatic nitrile 2 upon concentration of appropriate fractions.
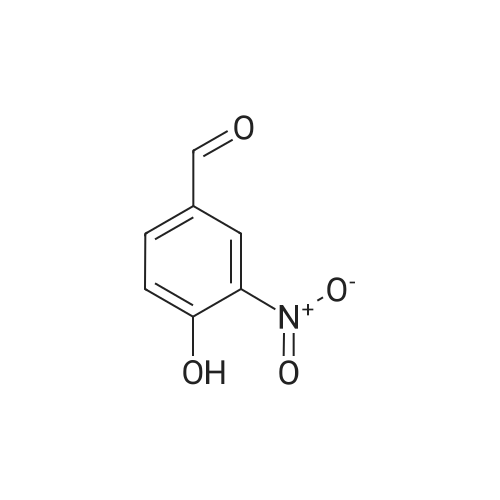
With hydroxylamine hydrochloride; sodium formate; In formic acid; water;
(1) A mixture of 5.0 g of 4-hydroxy-3-nitrobenzaldehyde with 2.5 g of hydroxylamine hydrochloride and 3.6 g of sodium formate was heated under reflux in 35 ml of formic acid for 5 hours. After the reaction mixture was cooled, water was added thereto. The precipitated crystal was collected by filtration to give 4.3 g of 4-hydroxy-3-nitrobenzonitrile.
3-Nitro-4-(2-oxiranylmethoxy)benzonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
65%
With potassium carbonate; In N,N-dimethyl-formamide; acetonitrile;
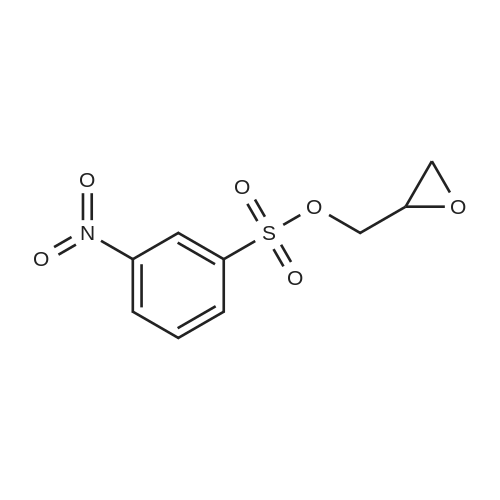
(a) 3-Nitro-4-(2-oxiranylmethoxy)benzonitrile <strong>[3272-08-0]4-Cyano-2-nitrophenol</strong> (0.80 g; 4.9 mmol) and K2CO3 (0.68 g; 4.9 mmol) were refluxed in MeCN (40 mL) for 1 h. The solvent was removed on a rotary evaporator and the residue dissolved in DMF (10 mL). 2-[(3-Nitrophenyl)sulfonyloxy]methyl}oxirane (1.2 g, 4.9 mmol; see Example B above) was added to the resultant solution. The solution was stirred at 40 C. for 12 h and filtered. The precipitate was washed with H2O to give the sub-title compound in a 65% yield.
With hydrogenchloride; In 1,4-dioxane; at 20℃;Cooling with ice;
EXAMPLE 18 <n="96"/>N-(4-chloro-3-(trifluoromethyI)phenyl)-5-(4,5-dihydro-lH-imidazol-2- yl)benzo[d]oxazol-2-amine (39) Step 1Synthesis of ethyl 4-hydroxy-3-nitrobenzimidate hydrochloride (34) A stirred mixture of <strong>[3272-08-0]4-hydroxy-3-nitrobenzonitrile</strong> 33 (5.2 g, 32 mmol) in a solution of abs. ethanol (250 ml) and dioxane (40 ml) was cooled in an ice bath as dry HCl gas was bubbled through it for lhr. The mixture was stirred overnight at room temperature and the resulting precipitate was collected, washed with ether and dried to give 6.2 g (79%) of the imidate ester HCl 34. 1H NMR (600MHz, DMSO-(I6) delta 1 1.8 (br s, 1 H), 8.64 (s, IH), 8.21 (d, IH, J= 9 Hz), 7.38 (d, IH, J= 9 Hz), 4.57 (q, 2H, J= 7.2 Hz), 1.47 (t, 3H, J= 7.2 Hz).
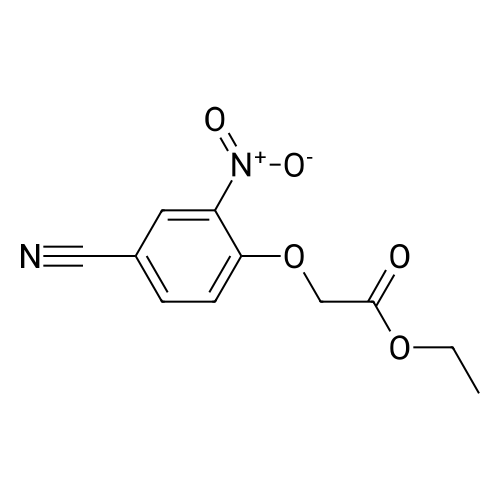
3-Oxo-3,4-dihydro-2H-benzo[1,4]oxazine-6-carbonitrile 24 mmol of 4-Hydroxy-3-nitrobenzonitrile were dissolved in DMF. 1.1 eq of Cs2CO3 were added and the reaction stirred at rt for 15 min. 1.5 eq of ethylbromo acetate were added and the reaction mixture heated to 50 C. for 2 h. The intermediate was isolated via extraction from ethylacetate/water. The organic layer was separated and dried over Na2SO4. After evaporation the resulting solid was redissolved in MeOH and sat. NH4Cl (1:1). 8 g of Zn powder were added and the suspension stirred at rt for 2 h. The solid was filtered off and the organic layer evaporated. Ethylacetate was added and the organic layer washed with sat. NaHCO3. The organic layer was separated again, dried over Na2SO4 and reduced resulting in a light brown solid. MS(ISO): 175.2 (MH+)
General procedure: Potassium carbonate (1-1.6 equiv) was added to a stirred solution of the phenol (1 equiv) in DMF. After 30 min the cyanobenzyl bromide (1-1.2 equiv) was added, and the mixture was stirred until completion (1-4 days). The reaction mixture was poured over ice or diluted with water. The precipitated product was filtered off and recrystallized if needed.
[00173] Intermediate 12A. 3-Amino-4-(benzyloxy)benzonitrile: Benzyl bromide (3.23 mL, 26.8 mmol) was added to a solution of potassium carbonate (3.40 g, 24.5 mmol) and <strong>[3272-08-0]4-hydroxy-3-nitrobenzonitrile</strong> (4.00 g, 24.4 mmol) in DMF (50 mL) and stirred at room temperature for 16 h. The reaction was diluted with water and EtOAc. The aqueous layer was extracted with EtOAc. The combined organics were washed with saturated NaHCC^ solution and brine, dried over MgS04, filtered, and concentrated. The crude was taken up in ethanol (200 mL) and zinc (15.9 g, 243 mmol) and ammonium chloride (13.0 g, 243 mmol) were added. The reaction was stirred at room temperature for 24 h. The reaction was diluted in EtOAc, filtered through CELITE, and concentrated to give Intermediate 12A (5.4 g, 100%), which was taken directly on to the next step. LCMS (ESI) m/z 225 (M+H)+, RT = 1.77 min (Method A).
Benzyl bromide (3.23 mL, 26.8 mmol) was added to a solution of potassium carbonate (3.4 g, 24 mmol) and 4- hydroxy-3-nitrobenzonitrile (4.0 g, 24 mmol) in DMF (50 mL) and stirred at rt for 16 h. The reaction was diluted with water and EtOAc. The aqueous layer was extracted with EtOAc and then the combined organics were washed with sat'd NaHC03 solution, brine, and then dried over MgS04, filtered and evaporated to give crude ether product. This crude was taken up in ethanol (200 mL) and zinc (15.9 g, 243 mmol) and ammonium chloride (13.0 g, 243 mmol) were added and stirred at rt for 24 hrs. The reaction was diluted in EtOAc and filtered through CELITE and then concentrated to give Intermediate 6A which was taken directly on to the next step. LCMS (ESI) m/z 225 (M+H)+, RT = 1.77 min (Method A).
With 1,4-diaza-bicyclo[2.2.2]octane; In neat (no solvent); at 55℃; for 12h;
General procedure: To 1.0 equiv. of phenol were added successively 1.0 equiv. of DABCO, 2.0 equiv. of aldehyde and 1.0 equiv. of isocyanide. The resulting mixture was stirred neat at 55 C for 12 h. The crude product was purified by flash chromatography on silica gel.
With potassium carbonate; In N,N-dimethyl-formamide; at 20℃; for 18h;
General procedure: To a solution of N-hydroxysalicylamide (0.5 g, 3.26 mmol) in anhydrous DMF (5 ml) was added 2a or 2b (3.26 mmol) and freshly calcinated K2CO3 (1.35 g, 9.78 mmol) and the mixture was kept, with stirring at the room temperature overnight. DMF was removed in vacuo and the residue was treated with water (10 ml) and then extracted with CH2Cl2 (5 ml). The organic layer was separated, washed with water, dried over anhydrous CaCl2, and purified by flesh chromatography on silica gel using CH2Cl2 as eluent. Dichloromethane was evaporated in vacuo. The resulting residue was benzo[d]oxazol-2(3H)-one 11. The water layer was acidify by hydrochloric acid, filtered and washed with water. The resulting residue was compound 12a(b).






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping