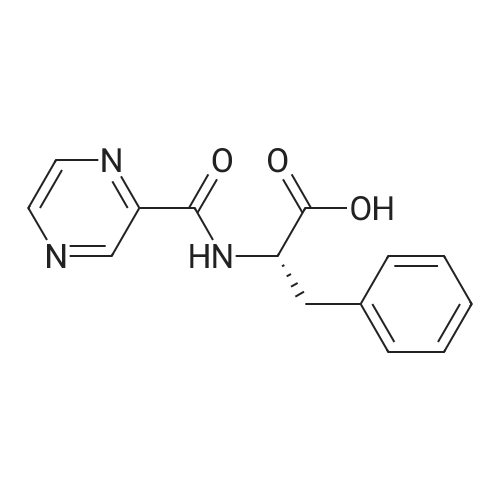
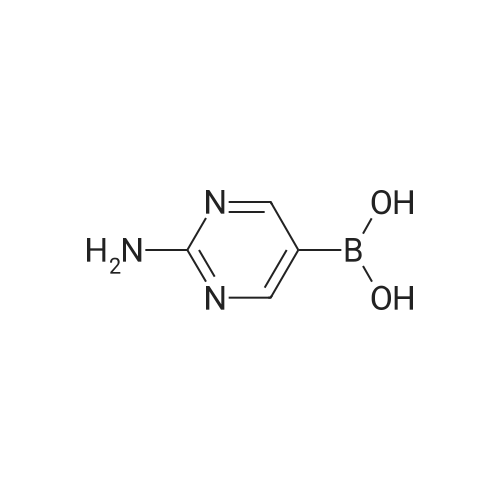
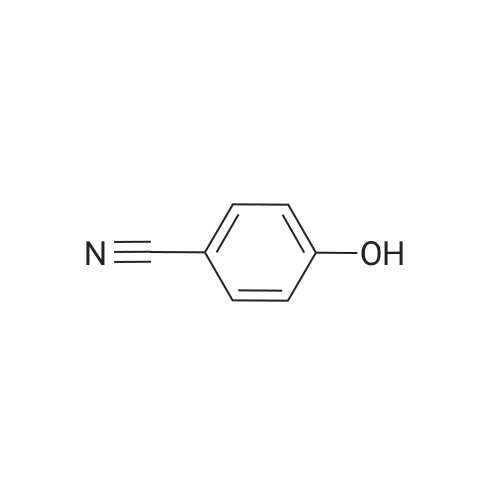
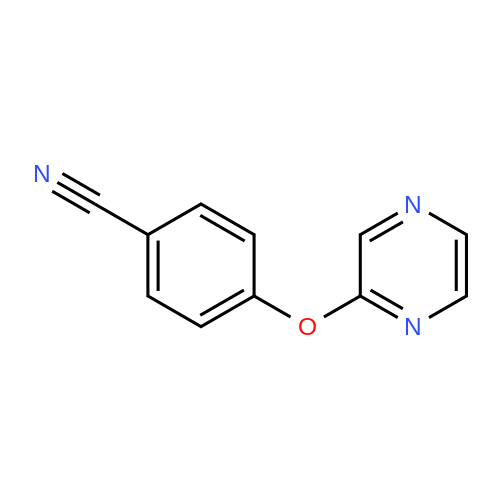
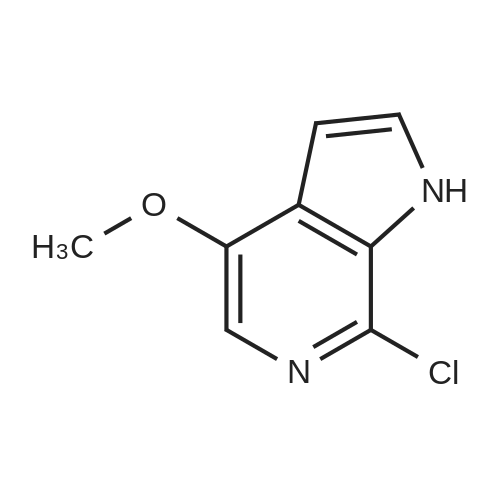
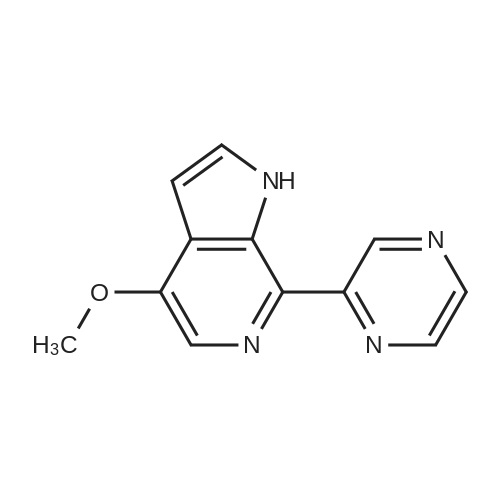
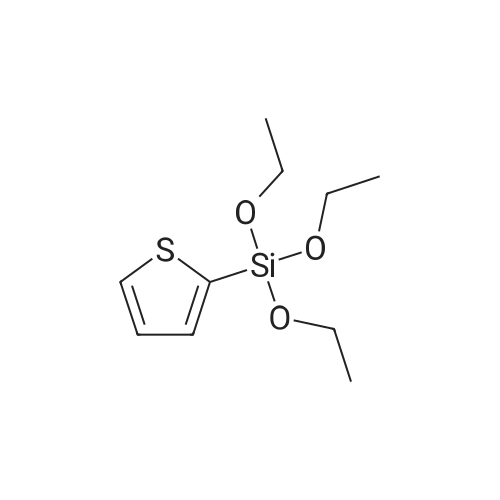
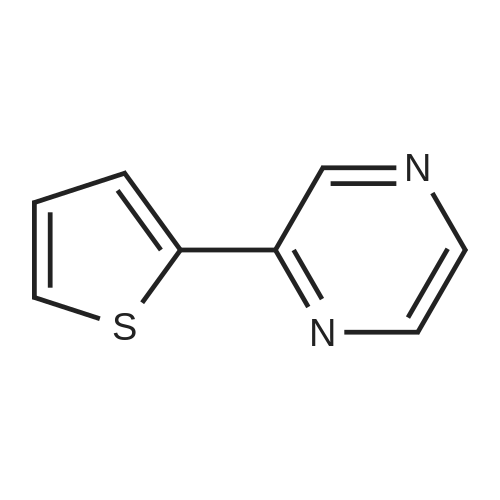
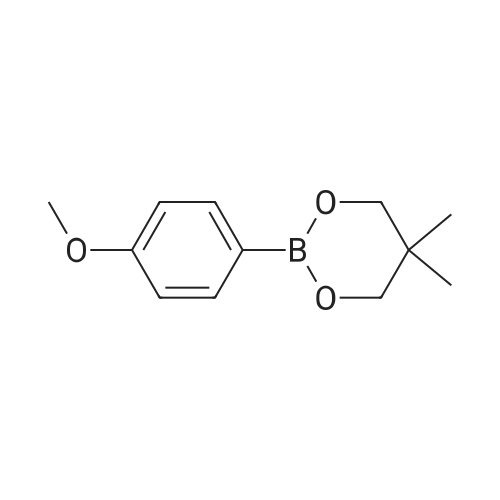
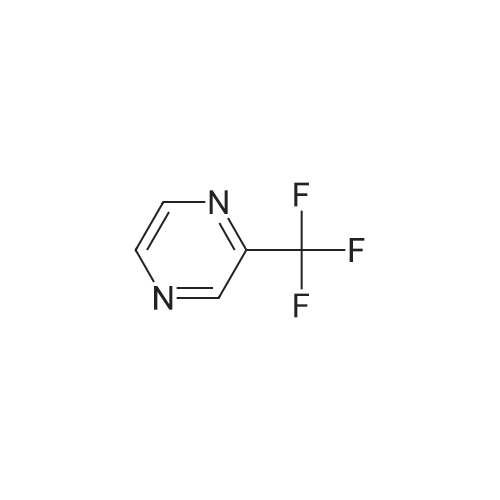
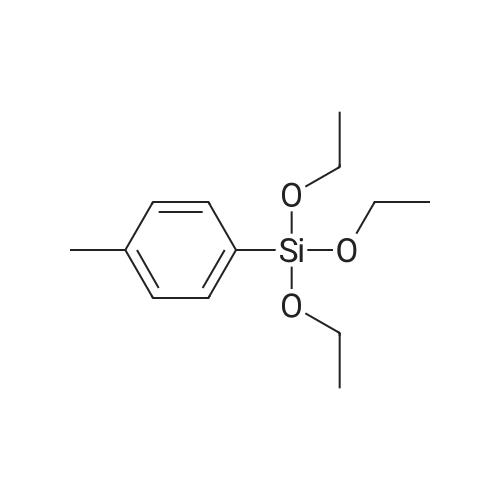
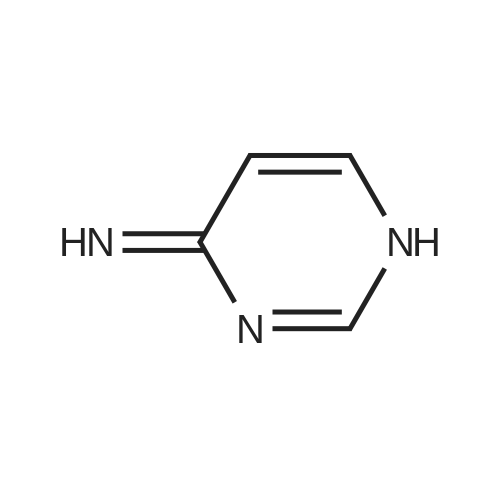
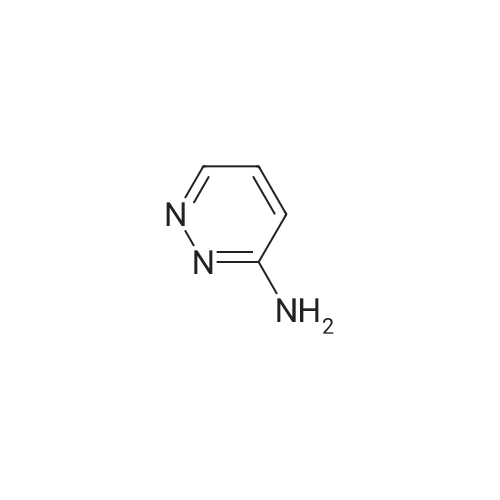
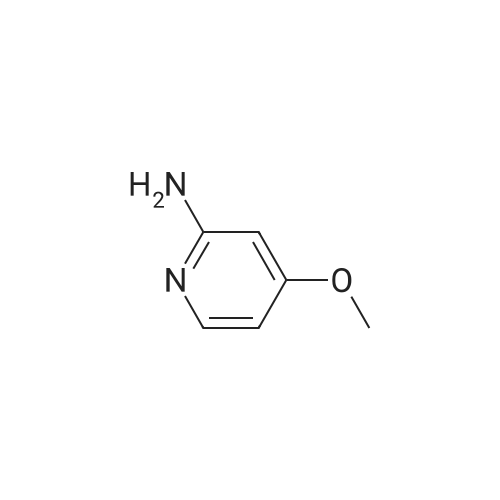
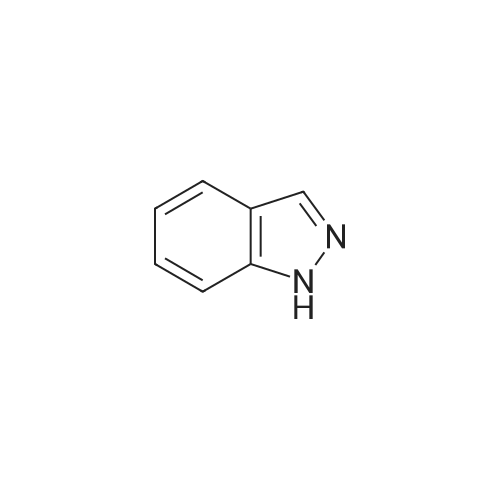
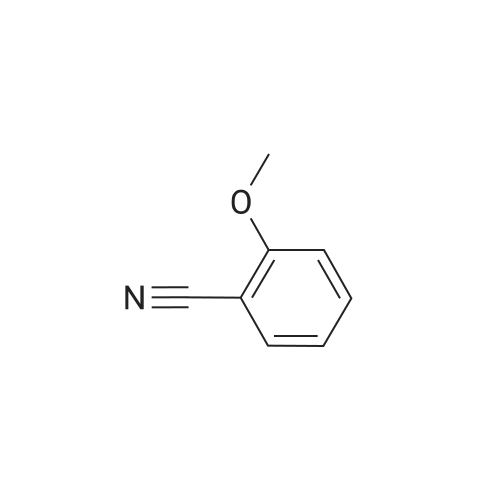
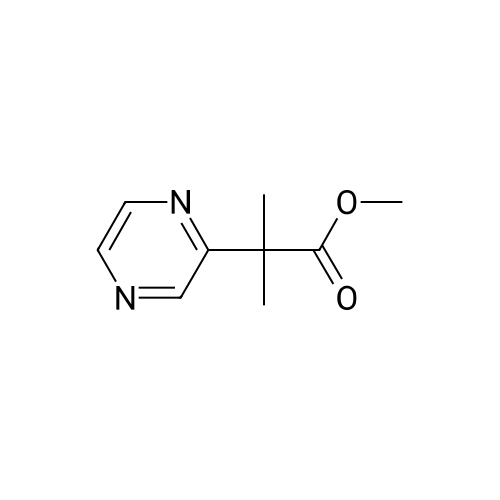
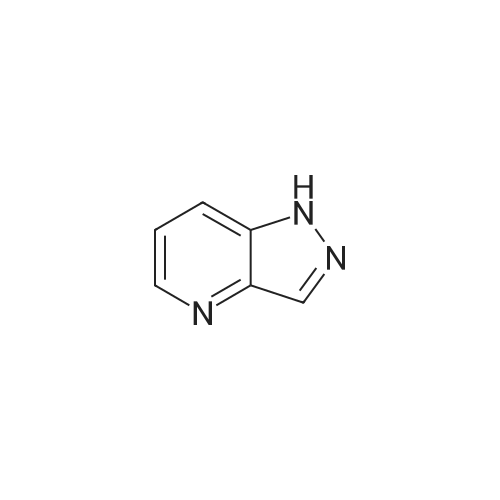
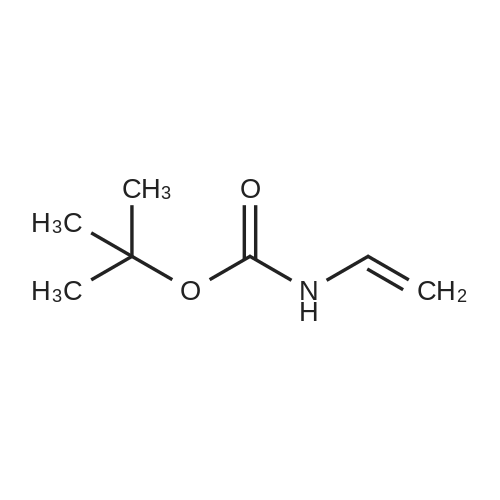
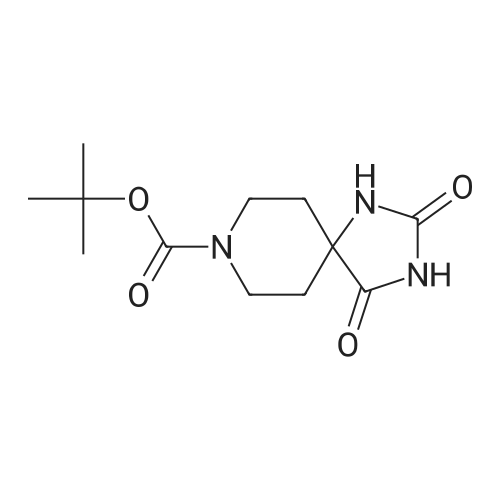
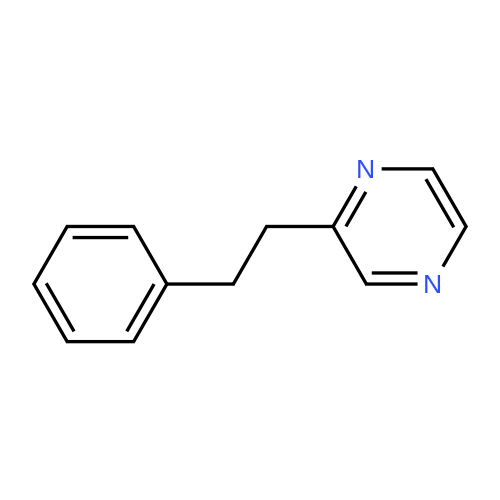
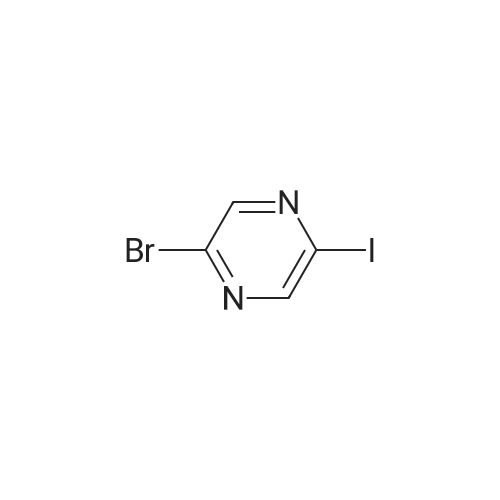
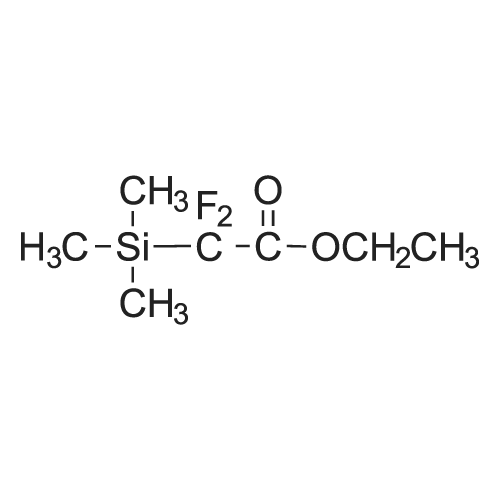| 78% |
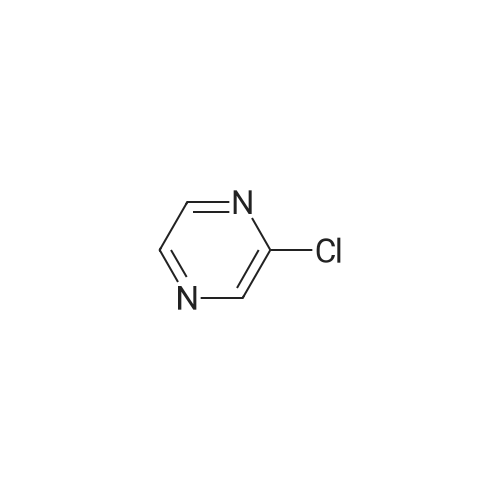
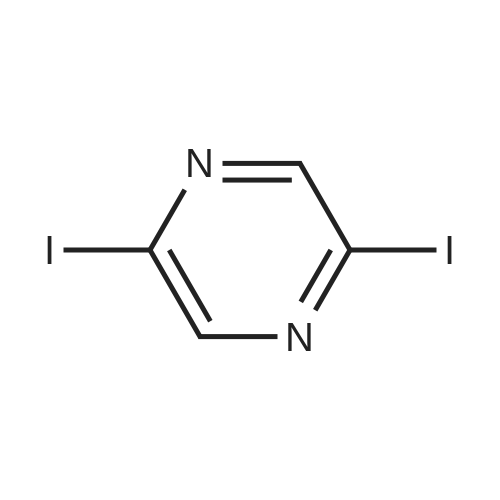
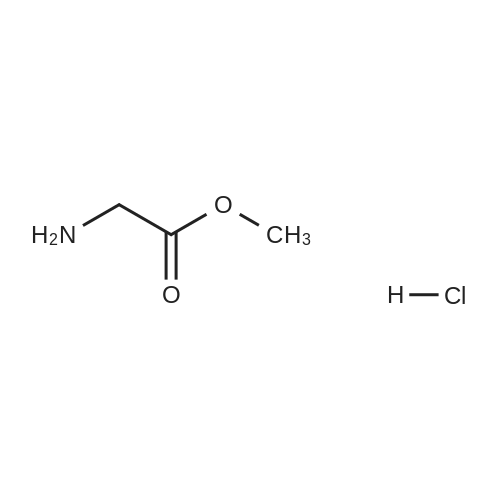
With caesium carbonate; thiourea; copper(II) oxide; In dimethyl sulfoxide; at 20 - 110℃;Inert atmosphere; |
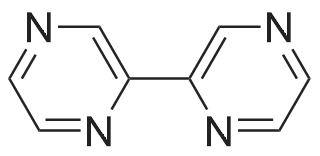
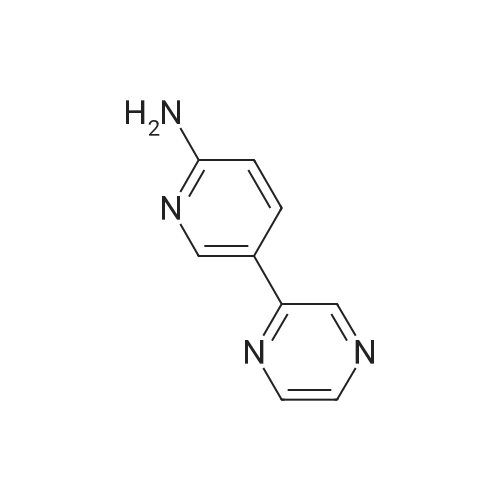
General procedure: To a stirred solution of aryl halides (2.0 mmol) and thiourea (1.2 equiv) in dry DMSO (2.0 mL) at rt was added nano CuO (5.0 mol %) followed by Cs2CO3 (2.0 equiv) and heated at 110 C for 15 h. The progress of the reaction was monitored by TLC. After the reaction was complete, the reaction mixture was allowed to cool, and a 1:1 mixture of ethyl acetate/water (20 mL) was added. The combined organic extracts were dried with anhydrous Na2SO4. The solvent and volatiles were completely removed under vacuum to give the crude product, which was purified by column chromatography on silica gel (petroleum ether/ethyl acetate, 9:1) to afford the corresponding coupling product in excellent yields.Recycling of the catalyst:after the reaction was complete, the reaction mixture was allowed to cool, and a 1:1 mixture of ethyl acetate/water (2.0 mL) was added and CuO was removed by centrifugation. After each cycle, the catalyst was recovered by simple centrifugation, washing with deionized water and ethyl acetate and then drying in vacuo. The recovered nano CuO was used directly in the next cycle.Data of representative examples:Dip-tolylsulfane (Table 3, entry 3): yellow oil;1H NMR (200 MHz, CDCl3, TMS): delta = 7.21 (d, 4H, J = 8.0 Hz), 7.06 (d, 4H, J = 8.0 Hz), 2.32 (s, 6H); 13C NMR (50 MHz, CDCl3, TMS): delta = 136.7, 132.81, 131.0, 129.8, 96.1.Table 3, entry 3): yellow oil;1H NMR (200 MHz, CDCl3, TMS): delta = 7.21 (d, 4H, J = 8.0 Hz), 7.06 (d, 4H, J = 8.0 Hz), 2.32 (s, 6H); 13C NMR (50 MHz, CDCl3, TMS): delta = 136.7, 132.81, 131.0, 129.8, 96.1.Bis(4-ethylphenyl)sulfane (Table 3, entry 4): colorless oil; 1HNMR (300 MHz, CDCl3, TMS): delta = 7.21(d, 4H, J = 7.8 Hz), 7.07 (d, 4H, J = 7.8 Hz), 2.62-2.52 (m, 4H), 1.26 (t, 6H, J = 7.8 Hz);13C NMR (75 MHz, CDCl3, TMS): delta = 143.1, 132.7, 131.0, 128.6, 28.3, 15.4; mass (EI): m/z 242 [M]+; Anal. calcd for: (C16H18S) C, 79.29; H, 7.49; S, 13.23; found: C,79.22; H,7.42; S,13.19.Table 3, entry 4): colorless oil; 1HNMR (300 MHz, CDCl3, TMS): delta = 7.21(d, 4H, J = 7.8 Hz), 7.07 (d, 4H, J = 7.8 Hz), 2.62-2.52 (m, 4H), 1.26 (t, 6H, J = 7.8 Hz);13C NMR (75 MHz, CDCl3, TMS): delta = 143.1, 132.7, 131.0, 128.6, 28.3, 15.4; mass (EI): m/z 242 [M]+; Anal. calcd for: (C16H18S) C, 79.29; H, 7.49; S, 13.23; found: C,79.22; H,7.42; S,13.19.Bis(3-nitrophenyl)sulfane (Table 3, entry 7): pale yellow oil; 1H NMR (300 MHz, CDCl3, TMS): delta = 8.19-8.15 (m, 4H), 7.65 (d, 2H, J = 8.3 Hz), 7.55 (t, 2H, J = 8.3 Hz); 13C NMR (75 MHz, CDCl3, TMS): delta = 148.8, 136.7, 130.7, 125.6, 122.7; mass (EI): m/z 276 [M]+; Anal. calcd for: (C12H8N2O4S) C, 52.17; H, 2.92; S, 11.61; N, 10.14; found: C, 52.12; H, 2.86; S, 11.55; N, 10.9.Table 3, entry 7): pale yellow oil; 1H NMR (300 MHz, CDCl3, TMS): delta = 8.19-8.15 (m, 4H), 7.65 (d, 2H, J = 8.3 Hz), 7.55 (t, 2H, J = 8.3 Hz); 13C NMR (75 MHz, CDCl3, TMS): delta = 148.8, 136.7, 130.7, 125.6, 122.7; mass (EI): m/z 276 [M]+; Anal. calcd for: (C12H8N2O4S) C, 52.17; H, 2.92; S, 11.61; N, 10.14; found: C, 52.12; H, 2.86; S, 11.55; N, 10.9.4,4'-Thiodianiline (Table 3, entry 11): brown solid; mp 104-105 C; 1H NMR (300 MHz, CDCl3, TMS): delta = 7.10 (d, 4H, J = 8.68 Hz), 6.52 (d, 4H, J = 8.68 Hz), 3.51 (br s, 4H); 13C NMR (75 MHz, CDCl3, TMS): delta = 145.5, 133.8, 132.6, 124.8, 115.6; mass (EI): m/z 216 [M]+; Anal. calcd for: (C12H12N2S) C, 66.63; H, 5.59; N, 12.95; S, 14.82; Found: C, 66.61; H, 5.58; N, 12.92; S, 14.81.Table 3, entry 11): brown solid; mp 104-105 C; 1H NMR (300 MHz, CDCl3, TMS): delta = 7.10 (d, 4H, J = 8.68 Hz), 6.52 (d, 4H, J = 8.68 Hz), 3.51 (br s, 4H); 13C NMR (75 MHz, CDCl3, TMS): delta = 145.5, 133.8, 132.6, 124.8, 115.6; mass (EI): m/z 216 [M]+; Anal. calcd for: (C12H12N2S) C, 66.63; H, 5.59; N, 12.95; S, 14.82; Found: C, 66.61; H, 5.58; N, 12.92; S, 14.81.Dithiophen-3-ylsulfane (Table 3, entry 15): yellow oil; 1H NMR (300 MHz, CDCl3, TMS): delta = 7.31-7.25 (m, 2H), 7.17-7.11(m, 2H), 6.96-6.94 (m, 2H); 13C NMR (75 MHz, CDCl3, TMS): delta = 129.6, 126.4, 124.7; mass (EI): m/z 197 [M]+; Anal. calcd for: (C8H6S3) C, 48.45; H, 3.05; S, 48.50; found: C,48.42; H,3.02; S,48.47.Table 3, entry 15): yellow oil; 1H NMR (300 MHz, CDCl3, TMS): delta = 7.31-7.25 (m, 2H), 7.17-7.11(m, 2H), 6.96-6.94 (m, 2H); 13C NMR (75 MHz, CDCl3, TMS): delta = 129.6, 126.4, 124.7; mass (EI): m/z 197 [M]+; Anal. calcd for: (C8H6S3) C, 48.45; H, 3.05; S, 48.50; found: C,48.42; H,3.02; S,48.47.Dipyrimidin-5-ylsulfane (Table 3, entry 17): colorless oil; 1H NMR (300 MHz, CDCl3, TMS): delta = 9.15 (s, 2H), 8.74(s, 4H); 13C NMR (75 MHz, CDCl3, TMS): delta = 158.6, 157.7, 129.8; mass (EI): m/z 190 [M]+; Anal. calcd for: (C8H6N4S) C, 50.51; H, 3.18; N, 29.45; S, 16.86; found: C, 50.45; H, 3.13; N, 29.41; S, 16.81.Table 3, entry 17): colorless oil; 1H NMR (300 MHz, CDCl3, TMS): delta = 9.15 (s, 2H), 8.74(s, 4H); 13C NMR (75 MHz, CDCl3, TMS): delta = 158.6, 157.7, 129.8; mass (EI): m/z 190 [M]+; Anal. calcd for: (C8H6N4S) C, 50.51; H, 3.18; N, 29.45; S, 16.86; f... |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping