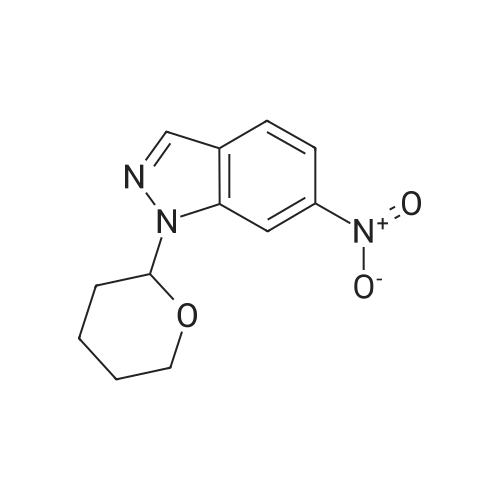
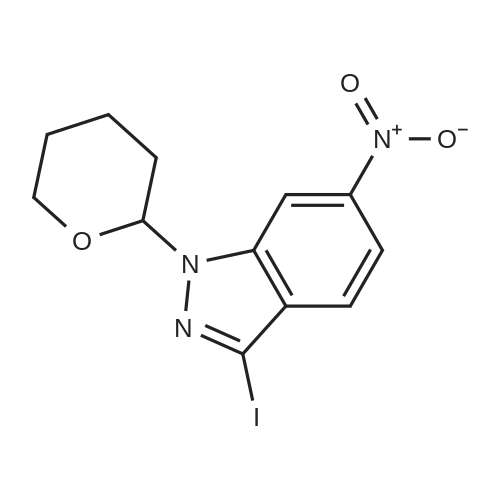
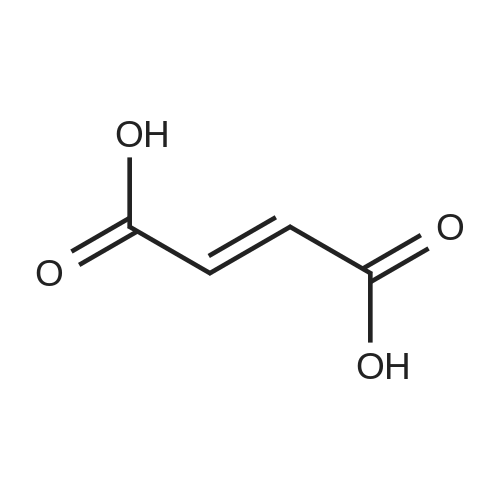
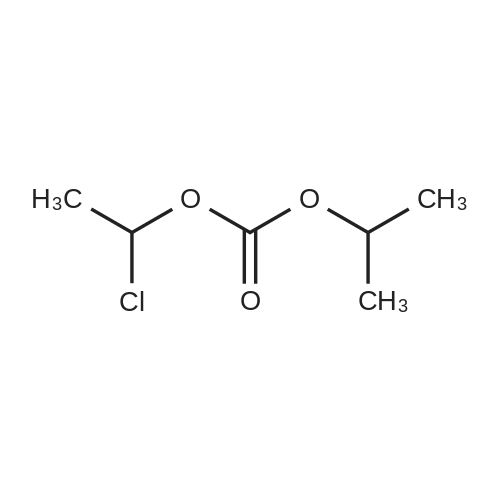
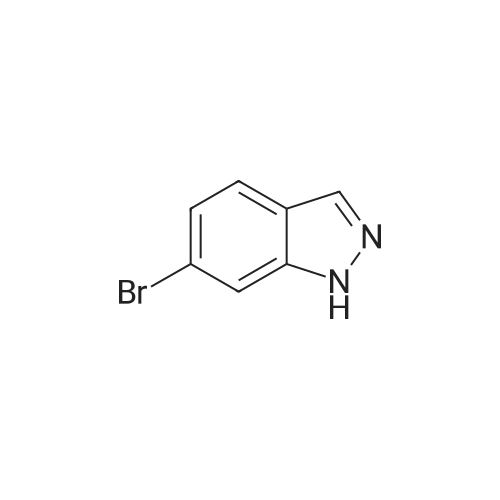
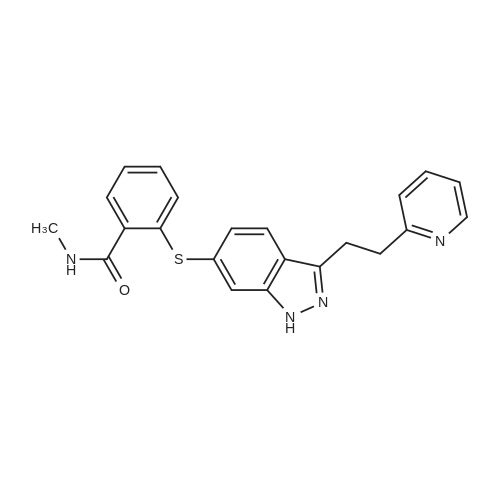
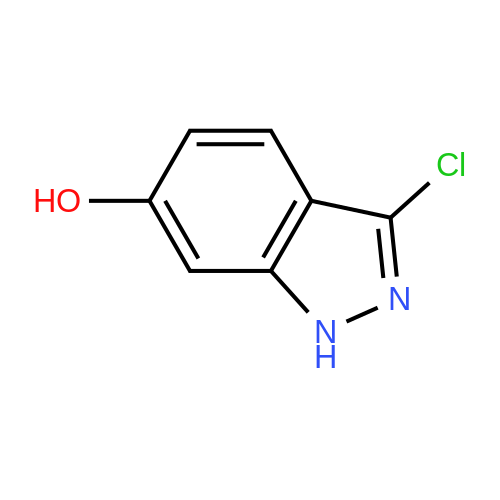
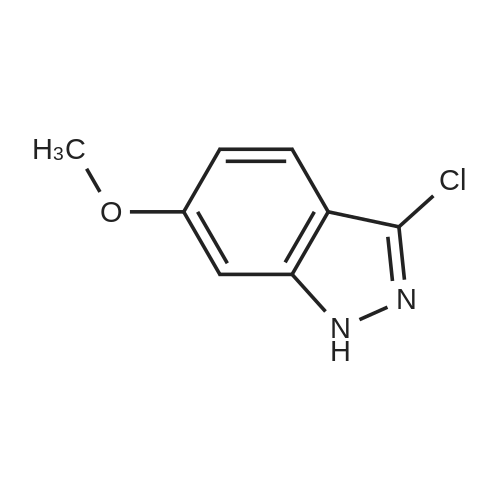
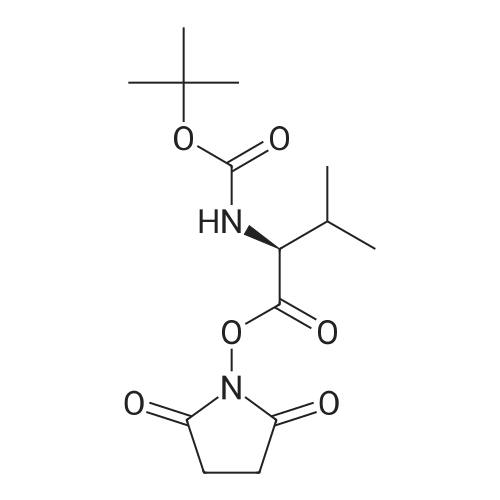
| 95.1% |
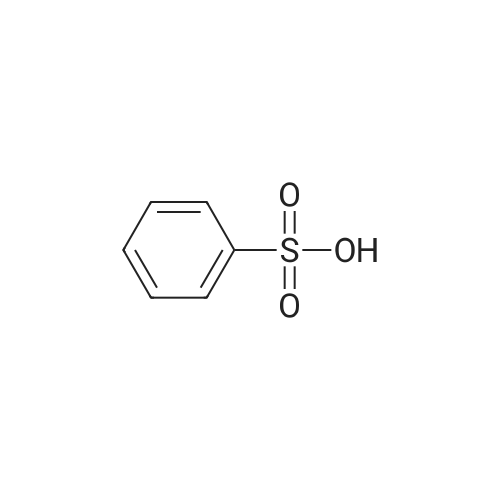
With toluene-4-sulfonic acid; In methanol; for 4h;Reflux; |
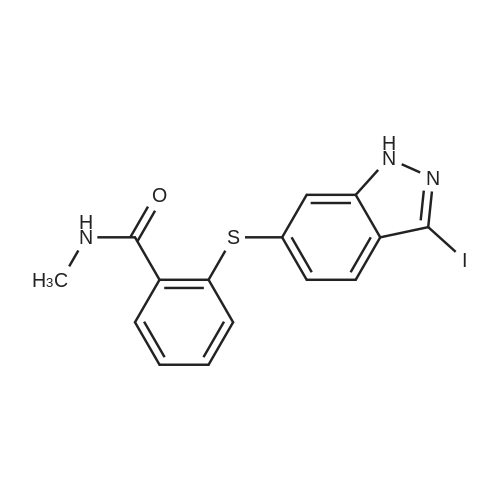
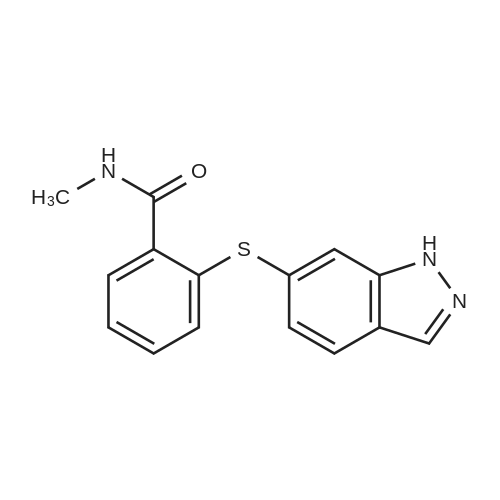
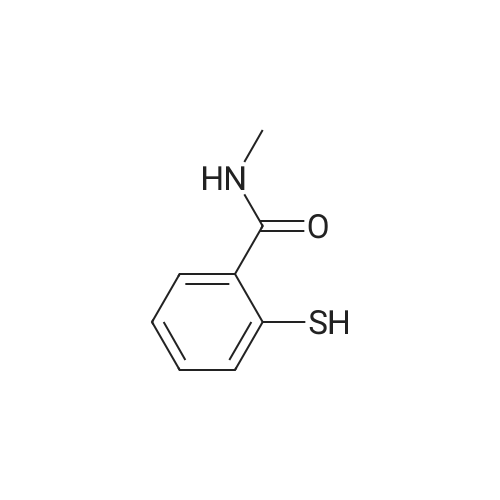
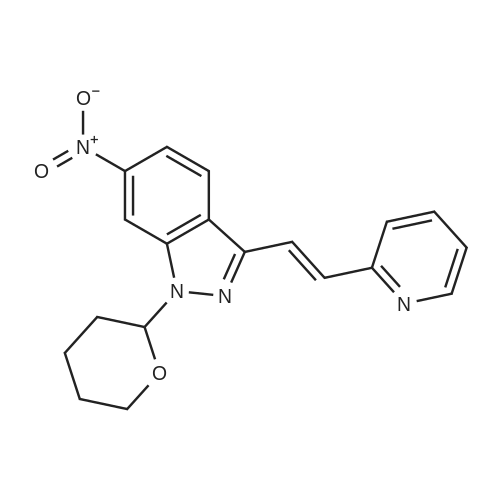
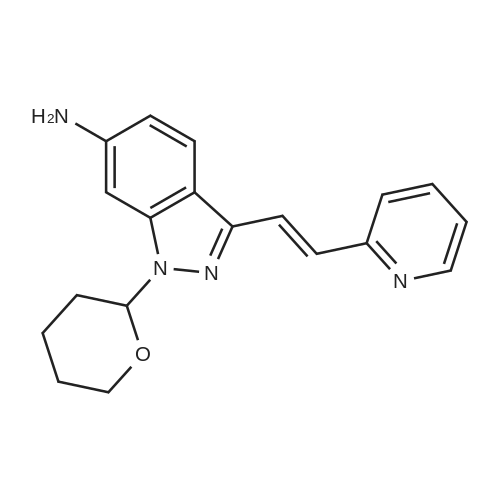
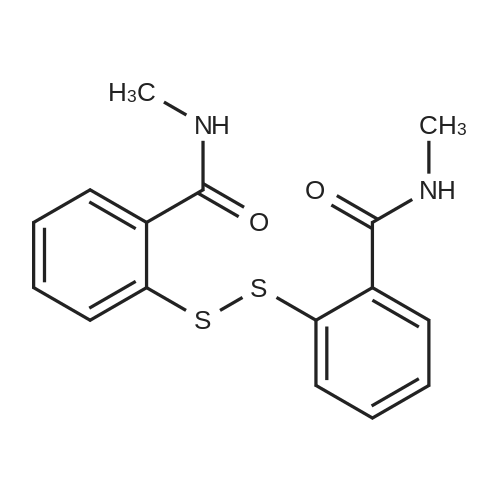
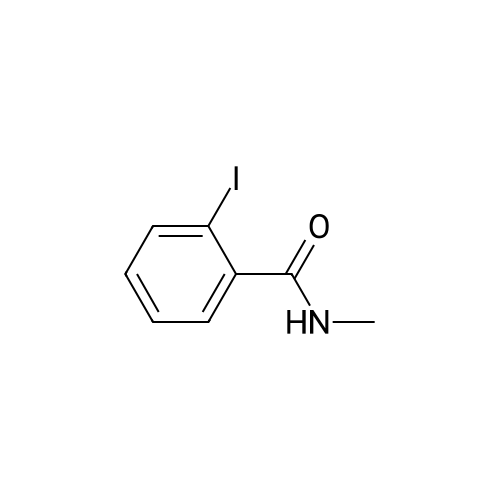
Example 2 The dried solid obtained in Example 1 was added into a flask with methanol (20 mL, 4 vol.) and p-toluenesulfonic acid (4.28 g, 2 eq.), and then heated at reflux for about 4 hours. After cooling, the resulting solid was isolated by filtration and washed with methanol (20 mL, 4 vol.) and water (20 mL, 4 vol.) to afford Axitinib (3.87 g, 95.1% weight yield). |
| 92.5% |
With toluene-4-sulfonic acid; In methanol; at 65℃; for 4h;Heating / reflux;Product distribution / selectivity; |
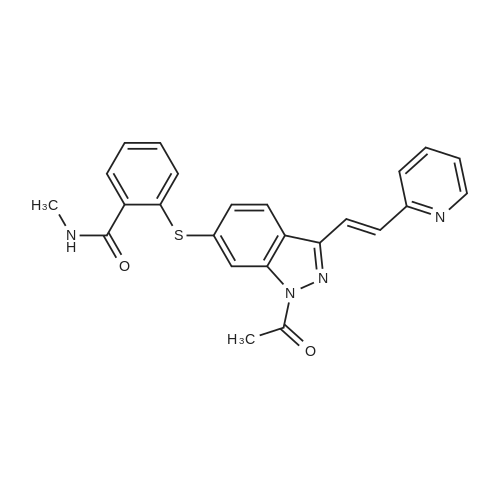
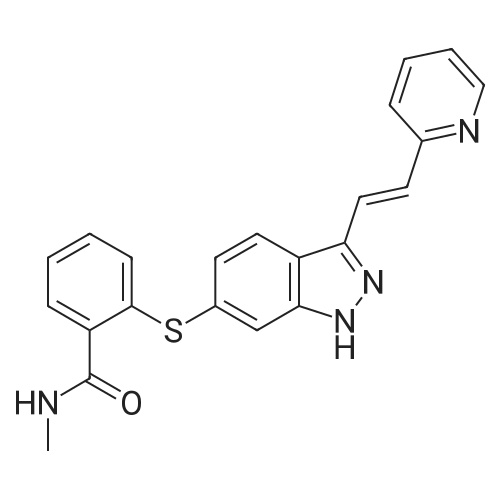
N-1 THP 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole (355 g) was suspended in 2,485 ml_ of methanol, after which p-toluenesulfonic acid monohydrate (718 g) was added. The mixture was then heated to 65 0C (hard reflux) for 4 hours under argon while the reaction was monitored by HPLC (gluco method). Heating continued until less than 1% of the N-1 THP protected starting material persisted. The heating was then removed and the reaction was cooled to room temperature. The solid was filtered and the wet cake was washed with methanol (2 volumes, 710 mL) then the solids were rinsed with ethyl acetate (2 volumes, 710 mL). The wet cake was transferred to a reactor containing sodium bicarbonate (126.84 g), deionized water (1800 mL), and ethyl acetate (975 mL), which was then stirred for 2 hours at 2O0C. The solids were filtered and washed with 5 volumes of deionized water (1800 mL), then with 2 volumes of ethyl acetate (760 mL), and then dried in a vacuum oven at 400C for 16 hours. The isolated yield for the reaction was 92.5% (274 g). The isolated material was identified as crystalline Form III free base (0.5 ethyl acetate solvate). 1H NMR, 300 MHz, (DMSO-D6), ppm; 13.35 (1 H, s), 8.60 (1 H, d, J=3.8 Hz), 8.39 (1 H, m), 8.23 (1 H, d, J=8.5 Hz), 7.95 (1 H, d, J=16.4 Hz), 7.82 (1 H, ddd, J=7.7, 7.6, 1.8 Hz), 7.67 (1 H, d, J=7.8 Hz), 7.60 (a H, s), 7.57 (1 H, d, J=16.4 Hz), 7.49 (1 H, dd, J=7.1 , 1.6 Hz), 7.35-7.26 (3 H, m), 7.19 (1 H, d, J=8.4 Hz), 7.04 (1 H, d, J=7.8 Hz), 2.77 (3 H, d, J=4.6 Hz). 13C NMR, 75 MHz, (DMSO-D6) ppm: 168.23, 155.18, 149.81 , 142.35, 142.22, 137.31 , 136.00, 132.89, 130.64, 130.36, 129.51 , 128.14, 126.50, 125.93, 124.08, 123.01 , 122.85, 122.12, 120.642, 115.08, 26.45. |
| 92.5% |
|
N-1 THP 6-[2-(methylcarbamoyl)phenylsulfanyl]-3-E-[2-(pyridine-2-yl)ethenyl]indazole (355 g) was suspended in 2,485 mL of methanol, after which p-toluenesulfonic acid monohydrate (718 g) was added. The mixture was then heated to 65 C. (hard reflux) for 4 hours under argon while the reaction was monitored by HPLC (gluco method). Heating continued until less than 1% of the N-1 THP protected starting material persisted. The heating was then removed and the reaction was cooled to room temperature. The solid was filtered and the wet cake was washed with methanol (2 volumes, 710 mL) then the solids were rinsed with ethyl acetate (2 volumes, 710 mL). The wet cake was transferred to a reactor containing sodium bicarbonate (126.84 g), deionized water (1800 mL), and ethyl acetate (975 mL), which was then stirred for 2 hours at 20 C. The solids were filtered and washed with 5 volumes of deionized water (1800 mL), then with 2 volumes of ethyl acetate (760 mL), and then dried in a vacuum oven at 40 C. for 16 hours. The isolated yield for the reaction was 92.5% (274 g). The isolated material was identified as crystalline Form III free base (0.5 ethyl acetate solvate). 1H NMR, 300 MHz, (DMSO-D6), ppm; 13.35 (1 H, s), 8.60 (1 H, d, J=3.8 Hz), 8.39 (1 H, m), 8.23 (1 H, d, J=8.5 Hz), 7.95 (1 H, d, J=16.4 Hz), 7.82 (1 H, ddd, J=7.7, 7.6, 1.8 Hz), 7.67 (1 H, d, J=7.8 Hz), 7.60 (1 H, s), 7.57 (1 H, d, J=16.4 Hz), 7.49 (1 H, dd, J=7.1, 1.6 Hz), 7.35-7.26 (3 H, m), 7.19 (1 H, d, J=8.4 Hz), 7.04 (1 H, d, J=7.8 Hz), 2.77 (3 H, d, J=4.6 Hz). 13C NMR, 75 MHz, (DMSO-D6) ppm: 168.23, 155.18, 149.81, 142.35, 142.22, 137.31, 136.00, 132.89, 130.64, 130.36, 129.51, 128.14, 126.50, 125.93, 124.08, 123.01, 122.85, 122.12, 120.642, 115.08, 26.45. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






 HazMat Fee +
HazMat Fee +

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping