| 75% |
With thionyl chloride; at 20℃; |
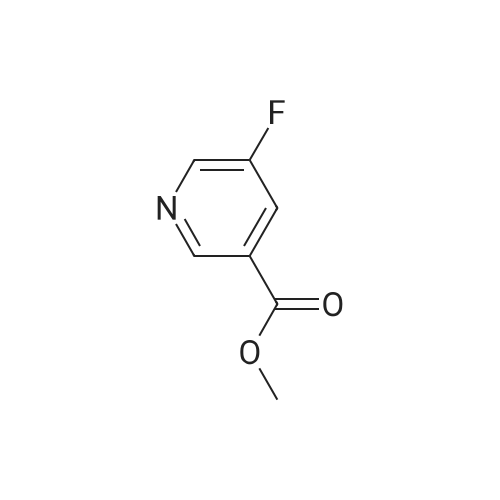
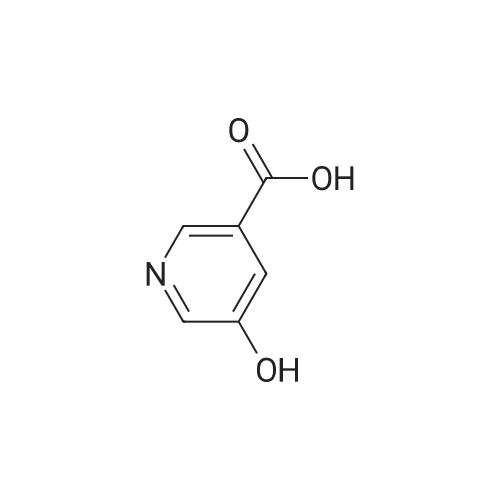
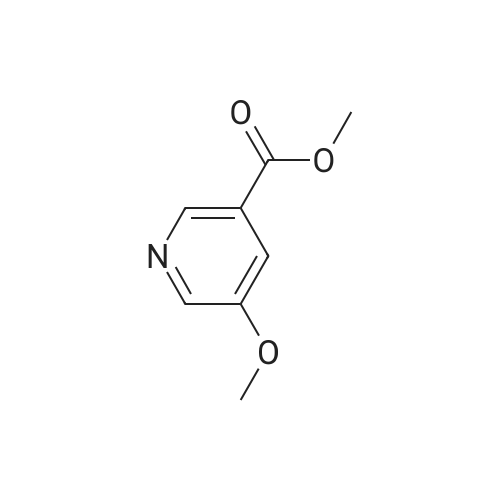
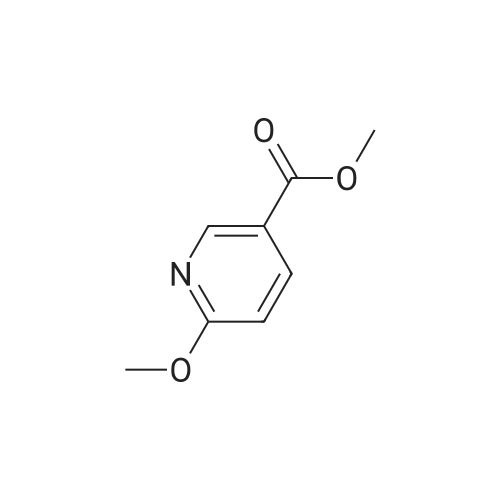
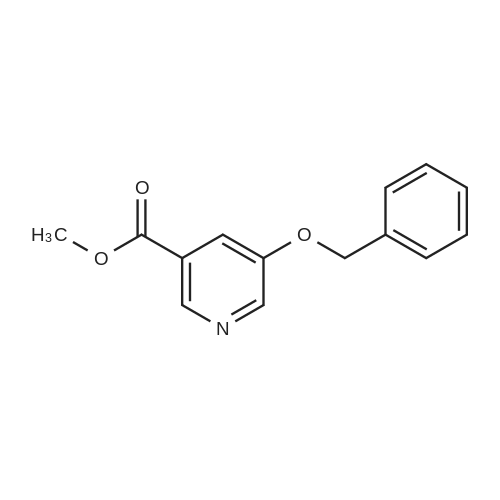
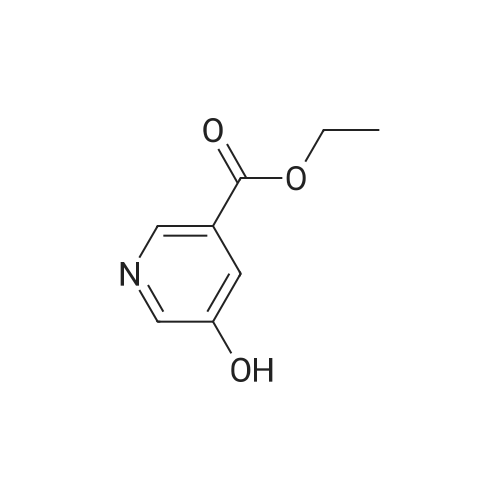
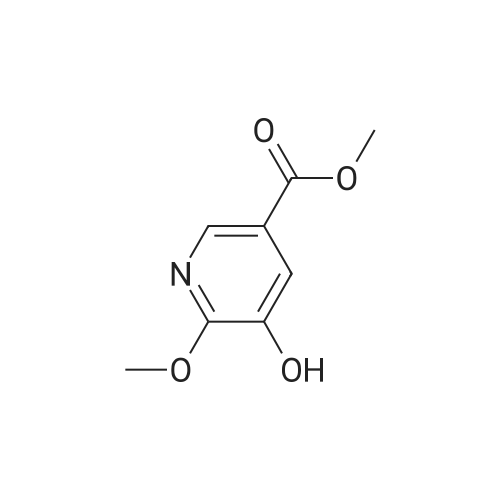
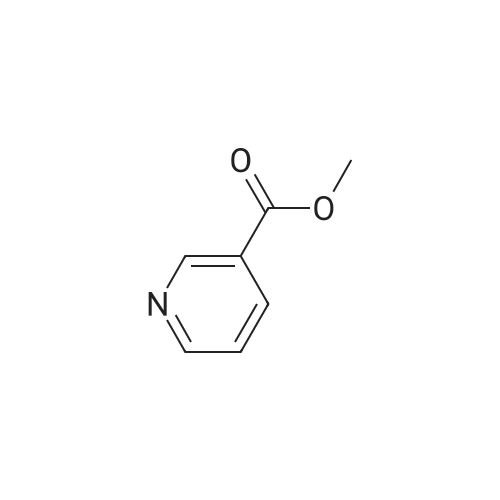
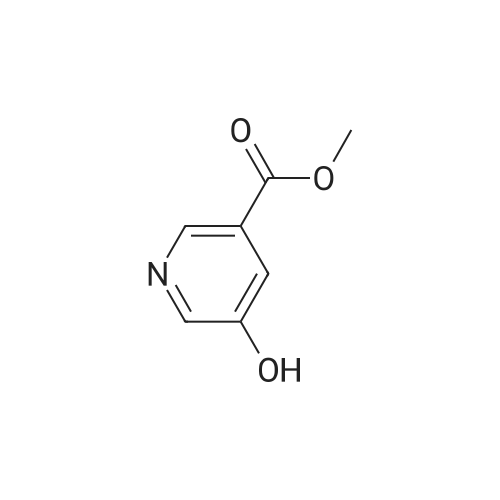
Synthesis of methyl 5-hydroxynicotinateO OHSOCI2 MeOHHO HO UNTo a stirred solution of <strong>[27828-71-3]5-hydroxynicotinic acid</strong> (10 g, 71 .9 mmol) in MeOH (100 mL)was added sulfurous dichloride (1 mL) drop wise over 5mm The resulting mixturewas stirred at room temperature overnight. The resulted solution was added 100 mLof NaHCO3.The precipitate was filtrated and washed with MeOH for several cycles togive 8.5 g of A031 -2 (75% yields), which was used for the nest step without furtherpurification. |
| 75% |
With thionyl chloride; at 20℃; for 0.0833333h; |
To a stirred solution of <strong>[27828-71-3]5-hydroxynicotinic acid</strong> (10 g, 71.9 mmol) in MeOH (100 mL) was added sulfurous dichloride (1 mL) drop wise over 5 min. The resulting mixture was stirred at room temperature overnight. The resulted solution was added 100 mL of NaHCO3. The precipitate was filtrated and washed with MeOH for several cycles to give 8.5 g of A031-2 (75% yields), which was used for the nest step without further purification. |
| 75% |
With thionyl chloride; at 20℃; |
To a stirred solution of <strong>[27828-71-3]5-hydroxynicotinic acid</strong> (10 g, 71 .9 mmol) in MeOH (100 mL) was added sulfurous dichloride (1 mL) drop wise over 5 min. The resulting mixture was stirred at room temperature overnight. The resulted solution was added 100 mL of NaHCOs.The precipitate was filtrated and washed with MeOH for several cycles to give 8.5 g (75% yields), which was used for the next step without further purification. |
| 75% |
With thionyl chloride; at 20℃; |
To a stirred solution of <strong>[27828-71-3]5-hydroxynicotinic acid</strong> (10 g, 71.9 mmol) in MeOH (100 mL) was added sulfurous dichloride (1 mL) drop wise over 5 min. The resulting mixture was stirred at room temperature overnight. The resulted solution was added 100 mL of NaHCO3. The precipitate was filtrated and washed with MeOH for several cycles to give 8.5 g of A031-2 (75% yields), which was used for the nest step without further purification. |
| 71% |
With sulfuric acid; at 70℃; |
A mixture of <strong>[27828-71-3]5-hydroxynicotinic acid</strong> (3 g, 21.6 mmol), cone H2SO4 (2.2 g, 21.6 mmol) in MeOH (50 mL) was heat at 70 C overnight. The mixture was concentrated, poured into water (100 mL) and extracted with EA (100 mL X 3). The combined organic layers were dried over anhydrous sodium sulfate and concentrated to afford methyl 5-hydro |
| 68% |
|
Preparation of Compound A29A mixture of compound A28 (25.0 g, 180 mmol) and concentrated H2SO4 (10 mL) in CH3OH (100 mL) was heated to reflux for overnight. The mixture was concentrated, the residue was washed with aqueous NaHCO3 (50 mL) and extracted with ethyl acetate (2×100 mL). The organic layer was dried over Na2SO4, filtered and concentrated to afford compound A29 (18.7 g, yield: 68%).1H NMR (300 MHz, DMSO-d6): delta: 10.42 (s, 1H), 8.60 (d, J=1.6 Hz, 1H), 8.36 (d, J=2.8 Hz, 1H), 7.60-7.61 (m, 1H), 3.87 (s, 3H). |
| 68% |
With sulfuric acid;Reflux; |
[0444] Preparation of compound A29: A mixture of compound A28 (25.0 g, 180 mmol) and concentrated H2SO4 (10 mL) in CH3OH (100 mL) was heated to reflux for overnight. The mixture was concentrated, the residue was washed with aqueous NaHC03 (50 mL) and extracted with ethyl acetate (2 x 100 mL). The organic layer was dried over a2S04, filtered and concentrated to afford compound A29 (18.7 g, yield: 68%). [0445] 1H NMR (300 MHz, DMSO-i 6): delta: 10.42 (s, 1H), 8.60 (d, J = 1.6 Hz, 1H), 8.36 (d, J = 2.8 Hz, 1H), 7.60-7.61 (m, 1H), 3.87 (s, 3H). [0446] Preparation of compound A30: BnOH (3.90 g, 36.1 mmol, 1.1 eq) and PPh3 (17.1 g, 65.4 mmol, 2.0eq) was added to a solution of compound A29 (5.00 g, 32.7 mmol) in THF (100 mL), then DEAD (6.80 g, 39.2 mmol, 1.2eq) was added at 0C. The mixture was stirred at room temperature for overnight. The solvent was evaporated, the residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate = 10: 1) to afford the compound A30 (5.70 g, yield: 71%) as white solid. [0447] 1H NMR (300 MHz, CDC13): delta: 8.83 (d, J = 1.6 Hz, 1H), 8.54(d, J = 2.8 Hz, 1H), 7.85-7.86 (m, 1H), 7.27-7.46 (m, 5H), 5.15 (s, 2H), 3.95 (s, 3H). [0448] Preparation of compound A31: A solution of compound A30 (12.8 g, 52.9mmol) in methylamine alcohol solution in sealed tube was stirred at 70 C for overnight. Then the mixture was cooled to room temperature and the solvent was evaporated to afford the compound A31 (12.0 g, yield: 100%). [0449] 1H NMR (300 MHz, CDC13): delta: 8.50 (d, J = 1.6 Hz, 1H), 8.48(d, J = 2.8 Hz, 1H), 7.73-7.74 (m, 1H), 7.73-7.74 (m, 5H), 6.16(s, 1H), 3.15 (s, 2H), 3.04 (d, J = 4.4 Hz, 3H). A32 A33 [0450] Preparation of compound A32: The solution of compound A31 (11.0 g, 45.5 mmol) in SOCl2 (100 mL) was heated to reflux for 4h. Then, SOCl2 was removed under vacuum and the residue was dissolved in MeCN (200 mL). TMSN3 (12.5 g, 90.0 mmol, 2.0eq) was added slowly and the mixture was stirred at 90C for 3h. Then the solvent was evaporated and the residue was purified by chromatography on silica gel (petroleum ether/ethyl acetate = 2: 3) to afford the compound A32 (9.50 g, yield: 78%). [0451] 1H NMR (300 MHz, CDC13): delta: 8.59 (d, J = 2.8 Hz, 1H), 8.56(d, J = 1.6 Hz, 1H), 7.68-7.69 (m, 1H), 7.3-7.46 (m, 5H), 5.21 (s, 2H), 4.17 (s, 3H). [0452] Preparation of compound A33: To a solution of compound A32 (5.00 g, 18.7 mmol) in CH3OH (100 mL) was added Pd(OH)2 ( 0.50 g), The mixture was stirred at room temperature under H2 atmosphere for 3h. The solid was filtered off and the filtrate was concentrated to get compound A33 (1.60 g, yield: 48%). [0453] 1H NMR (300 MHz, DMSO-i 6): delta: 10.56 (s, 1H), 8.49 (d, J = 1.6 Hz, 1H), 8.36(d, J = 2.8 Hz, 1H), 7.61-7.62 (m, 1H), 4.19(s, 3H). |
| 65% |
With thionyl chloride; In methanol; at 20 - 60℃; |
Thionyl chloride (780 jil, 10.7 mmol) was added drop-wise to a suspension of 5- hydroxynicotinic acid (497 mg, 3.57 mmol) in MeOH (5 ml) at ambient temperature. The mixture was heated at 60 C overnight. 0.1 M Potassium phosphate buffer (pH 7) (50 ml) was added and the mixture extracted with EtOAc. The combined organic layers were dried(Mg504). Yield: 354 mg (65%); white solid. |
|
|
A mixture of 1 g of <strong>[27828-71-3]5-hydroxynicotinic acid</strong>, 2 g Amberlyst 15 ion-exchange resin and 150 mL of methanol was heated to reflux with stirring for 24 hrs, cooled, basified with 10% ammonia in methanol, <n="35"/>filtered and concentrated under reduced pressure. The residue was taken up in 50 mL of chloroform and dried over MgSO4- Concentration under reduced pressure gave a tan crystalline solid: MS (m+1) = 154.1; 1H NMR (400 MHz, CDC13) 8.68 (s, IH), 8.32 (s, IH)3 7.72 (s, IH), 3.94 (s, 3H). |
|
With thionyl chloride; at 60℃; for 4h; |
10 mmol of <strong>[27828-71-3]5-hydroxy-3-carboxypyridine</strong> was dissolved in 100 mL of methanol. 5 mL of thionyl chloride was slowly added and reacted at 60 C for 4 hours. The solvent was distilled off under reduced pressure. The resulting product was used in the next reaction without purification. |
|
With sulfuric acid; at 70℃; for 12h;Inert atmosphere; |
Step 1 Methyl 5-hydroxynicotinate <strong>[27828-71-3]5-Hydroxynicotinic acid</strong> (2.50 g, 17.97 mmol) was dissolved in methanol (20.00 mL), added with sulfuric acid (18M, 47.90 uL), and then stirred at 70 C. under nitrogen atmosphere for 12 hours. After the reaction mixture was concentrated, the residue was dissolved in dichloromethane (50.00 mL) and the system was adjusted to pH=8 with saturated sodium bicarbonate solution. A white solid was slowly precipitated and filtered to give methyl 5-hydroxynicotinate (a white solid, 1.5 g, crude product). 1H NMR (400 MHz, CDCl3) 8.25 (s, 1H), 8.13 (d, J=1.8 Hz, 1H), 7.38 (br. s., 1H), 3.17 (s, 3H). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping