* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: at 100℃; for 48 h; Stage #2: With sodium hydroxide In water
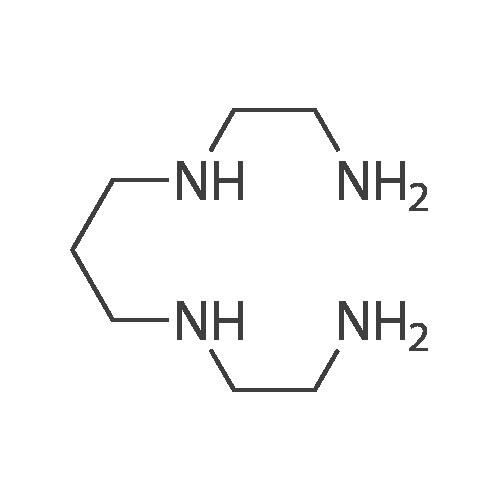
1,4,8,11-Tetra-p-tosyl-1,4,8,11-tetraazacyclotetradecane (13 mmol) was dissolved in 90percent concentrated sulfuric acid, allowed to react with stir at 100°C for 48 hours, and cooled to 0°C. To the resultant was dropwise added anhydrous ethanol (120 ml) and anhydrous ethyl ether (100 ml) successively. A solid was precipitated and filtrated. The filter cake was washed with a small amount of anhydrous ethanol and anhydrous ethyl ether, dried in vacuum. The resultant off white solid was dissolved with NaOH aqueous solution (90 ml, 1 mol/L), and extracted with chloroform (3x100 ml). The chloroform layer was dried over anhydrous sodium sulfate over night, reduced pressure distilled off chloroform, dried in vacuum to give a white solid product, recrystallized with toluene to obtain 2.2 g of 1,4,8,11-tetraazacyclotetradecane, yield 85percent, MS[M]+=200.3 m/e, 1H-NMP(400 M HZ, CDCl3)δppm: 1.72(t, 4H), 2.23 (s, 4H), 2.68 (s, 8H), 2.75 (t, 8H).
Reference:
[1] Patent: EP2163553, 2010, A1, . Location in patent: Page/Page column 18-19
[2] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1987, vol. 36, # 2, p. 372 - 376[3] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1987, # 2, p. 413 - 417
[4] Chemistry of Heterocyclic Compounds (New York, NY, United States), 1990, vol. 26, # 3, p. 346 - 349[5] Khimiya Geterotsiklicheskikh Soedinenii, 1990, # 3, p. 401 - 404
[6] Journal of Organic Chemistry USSR (English Translation), 1988, vol. 24, p. 1562 - 1571[7] Zhurnal Organicheskoi Khimii, 1988, vol. 24, # 8, p. 1731 - 1742
[8] Canadian Journal of Chemistry, 1995, vol. 73, # 5, p. 685 - 695
Reference:
[1] Journal of Organic Chemistry USSR (English Translation), 1988, vol. 24, p. 1562 - 1571[2] Zhurnal Organicheskoi Khimii, 1988, vol. 24, # 8, p. 1731 - 1742
9
[ 111514-29-5 ]
[ 295-37-4 ]
Reference:
[1] Bulletin of the Academy of Sciences of the USSR, Division of Chemical Science (English Translation), 1987, vol. 36, # 2, p. 372 - 376[2] Izvestiya Akademii Nauk SSSR, Seriya Khimicheskaya, 1987, # 2, p. 413 - 417
10
[ 917838-66-5 ]
[ 295-37-4 ]
[ 1571-33-1 ]
Reference:
[1] Collection of Czechoslovak Chemical Communications, 2006, vol. 71, # 3, p. 337 - 367
11
[ 4741-99-5 ]
[ 109-64-8 ]
[ 295-37-4 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1937, vol. 56, p. 343,347
[2] Inorg. Chem., 1963, vol. 4, p. 1102,1107
12
[ 131543-46-9 ]
[ 10563-26-5 ]
[ 295-37-4 ]
Reference:
[1] Canadian Journal of Chemistry, 1995, vol. 73, # 5, p. 685 - 695
13
[ 131543-46-9 ]
[ 10563-26-5 ]
[ 295-37-4 ]
[ 78303-77-2 ]
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1981, # 6, p. 302 - 304
14
[ 64-17-5 ]
[ 107-15-3 ]
[ 109-64-8 ]
[ 295-37-4 ]
[ 4741-99-5 ]
[ 62708-55-8 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1936, vol. 55, p. 835[2] Recueil des Travaux Chimiques des Pays-Bas, 1937, vol. 56, p. 345
15
[ 64-17-5 ]
[ 107-15-3 ]
[ 109-64-8 ]
[ 295-37-4 ]
[ 4741-99-5 ]
[ 62708-55-8 ]
Reference:
[1] Recueil des Travaux Chimiques des Pays-Bas, 1936, vol. 55, p. 835[2] Recueil des Travaux Chimiques des Pays-Bas, 1937, vol. 56, p. 345
16
[ 295-37-4 ]
[ 110078-46-1 ]
Reference:
[1] New Journal of Chemistry, 2001, vol. 25, # 9, p. 1168 - 1174
[2] Tetrahedron Letters, 2003, vol. 44, # 12, p. 2481 - 2483
[3] Patent: WO2017/37639, 2017, A1,
17
[ 623-24-5 ]
[ 295-37-4 ]
[ 110078-46-1 ]
Reference:
[1] Journal of the Chemical Society, Chemical Communications, 1991, # 4, p. 206 - 207
18
[ 295-37-4 ]
[ 24424-99-5 ]
[ 170161-27-0 ]
Yield
Reaction Conditions
Operation in experiment
30%
at 0℃;
,4,8-Tris(ter-butoxycarbonyl)-1,4,8,11-tetraazacyclotetradecane (1) was synthesized. Synthesis of 1 was performed by reported methods by Dessolin et al. (J. Dessolin, P. Galea, P. Vlieghe, J. Chermann, J. Kraus, J. Med. Chem. 1999,42, 229-241) with slight modifications. Typically, to a 1gm of cyclam (5.00 mmol, 1 equiv. ) in ice cold CH2C12 (lOOmL) was added to 2.0g of di-tert-butyl dicarbonate (9.00 mmol, 1.8 equiv. ). The solution was stirred overnight. After solvent evaporation, the crude pale yellow oil was {JND0255. DOC;1} - 34 - purified by flash column chromatography (MeOH/CH2Cl2 5: 95). The first fraction collected was the desired tri-boc protected cyclam (1) as the thick colorless oil (0.75g, 30percent yield). MS (electrospray) 523 (M+Na)+
Reference:
[1] Molecular Pharmaceutics, 2017, vol. 14, # 5, p. 1395 - 1404
[2] Chemistry (Weinheim an der Bergstrasse, Germany), 2002, vol. 8, # 21, p. 4965 - 4972
[3] Journal of the American Chemical Society, 2006, vol. 128, # 45, p. 14448 - 14449
[4] Chemical Communications, 2018, vol. 54, # 33, p. 4116 - 4119
[5] Bulletin de la Societe Chimique de France, 1996, vol. 133, # 1, p. 65 - 73
[6] European Journal of Inorganic Chemistry, 2017, vol. 2017, # 7, p. 1075 - 1086
[7] Chemical Communications, 2018, vol. 54, # 69, p. 9619 - 9622
[8] Bioorganic and Medicinal Chemistry Letters, 2008, vol. 18, # 9, p. 3007 - 3010
[9] European Journal of Inorganic Chemistry, 2006, # 12, p. 2357 - 2363
[10] Bioorganic and Medicinal Chemistry Letters, 2001, vol. 11, # 1, p. 71 - 74
[11] Patent: WO2005/117997, 2005, A1, . Location in patent: Page/Page column 34-35
[12] Nucleosides and Nucleotides, 1998, vol. 17, # 5, p. 957 - 968
[13] Tetrahedron Letters, 2002, vol. 43, # 13, p. 2463 - 2466
[14] Synlett, 2004, # 3, p. 453 - 456
[15] Bioorganic and Medicinal Chemistry Letters, 2006, vol. 16, # 23, p. 5988 - 5992
[16] Chemical Communications, 2003, # 15, p. 1812 - 1813
[17] ChemMedChem, 2010, vol. 5, # 8, p. 1272 - 1281
[18] Patent: WO2014/153624, 2014, A1, . Location in patent: Page/Page column 42; 43; 44
[19] Biomacromolecules, 2018, vol. 19, # 2, p. 392 - 401
[20] Patent: WO2010/126888, 2010, A1, . Location in patent: Page/Page column 12; 33
19
[ 295-37-4 ]
[ 24424-99-5 ]
[ 1007858-12-9 ]
[ 170161-27-0 ]
Yield
Reaction Conditions
Operation in experiment
35%
at 0 - 20℃;
Boc Cyclam. [0180] A solution of t-butyl dicarbonate (Boc2O) (2.96 g, 13.1 mmol) in 80 mL of methylene chloride (CH2Cl2) was added dropwise over a period of 2 hours to a solution of cyclam (0.88 g, 4.4 mmol) in CH2Cl2 at 0° C. The mixture was allowed to warm to room temperature and stirred overnight. The mixture was concentrated and purified by chromatography (ethyl acetate (AcOEt)→10:1 AcOEt:methanol (CH3OH)) to give cyclam-Boc4 (0.91 g, 35percent) as a white set foam and cyclam-Boc3 (1.22 g, 56percent) as a light yellow set foam. Tri Boc cyclam: 1H NMR (CDCl3) δ 1.46 (s, 27H), 1.65-1.76 (m, 2H), 2.00-1.80 (m, 2H), 2.62 (bt, J=5.6 Hz, 2H), 2.79 (t, J=4.8 Hz, 2H), 3.31 (t, J=6.4 Hz, 8H), 3.47-3.34 (m, 4H). (see Schickaneder, C.; Heinemann, F. W.; Alsfasser, R. “Copper II Complexes of the Tetraazamacrocyclic Tertiary Amide Ligand Alanyl-Cyclam,” Eur. J. Chem. 2006, 2357-2363)
With tetrabutylammomium bromide; sodium carbonate; In chloroform; water;Reflux;
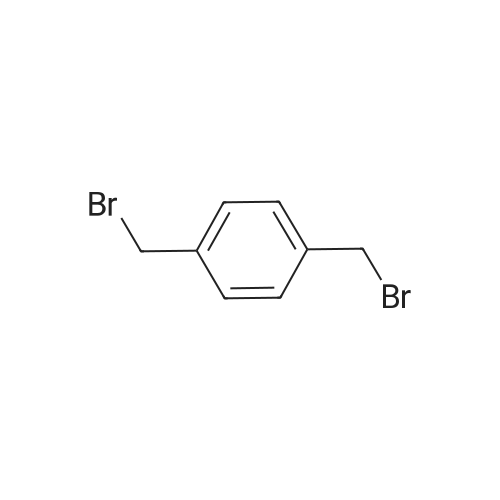
1,4,8,11-Tetraazacyclotetradecane 1 (0.205 mol) was dissolved in a mixture of water(160 mL) and chloroform (160 mL); sodium carbonate (0.205 mol) and a, a0-dibromop-xylene 2 (0.1mol) were added, followed by addition of 10% of TBAB catalyst(0.0205 mol). The solution was refluxed for 1-2 h, cooled to room temperature, pouredinto ice cold water and extracted with toluene (3x200 mL). The organic phases werecombined, dried over anhydrous MgSO4 and concentrated in vacuo to give a white solid3. The obtained solid material was dissolved in a minimum amount of dry methanol(250 mL) and dry HCl gas was gently passed through the solution to form a white precipitatewhich was filtered and dried to give 3 octahydrochloride.
With sodium hydroxide In chloroform; water Ambient temperature;
71%
With sodium hydroxide In water; acetonitrile for 6h;
General synthetic procedure A: N-tetraalkylation of macrocycles
General procedure: Macrocycle (1.0 equiv) and alkyl halide (4.1 equiv) were dissolved in a 1:1 mixture of aqueous 1 MNaOH and CH3CN (5-20 mL). The reaction mixture was shaken for 6 h. The resulting precipitate was collected by filtration, washed with hexane, and dried in vacuo to give the desired N-tetraalkyl derivative.
36%
With potassium carbonate In acetonitrile for 12h; Heating;
1.i
The azacrown ether (21 mmol) obtained from the step h was dissolved in dichloromethane (150 ml). To the system was added triethylamine (18 ml), and then dropwise added at room temperature p-tosyl chloride (43 mmol) in 300 ml dichloromethane solution. Upon the completion of dropwise addition, the mixture was stirred at room temperature over night. To the system was added water (40 ml). The organic phase was separated, dried over anhydrous sodium sulfate over night, reduced pressure distilled off dichloromethane, recrystallized with methanol to obtain 6.0g of a first solid; the mother liquor was distilled under reduced pressure and recrystallized, the resultant oily substance was dissolved with dicholormethane (100 ml) and triethylamine (10 ml). To this system was dropwise added p-tosyla chloride (2.0 g, 10 mmol) in dicholormethane (50 ml) solution. The mixture was allowed to react at room temperature for 2 hours, reduced pressure distilled off dichloromethane, recrystallized with methanol to obtain 2.6 g of a second batch of solid. The two solids were combined to provide 8.6 g of 1,4,8-tri(p-tosyl)-1,4,8,11-tetraazacyclotetradecane, yield 62%, MS[M]+=662.0 m/e.
51%
With triethylamine In dichloromethane at 20℃; for 3h;
50%
With triethylamine
36.4%
Stage #1: 1,4,8,11-Tetraazacyclotetradecane With triethylamine In dichloromethane at 20℃;
Stage #2: p-toluenesulfonyl chloride In dichloromethane at 20℃;
1 Example 1: Preparation of tris-(p-toluenesulfonyl)-1,4,8,11- tetraazacyclotetradecane (5)
Example 1: Preparation of tris-(p-toluenesulfonyl)-1,4,8,11- tetraazacyclotetradecane (5) -Cyclam (50g, 0.25 moles), methylene chloride (1250 mL), triethylamine (65.7g, 0.65 moles) were charged into 3L 3N RB flask and stirring was given. The solution of ptoluene sulfonyl chloride (95.3g, 0.50 moles) in methylene chloride (1250 mL) was added to the above reaction mass as dropwise during 50-60 minutes at room temperature. After addition reaction mass was stirred for another 4h then 2500 mL of water was added to the reaction mass and stirred for 1 5mm. layers were separated and aq. layer wasextracted with another 500 mL methylene chloride. The combined organic layer waswashed with brine solution and layers were separated. Organic layer was dried oversodium sulphate; solvent was distilled off under reduced pressure to give off whitecoloured solid. The solid was leached with methanol (600 mL) at room temperature andfiltcred. The resulting solid was recrystallized from the solvent mixture of methylenechloride: hexane (1:1) to afford pure tris-(p-toluenesulfonyl)- 1,4,8,11 -tetraazacyclotetradecane (5) (60g, 36.4%); purity: 98% (HPLC). ‘H NMR (400 MHzCDCI3): 0.85 (brs, 111), 1.71-1.74 (m, 2H), 1.96-2.03 (m, 2K), 2.42 (s, 2H), 2.45 (s,6H), 2.60-2.63 (t, 2H), 2.77-2.80 (t, 2H), 3.04- 3.10 (rn, 411), 3.16-3.19 (dd, 211). 3.23-3.27 (dd, 2H), 3.30-3.39 (rn, 4H). 7.30-7.32 (rn, 6H), 7.61-7.72 (rn, 611); ‘3C NMR (100MHz CDC13): 21.43, 29.55. 29.72, 45.48, 46.06, 47.38, 47.84, 48.22, 48.76, 49.23.51.36, 53.39, 127.08, 127.13, 129.64, 129.71, 134.96, 135.75, 136.72, 143.21. 143.34,+.14i.48; Mass (rnz): 66i.2 (M ion)
With triethylamine In chloroform
With triethylamine In acetonitrile at 70℃; for 18h;
1
The molar ratio of 1,4,8,11-tetraazacyclotetradecane, p-methylbenzenesulfonyl chloride and triethylamine was 1:3.5:4.1268 mg of 1,4,8,11-tetraazacyclotetradecane was weighed, and 4270 mg of p-toluenesulfonyl chloride was weighed, and both were poured into a 200 ml eggplant-shaped flask, and about 3.6 ml of triethylamine was added dropwise.Then 25 ml of acetonitrile was added to substantially dissolve the above solid.The condenser was placed on the condenser and condensed and refluxed for 18 hours in a 70 ° C oil bath.After the completion of the reaction, the solvent acetonitrile was completely evaporated, and the solid remaining in the eggplant-shaped flask was slightly yellow. 25 ml of a saturated sodium hydrogencarbonate solution was added, and the mixture was stirred at room temperature for 2 hours.The white solid is obtained by suction filtration, and is dried as a sulfur-containing small molecule compound, which is compound B, and its structural formula is as follows:
Stage #1: 1,4,8,11-tetrakis(p-toluenesulphonyl)-1,4,8,11-tetraazacyclotetradecane With sulfuric acid at 100℃; for 48h;
Stage #2: With sodium hydroxide In water
1.h
1,4,8,11-Tetra-p-tosyl-1,4,8,11-tetraazacyclotetradecane (13 mmol) was dissolved in 90% concentrated sulfuric acid, allowed to react with stir at 100°C for 48 hours, and cooled to 0°C. To the resultant was dropwise added anhydrous ethanol (120 ml) and anhydrous ethyl ether (100 ml) successively. A solid was precipitated and filtrated. The filter cake was washed with a small amount of anhydrous ethanol and anhydrous ethyl ether, dried in vacuum. The resultant off white solid was dissolved with NaOH aqueous solution (90 ml, 1 mol/L), and extracted with chloroform (3x100 ml). The chloroform layer was dried over anhydrous sodium sulfate over night, reduced pressure distilled off chloroform, dried in vacuum to give a white solid product, recrystallized with toluene to obtain 2.2 g of 1,4,8,11-tetraazacyclotetradecane, yield 85%, MS[M]+=200.3 m/e, 1H-NMP(400 M HZ, CDCl3)δppm: 1.72(t, 4H), 2.23 (s, 4H), 2.68 (s, 8H), 2.75 (t, 8H).
81%
With sulfuric acid
68%
With sulfuric acid at 100 - 105℃; Heating; 50-70 h;
With triethylamine; In dichloromethane;Inert atmosphere;
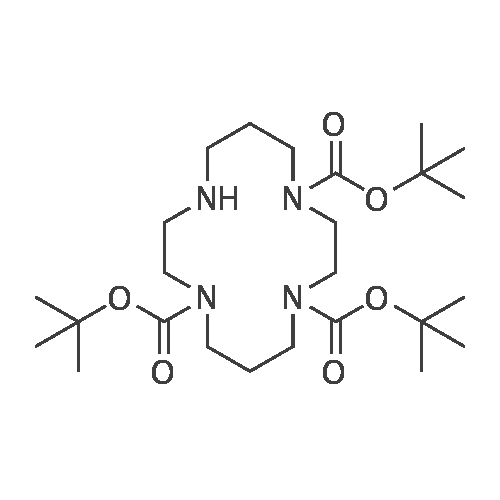
(5) Synthesis step of compound 5: Weigh 1.03 g (5.14 mmol) of 1,4,8,11-tetraazacyclotetradecane and 2.53 g (25.01 mmol) of triethylamine into 50 ml of dichloromethane and use Protected with argon, 20 ml of a solution of 1.86 g (8.52 mmol) of di-tert-butyl dicarbonate in dichloromethane were added dropwise to the reaction mixture.15 ml of a solution of 1.41 g (6.46 mmol) of di-tert-butyl dicarbonate in dichloromethane was slowly added dropwise under ice-water bath conditions, and the reaction mixture was stirred at room temperature overnight. Filtration, the organic phase was dried overnight and concentrated, and purified by silica gel chromatography to finally obtain compound 5 (2.12 g, 82%).
80%
In dichloromethane; at 0 - 20℃;
Weigh 2.5 g of compound 17-0 dissolved in 300 mL of dichloromethane and cooled to 0 C. A dichloromethane mixture containing 8.18 g of di-tert-butyl dicarbonate (8.18 g of di-tert-butyl dicarbonate dissolved in 160 mL of dichloromethane) was slowly added dropwise thereto. After the completion of the dropwise addition, the temperature was raised to room temperature and reacted overnight, and the reaction was completely monitored by TLC, concentrated, and dissolved in dichloromethane. Column chromatography (DCM: MeOH = 50:1 - 20:1) gave 5 g of colorless transparent liquid compound 17-1, yield: 80.0%.
30%
In dichloromethane; at 0℃;
,4,8-Tris(ter-butoxycarbonyl)-1,4,8,11-tetraazacyclotetradecane (1) was synthesized. Synthesis of 1 was performed by reported methods by Dessolin et al. (J. Dessolin, P. Galea, P. Vlieghe, J. Chermann, J. Kraus, J. Med. Chem. 1999,42, 229-241) with slight modifications. Typically, to a 1gm of cyclam (5.00 mmol, 1 equiv. ) in ice cold CH2C12 (lOOmL) was added to 2.0g of di-tert-butyl dicarbonate (9.00 mmol, 1.8 equiv. ). The solution was stirred overnight. After solvent evaporation, the crude pale yellow oil was {JND0255. DOC;1} - 34 - purified by flash column chromatography (MeOH/CH2Cl2 5: 95). The first fraction collected was the desired tri-boc protected cyclam (1) as the thick colorless oil (0.75g, 30% yield). MS (electrospray) 523 (M+Na)+
With triethylamine; In dichloromethane; at -15 - 20℃;Inert atmosphere;
To a solution of cyclam (S5, 1.51 g, 7.54 mmol) and triethylamine (5.20 mL, 37.3 mmol) in anhydrous DCM (300 mL) was added dropwise di-tert-butyl dicarbonate (2.95 g, 13.5 mmol) in anhydrous DCM (90 mL) under N2. After the addition was complete, the reaction mixture was cooled to -15 C, and a second portion of di-tert-butyl dicarbonate (1 .96 g, 8.98 mmol) in anhydrous DCM (60 mL) was added. The reaction mixture wasstirred at room temperature overnight and washed with 0.5 M Na2CO3 (2 x 150 mL).The organic phase was dried over Na2SO4 and concentrated under reduced pressure.The residue was purified by flash column chromatography (silica gel, EtOAc ramping toEtOAc:CH3OH = 9:1) to give S7 as a white foam (2.91 g, 77%). RF (EtOAc:CH3OH =9:1) 0.54. m.p. 46-47 C. IR Vmax/cm1 2973, 2932, 2818, 1681, 1464, 1409, 1 389, 1 364,1239, 1158. 1H NMR (200 MHz, CDCI3) 51.46 (5, 27H, 3 x C(CH3)3), 1.60-1.80 (m, 2H, CH2CH2CH2), 1.80-2.10 (m, 2H, CH2CH2CH2), 2.62 (t, 2H, J5.6, CH2NHCH2), 2.78 (t, 2H, J 5.4, CH2NHCH2), 3.20-3.50 (m, 1 2H, 3 x CH2N(Boc)CH2) (one secondary amine proton signal (NH) not observed). MS (ESI) m/z 501 .3 ([M+H], 100%), 523.5 ([M+Na], 17%). The spectroscopic data were in agreement with those in the literature.40
In dichloromethane; at 20℃;
Commercially available (Aldrich Chemical Co., Milwaukee, WI) compound 20 (1.47 g) is suspended in CH2Cl2 (70 ml). To this suspension is added a solution of (BOC)2O (3.86 g in 70 ml of CH2Cl2) dropwise with stirring at RT. The stirring is continued for 2h. The reaction mixture is concentrated to dryness and purified by column chromatography (CombiFlash) to give compound 21(1.8 g)
With triethylamine In ethanol for 15h; Ambient temperature;
56.8%
With potassium carbonate In acetonitrile for 40h; Reflux;
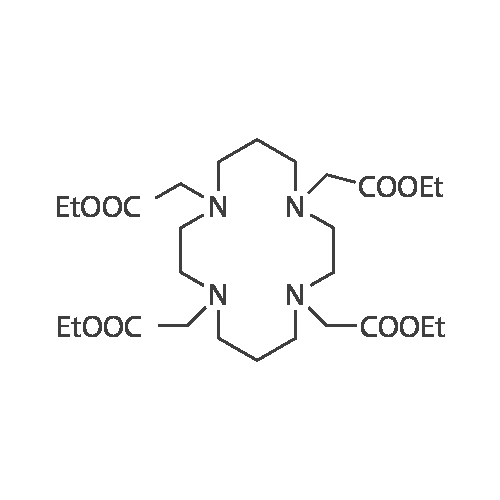
9 Example 8: Preparation of 2,2',2'',2'''-(1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetraacetic acid(Chemical formula 3e)
Cyclam (10.0 g) and acetonitrile (400.0 ml) were added and stirred, and K2CO3 (34.5 g) and Ethyl bromoacetate (36.7 g) were added. The mixture was heated and refluxed for 40 hours to complete the reaction. Cool to room temperature and filter, discard the solid and concentrate the filtrate in vacuo. Add 200 ml of MC and 300 ml of purified water to the concentrate, stir for 30 minutes, and separate. The organic layer was MgSO4-treated, concentrated in vacuo and column-purified with MC-MeOH to obtain tetraethyl 2,2 ', 2' ', 2' '- (1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl ) tetraacetate. (Yield: 56.8%)
56.8 %
With tri-tert-butyl phosphine; potassium carbonate In acetonitrile Reflux;
9 Preparation of 2,2',2",2",2"'-(1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetraacetic acid (chemical formula 3e)
Add 10.0 g of Cyclam and 400.0 ml of acetonitrile for stirring, and add 34.5 g of K2CO3 and 36.7 g of ethyl bromoacetate heated and refluxed for 40 hours to end the reaction. After cooling to room temperature, it is filtered, and the filtrate is vacuum concentrated after the exhaust gas is solid. Add 200ml of dichloromethane (MC) and 300ml of purified water to the concentrate, stir for 30 minutes, and stand for layer separation. After vacuum concentration of the organic layer treated with MgSO4, column purification with MC-methanol (MC-MeOH) yielded 15.4 g of tetraethyl 2,2',2",2",2"-(1,4, 8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetraacetate (yield: 56.8%).
56.8 %
With tri-tert-butyl phosphine; potassium carbonate In acetonitrile Reflux;
9 Preparation of 2,2',2",2",2"'-(1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetraacetic acid (chemical formula 3e)
Add 10.0 g of Cyclam and 400.0 ml of acetonitrile for stirring, and add 34.5 g of K2CO3 and 36.7 g of ethyl bromoacetate heated and refluxed for 40 hours to end the reaction. After cooling to room temperature, it is filtered, and the filtrate is vacuum concentrated after the exhaust gas is solid. Add 200ml of dichloromethane (MC) and 300ml of purified water to the concentrate, stir for 30 minutes, and stand for layer separation. After vacuum concentration of the organic layer treated with MgSO4, column purification with MC-methanol (MC-MeOH) yielded 15.4 g of tetraethyl 2,2',2",2",2"-(1,4, 8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetraacetate (yield: 56.8%).
With sodium hydroxide In water; acetonitrile for 6h;
General synthetic procedure A: N-tetraalkylation of macrocycles
General procedure: Macrocycle (1.0 equiv) and alkyl halide (4.1 equiv) were dissolved in a 1:1 mixture of aqueous 1 MNaOH and CH3CN (5-20 mL). The reaction mixture was shaken for 6 h. The resulting precipitate was collected by filtration, washed with hexane, and dried in vacuo to give the desired N-tetraalkyl derivative.
79%
With triethylamine In dichloromethane for 72h; Heating;
26%
With triethylamine In tetrahydrofuran for 6h; Heating;
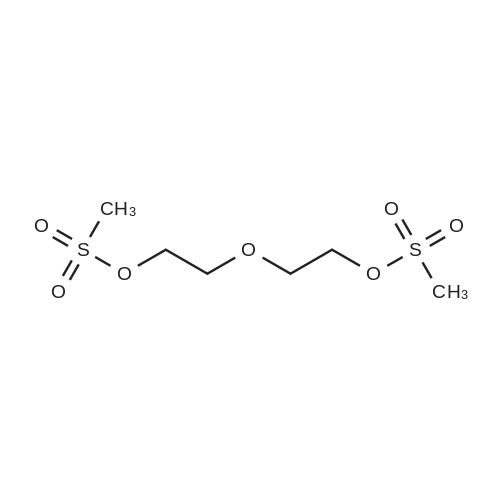
Stage #1: 1,4,8,11-Tetraazacyclotetradecane; diethylene glycol dimesylate With sodium carbonate In tetrahydrofuran for 240h; Heating;
Stage #2: With copper(II) perchlorate In acetonitrile for 1h; Heating;
Stage #3: With sodium sulfide In water for 0.5h;
With triethylamine In methanol at 20℃; for 5.08333h;
1.A; 18
4.006 g (20 mmol) of cyclam (1,4,8,11-tetraazacyclotetradecane) was placed into a solution of 2.79 ml of triethylamine in 15 ml of dried methanol. 6.92 ml of ethyl trifluoroacetate was added dropwise to the upper solution at room temperature with stirring. The addition continued over a period of 5 min. The homogeneous reaction mixture was cooled with an ice-water bath to control the mild exothermicity. Stirring was continued under nitrogen for 5 hours. Volatiles were removed in vacuo. The residue was passed through a small silica-gel plug (25 g) and eluted with 100% ethyl acetate. The eluted solvent was concentrated to give the product as a white foam (8.972 g, 95% yield).
93%
With triethylamine In methanol at 25℃; for 5h; Inert atmosphere;
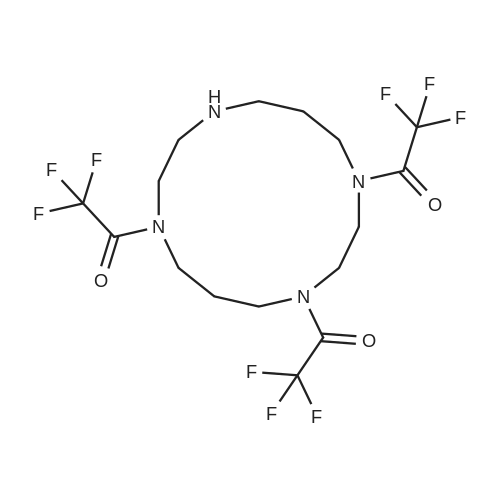
1 VYR2O7 can be synthesized in several ways. Shown below is Scheme 1 for the synthesis of 1,4, 8-Tris(trifluoroacetyl)- 1,4,8,11 -tetraazacyclotetradecane (N4-TFA3; TFA3 -cyclam).
Cyclam (N4;1,4,8,11-tetraazacyclotetradecane; N4; 7.53 g, 37.58 mmol) was dissolved in 30 mL of methanol. To this clear solution, NEt3 (5.20 mL, 37.58 mmol) was added in one portion. Then, ethyl trifluoroacetate (18.0 mL, 150.3 mmol) was added dropwise over a period of 5 minutes while stirring. The homogeneous reaction mixture was cooled with ice-water bath to control the mild exothermic reaction and stirred at 25 °C under the atmosphere of N2 for 5 h. Volatiles were then removed under vacuum. The residue was dissolved in minimum amount of CH2C12 (2.0 mL) and passed through a short silica gel pad (25 g), eluted with 100% EtOAc. The eluent was concentrated to give the product a white semi solid form (17.1 g, 92.5%). The resulting compound was analyzed by proton nuclear magnetic resonance (‘H NMR), carbon-13 nuclear magnetic resonance (‘3C NMR), and high-resolution mass spectrometry (HRMS) by electrospray ionization (ESI). The ‘H NMR data, ‘3C NMR data, and the HRMS-ESI determination of mass of VYR2O7 are as follows. [0074] The ‘H NMR data (200 MHz, CDC13): 3.85 - 3.25 (multiplet, 12 H), 2.80 (broad singlet, 2 H), 2.74-2.50 (broad singlet, 2H), 2.30-1.90 (multiplet, 2 H), 1.85-1.63 (multiplet, 2 H), 1.25- 0.60 (multiplet, 1 H), and the ‘3C NMR data (75.5 MHz, CDC13): 158.74-157.31 (multiplet, C=0, multiplets due to existence of conformers), 122.84-11.32 (quartet, CF3, due to C-F coupling, JC-F - 264 Hz, further split due to existence of conformers), 51.2 - 46.2 (multiplet, CH2 next to N), 29.4 - 27.8 (multiplet, CH2); The HRMS-ESI mass calculated for C,6H21F9N403 was C 39.35; H 4.33; N 11.47; 09.83; and the mass found was: C 39.19; H 4.36; N 11.33; 0 10.04.
93%
With triethylamine In methanol at 20℃; Inert atmosphere;
92.5%
With triethylamine In methanol at 20℃; for 5h;
87.17%
With triethylamine In methanol at 30℃; for 8h; Cooling with ice;
2.2
(2) Take 40.0 g of 1,4,8,11-tetraazacyclotetradecane in 200 mL of methanol and add 40 mL of triethylamine.79.4 g of ethyl trifluoroacetate was added dropwise using a constant pressure dropping funnel under an ice bath, and the solution was clarified.Reaction at 30 ° C for 8 hours, the reaction solution was spin-dried by column chromatography to give a colorless oil compound III, 85 g, yield 87.17%;
86%
With triethylamine In methanol at 20℃; for 5h; Inert atmosphere;
In methanol at 0℃;
With triethylamine In methanol; dichloromethane
3 Preparation of 1,4,8-tris (trifluoroacetyl)-1,4,8,11-tetraazacyclotetradecane
Example 3 Preparation of 1,4,8-tris (trifluoroacetyl)-1,4,8,11-tetraazacyclotetradecane Cyclam (7.53 g, 37.58 mmol) was dissolved in bench MeOH (30 mL). To this clear solution was added NEt3 (5.20 mL, 37.58 mmol) in one portion, followed by portional addition of ethyl trifluoroacetate (18.0 mL, 150.3 mmol) during a period of 5 minutes. The reaction may be chilled to keep temperature under 25° C. Stirring was continued under N2 for 5 h. Volatiles were then removed under vacuum. The residue was dissolved in minimum amount of CH2Cl2 (~2.0mL) and passed through a short silica gel pad (~25 g), eluted with 100% EtOAc. The eluent was concentrated to give the product as a white semi solid (17.05 g, 92.5%), 1H NMR (200 MHz, CDCl3): δ3.85-3.25 (multiplet, 12 H), 2.80 (broad singlet, 2 H), 2.74-2.50 (broad singlet, 2H), 2.30-1.90 (multiplet, 2 H), 1.85-1.63 (multiplet, 2 H), 1.25-0.60 (multiplet, 1 H). 13C NMR (75.5 MHz, CDCl3): δ158.74-157.31 (multiplet, C=O, muitiplets due to existence of conformers), 122.84-11.32 (quartet, CF3, due to C-F coupling, JC-F ~264 Hz, further split due to existence of conformers), 51.2-46.2 (multiplet, CH2 next to N), 29.4-27.8 (multiplet, CH2); Mass of C16H21F9N4O3 requires: C 39.35; H 4.33; N 11.47; O 9.83; found: C 39.19; H 4.36; N 11.33; O 10.04.
With triethylamine In methanol at 20℃; for 5h; Inert atmosphere; Cooling with ice;
2.8 g
With triethylamine In methanol at 20℃; for 5h;
With triethylamine In methanol at 20 - 30℃; for 4h;
7 Example 7: Preparation of crystalline form S of plerixafor intermediate (compound shown in formula II)
In a 2L three-neck flask, add 100.0g of 1,4,8,11-tetraazacyclotetradecane, add 400mL methanol, 50.5g triethylamine, drop 283.7g ethyl trifluoroacetate, 20~30 React for 4h, concentrate to dryness under reduced pressure to obtain an oily substance, add 970mL acetonitrile to the oily substance intermediate to dissolve, add 43.7g p-dichlorobenzyl, 20.7g potassium iodide, heat to reflux reaction; after the reaction is completed, cool down to 20~30, pump After filtering, the filtrate was concentrated to dryness to obtain an oily substance, and then 1.0L of ethanol was added to the oily substance to dissolve it by heating, 4.0L of purified water was added dropwise to crystallize, suction filtration, the filter cake was rinsed with water, and air-dried to obtain the Prosafo intermediate ( The crude product of the compound represented by formula II) is 270.00 g, ready for use, and the HPLC purity is 94.1%. The HPLC spectrum is shown in FIG. 7.Put 10.0g of the crude product of plerixafor intermediate (II) prepared above into a 250ml three-necked flask, add 80mL ethyl acetate, stir and heat to reflux to clear, stop heating, slowly lower the temperature, and lower the temperature by 5°C every hour. To 25~30°C , keep at 25~30°C , crystallize for 8~12h, filter with suction, filter cake 45~55°C and air-dried to obtain 7.20g off-white solid, HPLC purity 99.1%, yield 72.0%, it is plerixafor intermediate compound (II) crystal form S.
With potassium carbonate In chloroform at 20℃; for 48h;
5
In a first phase, ethyl (1,4,8,11-tetraazacyclotetradec-1-yl)ethanoate is obtained by slowly adding 6.68 g (40.0 mmol) of ethyl bromoacetate onto a solution of 40 g (200.0 mmol) of cyclam and 16 g (116 mmol) of potassium carbonate in 1 L of chloroform. The reaction mixture is maintained under stirring at room temperature for 48 h. After filtration on celite and evaporation of the solvent, the residue is taken up in petroleum ether. The excess cyclam is filtered off, the filtrate is concentrated and ethyl (1,4,8,11-tetraazacyclotetradec-1-yl)ethanoate (so-called compound 5.0) is thereby obtained, as 11.21 g of a slightly yellow oil (yield=98%) which is used without any purification.
With potassium carbonate In chloroform at 20℃; for 48h;
With triethylamine; In N,N-dimethyl-formamide; at 20℃; for 16h;
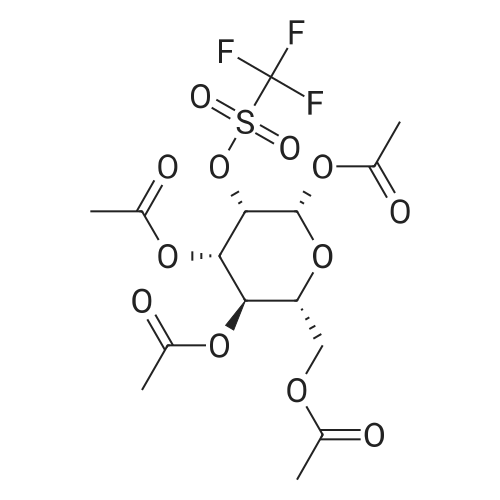
1,3,4,6-Tetra-O-acetyl-2-O-trifluoromethanesulfonyl-beta-D-mannopyranose (300 mg, 0.625 mmol) in 5 ml DMF was added to the mixture of 1,4,8,12-tetraazacyclopentacecane (N4) (250.2 mg, 1.237 mmol) and triethylamine (174 muL, 1.249 mmol) in 5 ml of DMF. The reaction mixture was stirred at room temperature for 6 hrs. The reaction solvent was evaporated to dryness at 40-45 C. under high vacuum. 1,4-Dioxane (10 ml) was then added. The precipitate was filtered. Hydrochloric acid (4N) in 1,4-dioxane (2 ml, 8 mmol) was added. The mixture was cooled in an ice-bath. The mixture was filtered through a Buechner funnel and washed with diethyl ether (2×5 ml). The filtrate was evaporated to dryness, yielding a white solid (383.1 mg, 90.8%). 1H-NMR of N4-DG delta (ppm) 8.50 (s, 1H), 3.98-4.01 (m, 1H), 3.76 (s, 2H), 3.54-3.60 (m, 9H), 3.38-3.45 (m, 8H), 3.31-3.37 (m, 1H), 3.18-3.22 (m, 1H), 2.02-2.31 (m, 4H), 2.15 (s, 12H). 13C-NMR of N4-DG 6 (ppm) 197.3, 175.2, 170.4, 165.6, 67.0, 66.8, 66.4, 51.7, 45.3, 44.0, 43.6, 43.2, 42.9, 42.5, 41.9, 38.6, 37.5, 37.3, 31.8, 19.5, 19.3, 14.5. The synthetic scheme is shown in FIG. 1.
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; N-ethyl-N,N-diisopropylamine In dichloromethane at 20℃;
9D
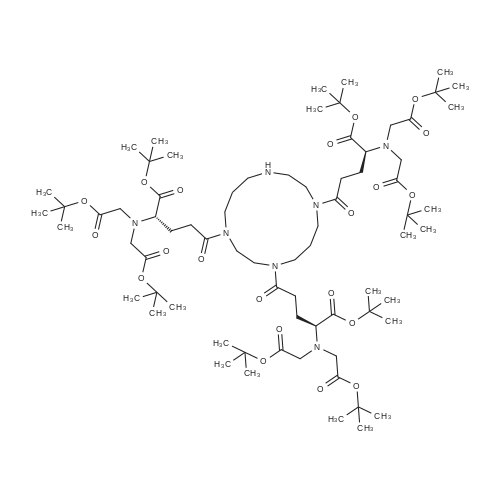
D) Synthesis von Tris-NTA-OtBu (17 or 18) (see also Figure 26); 15 (431 mg; 1.0 mmol) is dissolved in dry dichloromethane (30 ml), and then 16 (67 mg; 0.33 mmol), TBTU (417 mg; 1.3 mmol), and DIPEA (0.3 ml; 1.75 mmol) are added. The obtained reaction mixture is rinsed with nitrogen and stirred over night at room temperature. The volatile components are then drawn off in vacuo, the residue is slurried with dichloromethane (50 ml) and washed 3x with MQ water (15 ml each). The organic phase is dried over anhydrous sodium sulfate, and concentrated in vacuo. The oily residue is purified over a silica column with a gradient of 100% ethyl acetate to 50% ethyl acetate in methanol in 5 volumesof the column.Yield: 0.37 g (0.26 mmol) Tris-NTA-OtBu (17), 79 % o.th. TLC: Rf = 0.5 in ethylacetate/methanol (3:1)MS (MALDI, ESI, CraH^NyCfei); MH+ 1441
With O-(benzotriazol-1-yl)-N,N,N',N'-tetramethyluronium tetrafluoroborate; N-ethyl-N,N-diisopropylamine In dichloromethane
With sodium hydroxide; formaldehyd; formic acid; In water;
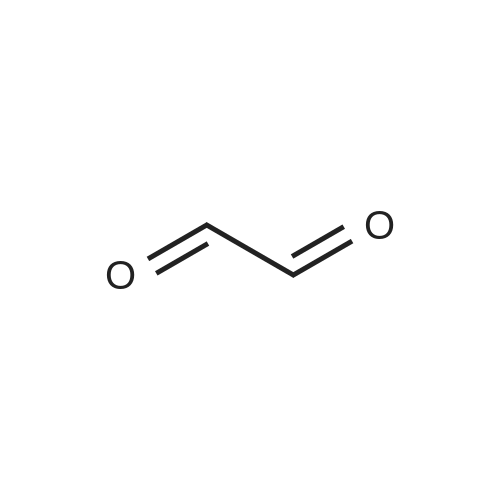
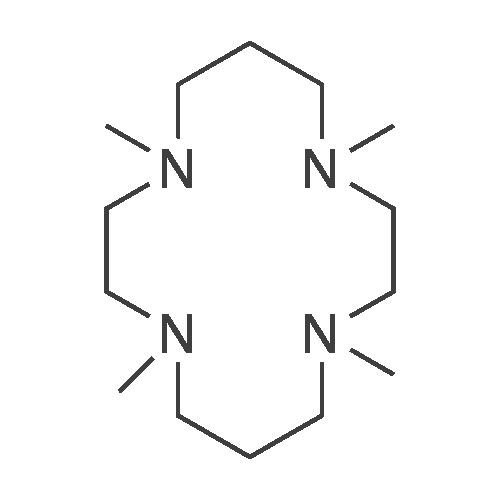
EXAMPLE 12 1,4,8,11-tetraaza-1,4,8,11-tetramethylcyclotetradecane [FIG. 12]. A solution consisting of 1.0 g (0.005 mol) of cyclam, 5.3 mL of formic acid, 4.5 mL of 37% formaldehyde and 1 mL of water was refluxed for 18 h. The reaction mixture was transferred with 6 mL of water to a beaker and cooled to 5 C. in an ice bath. While stirring, a concentrated solution of NaOH was slowly added to pH>12, The temperature was kept below 25 C. during the addition and then extracted with 3*30 mL of CH2Cl2., dried over Na2SO4, and evaporated to dryness. The resultant oil (0.98 g, 71%) was analyzed. Mass spectral analysis showed a m/e=256. 1H NMR (CDCl3): δ 1.68 (4H, quin), 2.22 (12H, s), 2.64 (8H, t), 2.75 (8H, t).
With sodium hydroxide; formaldehyd; formic acid; In water;
EXAMPLE 12 1,4,8,11-tetraaza-1,4,8,11-tetramethylcyclotetradecane [FIG. 12] A solution consisting of 1.0 g (0.005 mol) of cyclam, 5.3 mL of formic acid, 4.5 mL of 37% formaldehyde and 1 mL of water was refluxed for 18 h. The reaction mixture was transferred with 6 mL of water to a beaker and cooled to 5 C. in an ice bath While stirring, a concenrated solution of NaOH was slowly added to pH>12, The temperature was kept below 25 C. during the addition and then extracted with 3*30 mL of CH2Cl2., dried over Na2SO4, and evaporated to dryness. The resultant oil (0.98 g, 71%) was analyzed Mass spectral analysis showed a m/e=256. 1H NMR (CDCl3): δ 1.68 (4H, quin), 2.22 (12H, s), 2.64 (8H, t), 2.75 (8H, t).
With hydrogenchloride In methanol methanolic solution of ligand was added dropwise to a suspension of K2<RuCl5(OH2)> in methanol under reflux, filtered, evapd., the brown solid was dissolved in dil. boiling HCl; the hot soln. was filtered, concd. HCl was added, the formed leaflets were filtered off, washed with ethanol and diethyl ether, dried in vac. at 78°C;
With HCl In ethanol reacted for 24 h, under Ar; recrystd. from 1.0 M HCl, purified by Dowex AG 50W-X2, recrystd. from 0.1 M HCl, washed with acetone, dried in vac.;
With sodium hydroxide; In water; at 20 - 80℃; for 4.25h;
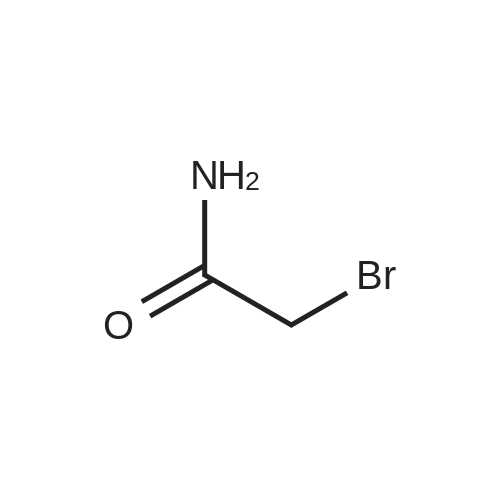
In a typicalpreparation, 1,4,8,11-tetraazacyclotetradecane (Cyclam) (0.44 g, 2.20 mmol) was added to a round bottom flask. Water (4 mL) and 2 M NaOH (0.5 mL) were added to dissolve the Cyclam. 2-Bromoacetamide (1.39 g, 10.1 mmol) was added and additional 2 M NaOH was added to maintain pH 10-11. Upon reaction, the solution became opaque white. The solution was stirred for 15 minutes at 80 C, and then stirred for an additional 4 hours at roomtemperature. The precipitate was isolated by filtration and rinsed with 0.20 M NaOH, thenrinsed with acetone. The precipitate was dried under vacuum. Yield: 76 %. ESI-MS m/z:215.3 (100 %), 216.1 (<10%) [M/2]+; 429.4 (70 %), 430.4 (10%) [M + H+]; 451.4 (100%), 452.4 (20 %) [M + Na+]. ?H NMR (300 MHz, D20 + DC1): 3.39 (s, 8H, pendent CH2),2.90 (s, 8H, ring CH2 [NCH2CH2N]), 2.82 (s, 8H, ring CH2 [NCH2CH2CH2N]), 1.73 (t,4H, ring CH2 [NCH2CH2CH2N], J= 7). ?3C NMR (75 MHz, D20 + DC1): 173.34, 55.22,52.13, 51.35, 22.56.
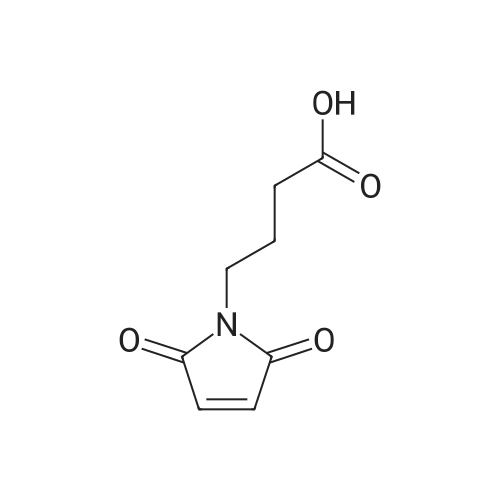
Stage #1: 4-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)butanoic acid With 4-methyl-morpholine; HATU In N,N-dimethyl-formamide at 0℃; for 0.166667h;
Stage #2: 1,4,8,11-Tetraazacyclotetradecane In N,N-dimethyl-formamide at 20℃; for 24h;
4.4. General procedure for the synthesis of PWT1, PWT2 and PWT3 cores
General procedure: To a stirred solution of 4-(2,5-dioxo-2,5-dihydro-1Hpyrrol-1-yl)butanoic acid (1.1 mmol) in (DMF) (5 mL) at 0°C, [O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] (HATU) (1.1 mmol) and 4-methylmorpholine (1.1 mmol) were added. After 10 min a solution of compound 3,4 or cyclam (0.25 mmol) in DMF (3 mL) deprotonated with 4-methylmorpholine was added and the reaction mixture stirred at room temperature for 24 h. After evaporation of the solvent, the oil was treated twice with H2O, dried in vacuum and purified by flash chromatography (eluent: CH2Cl2/MeOH 9.5:0.5) to give a pale yellow solid that was further purified by preparative HPLC using a Water Delta Prep 3000 system with a Jupiter column C18 (250 x 30 mm, 300 A, 15 μm spherical particle size). The column was perfused at a flow rate of 20 mL/min with solvent A (water, 0.1% TFA), and a linear gradient from 10% to 70% of solvent B (60% CH3CN, 0.1% TFA) over 30 min was adopted for the elution of compounds. Final purity was determined as >=95% by analytical HPLC (0-50% CH3CN in 25 min) with a Luna C18 column (4.6 x 100 mm, 3 μm particle size) at a flow rate of 0.5 mL/min.
Stage #1: 4-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1-yl)butanoic acid With 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride In N,N-dimethyl-formamide at 0℃; for 0.166667h;
Stage #2: 1,4,8,11-Tetraazacyclotetradecane In N,N-dimethyl-formamide for 4h;
1 Detailed procedure for the synthesis of PWT2 (reference to Figure 1 ) PWT2
To a stirred solution of compound 6 (1 .1 mmol) in (DMF) (5 mL) at 0 °C, WSC (1 .2 mmol) and Oxp (1 .2 mmol) were added. After 10 min a solution of compound 7 (0.25 mmol) in DMF (3 mL) was added and the reaction mixture stirred for 4 h. After evaporation of the solvent, the residue was purified by preparative HPLC to give a highly hygroscopic white solid after lyophilization.
Boc Cyclam. [0180] A solution of t-butyl dicarbonate (Boc2O) (2.96 g, 13.1 mmol) in 80 mL of methylene chloride (CH2Cl2) was added dropwise over a period of 2 hours to a solution of cyclam (0.88 g, 4.4 mmol) in CH2Cl2 at 0 C. The mixture was allowed to warm to room temperature and stirred overnight. The mixture was concentrated and purified by chromatography (ethyl acetate (AcOEt)?10:1 AcOEt:methanol (CH3OH)) to give cyclam-Boc4 (0.91 g, 35%) as a white set foam and cyclam-Boc3 (1.22 g, 56%) as a light yellow set foam. Tri Boc cyclam: 1H NMR (CDCl3) delta 1.46 (s, 27H), 1.65-1.76 (m, 2H), 2.00-1.80 (m, 2H), 2.62 (bt, J=5.6 Hz, 2H), 2.79 (t, J=4.8 Hz, 2H), 3.31 (t, J=6.4 Hz, 8H), 3.47-3.34 (m, 4H). (see Schickaneder, C.; Heinemann, F. W.; Alsfasser, R. ?Copper II Complexes of the Tetraazamacrocyclic Tertiary Amide Ligand Alanyl-Cyclam,? Eur. J. Chem. 2006, 2357-2363)
With potassium carbonate In acetonitrile at 80℃; for 12h; Schlenk technique; Inert atmosphere;
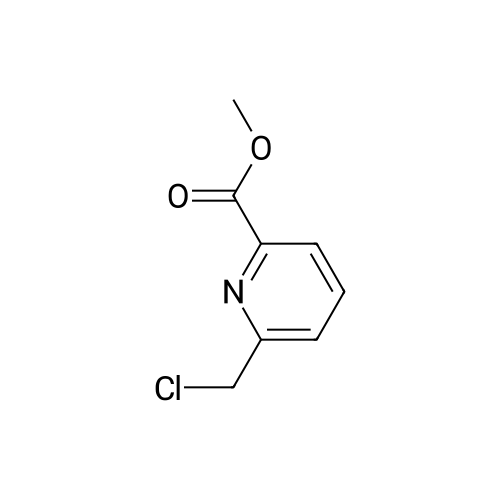
3 General procedure for H4tpaen derivatives (6a-e). Coupling step (5a-e)
General procedure: Methyl 6-chloromethyl-pyridine-2-carboxylate 3 (500 mg, 2.7 mmol), K2CO3 (391 mg, 2.8 mmol) followed by freshly distilled acetonitrile (10 mL) were added to a Schlenk flask under argon atmosphere. Diamine (0.67 mmol) was then added. The mixture was stirred at 80 °C for 12 h. Acetonitrile was then removed under vacuum. The obtained residue was diluted with dichloromethane and washed with water. The organic layers were dried on Na2SO4, filtered and concentrated under vacuum. The resulting residue was recrystallized in i-PrOH.
With triethylamine In chloroform at 45℃; for 72h; Inert atmosphere;
2.2 4.2.1 N-3-Propyl-1,4,8,11-tetraazacyclotetradecane (C3H7-cyclam)
The ligand was prepared based on the method used to prepare similar compounds [19]. Bromopropane (92 mg, 0.75 mmol) was added to a solution of cyclam (600mg, 3 mmol) and trimethylamine (0.13 mL, 0.90 mmol) in chloroform (60 mL), and the solution was stirred at 45 °C under nitrogen gas for 3 d. The resulting mixture was washed ten times with 1M sodium hydroxide aqueous solution and then washed three times with water, dried over MgSO4, and the solvent was evaporated at 50 °C to yield 150 mg of a yellow oil (80%, versus bromopropane). Unreacted cyclam could be recovered from the water layer [32]. 1H NMR (400 MHz, CDCl3, TMS): δ=0.87 (m, 3H), 1.47 (m, 2H), 1.71 (m, 3H), 2.36 (m, 3H), 2.49 (m, 4H), 2.60 (m, 4H), 2.67 (m, 3H), 2.73 (m, 7H).
Stage #1: 1,4,8,11-Tetraazacyclotetradecane With zinc diacetate In 5,5-dimethyl-1,3-cyclohexadiene at 110℃; for 0.166667h; Inert atmosphere;
Stage #2: bromobenzene With copper(I) oxide; potassium <i>tert</i>-butylate In 5,5-dimethyl-1,3-cyclohexadiene at 110℃; for 26h; Inert atmosphere;
3 Synthesis of 1-phenyl-1 .4,8,1 1-tetraazacyclotetradecane having formula (1b1)
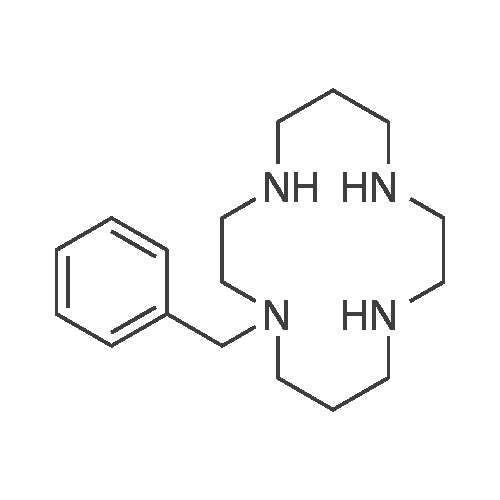
All glassware used was previously furnace-dried at 130°C under vacuum for one night, rapidly assembled under heating, and cooled to room temperature (25°C), under argon flow (Ar). Into a 50 ml long-necked test tube, provided with a magnetic stir bar and a tap, were introduced, under argon (Ar) flow, 255 mg (1.27 χ 10"3 mol) of 1 ,4,8,11- tetraazacyclotetradecane (Aldrich) having formula (Mb), 250 mg (1.36 * 10~3 mol) of anhydrous zinc acetate [(CH3COO)2Zn(anhyCirous)] and 10.0 ml of anhydrous xylene [C6H4(CH3)2(anhydrous)] (Aldrich). The long-necked test tube was subsequently immersed in a bath which had been preheated to 110°C, and the whole was left under stirring (700 rpm) for 10 minutes. The long-necked test tube was subsequently removed from the heating bath and brought to room temperature (25°C) using compressed air, and subsequently, under argon flow (Ar), 320 mg (2.24 * 10"2 mol) of cuprous oxide (Cu20) (Aldrich), 747 mg (6.66 * 10"3 mol) of potassium f-butoxide (f-BuOK) (Aldrich), 147 μΙ (1.40 χ 10"3 mol) of bromobenzene (Aldrich) having formula (Ilia), and a further 10.0 ml of anhydrous xylene [C6H4(CH3)2(an ydrous)] (Aldrich) were added. The long-necked test tube was subsequently again immersed in a bath which had been preheated to 110°C, and the whole was left under stirring (700 rpm) for 26 hours, working as follows: when 110°C was reached, the long-necked test tube was allowed to vent by opening the tap, still under argon (Ar) flow, for approximately 2 seconds, so as to avoid the pressure increase resulting from the temperature increase, and subsequently the tap was closed again and the long-necked test tube was kept at said temperature for 26 hours. At the end, the progress of the reaction was controlled, working as follows: 0.1 ml of reaction mixture were removed from the long-necked test tube and placed in a test tube containing 1 ml of toluene [C6H5CH3] (Aldrich) and 2 ml of an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq )], and the whole was put under stirring to obtain a biphasic system comprising a predominantly organic phase and a predominantly aqueous phase. The predominantly organic phase was separated and subjected to Thin Layer Chromatography (TLC) on neutral alumina, using a methanol [CH3OH] (Aldrich )/dichloromethane [CH2CI2] (Aldrich) mixture (1/1 , v/v) as eluent and a 254 nm ultraviolet (UV) lamp as detector: said analysis indicated that 1-phenyl-1 ,4,8,1 1 - tetraazacyclotetradecane having formula (Ib-i ) had been formed. The reaction mixture was subsequently submerged in a separating funnel containing toluene [C6H5CH3] (Aldrich) and an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq.)]: the whole was left under stirring to obtain a biphasic system comprising a predominantly organic phase and a predominantly aqueous phase. The predominantly organic phase was separated and extracted three times using an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq )]: the aqueous phases obtained were put together, and extracted twice using toluene [C6H5CH3] (Aldrich). The organic phases obtained at the end of the extractions were put together and dried using a rotary evaporator, and the resulting residue was dissolved in the minimum possible volume of dichioromethane (CH2CI2) (Aldrich); subsequently 2 g of basic alumina (Aldrich) were added thereto and subsequently it was dried again using a rotary evaporator. The powder obtained was placed over a basic alumina panel (Aldrich) in order to be filtered, initially, using n-heptane (Aldrich), so as to remove all the low-polarity impurities, and subsequently, working in gradient elution towards a methanol [CH3OH] (Aldrich dichloromethane [CH2CI2] (Aldrich) mixture (1/1 , v/v), so as to elute the 1-phenyl- 1 ,4,8,11-tetraazacyclotetradecane having formula (lb,). The filtrate obtained was evaporated until dry using a rotary evaporator to obtain 281 mg of a white solid of pure 1- phenyl-1 ,4,8,11-tetraazacyclododecane having formula (Ibi) (80% yield), which was subjected to the following characterizations. DCI/MS (m/z): [M+Hf: 277.2 (100%); [M+H+gas]+: 333.3 (18%). Elemental analysis [found (calculated)]: C: 69.60% (69.52%); H: 10.12 (10.21 %); N: 20.30% (20.27%).
Stage #1: 1,4,8,11-Tetraazacyclotetradecane With zinc diacetate In 5,5-dimethyl-1,3-cyclohexadiene at 110℃; for 0.166667h; Inert atmosphere;
Stage #2: bromobenzene With copper(I) oxide; potassium <i>tert</i>-butylate In 5,5-dimethyl-1,3-cyclohexadiene at 110℃; for 26h; Inert atmosphere;
4 Synthesis of I ,4,8-triphenyl- 1,4,8,11 -tetraazacyclotetradecane havinQ formula (1b2)
All glassware used was previously furnace-dried at 130°C under vacuum for one night, rapidly assembled under heating, and cooled to room temperature (25°C), under argon flow (Ar). Into a 50 ml long-necked test tube, provided with a magnetic stir bar and a tap, were introduced, under argon (Ar) flow, 250 mg (1.25 χ 103 mol) of 1 ,4,8,11- tetraazacyclotetradecane (Aldrich) having formula (lib), 247 mg (1.35 χ 10"3 mol) of anhydrous zinc acetate [(CH3COO)2Zn(anhydrous)] and 10.0 ml of anhydrous xylene [C6H4(CH3)2(anhyclro.s)] (Aldrich). The long-necked test tube was subsequently immersed in a bath which had been preheated to 1 10°C, and the whole was left under stirring (700 rpm) for 10 minutes. The long-necked test tube was subsequently removed from the heating bath and brought to room temperature (25°C) using compressed air, and subsequently, under argon flow (Ar), 289 mg (2.02 * 10"3 mol) of cuprous oxide (Cu20) (Aldrich), 1.82 g (1.62 * 10"2 mol) of potassium i-butoxide (f-BuOK) (Aldrich), 421 μΙ (4.01 * 10 3 mol) of bromobenzene (Aldrich) having formula (Ilia), and a further 10.0 ml of anhydrous xylene [C6H4(CH3)2(anhydrous)] (Aldrich) were added. The long-necked test tube was subsequently again immersed in a bath which had been preheated to 1 10°C, and the whole was left under stirring (700 rpm) for 26 hours, working as follows: when 110°C was reached, the long-necked test tube was allowed to vent by opening the tap, still under argon (Ar) flow, for approximately 2 seconds, so as to avoid the pressure increase resulting from the temperature increase, and subsequently the tap was closed again and the long-necked test tube was kept at said temperature for 26 hours. At the end, the progress of the reaction was controlled, working as follows: 0.1 ml of reaction mixture were removed from the long-necked test tube and placed in a test tube containing 1 ml of toluene [C6H5CH3] (Aldrich) and 2 ml of an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq )], and the whole was left under stirring to obtain a biphasic system comprising a predominantly organic phase and a predominantly aqueous phase. The predominantly organic phase was separated and subjected to Thin Layer Chromatography (TLC) on neutral alumina, using dichloromethane [CH2CI2] (Aldrich) as eluent and a 254 nm ultraviolet (UV) lamp as detector: said analysis indicated that 1 ,4,8-triphenyl-1 ,4,8,1 1-tetraazacyclotetradecane having formula (lb2) had been formed. The reaction mixture was subsequently submerged in a separating funnel containing toluene [C6H5CH3] (Aldrich) and an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq >]: the whole was put under stirring to obtain a biphasic system comprising a predominantly organic phase and a predominantly aqueous phase. The predominantly organic phase was separated and extracted three times using an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq )]: the aqueous phases obtained were put together, and extracted twice using toluene [C6H5CH3] (Aldrich). The organic phases obtained at the end of the extractions were put together and dried using a rotary evaporator, and the resulting residue was dissolved in the minimum possible volume of dichloromethane (CH2CI2) (Aldrich); subsequently 2 g of basic alumina (Aldrich) were added thereto and subsequently it was dried again using a rotary evaporator. The powder obtained was placed over a basic alumina panel (Aldrich) in order to be filtered, initially, using n-heptane (Aldrich), so as to remove all the low-polarity impurities, and subsequently, working in gradient elution towards dichloromethane [CH2CI2] (Aldrich), so as to elute the 1 ,4,8-triphenyl-1 ,4,8,11- tetraazacyclotetradecane having formula (lb2). The filtrate obtained was evaporated until dry using a rotary evaporator to obtain 423 mg of a white solid of pure 1 ,4,8-triphenyl-1 ,4,8, 1 1- tetraazacyclotetradecane having formula (lb2) (79% yield), which was subjected to the following characterizations. DCI/MS (m/z): [ +ΗΓ: 429.3 (100%); [M+H+gas : 485.4 (18%). HRMS (m/z): 429.3017±1.0029 ppm [C28H37N4]+. Elemental analysis [found (calculated)]: C: 78.51% (78.46%); H: 8.50 (8.47%); N: 13.01 % (13.07%).
Stage #1: 1,4,8,11-Tetraazacyclotetradecane With zinc diacetate In 5,5-dimethyl-1,3-cyclohexadiene at 110℃; for 0.166667h; Inert atmosphere;
Stage #2: bromobenzene With copper(I) oxide; potassium <i>tert</i>-butylate In 5,5-dimethyl-1,3-cyclohexadiene at 110℃; for 26h; Inert atmosphere;
5 Synthesis of 1.4.8,11 -tetraphenyl-1 .4.8,11 -tetraazacyclotetradecane having formula (1b3)
All glassware used was previously furnace-dried at 130°C under vacuum for one night, rapidly assembled under heating, and cooled to room temperature (25°C), under argon flow (Ar). Into a 500 ml three-necked flask, provided with a magnetic stir bar, a thermometer having a ground glass joint, an insufflator having a tap, and a plug, were introduced, under argon (Ar) flow, 2.55 g (1.27 * 10"2 mol) of 1 ,4,8,1 1 -tetraazacyclotetradecane (Aldrich) having formula (lib), 2.49 g (1.36 * 10~2 mol) of anhydrous zinc acetate [(CH3COO)2Zn(anhydrous)] and 100 ml of anhydrous xylene [C6H4(CH3)2(anhydrous)] (Aldrich). The flask was subsequently immersed in a bath which had been preheated to 119°C, and the whole was left under stirring (700 rpm) for 10 minutes. The flask was subsequently removed from the heating bath and brought to room temperature (25°C) using compressed air, and subsequently, under argon flow (Ar), 3.10 g (2.17 * 10"2 mol) of cuprous oxide (Cu20) (Aldrich), 27.4 g (0.244 mol) of potassium f-butoxide (f-BuOK) (Aldrich), 5.60 ml (5.33 * 10"2 mol) of bromobenzene (Aldrich) having formula (Ilia), and a further 80.0 ml of anhydrous xylene [C6H4(CH3)2(an ydrous)] (Aldrich) were added. The flask was subsequently again immersed in a bath which had been preheated to 119°C, and the whole was left under stirring (700 rpm) for 26 hours, working as follows: when 110°C was reached (as shown by the thermometer having a ground glass joint), the tap of the insufflator was opened, still under argon (Ar) flow, for approximately 5 seconds, so as to avoid the pressure increase resulting from the temperature increase, and subsequently the tap was closed again and the long-necked test tube was kept at said temperature for 26 hours. At the end, the progress of the reaction was controlled, working as follows: 0.1 ml of reaction mixture were removed from the flask and placed in a test tube containing 1 ml of toluene [C6H5CH3] (Aldrich) and 2 ml of an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq.)], and the whole was put under stirring to obtain a biphasic system comprising a predominantly organic phase and a predominantly aqueous phase. The predominantly organic phase was separated and subjected to Thin Layer Chromatography (TLC) on neutral alumina, using dichloromethane [CH2CI2] (Aldrich) as eluent and a 254 nm ultraviolet (UV) lamp as detector: said analysis indicated that 1 ,4,8,1 1-tetra phenyl- 1 , 4,8,1 1 -tetraazacyclotetradecane having formula (lbs) had been formed. The reaction mixture was subsequently submerged in a separating funnel containing toluene [C6H5CH3] (Aldrich) and an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq.)]: the whole was put under stirring to obtain a biphasic system comprising a predominantly organic phase and a predominantly aqueous phase. The predominantly organic phase was separated and extracted three times using an aqueous solution at pH 14 prepared using potassium hydroxide (Aldrich) [KOH(aq )]: the aqueous phases obtained were put together, and extracted twice using toluene [C6H5CH3] (Aldrich). The organic phases obtained at the end of the extractions were put together and dried using a rotary evaporator, and the resulting residue was dissolved in the minimum possible volume of dichloromethane (CH2CI2) (Aldrich); subsequently 10 g of basic alumina (Aldrich) were added thereto and subsequently it was dried again using a rotary evaporator. The powder obtained was placed over a basic alumina panel (Aldrich) in order to be filtered, initially, using n-heptane (Aldrich), so as to remove all the low-polarity impurities, and subsequently, working in gradient elution towards dichloromethane [CH2CI2] (Aldrich), so as to elute the 1 , 4,8,1 1 -tetraphenyl-1 , 4,8,11-tetraazacyclotetradecane having formula (lb3). The filtrate obtained was evaporated until dry using a rotary evaporator to obtain 6.09 g of a white solid of pure 1 ,4,8,1 1 -tetraphenyl-1 , 4,8, 1 1-tetraazacyclotetradecane having formula (lb3) (95% yield), which was subjected to the following characterisations. DCI/MS (m/z): [M+H]+: 505.3 (100%); [M+H+gasf: 561.3 (18%). HRMS (m/z): 505.3332±1.3196 ppm [C3,H4^A] Elemental analysis [found (calculated)]: C: 80.95% (80.91 %); H: 8.05% (7.99%); N: 1 1.08% (11.10%). 1H-MNR (400 MHz, DMSOd6l) δ (ppm): 7.20 (dd, J2 1 = 7.3 Hz, J2,3 = 8.0 Hz, 8H, H2); 6.80 (d, J3,2 = 8.0 Hz, 8H, H3); 6.67 (t, J1 2 = 7.3 Hz, 4H, H,); 3.55 (s, 8H, H6); 3.49 (br. t, J ,S = 6.7 Hz, 8H, H4); 1.93 (br. m, J5A = 6.7 Hz, 4H, H5). ICP (ppm): B < 30; Na = 20*102; Mg = 50; Al = 80 01; P < 20; K < 10*101; Sc < 5; Ti = 3; V < 10; Cr = 7; Mn = 0.4; Fe = 90; Ni < 30; Cu = 7.0; Zn < 10 01; As = 0.6; Sr = 1 ; Nb < 3; Mo < 5; Pd = 5; Sn < 1; Ba < 1001; W
95%
Stage #1: 1,4,8,11-Tetraazacyclotetradecane With zinc diacetate In 5,5-dimethyl-1,3-cyclohexadiene at 119℃; for 0.166667h; Inert atmosphere;
Stage #2: bromobenzene With copper(I) oxide; potassium <i>tert</i>-butylate In 5,5-dimethyl-1,3-cyclohexadiene at 25 - 119℃; for 26h; Inert atmosphere;
1 Synthesis of 1,4,8,11-tetraphenyl-1 ,4,8,11-tetraazacyclotetradecane
All glassware used was previously oven-dried at 130°C under vacuum for one night, rapidly assembled under heating, and cooled to room temperature (25°C), under argon flow (Ar). 2.55 g (1.27x102mo1)of 1,4,8,11-tetraazacyclotetradecane (Aldrich) having the formula (Ia3), 2.49 g (1.36 xl 0.2 mol) of anhydrous zinc acetate [(CH3COO)2Zfl(anhydrous)] and 100 ml of anhydrous xylene [C6H4(CH3)2(aflhydrous)] (Aldrich) were introduced, under argon flow, into a 500 ml, three-necked flask equipped with a magnetic stirrer bar, thermometer with ground glass cone, insufflator with tap and stoppers.Said flask was stoppered, immersed in a bath preheated to 119°C and left at said temperature under magnetic stirring (700 rpm) for 10 minutes. The flask was then removed from the heating bath and adjusted to ambient temperature (25°C) with compressed air, after which 3.10 g (2.17x102 mol) of copper(l) oxide (Cu20) (Aldrich), 27.4 g (0.244 mol) of potassium t-butoxide (t-BuOK) (Aldrich), 5.60 ml (5.33x102mol) of bromobenzene (Aldrich) having the formula (lila) and a further 80.0 ml of anhydrous xylene [C6H4(CH3)2(aflhydrous)] (Aldrich) were added under argon (Ar) flow.The flask was then reimmersed in the bath preheated to 119°C and the whole was left under stirring (700 rpm) for 26 hours, operating as follows: when 110°C was reached (indicated by the thermometer with ground glass cone), the insufflator tap was opened, still under argon (Ar) flow, for approximately 5 seconds, in order to avoid any increase in pressure due to the increase in temperature, and the tap was subsequently closed again and the flask was kept at said temperature for 26 hours.At the end, the degree of progress of the reaction was checked, operating as follows:0.1 ml of reaction mixture were taken from the flask and placed in a test tube containing 1 ml of toluene [C6H5CH3] (Aldrich) and 2 ml of an aqueous solution at pH 14 prepared with potassium hydroxide (Aldrich) [KOH(aq)] and the whole was placed under stirring, a two-phase system comprising a predominantly organic phase and a predominantly aqueous phase being obtained. The predominantly organic phase was separated and subjected to thin-layer chromatography (TLC) on neutral alumina using dichloromethane [CH2CI2J (Aldrich) as eluent and a 254 nm ultraviolet (UV) lamp as detector: said analysis revealed that 1,4,8,1 1-tetraphenyl-1 ,4,8,11- tetraazacyclotetradecane having the formula (laia) had formed.The reaction mixture was then drowned in a separating funnel containing toluene [C6H5CH3] (Aldrich) and an aqueous solution at pH 14 prepared with potassium hydroxide (Aldrich) [KOH(aq)]: the whole was placed under stirring, a two-phase system comprising a predominantly organic phase and a predominantly aqueous phase being obtained. The predominantly organic phase was separated and extracted three times with an aqueous solution at pH 14 prepared with potassium hydroxide (Aldrich) [KOH(aq,)]: the aqueous phases obtained were combined and extracted twice with toluene [C6H5CH3] (Aldrich). The organic phases obtained at the end of the extractions were combined and evaporated to dryness with a rotary evaporator and the resultant residue was dissolved in the minimum possible volume of dichloromethane (CH2CI2) (Aldrich), after which 10 g of basic alumina (Aldrich) were added and the mixture was again evaporated to dryness with the rotary evaporator.The powder obtained was placed on a basic alumina plate (Aldrich) so it could be filtered, initially, with n-heptane (Aldrich) to carry away any low-polarity impurities and, subsequently, with an elution gradient up to a 1/1 vol./vol. mixture of n-heptane (Aldrich)/dichloromethane [CH2CI2] (Aldrich) to elute the 1,4,8,11 -tetraphenyl-1 ,4,8, 11- tetraazacyclotetradecane having the formula (laia). The filtrate obtained was evaporated to dryness with a rotary evaporator, 6.09 g of a white solid of pure 1,4,8,11- tetraphenyl-1 ,4,8, 11 -tetraazacyclotetradecane having the formula (laia) (95% yield) being obtained which was subjected to the following characterisations.DCI/MS (m/z): [M+H}: 505.3 (100%); [M+H+gasj: 561.3 (18%). HRJMS (m/z): 505.3332±1.3196 ppm [C34H41N4}. Elemental analysis [found (calculated)]: C: 80.95% (80.91%); H: 8.05% (7.99%); N:11.08% (11.10%). 1H MNR (400 MHz, DMSOd6,) 6 (ppm): 7.20 (dd, J21 7,3 Hz, J23 8.0 Hz, 8H, H2);6.80 (d, J3,2 8.0 Hz, 8H, H3); 6.67 (t, J12= 7.3 Hz, 4H, H1); 3.55 (s, 8H, H6); 3.49 (br. t, J45 6.7 Hz, 8H, H4); 1.93 (br. m, J54= 6.7 Hz, 4H, H5).ICPMS (ppm): B < 30; Na 20*102; Mg = 50; Al 80*101; P < 20; K < 10*101; Sc < 5; Ti = 3; 10 V; Cr= 7; Mn = 0,4; Fe = 90; Ni <30; Cu = 7.0; Zn <10*101; As = 0.6; 5r 1; Nb <3; Mo <5; Pd = 5; Sn <1; Ba <10*101; W <5; Ir <3; Pt <3; Au <5; Pb = 0.6; Bi <1.
In dichloromethane; at 20℃; for 4h;Cooling with ice;
To 10. 0g (50. 0mmol) of 1,4,8,11-tetraazacyclotetradecane 1250mL of methylene chloride solution in an ice water bath was added 20. 0g (90.0mmol, 1. 8 equiv. ) di-t-butyl dicarbonate, and the solution was stirred at room temperature for 4 hours. The solvent was evaporated to dryness, purified by silica gel column chromatography (MeOH / CH2Cl2 5:95) to give "two of Boc" compound "three of Boc" compounds.
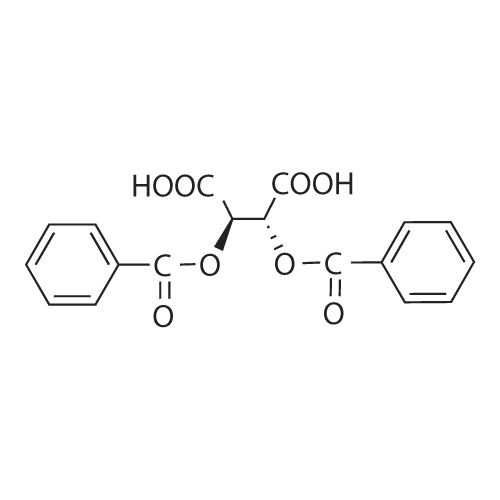
(2R,3R)-2,3-bis(benzoyloxy)butanedioic acid decahydro-1H,6H-3a,5a,8a,10a-tetraazapyrene[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
78.9%
To a stirred solution of 1,4,8,11-tetraazacyclotetradecane (50.Og, 0.249 mole) in methanol (1625 ml) at -5C was added a solution of glyoxal (44.2 g, 0.304 mole) in methanol (250 ml) dropwise. After complete addition the reaction temperature was raised to 30C and stirred for 4 hrs. Methanol was distilled off and the residue was stirred with ethyl acetate (750 ml) and filtered through celite bed. The filtrate was added dropwise to a stirred solution of dlbenzoyl-L-tartaric acid (205.7g,0.574 mole) in ethyl acetate (1500 ml) and the reaction mixture was stirred for 3 hrs at 30C. The reaction mixture was filtered and solid obtained was washed with ethyl acetate. The crude product was dried under vacuum at 55C for 20 hrs. The crude product was again slurry washed with water and dried to give the title compound.Yield: 78.9% (185g)GC Purity: 99.87%
With triethylamine In N,N-dimethyl-formamide at 20℃; for 12h;
N-(3,5-Dimethyladamantan-1-yl)-1,4,7,10-tetraazacyclododecan-1-carboxamide (9)
General procedure: 1,4,7,10-Tetraazacyclododecane (860 mg, 5 mmol) and triethylamine (200 mg, 2 mmol) were added to a solution of 1-isocyanato-3,5-dimethyladamantane (205 mg, 1 mmol) in DMF (5 mL). The mixture was stirred for 12 h at ~ 20 °C. A crystalline precipitate was collected by filtration, washed with water (25 mL), and dried in vacuo
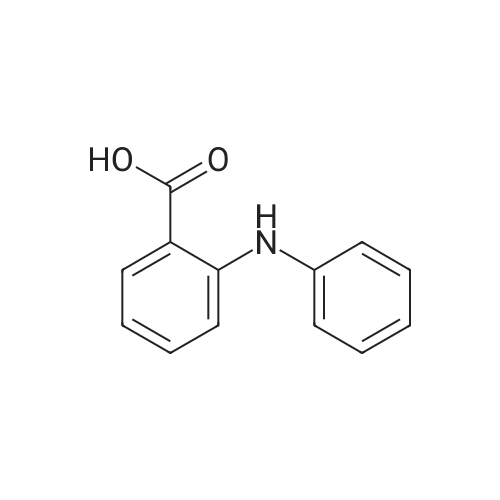
Fenamic acid (0.5mmol, 107mg), potassium hydroxide (0.5mmol, 28mg) and zinc chloride (0.25mmol, 34mg) were dissolved in 20mL of methanol. After 1h stirring methanolic solution of tmen (0.25mmol, 29mg), bapen (0.25mmol, 44mg) or cyclam (0.25mmol, 50mg) was added to the mixture, respectively. The mixture was transferred into a Teflon-lined stainless-steel autoclave and solvothermaly treated at 100C for 24h. Resultant pink solutions were left aside for crystallization and after few days colorless air stable crystals of 2-4 were isolated from respective solutions.
With triethylamine In dichloromethane at 30℃; for 10h; Cooling with ice;
2.1
(1) 30.0 g of 1,4,8,11-tetraazacyclotetradecane was dissolved in 1 L of dry methylene chloride, and 52 mL of triethylamine and 48.5 g of p-toluenesulfonyl chloride were added under an ice bath to obtain a colorless solution. The solution was reacted at 30°C for 10 hours.The reaction was directly spin-dried and purified by column chromatography to give Compound IV as a white foamy solid 62g, yield 81.39%;
tetraethyl 5,5',5'',5'''-(1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl)tetrakis(furan-2-carboxylate)[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With tri-tert-butyl phosphine; bis(dibenzylideneacetone)-palladium(0); sodium t-butanolate; In toluene; at 35 - 50℃; for 0.5h;
Cyclam 10.0g,<strong>[6132-37-2]ethyl 5-bromofuran-2-carboxylate</strong> , 24.0 g of t-BuONa and 400 ml of toluene are stirred and stirred. The temperature was raised to 35 ° C and 2.0 g of 50percent (t-Bu) 3P toluene solution was added. After stirring for 30 minutes, the temperature was raised to 50 ° C. and 1.5 g of Pd (dba) 2 was added.After cooling to room temperature, 1000 ml of purified water is added, and the mixture is left to stand for 30 minutes to separate the layers, and the aqueous layer is discarded.The organic layer was MgSO4-treated, concentrated in vacuo and purified by MC-MeOH column to obtain tetraethyl 5,5 ', 5' ', 5' '- (1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetrayl) tetrakis (furan-2-carboxylate)14.1 g. (Yield: 37.5percent).
[Zn(1,4,8,11-tetraazacyclotetradecane)(niflumicato)2][ No CAS ]
Yield
Reaction Conditions
Operation in experiment
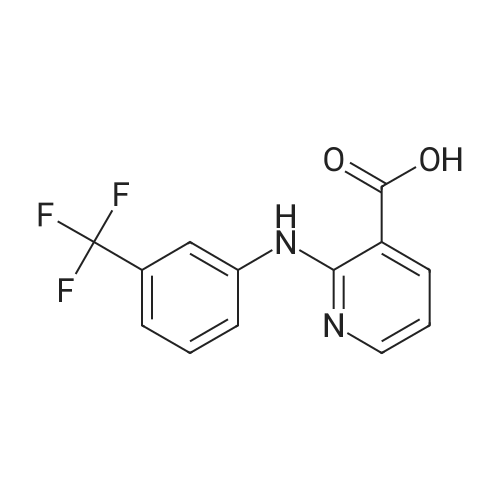
General procedure: Niflumic acid (0.5 mmol, 141 mg), potassium hydroxide (0.5 mmol, 28 mg) and zinc chloride (0.25 mmol, 34 mg) were dissolved in 15 mL of methanol. After 1 h stirring, a methanolic solution (5 mL) of cyclam (0.25 mmol, 50 mg) or tmen (0.25 mmol, 29 mg) was added to the mixture. The mixture was subsequently transferred into a Teflon-lined stainless steel autoclave and solvothermally treated at 100 C for 24 h. The colorless homogenous solution was filtered and left aside for crystallization. After few days, the colorless crystals of 2 and 3 were isolated from respective solutions by filtration. Elemental Anal. Calc. for 2 (C36H40F6N8O4Zn); MW 828.13): C,52.21%; H, 4.87%; N, 13.53%; found: C, 52.49%; H, 5.06%; N, 13.66%.Elemental Anal. Calc. for 3 (C32H32F6N6O4Zn; MW 744.00): C,51.66%; H, 4.34%; N, 11.30%; found: C, 51.87%; H, 4.66%; N 11.54%.IR (cm1): (2) n(N-H), 3262 (m); n(N-H)cyclam, 3195 (m); n(C-H)ar,3047 (m); n(C-H)cyclam, 2955 (m), 2854 (m); d(N-H)(cyclam), 1620 (s);nas(COO), 1594 (s); (C-C, C-N)ar, 1517 (s), 1447 (s); ns(COO), 1380 (s);n n(C-F), 1333 (s); n(C-N), 1236 (s); n(CCF), 1112 (s); d(C-F), 779 (s);n(Zn-O), 463 (m).
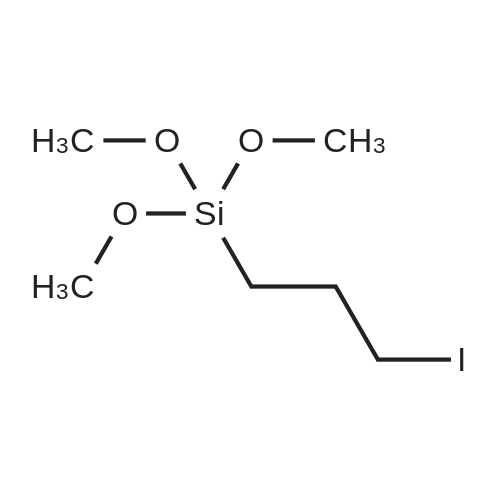
1-(3-(trimethoxysilyl)propyl)-1,4,8,11-tetraazacyclotetradecane[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
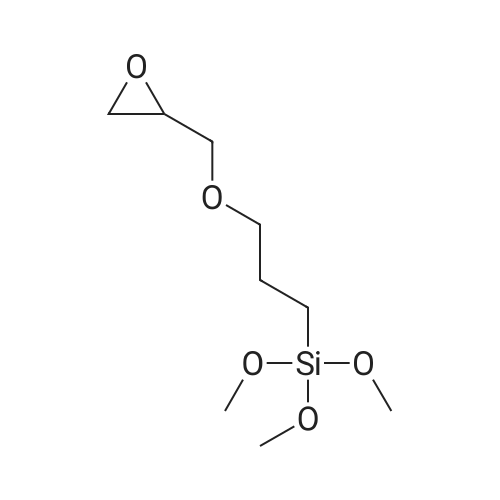
Stage #1: 1,4,8,11-Tetraazacyclotetradecane With sodium hydride In acetonitrile for 1h; Inert atmosphere; Reflux;
Stage #2: 3-iodopropyltrimethoxysilane In acetonitrile Reflux;
3.3 1-(3-(trimethoxysilyl)propyl)-1,4,8,11-tetraazacyclotetradecane (silylcyclam)
Cyclam (2.50g, 12.5mmol) was suspended in 200mL of 74 acetonitrile under argon atmosphere. 63 NaH (300mg, 12.5mmol) was added in small portions and resulted in the evolution of hydrogen. The resulting mixture was heated to reflux for 1h. A solution of 10 3-iodopropyltrimethoxysilane in 100mL of acetonitrile was subsequently added dropwise and the reaction mixture heated to reflux overnight. The solvent was removed under reduced pressure and the residue washed three times with CH2Cl2 (∼60mL) and filtered. To obtain the modified cyclam as a brownish 75 oil (4.13g, 11.45mmol, 92%) CH2Cl2 was removed under reduced pressure. 1H NMR (CDCl3): δ=0.44-0.70 (m, 2 H, CH2Si), 1.36-1.88 (m, 6 H, CH2CH2Si, CH2(CH2N)2), 2.26-3.17 (m, 18 H, CH2N), 3.47-3.58 (s, 9 H, CH3OSi) ppm, broad peaks indicating mixture of multiply substituted derivatives; 13C NMR (CDCl3): δ=8.6 (CH2Si), 18.9 (CH2CH2Si), 26.9, 29.6 (CH2(CH2NH)2), 48.4, 48.7, 49.6, 50.0, 50.1, 51.6, 53.9, 55.2, 56.1 (CH2N, CH3OSi), 58.9 (NCH2CH2CH2Si) ppm; IR: 695 (w), 814 (s), 1072 (s), 1189 (m), 1300 (w), 1364 (w), 1461 (m), 1600 (m), 1638 (w), 2186 (w), 2810 (w), 2837 (w), 2939 (s), 3201 (w), 3305 (w) cm-1
2.3. Preparation of solid CT-complexes
General procedure: The three solid CT-complexes formed in the reaction of the donorTACTD with each of TFQ, DDQ and TBCHD were isolated in CHCl3 bythe drop wise addition of a saturated solution (70 ml) of the donor toa saturated solution (85 ml) of each of the acceptors. The mixture ineach case was stirred for about 10-15 min. The mixing of reactantswas associated with a strong change in color. The resulting precipitatein each case was filtered immediately, washed several times with minimumamountsof CHCl3, and dried under vacuum. The three complexesand adduct were characterized using spectroscopic techniques (FTIRand UV-Vis) and by elemental analysis (theoretical values are shownin brackets): [(TACTD)(TFQ)2] dark brown complex (M/W: 560.44 g);C, 34.30% (34.26%) H, 4.25% (4.28%); N, 9.89% (9.99%); [(TACTD)(DDQ)2] brown complex (M/W: 654.32 g); C, 44.10% (44.02%); H,3.61% (3.67%); N, 12.79% (12.84%) and [(TACTD)(TBCHD)2] reddishbrown complex (M/W: 1019.72.87.02 g); C, 25.81% (25.89%); H, 2.52%(2.55%); N, 5.42% (5.49%).
With sodium hydroxide In water; acetonitrile for 4h;
1,4,8,11-Tetra(p-toluenesulfonyl)-1,4,8,11-tetraazacyclotetradecane (3)
Cyclam (1) (0.20 g, 1.00 mmol) wasdissolved in 1 M aq NaOH solution (15 ml). A solution ofp-toluenesulfonyl chloride (0.78 g, 4.09 mmol) in MeCN(60 ml) was added, and the reaction mixture was stirred for4 h. The white precipitate was filtered off, washed withMeCN, and dried under reduced pressure. Yield 0.51 g(62%), colorless crystals, mp 278°C (decomp.). IR spectrum,ν, cm-1: 1338, 1326, 1154 (S=O). 1H NMR spectrum (400MHz, CDCl3), δ, ppm (J, Hz): 1.85-1.91 (4H, m,2NCH2CH2CH2N); 2.44 (12H, s, 4CH3); 3.12-3.15 (8H, m,2NCH2CH2CH2N); 3.22 (8H, br. s, 2NCH2CH2N); 7.34(8H, d, 3JHH = 8.0, H-2,6 or H-3,5 Ar); 7.70 (8H, d,3JHH = 8.0, H-3,5 or H-2,6 Ar). 13C NMR spectrum (101 MHz,CDCl3), δ, ppm: 21.7 (CH3); 27.9 (CH2CH2CH2); 48.1(NCH2CH2CH2N); 50.1 (NCH2CH2N); 127.6 (C-2,6 orC-3,5 Ar); 130.1 (C-3,5 or C-2,6 Ar); 134.5 (C-4 Ar);144.0 (C-1 Ar). Found, %: C 54.63; N 6.95; H 6.11.C38H48N4O8S4·H2O. Calculated, %: C 54.66; N 6.71; H 6.04.
With triethylamine In acetonitrile at 70℃; for 24h;
1
The molar ratio of 1,4,8,11-tetraazacyclotetradecane, p-methylbenzenesulfonyl chloride and triethylamine was 1:6:6.5.A weighed 1030 mg of 1,4,8,11-tetraazacyclotetradecane and 5946 mg of p-toluenesulfonyl chloride were added to a 250 ml eggplant-shaped flask.25 ml of acetonitrile was added to substantially dissolve the above solid.The condenser was placed on a condensing tube, and 4.7 mL of triethylamine was added dropwise thereto, and condensed and refluxed in an oil bath at 70 ° C for 24 hours.After the reaction is completed, the condenser is removed and the product emits a thick pungent odor.Add 35 ml of saturated sodium bicarbonate solution, stir at room temperature for 2 h, and suction to obtain a solid. Dry the solid and add it to a 200 ml eggplant-shaped bottle, add 15 ml of tetrahydrofuran, recrystallize in a 68 ° C oil bath, and recrystallize. A large amount of solid is precipitated by cooling, and a white solid is obtained by suction filtration, and dried again to be a sulfur-containing small molecule compound, which is compound C, and its structural formula is as follows:
1,4,8,11-tetrakis(diphenylphosphoryl)-1,4,8,11-tetraazacyclotetradecane (2)
0.050 g Cyclam (0.25 mmol) and0.202 g triethylamine (2.00 mmol) were mixed in a round-bottomflask with 50 mL chloroform. 0.468 g diphenyl phosphoryl chloride(1.75 mmol) was then added slowly to the solution. The reactionmixture was stirred for 4 h at rt. At the beginning, the reactionmixture produced lots of cloudy white fume, which disappearedafter 10 min of stirring. The resulting clear fainted yellow solutionwas then brought to dryness using a rotavapor. The acquired residuewas washed thoroughly with water and then pumped to dry.Crystallization was set up with hexanes evaporating into a chloroformsolution of 2 at rt. Big, fluffy white needle-like crystalsformed the next morning. After the crystals were obtained and dried, 0.24 g of 2 was obtained (84%). 1H NMR (CDCl3, 600 MHz, d,ppm): 7.31-7.13 (m, 40H, aromatic C-H), 3.26-3.24 (m, 8H, N-CH2-CH2-N), 3.20 (dd, J 15.5, 8.8 Hz, 8H, N-CH2-CH2-CH2-N), 1.70(quint, J 7.6 Hz, 4H, N-CH2-CH2-CH2-N). LR-MS: [MH], 1129.4(Calcd 1129.3); [M NH4], 1146.4 (Calcd 1146.4); [MNa], 1151.3(Calcd 1151.3).
Stage #1: 1,4,8,11-Tetraazacyclotetradecane; 1.3-chlorobromopropane; N,N-bis-(3-trimethoxysilylpropyl)amine With tert-butylmethyl ether; potassium carbonate In cyclohexane at 0 - 20℃; for 1h; Inert atmosphere;
Stage #2: With triethylamine In acetonitrile at 40℃; for 4h; Inert atmosphere;
4 Preparation Example 1
General procedure: Put 8 mol (1105.68 g) of potassium carbonate into a 3L 4-necked round flask connected with a stirrer, thermometer, dropping funnel and shrink line, reduce the pressure to remove moisture completely, and then under an argon atmosphere. 4 mol (629.76 g) of 1-bromo-3-chloropropane, 1 mol (88.15 g) of t-butylmethyl ether, and 1 mol (84.16 g) of cyclohexane were added and stirred at 0° C. at 300 rpm. Here, 1 mol (172,27 g) of 1,4,7,10-tetraazacyclododecane (1,4,7,10-tetraazacyclododecane) was added over 1 hour through a dropping funnel, After completion of the input, the temperature was raised to room temperature to proceed with the reaction. Thereafter, 400 ml of cyclohexane was added, and after sufficient stirring, the remaining HBr was removed using a saturated aqueous sodium hydrogen carbonate solution. Thereafter, the salt was removed using distilled water and brine, and the remaining moisture was removed with sodium sulfate. Sodium sulfate was removed using a filter, the solvent was removed with a rotary concentrator, and then distilled to prepare a purified intermediate. Then, put the intermediate in a 5 L round-bottom flask connected to a shrink line and completely remove moisture under reduced pressure, and then put 1000 ml of acetonitrile under an irgon atmosphere, bis(3-triethoxysilyl)propylamine (bis(3-triethoxysilyl)propylamine) 4 mol (1702.2 g) and triethylamine (triethylamine) 10.12 g were added, heated to 40°C and stirred for 4 hours. Thereafter, the solvent and triethylamine were removed using a rotary concentrator to prepare a compound represented by the following Chemical Formula 1-1. It was confirmed that the prepared compound was synthesized by 1H-NMR (nuclear magnetic resonance spectroscopy).
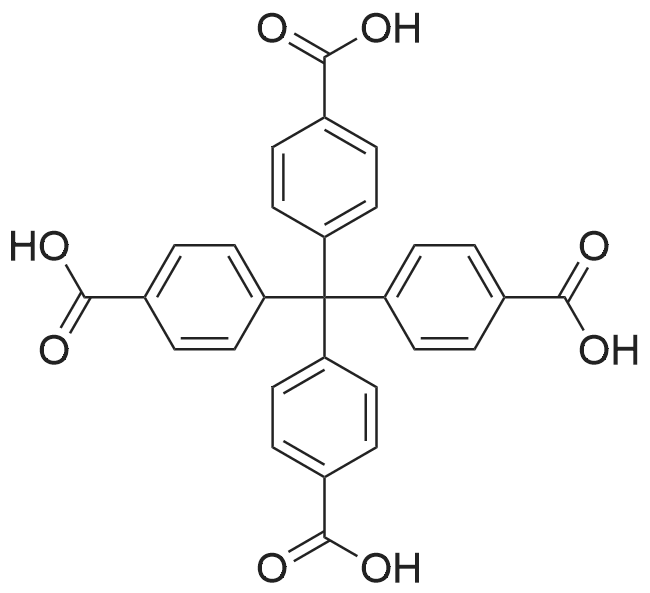
2.2.1. [( Zn(cyclam)(H 2 BPTC)] n (1)
A 10 mL of aqueous solution containing cyclam (0.024 g, 0.12 mmol) and Zn(NO 3 ) 2 •6H 2 O (0.035 g, 0.12 mmol) was added dropwise to 10 mL of DMSO solution containing H 4 BPTC ligand (0.01 g, 0.03 mmol). The mixture was placed in a 25 mL vial then kept in an oven at 90 °C. After 48 h followed by slow cooling to room temperature, colorless block-like crystals have been obtained prior for structure characterization. Yield 42% (0.0075 g). Elemental Analysis (%); Calcd. (found) for ZnC 26 H 32 N 4 O 8 : C, 52.58 (52.34); H, 5.43 (5.28); N, 9.43 (9.32). FT-IR (ATR, cm -1 ): 3385 (m), 3237 (s), 3159 (m), 2995 (w), 2917 (s), 2865 (m), 2612 (m), 1558 (s), 1461 (w), 1431 (s), 1406 (s), 1355 (s), 1311 (m), 1102 (m), 1019 (s), 952 (m), 904 (w), 875 (w), 827 (w), 770 (m), 732 (w), 658 (m).
2.2.2. [Ni 2 (cyclam) 2 (BPTC)] n (2)
A 10 mL of aqueous solution containing cyclam (0.024 g, 0.12 mmol) and Ni(NO 3 ) 2 •6H 2 O (0.035 g, 0.12 mmol) was added dropwise to 10 mL of DMF solution containing H 4 BPTC ligand (0.01 g, 0.03 mmol). The mixture was placed in a 25 mL vial then kept in an oven at 90 °C. After 48 h followed by slow cooling to room temperature, block-like crystals of purple color have been ob- tained prior for structure characterization. Yield 78% (0.02 g). El- emental Analysis (%); Calcd. (found) for Ni 2 C 36 H 54 N 8 O 8 : C, 51.22 (51.07); H, 6.45 (6.29); N, 13.27 (13.16). FT-IR (ATR, cm -1 ): 3208 (m), 2914 (m), 2853 (m), 1608 (m), 1560 (m), 1467 (w), 147 (w), 1350 (m), 1309 (w), 1240 (w), 1097 (w), 1065 (w), 1041 (w), 990 (w), 912 (w), 880 (m), 774 (w), 716 (w), 656 (w).
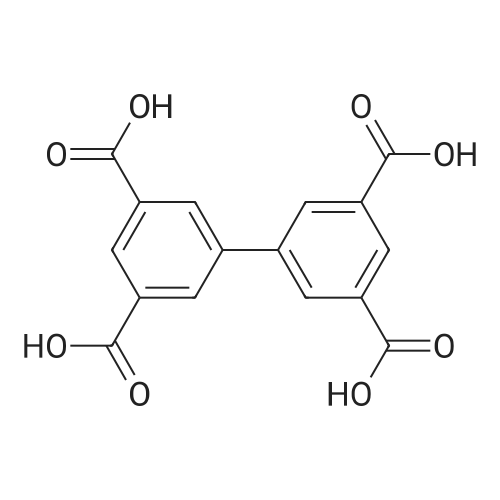
1.5Zn(2+)*1.5C10H24N4*0.5C22H10O8(4-)*0.5C22H12O8(2-)*H2O[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
60%
In water; N,N-dimethyl-formamide at 90℃; for 48h;
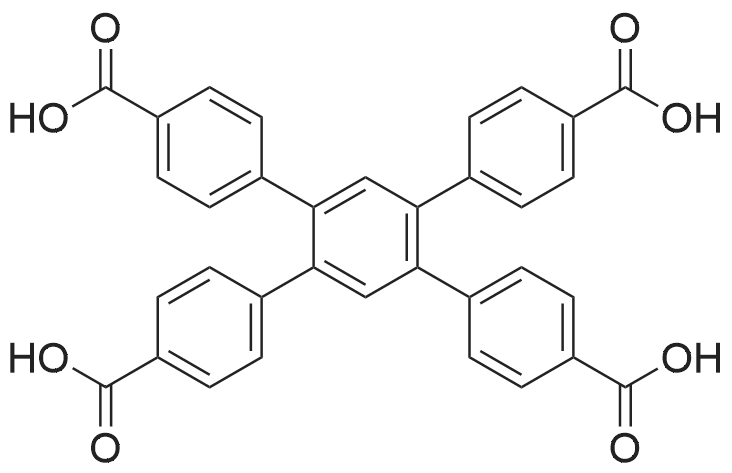
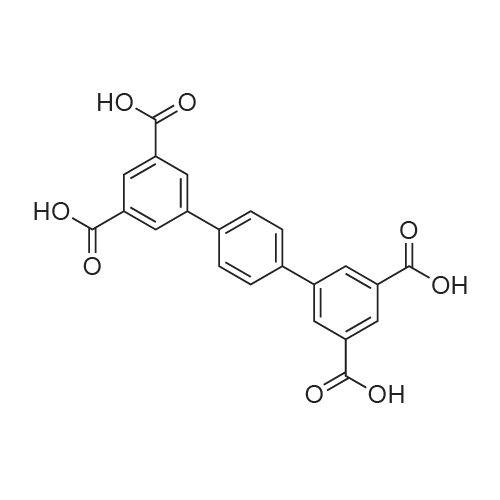
2.2.3. [Ni(cyclam)(H 2 TPTC)] n (3)
General procedure: To a 10 mL of DMF solution containing H 4 TPTC ligand (0.01 g, 0.024 mmol) was added dropwise a 10 mL of aqueous solution containing cyclam (0.02 g, 0.1 mmol) and Ni(NO 3 ) 2 •6H 2 O (0.029 g, 0.1 mmol). Block-like crystals of light-pink color prior for struc- ture characterization have been isolated after 2 days. Same phase has also been obtained by using DMSO/H 2 O medium (1:1, 20 mL) instead of DMF/H 2 O. Yield 69% (0.011 g).
2.2.3. [Ni(cyclam)(H 2 TPTC)] n (3)
To a 10 mL of DMF solution containing H 4 TPTC ligand (0.01 g, 0.024 mmol) was added dropwise a 10 mL of aqueous solution containing cyclam (0.02 g, 0.1 mmol) and Ni(NO 3 ) 2 •6H 2 O (0.029 g, 0.1 mmol). Block-like crystals of light-pink color prior for struc- ture characterization have been isolated after 2 days. Same phase has also been obtained by using DMSO/H 2 O medium (1:1, 20 mL) instead of DMF/H 2 O. Yield 69% (0.011 g). Elemental Analysis (%); Calcd. (found) for NiC 32 H 36 N 4 O 8 : C, 57.94 (57.88); H, 5.47 (5.33); N, 8.45 (8.36). FT-IR (ATR, cm -1 ): 3402 (s), 3268 (s), 3228 (w), 3169 (s), 2940 (s), 2857 (m), 2585 (w), 1712 (s), 1603 (m), 1564 (s), 1454 (w), 1417 (w), 1436 (s), 1366 (s), 1300 (m), 1215 (s), 1156 (w), 1104 (m), 1076 (w), 1018 (s), 952 (s), 872 (m), 845 (m), 793 (m), 760 (m), 735 (m), 673 (s), 607 (w).
With sodium hydroxide In lithium hydroxide monohydrate at 30 - 80℃; for 6h; Inert atmosphere; Green chemistry;
1.3 Step (3): Preparation of compound of formula 4 1,4,8,11-tetraazacyclotetradecane
Keeping a slight positive pressure of nitrogen, add 2000 mL of water and 200 g of sodium hydroxide to the 5L reaction flask, add 357.61 g (0.941 mol) of the compound of formula 3 in batches under stirring, and control the temperature not to exceed 30 °C and stir well.After the addition, the temperature was raised to 80° C. for the reaction. During the reaction, the pH value of the system was monitored and kept at 9-14. If the pH was lower than 9, solid sodium hydroxide should be added thereto.The reaction was maintained at 80°C for 6 hours, and the reaction was completed.The reaction solution was cooled to room temperature, filtered, an appropriate amount of dichloromethane was added to the filtrate for extraction, and the product in the filtrate was completely extracted.The extracts were combined, and an appropriate volume of dichloromethane was recovered by atmospheric distillation.An appropriate amount of petroleum ether was added to the remaining solution, and the mixture was cooled to 0°C for crystallization.The crystallization was kept overnight, and a large amount of white solid was precipitated.Filter, collect the filter cake, wash the filter cake with an appropriate amount of cold petroleum ether, and drain to obtain 177.69 g (0.887 mol) of white crystal product.The yield is 94.3%, the total yield of the three-step reaction is 88.7%,
94.3%
With sodium hydroxide In lithium hydroxide monohydrate at 30 - 80℃; for 6h; Inert atmosphere;
1.3 Step (3): Preparation of compound 1,4,8,11-tetraazacyclotetradecane of formula 5
Keeping a slight positive pressure of nitrogen, add 2000 mL of water and 200 g of sodium hydroxide to the 5L reaction flask, add 357.61 g (0.941 mol) of the compound of formula 3 in batches under stirring, and control the temperature not to exceed 30 °C and stir well. After the addition, the temperature was raised to 80° C. for the reaction. During the reaction, the pH value of the system was monitored and kept at 9-14. If the pH was lower than 9, solid sodium hydroxide should be added thereto. The reaction was maintained at 80°C for 6 hours, and the reaction was completed. The reaction solution was cooled to room temperature, filtered, an appropriate amount of dichloromethane was added to the filtrate for extraction, and the product in the filtrate was completely extracted. The extracts were combined, and an appropriate volume of dichloromethane was recovered by atmospheric distillation. An appropriate amount of petroleum ether was added to the remaining solution, and the mixture was cooled to 0°C for crystallization. The crystallization was kept overnight, and a large amount of white solid was precipitated. Filter, collect the filter cake, wash the filter cake with an appropriate amount of cold petroleum ether, and drain to obtain 177.69 g (0.887 mol) of white crystal product. The yield is 94.3%, the total yield of the three-step reaction is 88.7%, Product titration purity: 99.3% (by HClO4),
With lithium hydroxide monohydrate In methanol; water monomer at 30 - 80℃; for 6h; Inert atmosphere; Green chemistry;
2.3 Step (3): Preparation of compound of formula 4 1,4,8,11-tetraazacyclotetradecane
Maintain a slight positive pressure of nitrogen, add 500 mL of methanol, 1000 mL of water and 265.66 g of lithium hydroxide to the 5L reaction flask, add 265.66 g (0.912 mol) of the compound of formula 3 in batches under stirring, and control the temperature not to exceed 30 ° C. Stir uniform.After the addition is completed, the temperature is raised to 80°C for the reaction. During the reaction, the pH value of the system is monitored and kept at 9-14. If the pH is lower than 9, lithium hydroxide solid should be added therein.The reaction was maintained at 80°C for 6 hours, and the reaction was completed.The reaction solution was cooled to room temperature, filtered, an appropriate amount of dichloromethane was added to the filtrate for extraction, and the product in the filtrate was completely extracted.The extracts were combined, and an appropriate volume of dichloromethane was recovered by atmospheric distillation.An appropriate amount of petroleum ether was added to the remaining solution, and the mixture was cooled to 0°C for crystallization.The crystallization was kept overnight, and a large amount of white solid was precipitated.Filter, collect the filter cake, wash the filter cake with an appropriate amount of cold petroleum ether, and drain to obtain 173.20 g (0.865 mol) of white crystal product.The yield is 94.8%, the total yield of the three-step reaction is 86.5%,
With potassium hydroxide In ethanol; lithium hydroxide monohydrate at 30 - 80℃; for 6h; Inert atmosphere; Green chemistry;
3.3 Step (3): Preparation of compound of formula 4 1,4,8,11-tetraazacyclotetradecane
Maintain a slight positive pressure of nitrogen, add 500 mL of ethanol, 1000 mL of water and 200 g of potassium hydroxide to the 5L reaction flask, add 430.89 g (0.909 mol) of the compound of formula 3 in batches under stirring, and control the temperature not to exceed 30 ° C. Stir well .After the addition, the temperature was raised to 80°C for the reaction. During the reaction, the pH value of the system was monitored and kept at 9-14. If the pH was lower than 9, potassium hydroxide solid should be added therein.The reaction was maintained at 80°C for 6 hours, and the reaction was completed.The reaction solution was cooled to room temperature, filtered, an appropriate amount of dichloromethane was added to the filtrate for extraction, and the product in the filtrate was completely extracted.The extracts were combined, and an appropriate volume of dichloromethane was recovered by atmospheric distillation.An appropriate amount of petroleum ether was added to the remaining solution, and the mixture was cooled to 0°C for crystallization.The crystallization was kept overnight, and a large amount of white solid was precipitated.Filter, collect the filter cake, wash the filter cake with an appropriate amount of cold petroleum ether, and drain to obtain 173.36 g (0.865 mol) of white crystal product.The yield is 95.2%, the total yield of the three-step reaction is 86.5%,
95.2%
With potassium hydroxide In ethanol; lithium hydroxide monohydrate at 30 - 80℃; for 6h; Inert atmosphere;
2.3 Step (3): Preparation of compound 1,4,8,11-tetraazacyclotetradecane of formula 5
Maintain a slight positive pressure of nitrogen, add 500 mL of ethanol, 1000 mL of water and 200 g of potassium hydroxide to the 5L reaction flask, add 430.89 g (0.909 mol) of the compound of formula 3 in batches under stirring, and control the temperature not to exceed 30 ° C. Stir well . After the addition, the temperature was raised to 80°C for the reaction. During the reaction, the pH value of the system was monitored and kept at 9-14. If the pH was lower than 9, potassium hydroxide solid should be added therein. The reaction was maintained at 80°C for 6 hours, and the reaction was completed. The reaction solution was cooled to room temperature, filtered, an appropriate amount of dichloromethane was added to the filtrate for extraction, and the product in the filtrate was completely extracted. The extracts were combined, and an appropriate volume of dichloromethane was recovered by atmospheric distillation. An appropriate amount of petroleum ether was added to the remaining solution, and the mixture was cooled to 0°C for crystallization. The crystallization was kept overnight, and a large amount of white solid was precipitated. Filter, collect the filter cake, wash the filter cake with an appropriate amount of cold petroleum ether, and drain to obtain 173.36 g (0.865 mol) of white crystal product. The yield is 95.2%, the total yield of the three-step reaction is 86.5%, Product titration purity: 99.2% (by HClO4),
With potassium hydroxide In isopropanol at 30 - 80℃; for 6h; Inert atmosphere; Green chemistry;
4.3 Step (3): Preparation of compound of formula 4 1,4,8,11-tetraazacyclotetradecane
Maintain a slight positive pressure of nitrogen, add 500ml of isopropanol, 1000ml of water and 200g of potassium hydroxide to the 5L reaction flask, add 369.68g (0.901mol) of the compound of formula 3 in batches under stirring, and control the temperature not to exceed 30°C, Stir well.After the addition, the temperature was raised to 80°C for the reaction. During the reaction, the pH value of the system was monitored and kept at 9-14. If the pH was lower than 9, potassium hydroxide solid should be added therein.The reaction was maintained at 80°C for 6 hours, and the reaction was completed.The reaction solution was cooled to room temperature, filtered, an appropriate amount of dichloromethane was added to the filtrate for extraction, and the product in the filtrate was completely extracted.The extracts were combined, and an appropriate volume of dichloromethane was recovered by atmospheric distillation.An appropriate amount of petroleum ether was added to the remaining solution, and the mixture was cooled to 0°C for crystallization.The crystallization was kept overnight, and a large amount of white solid was precipitated.Filtration, collecting the filter cake, washing the filter cake with an appropriate amount of cold petroleum ether, and sucking dry to obtain 172.74 g (0.862 mol) of white crystal product.The yield is 95.7%, the total yield of the three-step reaction is 86.2%,






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science








 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping