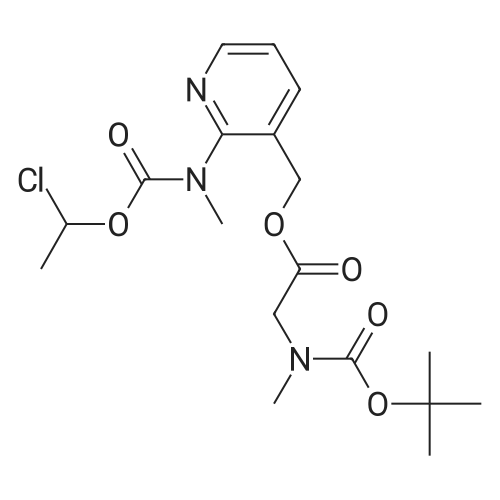
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
With hydrogenchloride; sodium hydroxide In methanol; water
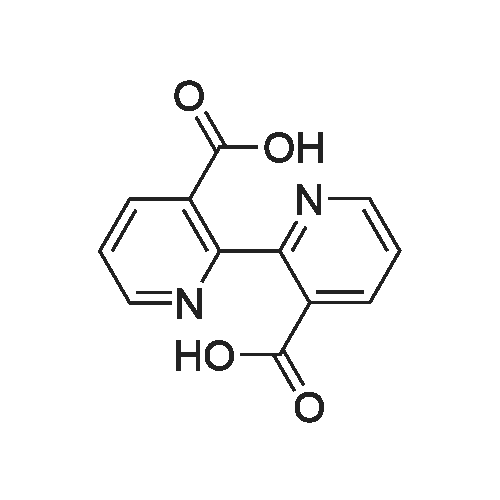
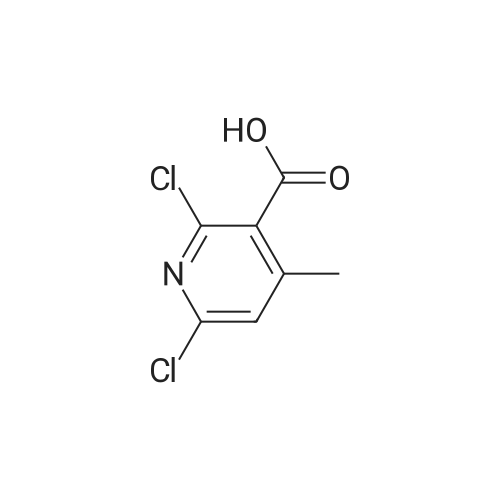
Example 4 9.50 g of 2-chloronicotinic acid and 5.30 g of sodium hydroxide are dissolved in a mixture of 40 ml of water and 40 ml of methanol. Following the addition of 4 g of palladium (5percent by weight on activated carbon) as catalyst, the mixture is stirred for 30 h at 80-85° C. at 0.1 MPa. The catalyst is then filtered off. Following acidification to pH 1 using hydrochloric acid, the product, 2,2'-bipyridyl-3,3'-dicarboxylic acid, precipitates out as a white solid. This gives 3.5 g (48percent yield).
Reference:
[1] Patent: US6500956, 2002, B1,
2
[ 2942-59-8 ]
[ 52200-48-3 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2011, vol. 353, # 8, p. 1359 - 1366
3
[ 2942-59-8 ]
[ 10366-35-5 ]
Reference:
[1] Indian Journal of Chemistry - Section B Organic and Medicinal Chemistry, 2008, vol. 47, # 2, p. 315 - 318
[2] Tetrahedron, 1995, vol. 51, # 48, p. 13177 - 13184
[3] Journal of Organometallic Chemistry, 2012, vol. 713, p. 42 - 50
[4] Bioorganic and Medicinal Chemistry Letters, 2013, vol. 23, # 5, p. 1424 - 1427
4
[ 2942-59-8 ]
[ 38521-46-9 ]
Yield
Reaction Conditions
Operation in experiment
84%
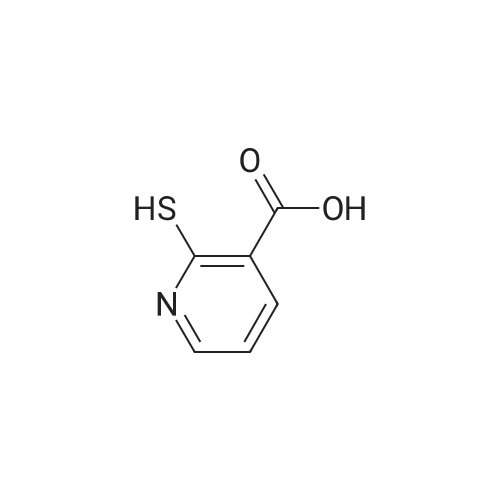
With thiourea In water at 90℃; for 8 h;
A suspension of 2-chloronicotinic acid (790 mg, 5 mmol) and thiourea (457 mg, 6 mmol) was suspended in water (15 mL), refluxed at 90°C for 8 h, cooled to room temperature to give a pale yellow suspension. For 8 or so, stirring 10min, adding dilute hydrochloric acid, precipitation of light yellow solid, filtered out drying, the target compound (590mg, 84percent).
Reference:
[1] Patent: CN103626693, 2016, B, . Location in patent: Paragraph 0157-0159
[2] Roczniki Chemii, 1932, vol. 12, p. 493,495[3] Chem. Zentralbl., 1932, vol. 103, # II, p. 3400
[4] Journal of the American Chemical Society, 1948, vol. 70, p. 3908,3910
5
[ 2942-59-8 ]
[ 609-71-2 ]
[ 5345-47-1 ]
Reference:
[1] Die Pharmazie, 1980, vol. 34, # 5-6, p. 253 - 256
6
[ 2942-59-8 ]
[ 101012-32-2 ]
Reference:
[1] Chinese Chemical Letters, 2011, vol. 22, # 9, p. 1009 - 1012
7
[ 67-56-1 ]
[ 2942-59-8 ]
[ 40134-18-7 ]
Yield
Reaction Conditions
Operation in experiment
93%
for 8 h; Reflux
To the 250 ml flask is added in three 15.76g (0.1 µM) 2 - chloro -3 - picolinic acid, 100 ml methanol, under the ice-bath adds by drops 17.85g (0.15 µM) thionyl chloride, stirring and heating to reflux, the stirring reaction 8h. Thin layer chromatography tracking response, the raw materials point disappears, stopping the reaction. Reduced pressure distillation to remove the solvent to obtain light yellow 15.95g, yield 93percent
86%
Stage #1: With oxalyl dichloride In dichloromethane at 20℃; for 3 h; Stage #2: With triethylamine In dichloromethane for 0.5 h; Cooling with ice Stage #3: With sodium hydrogencarbonate In water
(1) Production of methyl 2-chloronicotinate 15.0 g (95.2 mmol) of 2-chloronicotinic acid was dissolved in dichloromethane (150 mL), and 8.37 mL (94.5 mmol) of oxalyl chloride was added thereto. Six droplets of N,N-dimethylformamide were added to the above mixture, and the resulting mixture was stirred for 3 hours at room temperature. 40.1 mL (286 mmol) of triethylamine and 37 mL (914 mmol) of methanol were sequentially added dropwise to the above reaction mixture under ice cooling, and the resulting mixture was stirred for 30 minutes under ice cooling. The solvent was distilled off under reduced pressure from the reaction mixture, and then an aqueous solution of sodium hydrogen carbonate was added to the residue. The resulting mixture was extracted with diethyl ether. The organic phase was washed with water, dried, and concentrated, and the residue was purified by column chromatography (ethyl acetate:n-hexane=1:4). 13.9 g of the title compound was obtained as a pale yellow liquid (yield: 86percent). 1H-NMR data (CDCl3/TMS δ (ppm)): 3.97 (3H, s), 7.33 (1H, dd), 8.17 (1H, dd), 8.53 (1H, dd)
67%
at 150℃; for 0.333333 h; Microwave irradiation
Example 136; Preparation of 2-chloro-pyridine-3-carboxylic acid methyl esterA mixture a 2-chloro nicotinic acid (2.0 g, 12.7 mmol), trimethyl ortho formate (8 ml) and methanol (10 drops) was heated to 15O0C for 20 minutes in a microwave. The solution was allowed to cool to room temperature, diluted with ethyl acetate and washed with aqueous sodium hydroxide (2M). The organic extract was dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (eluent: 0-40percent ethyl acetate in hexane) to give 2-chloro-pyridine-3-carboxylic acid methyl ester as a colourless oil (1.453 g, 67percent yield).1H-NMR (400 MHz, CDCl3): 3.97 (3H5 s, Me), 7.33 (IH, m, CH), 8.19 (IH, m, CH), 8.52 (IH, m, CH) ppm.
With thionyl chloride; triethylamine In methanol; toluene
EXAMPLE 3: Preparation of methyl 2-chloronicotinate (6) 81,9 g of thionyl chloride are slowly added to 20 g of 2-chloronicotinic acid (1), under stirring, and the suspension is stirred for 5 minutes. The mixture is heated to reflux for 60 minutes till obtaining a clear solution which is evaporated under vacuum. The residue is taken up into 200 ml of anhydrous toluene and concentrated under vacuum to 100 ml. The obtained solution is slowly dropped into a solution of 9 g of methanol and 14,6 g of triethylamine in 100 ml of anhydrous toluene, keeping temperature at about 20°C, under stirring. The mixture is left to stand for 5 hours, then cooled to +5°C and the precipitated triethylamine hydrochloride is filtered and washed with 20 ml of toluene. The organic phase is washed with 50 ml of 5percent p/v aqueous sodium hydrogen carbonate, then with 2 x 200 ml of water, dried over sodium sulfate, filtered and concentrated under vacuum to remove the solvent. 21,35 g of methyl 2-chloronicotinate are obtained in form of a slightly yellow oil, having b.p. 225-230°C, the structure of which is confirmed by the IR and NMR spectra. Yield about 98percent by mole.
Reference:
[1] Patent: EP349902, 1991, A3,
[2] Tetrahedron, 1997, vol. 53, # 47, p. 16061 - 16082
[3] Synthetic Communications, 1996, vol. 26, # 12, p. 2257 - 2272
[4] Journal of Medicinal Chemistry, 1993, vol. 36, # 18, p. 2676 - 2688
[5] Patent: US5767134, 1998, A,
9
[ 2942-59-8 ]
[ 40134-18-7 ]
Yield
Reaction Conditions
Operation in experiment
86%
With thionyl chloride; triethylamine In 1,4-dioxane; methanol; ethyl acetate
EXAMPLE 1 Preparation of 2-chloronicotinic acid methyl ester (18) To a mixture of 2-chloronicotinic acid (Aldrich, 100.0 g, 0.63 mol) in 1,4-dioxane (500 mL) was added thionyl chloride (70 mL, 0.96 mol). The suspension was heated under reflux for 22 h with a gas trap to absorb hydrogen chloride gas. After evaporation of the solvent, the residue was dissolved in methanol (300 mL). To the solution was added dropwise triethylamine (TEA, 120 mL, 1.26 mol) at 0° C. over 2 h. The solvents were evaporated and the residue was suspended in ethyl acetate. The precipitate was removed by filtration. The filtrate was concentrated to afford the ester 18 (92.3 g, 86percent) as an oil: Rf(1:5 v/v ethyl acetate-hexane) 0.38. 1H NMR (300 MHz, CDCl3) δ8.53 (dd, 4.8 Hz, 1H), 8.19 (dd, 7.6 Hz, 1H), 7.37 (dd, 7.7 Hz, 1H) and 3.97 (s, 3H). 13C NMR (75 MHz, CDCl3) δ164.5, 151.6, 149.6, 140.0, 126.4, 121.9 and 52.5.
86%
With thionyl chloride; triethylamine In 1,4-dioxane; methanol; ethyl acetate
Example 1 Preparation of 2-chloronicotinic acid methyl ester (18) To a mixture of 2-chloronicotinic acid (Aldrich, 100.0 g, 0.63 mol) in 1,4-dioxane (500 mL) was added thionyl chloride (70 mL, 0.96 mol). The suspension was heated under reflux for 22 h with a gas trap to absorb hydrogen chloride gas. After evaporation of the solvent, the residue was dissolved in methanol (300 mL). To the solution was added dropwise triethylamine (TEA, 120 mL, 1.26 mol) at 0° C. over 2 h. The solvents were evaporated and the residue was suspended in ethyl acetate. The precipitate was removed by filtration. The filtrate was concentrated to afford the ester 18 (92.3 g, 86percent) as an oil: Rf (1:5 v/v ethyl acetate-hexane) 0.38. 1H NMR (300 MHz, CDCl3) δ 88.53 (dd, 4.8 Hz, 1H), 8.19 (dd, 7.6 Hz, 1H), 7.37 (dd, 7.7 Hz, 1H) and 3.97 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 164.5, 151.6, 149.6, 140.0, 126.4, 121.9 and 52.5.
Intermediate 3. 2-Chloronicotinic acid methyl ester.; To a room temperature solution of2-chloronicotinic acid (5.00 g, 31.7 mmol) in 20percent MeOH/toluene (100 mL) was added TMS diazomethane (trimethylsilyl diazomethane) (2.0M in hexanes, 31.7 mL) dropwise and the reaction was monitored by TLC (thin-layer chromatography) until completion. The reaction was concentrated down under reduced pressure. The residue was purified by silica chromatography using a gradient of hexanes:EtOAc (100:0) to hexanes:EtOAc (80:20) to yield 4.65 g (85.42percent) of Intermediate 3 as a yellow oil. EPO <DP n="36"/>
Stage #1: Heating / reflux Stage #2: With sodium hydroxide In water Stage #3: With hydrogenchloride In water
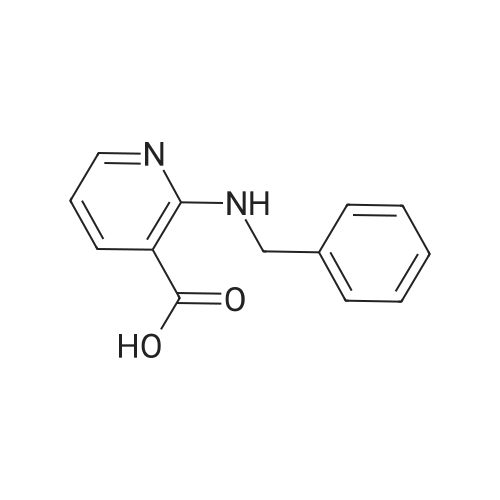
Benzylamine (14 mL, 126. 8 mmol) was added to a solution of chloronicotinic acid (10 g, 63.4 mmol) in pyridine and refluxed overnight. The pyridine was distilled and the residue was dissolved in IN NaOH. The solution was diluted with water to adjust the pH to 10 to 11 and washed by dichloromethane. The aqueous phase was neutralized with cold aqueous 10 percent HC1 solution to adjust the pH to 6 to 7. The solids formed were filtered, washed with cold water, and dried in a vacuum oven to yield 12.2 g (84 percent) OF 2-BENZYLAMINO nicotinic acid (1) as white solids. MP: 148 °C ; 1H-NMR (DMSO-d6): d 4.69 (d, J= 3.6 Hz, 2H), 6.61 (dd, J= 4.9, 7.7 Hz, 1H), 7.23 (m, 1H), 7.29 (m, 4H), 8.08 (dd, J = 1.8, 7.0 Hz, 1H), 8. 28 (DD, J = 1.8, 7.0 Hz, 1H), 8.47 (br. s, 1H), 13.10 (s, 1H) ; EIMS: 229 (M+1), 251 (M+23).
Reference:
[1] Journal of Medicinal Chemistry, 1988, vol. 31, # 11, p. 2108 - 2121
[2] Patent: WO2005/21546, 2005, A1, . Location in patent: Page/Page column 111; 159-160
[3] Tetrahedron Letters, 2003, vol. 44, # 32, p. 6021 - 6023
[4] Tetrahedron, 2005, vol. 61, # 42, p. 10081 - 10092
[5] Tetrahedron Letters, 2009, vol. 50, # 21, p. 2481 - 2483
[6] Journal of Medicinal Chemistry, 1990, vol. 33, # 10, p. 2697 - 2706
[7] Patent: US5854257, 1998, A,
[8] Patent: WO2004/20414, 2004, A1, . Location in patent: Page 97
29
[ 2942-59-8 ]
[ 87-60-5 ]
[ 17737-65-4 ]
Reference:
[1] Journal of Medicinal Chemistry, 1990, vol. 33, # 10, p. 2697 - 2706
30
[ 2942-59-8 ]
[ 98-16-8 ]
[ 4394-00-7 ]
Reference:
[1] Journal of Medicinal Chemistry, 1990, vol. 33, # 10, p. 2697 - 2706
[2] Medicinal Chemistry Research, 2013, vol. 22, # 5, p. 2411 - 2420
31
[ 2942-59-8 ]
[ 75-16-1 ]
[ 55676-21-6 ]
Reference:
[1] Journal of Medicinal Chemistry, 2014, vol. 57, # 18, p. 7577 - 7589
[2] Patent: WO2011/79804, 2011, A1, . Location in patent: Page/Page column 24
[3] Patent: US2012/245178, 2012, A1, . Location in patent: Page/Page column 13
32
[ 49609-84-9 ]
[ 2942-59-8 ]
[ 79-37-8 ]
[ 594-27-4 ]
[ 144-55-8 ]
[ 55676-21-6 ]
Reference:
[1] Patent: US5688795, 1997, A,
33
[ 2942-59-8 ]
[ 55676-21-6 ]
Reference:
[1] ACS Medicinal Chemistry Letters, 2012, vol. 3, # 9, p. 764 - 768
[2] Patent: US2012/258982, 2012, A1,
[3] European Journal of Medicinal Chemistry, 2012, vol. 57, p. 311 - 322
[4] Bulletin of the Korean Chemical Society, 2013, vol. 34, # 4, p. 1253 - 1256
[5] Bulletin of the Korean Chemical Society, 2015, vol. 36, # 7, p. 1908 - 1911
[6] Patent: WO2006/123145, 2006, A1,
[7] Patent: WO2016/132134, 2016, A1,
[8] Patent: KR101766414, 2017, B1,
34
[ 2942-59-8 ]
[ 42330-59-6 ]
Yield
Reaction Conditions
Operation in experiment
68%
Stage #1: With lithium aluminium tetrahydride In tetrahydrofuran at 0 - 50℃; for 1 h; Stage #2: With sodium hydroxide; water In tetrahydrofuran at 0℃;
(Step 1) To a suspension of lithium aluminum hydride (8.60 g) in THF (200 mL) was added a solution of 2-chloronicotinic acid (22.4 g) in THF (30 mL) at 0°C, and the mixture was stirred at 50°C for 1 hr. After the reaction mixture was cooled to 0°C, 1N aqueous sodium hydroxide solution (9 mL) was added. The precipitate was filtered off, and the filtrate was concentrated. The residue was purified by silica gel column chromatography (solvent gradient; 10-->25percent ethyl acetate/hexane) to give (2-chloropyridin-3-yl)methanol (14.0 g, 68percent) as a colorless oil. 1H-NMR(CDCl3): δ 2.42-2.75(1H,m), 4.80(2H,d,J=4.8Hz), 7.29(1H,dd,J=7.8,4.8Hz), 7.88-7.93(1H,m), 8.29-8.32(1H,m)
87%
With sodium hydroxide; lithium aluminium tetrahydride In tetrahydrofuran; water
EXAMPLE 35A (2-chloro-3-pyridinyl)methanol A solution of 2-chloronicotinic acid (2.0 g, 12.6 mmol) in THF (15 mL) at 0° C. was treated dropwise with 1M LAH in THF (15 mL, 15.0 mmol), warmed to room temperature, stirred for 5 hours, treated sequentially with water (0.5 mL), 40percent NaOH solution (0.5 mL) and water (1.5 mL), stirred for 2 hours, filtered through a pad of diatomaceous earth (Celite.(R).), and extracted with dichloromethane. The combined extracts were dried (MgSO4), filtered, and concentrated to provide 1.56 g (87percent) of the desired product of sufficient purity for subsequent use. MS (DCI/NH3) m/z 144 (M+H)+; 1H NMR (CDCl3) δ8.34 (dd, 1H), 7.90 (m, 1H), 7.29 (dd, 1H), 4.80 (s, 2H).
87%
With sodium hydroxide; lithium aluminium tetrahydride In tetrahydrofuran; water
EXAMPLE 35A (2-chloro-3-pyridinyl)methanol A solution of 2-chloronicotinic acid (2.0 g, 12.6 mmol) in THF (15 mL) at 0° C. was treated dropwise with 1M LAH in THF (15 mL, 15.0 mmol), warmed to room temperature, stirred for 5 hours, treated sequentially with water (0.5 mL), 40percent NaOH solution (0.5 mL) and water (1.5 mL), stirred for 2 hours, filtered through a pad of diatomaceous earth (Celite.(R).), and extracted with dichloromethane. The combined extracts were dried (MgSO4), filtered, and concentrated to provide 1.56 g (87percent) of the desired product of sufficient purity for subsequent use. MS (DCI/NH3) m/z 144 (M+H)+; 1H NMR (CDCl3) δ 8.34 (dd, 1H), 7.90 (m, 1H), 7.29 (dd, 1H), 4.80 (s, 2H).
Reference:
[1] Synthesis, 2010, # 20, p. 3439 - 3448
[2] Synthetic Communications, 2008, vol. 38, # 6, p. 889 - 904
[3] Patent: EP2018863, 2009, A1, . Location in patent: Page/Page column 73-74
[4] Chemical and Pharmaceutical Bulletin, 2000, vol. 48, # 5, p. 694 - 707
[5] Journal of Heterocyclic Chemistry, 1995, vol. 32, # 5, p. 1595 - 1597
[6] Patent: US2003/87940, 2003, A1,
[7] Patent: US2002/115640, 2002, A1,
[8] Patent: US5739326, 1998, A,
[9] Chinese Chemical Letters, 2011, vol. 22, # 9, p. 1009 - 1012
35
[ 2942-59-8 ]
[ 78607-36-0 ]
Reference:
[1] Advanced Synthesis and Catalysis, 2011, vol. 353, # 8, p. 1359 - 1366
36
[ 2942-59-8 ]
[ 77152-08-0 ]
[ 131747-43-8 ]
Reference:
[1] Journal of Organic Chemistry, 2013, vol. 78, # 22, p. 11126 - 11146
37
[ 2942-59-8 ]
[ 133627-47-1 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 1993, vol. 30, # 3, p. 771 - 779
Reference:
[1] Patent: WO2011/79804, 2011, A1,
[2] Patent: US2012/245178, 2012, A1,
[3] European Journal of Medicinal Chemistry, 2012, vol. 57, p. 311 - 322
[4] Journal of Medicinal Chemistry, 2014, vol. 57, # 18, p. 7577 - 7589
40
[ 2942-59-8 ]
[ 112006-75-4 ]
Reference:
[1] Patent: CN106749183, 2017, A,
41
[ 2942-59-8 ]
[ 338990-31-1 ]
Reference:
[1] Bioorganic and medicinal chemistry letters, 2003, vol. 13, # 2, p. 191 - 196
42
[ 2942-59-8 ]
[ 155058-02-9 ]
[ 40134-18-7 ]
Reference:
[1] Patent: US5391554, 1995, A,
43
[ 2942-59-8 ]
[ 155058-02-9 ]
Reference:
[1] Journal of Heterocyclic Chemistry, 2006, vol. 43, # 5, p. 1311 - 1317
44
[ 3731-53-1 ]
[ 2942-59-8 ]
[ 854382-06-2 ]
Yield
Reaction Conditions
Operation in experiment
89%
Stage #1: at 20℃; for 0.333333 h; Stage #2: at 110℃; for 2 h;
2 - chloro nicotinic acid (25.3mmol), copper oxide (catalytic amount), potassium carbonate (25.3mmol) is added to the 100 ml round bottom flask, under normal temperature 20min, adding 4 - dimethylamino pyridine (50.6mmol), 110 °C heating 2h. After adding the ethyl acetate to stir to room temperature, filtered, the filter cake washed first with ethyl acetate for 2 times, water (20 ml) dissolved, 4N hydrochloric acid adjusted to pH 5 - 6, standing out, filtered, the filter cake drying, to obtain crude, ethanol heat beating further purification, filtration, forms offwhite solid 5.16g, yield 89percent
Reference:
[1] Journal of Medicinal Chemistry, 2017, vol. 60, # 7, p. 2930 - 2943
[2] Patent: CN106565599, 2017, A, . Location in patent: Paragraph 0082-0086
[3] Bioorganic and Medicinal Chemistry Letters, 2007, vol. 17, # 21, p. 6003 - 6008
45
[ 2942-59-8 ]
[ 884049-52-9 ]
Reference:
[1] Chinese Chemical Letters, 2011, vol. 22, # 9, p. 1009 - 1012
46
[ 2942-59-8 ]
[ 896705-16-1 ]
Reference:
[1] Journal of Medicinal Chemistry, 2014, vol. 57, # 11, p. 4950 - 4961
[2] Patent: WO2015/143293, 2015, A1,
A 250 mL four-necked flask equipped with a mechanical stirrer and a thermometer was charged with 25.5 g of 2-chloro-3-methylpyridine (0.2Mol, 100 mL of glacial acetic acid and 2.5 g (0.01 mol) of manganese acetate. Stir at room temperature, into the ozone generated by the ozone generator5.4L (0.24mol), 5h after the reaction solution in a large number of white solid production, TLC monitoring reaction process, after the end of the reaction,The acetic acid was decompressed and reduced to 1 volume of the original volume, filtered and dried to obtain 23.5 g of a white solid product, 98percent yield
To the 250 ml flask is added in three 15.76g (0.1 muM) 2 - chloro -3 - picolinic acid, 100 ml methanol, under the ice-bath adds by drops 17.85g (0.15 muM) thionyl chloride, stirring and heating to reflux, the stirring reaction 8h. Thin layer chromatography tracking response, the raw materials point disappears, stopping the reaction. Reduced pressure distillation to remove the solvent to obtain light yellow 15.95g, yield 93percent
86%
(1)Production of methyl 2-chloronicotinate15.0 g (95.2 mmol) of 2-chloronicotinic acid was dissolved in dichloromethane (150 mL), and 8.37 mL (94.5 mmol) of oxalyl chloride was added thereto.Six droplets of N,N-dimethylformamide were added to the above mixture, and the resulting mixture was stirred for 3 hours at room temperature. 40.1 mL (286 mmol) of triethylamine and 37 mL (914 mmol) of methanol were sequentially added dropwise to the above reaction mixture under ice cooling, and the resulting mixture was stirred for 30 minutes under ice cooling.The solvent was distilled off under reduced pressure from the reaction mixture, and then an aqueous solution of sodium hydrogen carbonate was added to the residue.The resulting mixture was extracted with diethyl ether.The organic phase was washed with water, dried, and concentrated, and the residue was purified by column chromatography (ethyl acetate:n-hexane=1:4).13.9 g of the title compound was obtained as a pale yellow liquid (yield: 86percent).1H-NMR data (CDCl3/TMS delta (ppm)):3.97 (3H, s), 7.33 (1H, dd), 8.17 (1H, dd), 8.53 (1H, dd)
67%
With trimethyl orthoformate; at 150℃; for 0.333333h;Microwave irradiation;
Example 136; Preparation of 2-chloro-pyridine-3-carboxylic acid methyl esterA mixture a 2-chloro nicotinic acid (2.0 g, 12.7 mmol), trimethyl ortho formate (8 ml) and methanol (10 drops) was heated to 15O0C for 20 minutes in a microwave. The solution was allowed to cool to room temperature, diluted with ethyl acetate and washed with aqueous sodium hydroxide (2M). The organic extract was dried over magnesium sulfate and concentrated. The residue was purified by column chromatography on silica gel (eluent: 0-40percent ethyl acetate in hexane) to give 2-chloro-pyridine-3-carboxylic acid methyl ester as a colourless oil (1.453 g, 67percent yield).1H-NMR (400 MHz, CDCl3): 3.97 (3H5 s, Me), 7.33 (IH, m, CH), 8.19 (IH, m, CH), 8.52 (IH, m, CH) ppm.
Example 34; 1-(2-fluorobenzyl)-3-[(5-nitro-2-pyridinyl)oxy]-1H-pyrazolo[3,4-b]pyridine; Example 34A; methyl 2-chloronicotinate; 2-Chloronicotinic acid (4.73 g, 30 mmol) was dissolved in methanol (100 mL) and treated with concentrated H2SO4 (2.5 mL). The mixture was refluxed for 4 hours, allowed to cool to room temperature, and concentrated under reduced pressure. The residue was partitioned between a saturated solution of NaHC03 and ethyl acetate. The ethyl acetate layer was separated, washed with water, brine, dried over anhydrous MgS04, filtered, and the filtrate was concentrated under reduced pressure to provide the title compound.
2-chloro-nicotinic acid methyl ester (52): 52 was prepared according to the method of Synth. Comm. 26 (12), 2257-2272 (1996). To a nitrogen purged flask was charged 2-chloro-nicotinic acid (1000.0 g, 6.0 moles, 1.0 eq) followed by 9L methylene chloride. To this was added thionyl chloride (1.4 L, 19.7 moles, 3.2 eq. ) and the reaction was heated to 40oC with vigorous stirring under nitrogen overnight. The acid chloride solution was cooled in an ice bath and methanol (3L, 74 moles, 12 eq. ) was slowly added while keeping the temperature at 20 C. The rate limiting parameter is the vigorous evolution of copious quantities of HC1 gas. After the addition, HPLC analysis [Tret starting material = 7.5 min, Tret 52 = 11 min] showed the product had formed immediately. The volatiles were removed in vacuo and the residue extracted from 10percent Na2CO3 with EtOAc. The combined organics were dried (MgS04), filtered, and concentrated to a pale yellow oil.
Preparation 32; 2-Chloro-nicotinic acid methyl ester; Oxalyl chloride (13. 28ml, 0. 15MMOL) was added dropwise to a solution of 2- chloronicotinic acid (24g, 0. 153MMOL) in dichloromethane (250ml) containing DIMETHYLFORMAMIDE (10DROPS) under nitrogen at room temperature. The reaction was stirred at room temperature for 1.5h and was then cooled to 0°C. Triethylamine (63. 7ml, 0. 457MMOL) was added slowly followed by METHANOL (60MI). After stirring at 0°C for 30min, the solvent was removed under reduced pressure and the residue was dissolved in sat. sodium bicarbonate solution (200ml). This was extracted with DIETHYLETHER (5X100ML) and the combined organic extracts were dried over MgS04 and the solvent was removed under reduced pressure. The residue was purified by flash column chromatography on silica gel eluting with diethylether to give 2- chloronicotinic acid methyl ester (20.7g) as a pale yellow oil. 1H NMR (400MHZ, CDC13) : A = 8.46-8. 52 (1H, m), 8.12-8. 17 (1H, m), 7.28-7. 32 (1H, m), 3.92 (3H, s) ppm.
A suspension of 2-chloronicotinic acid (790 mg, 5 mmol) and thiourea (457 mg, 6 mmol) was suspended in water (15 mL), refluxed at 90C for 8 h, cooled to room temperature to give a pale yellow suspension. For 8 or so, stirring 10min, adding dilute hydrochloric acid, precipitation of light yellow solid, filtered out drying, the target compound (590mg, 84%).
Benzylamine (14 mL, 126. 8 mmol) was added to a solution of chloronicotinic acid (10 g, 63.4 mmol) in pyridine and refluxed overnight. The pyridine was distilled and the residue was dissolved in IN NaOH. The solution was diluted with water to adjust the pH to 10 to 11 and washed by dichloromethane. The aqueous phase was neutralized with cold aqueous 10 % HC1 solution to adjust the pH to 6 to 7. The solids formed were filtered, washed with cold water, and dried in a vacuum oven to yield 12.2 g (84 %) OF 2-BENZYLAMINO nicotinic acid (1) as white solids. MP: 148 C ; 1H-NMR (DMSO-d6): d 4.69 (d, J= 3.6 Hz, 2H), 6.61 (dd, J= 4.9, 7.7 Hz, 1H), 7.23 (m, 1H), 7.29 (m, 4H), 8.08 (dd, J = 1.8, 7.0 Hz, 1H), 8. 28 (DD, J = 1.8, 7.0 Hz, 1H), 8.47 (br. s, 1H), 13.10 (s, 1H) ; EIMS: 229 (M+1), 251 (M+23).
a) A mixture of 2-chloronicotinic acid (10.0 g) and benzylamine (13.34 ml) was heated at 130 C. for 2 hours. The mixture was cooled and the resulting solid was broken up, triturated with water, acidified to pH 6 and filtered. The solid obtained was boiled up in IMS (200 ml), cooled and filtered to give 2-benzylaminonicotinic acid, m.p. 223-225 C.
With potassium carbonate; copper(I) bromide; In DMF (N,N-dimethyl-formamide); at 100.0℃; for 2h;
Potassium carbonate(4. 82g, 34.9mmol) was added to a mixture of 2- chloronicotinic acid (5. 0g, 31.7mol), benzylamine (3. 47ml, 31.7mmol) and copper(t) bromide (450mg, 3.17mmol) in N, N-dimethytformamide (50ml), and the reaction heated at100 C for 2 hours, then cooled. The resulting solid was filtered off and the filtrate evaporated under reduced pressure. The residue was partitioned between 4N sodium hydroxide solution (25ml) and dichloromethane (25ml) and the layers separated. The organic solution was extracted with water (2x), and the combined aqueous solutions, neutralised using concentrated hydrochloric acid. The resulting solid was filtered off, washed with cold water anddried in vacuo at50 C, to afford the title compound,1.6g. H-nmr (DMSOd6, 300MHz) 8 : 4. 69 (d, 2H), 6.62 (dd, 1H), 7.31 (m, 5H), 8.10 (dd, 1H), 8.24 (dd, 1H), 8. 48 (bs, 1H).
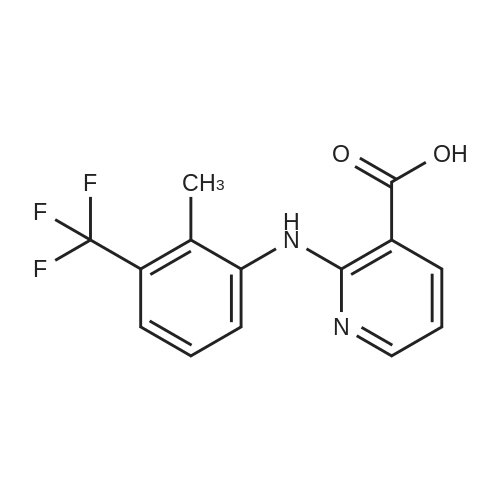
General procedure: To the mixture of an appropriately substituted phenylamine 16a-h (0.054mol) and glacial acetic acid (100mL), 2-chloronicotinic acid 15 (0.032mol) was added at room temperature. Upon the completion of addition, the reaction mixture was stirred at 100C for 3-6h and monitored by thin-layer chromatography (TLC). The reaction mixture was cooled to room temperature and basified with potassium hydroxide to pH 12, and filtered. The filtrate was acidified with hydrochloric acid to pH 3, and stirred for 0.5h. The precipitated was collected by filtration and dried to give the corresponding 2-(substitutedphenylamino)nicotinic acid 17a-h as white solids.
With acetic acid; at 100℃; for 5h;
General procedure: Corresponding anilines 18a-h (0.112 mol) and 2-chloronicotinic acid (0.064 mol) in glacial acetic acid (75 mL) were added at roomtemperature and the reaction mixture was stirred at 100 C for 3-6 h and analyzed by thin layer chromatography (TLC). The reactionmixture was cooled to room temperature, added with theappropriate amount of water, basified with potassium hydroxide topH = 12 and filtered. The filtrate was acidified to pH 3 with hydrochloricacid and stirred for 0.5 h. The precipitate was collectedby filtration and dried to give the corresponding products 19a-h asa white solid.
With potassium carbonate; In acetone; at 20℃; for 20h;
Protocol using trimethylsilyldiazomethane: To a solution of 2-chloronicotinic acid 5 (2.0 g, 12.7 mmol) in a mixture THF/MeOH (3:1) (100 mL) was added the solution of trimethylsilyldiazomethane in hexane (2.0 M, 7.6 ml, 15.2 mmol). After 1.5 h at room temperature, 0.2 equiv. of acetic acid was added, then the reaction mixture was concentrated under reduced pressure to give the crude product, which was engaged directly to the following step without any purification. The ester 14 was isolated as yellow oil (2.16 g, 99%). Protocol using dimethyl sulfate: To a suspension of 2-chloronicotinic acid 5 (5.0 g, 32 mmol) in acetone (40 mL) were added the potassium carbonate (6.63 g, 48 mmol, 1.5 equiv.) and the dimethyl sulfate (3.35 mL, 35 mmol, 1.1 equiv.). After 20 h at room temperature, the reaction mixture was filtered and rinsed with acetone. The filtrate was then concentrated under reduced pressure and diluted with CH2Cl2. The solution was then washed with a saturated aqueous solution of sodium carbonate, dried over Na2SO4, filtered and concentrated under reduced pressure, to give the almost clean crude mixture which was purified by distillation under reduce pressure (4.0 g, 71%). 1H NMR (300 MHz, CD3OD) delta 8.50 (dd, J = 4.8, 2.0 Hz, 1H), 8.23 (dd, J = 7.7, 2.0 Hz, 1H), 7.48 (dd, J = 7.7, 4.8 Hz, 1H), 3.94 (s, 3H); 13C NMR (75 MHz, CD3OD) delta 166.3, 153.0, 150.4, 141.8, 128.6, 124.0, 53.4.
2 - chloro nicotinic acid (25.3mmol), copper oxide (catalytic amount), potassium carbonate (25.3mmol) is added to the 100 ml round bottom flask, under normal temperature 20min, adding 4 - dimethylamino pyridine (50.6mmol), 110 C heating 2h. After adding the ethyl acetate to stir to room temperature, filtered, the filter cake washed first with ethyl acetate for 2 times, water (20 ml) dissolved, 4N hydrochloric acid adjusted to pH 5 - 6, standing out, filtered, the filter cake drying, to obtain crude, ethanol heat beating further purification, filtration, forms offwhite solid 5.16g, yield 89%
(Step 1) To a suspension of lithium aluminum hydride (8.60 g) in THF (200 mL) was added a solution of 2-chloronicotinic acid (22.4 g) in THF (30 mL) at 0C, and the mixture was stirred at 50C for 1 hr. After the reaction mixture was cooled to 0C, 1N aqueous sodium hydroxide solution (9 mL) was added. The precipitate was filtered off, and the filtrate was concentrated. The residue was purified by silica gel column chromatography (solvent gradient; 10?25% ethyl acetate/hexane) to give (2-chloropyridin-3-yl)methanol (14.0 g, 68%) as a colorless oil. 1H-NMR(CDCl3): delta 2.42-2.75(1H,m), 4.80(2H,d,J=4.8Hz), 7.29(1H,dd,J=7.8,4.8Hz), 7.88-7.93(1H,m), 8.29-8.32(1H,m)
1.56 g (87%)
With sodium hydroxide; lithium aluminium tetrahydride; In tetrahydrofuran; water;
EXAMPLE 35A (2-chloro-3-pyridinyl)methanol A solution of 2-chloronicotinic acid (2.0 g, 12.6 mmol) in THF (15 mL) at 0 C. was treated dropwise with 1M LAH in THF (15 mL, 15.0 mmol), warmed to room temperature, stirred for 5 hours, treated sequentially with water (0.5 mL), 40% NaOH solution (0.5 mL) and water (1.5 mL), stirred for 2 hours, filtered through a pad of diatomaceous earth (Celite), and extracted with dichloromethane. The combined extracts were dried (MgSO4), filtered, and concentrated to provide 1.56 g (87%) of the desired product of sufficient purity for subsequent use. MS (DCI/NH3) m/z 144 (M+H)+; 1H NMR (CDCl3) delta8.34 (dd, 1H), 7.90 (m, 1H), 7.29 (dd, 1H), 4.80 (s, 2H).
1.56 g (87%)
With sodium hydroxide; lithium aluminium tetrahydride; In tetrahydrofuran; water;
EXAMPLE 35A (2-chloro-3-pyridinyl)methanol A solution of 2-chloronicotinic acid (2.0 g, 12.6 mmol) in THF (15 mL) at 0 C. was treated dropwise with 1M LAH in THF (15 mL, 15.0 mmol), warmed to room temperature, stirred for 5 hours, treated sequentially with water (0.5 mL), 40% NaOH solution (0.5 mL) and water (1.5 mL), stirred for 2 hours, filtered through a pad of diatomaceous earth (Celite), and extracted with dichloromethane. The combined extracts were dried (MgSO4), filtered, and concentrated to provide 1.56 g (87%) of the desired product of sufficient purity for subsequent use. MS (DCI/NH3) m/z 144 (M+H)+; 1H NMR (CDCl3) delta 8.34 (dd, 1H), 7.90 (m, 1H), 7.29 (dd, 1H), 4.80 (s, 2H).
In tetrahydrofuran; water;
Step A 2-Chloro-3-pyridinemethanol To a stirred solution of 2-chloronicotinic acid (10.0 g, 63.4 mmol) in anhydrous THF (150 mL) at 0 C. under N2 was added lithium aluminum hydride (63 mL of a 1.0M solution in THF, 63 mmol) over 10 minutes. The solution was allowed to warm to room temperature over 2 h. Small quantities of ice were carefully added to quench the reaction followed by water. The aqueous solution was extracted with ether. The combined organic extracts were washed with water and saturated aqueous NaCl, dried over MgSO4, and concentrated in vacuo to give the title compound of Step A as an orange solid (6.18 g). 1 H NMR (CDCl3, 300 MHz) delta8.30 (dd, 1H), 7.90 (dd, 1H), 7.29 (dd, 1H), 4.80 (s,2H), 2.42 (br s,1H).
With thionyl chloride; triethylamine; In methanol; toluene;
EXAMPLE 3: Preparation of methyl 2-chloronicotinate (6) 81,9 g of thionyl chloride are slowly added to 20 g of 2-chloronicotinic acid (1), under stirring, and the suspension is stirred for 5 minutes. The mixture is heated to reflux for 60 minutes till obtaining a clear solution which is evaporated under vacuum. The residue is taken up into 200 ml of anhydrous toluene and concentrated under vacuum to 100 ml. The obtained solution is slowly dropped into a solution of 9 g of methanol and 14,6 g of triethylamine in 100 ml of anhydrous toluene, keeping temperature at about 20°C, under stirring. The mixture is left to stand for 5 hours, then cooled to +5°C and the precipitated triethylamine hydrochloride is filtered and washed with 20 ml of toluene. The organic phase is washed with 50 ml of 5percent p/v aqueous sodium hydrogen carbonate, then with 2 x 200 ml of water, dried over sodium sulfate, filtered and concentrated under vacuum to remove the solvent. 21,35 g of methyl 2-chloronicotinate are obtained in form of a slightly yellow oil, having b.p. 225-230°C, the structure of which is confirmed by the IR and NMR spectra. Yield about 98percent by mole.
With thionyl chloride; In methanol;
Synthesis of 2-Chloronicotinic Methyl Ester Compound 4 To a suspension of 2-chloronicotinic acid 3 (15.8 g, 0.1 mol) in 100 ml of dry MeOH at 0° C. was added thionyl chloride (17.8 g, 0.15 mol) dropwise. The reaction was then slowly warmed to room temperature and stirred for 2 days. The solvent was then removed via vacuum and the residue was dissolved in sat. NaHCO3. The resulted basic solution was then extracted with EtOAc (3*100 ml). The combined organic phases were washed with brine, dried with NaSO4, concentrated via vacuum to give 2-chloronicotinic methyl ester 4(15.3 g, 89percent) as an oil: 1 H NMR (CDCl3, 300 MHZ) delta 8.52 (dd, J=1.8, 4.5 Hz, 1H), delta 8.19 (dd, J=2.1, 7.8 Hz, 1H), delta 7.38 (dd, J=4.8, 7.8 Hz, 1H), delta 3.97 (s, 3H)
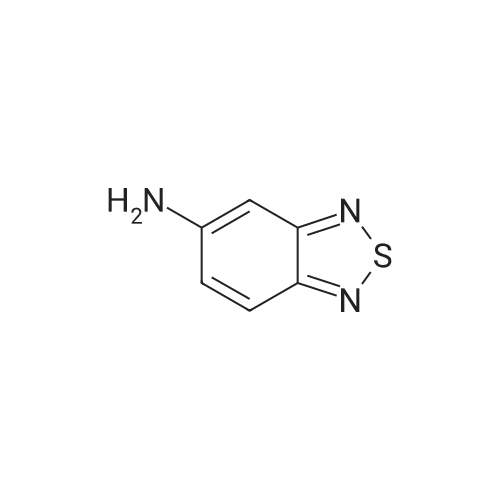
With potassium carbonate; copper(II) oxide; In water; at 140℃; for 4h;
(1) A mixture of 2-chloronicotinic acid (14.18g, 90mmol), 5-amino-2,1,3-benzothiadiazole (27.34g, 180mmol; synthesized according to Chem. Abst., 49, 3170a), potassium carbonate (14.41g, 104mmol) and cupric oxide (358mg, 4.5mmol) was heated at 140C for 4 hours. After admixing with water, the mixture was heated while stirring, then cooled to room temperature, and filtered to give a precipitate. After washing with ether, the resultant crude crystal was dissolved in ethanol-water, and filtered while it was hot. The resulting filtrate was acidified by addition of hydrochloric acid and a deposited precipitate was filtered off. The resultant precipitate was washed with water, and dried under reduced pressure to give 2-(2,1,3-benzothiadiazol-5-ylamino)nicotinic acid (14.52g, 59%).1H NMR(DMSO-d6) d: 7.03-7.07(1H, app-dd, J=4.6Hz, 7.9Hz), 7.67-7.71(1H, app-dd, J=2.3Hz, 9.6Hz), 7.98-8.02(1H, app-d, J=9.9Hz), 8.33-8.37(1H, app-dd, J=2.0Hz, 7.9Hz), 8.57-8.59(1H, app-dd, J=2.0Hz, 4.9Hz), 8.94-8.95(1H, app-ddd, J=2.0Hz), 10.93(1H, brs)
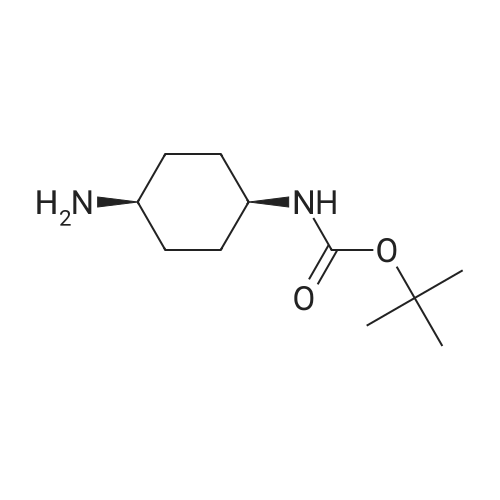
Preparation 10: Syn-{4-[(2-Chloro-pyridine-3-carbonyl)-amino]-cyclohexyl}-carbamic acid tert-butyl ester Oxalyl chloride (8ml, 90mmol) was added over 10 minutes to an ice-cooled suspension of 2-chloronicotinic acid (10g, 57 mmol) and N,N-dimethylformamide (5 drops) in dichloromethane (200ml). The suspension was then stirred at room temperature for 3 hours, and concentrated under reduced pressure. The residue was azeotroped with dichloromethane to give the intermediate acid chloride as a white solid. This was dissolved in dichloromethane (200ml), the solution cooled in a water bath, then N-diisopropylethylamine (20ml, 115mmol) and the amine from preparation 5 (13.4g, 62mmol) added. The reaction mixture was stirred for 18 hours, diluted with dichloromethane (100ml) and washed sequentially with 10% citric acid solution, saturated sodium bicarbonate solution (x2), water then brine. The organic solution was dried (MgSO4) and evaporated under reduced pressure to afford the title compound in 97% yield.
With thionyl chloride; triethylamine; In 1,4-dioxane; methanol; ethyl acetate;
EXAMPLE 1 Preparation of 2-chloronicotinic acid methyl ester (18) To a mixture of 2-chloronicotinic acid (Aldrich, 100.0 g, 0.63 mol) in 1,4-dioxane (500 mL) was added thionyl chloride (70 mL, 0.96 mol). The suspension was heated under reflux for 22 h with a gas trap to absorb hydrogen chloride gas. After evaporation of the solvent, the residue was dissolved in methanol (300 mL). To the solution was added dropwise triethylamine (TEA, 120 mL, 1.26 mol) at 0° C. over 2 h. The solvents were evaporated and the residue was suspended in ethyl acetate. The precipitate was removed by filtration. The filtrate was concentrated to afford the ester 18 (92.3 g, 86percent) as an oil: Rf(1:5 v/v ethyl acetate-hexane) 0.38. 1H NMR (300 MHz, CDCl3) delta8.53 (dd, 4.8 Hz, 1H), 8.19 (dd, 7.6 Hz, 1H), 7.37 (dd, 7.7 Hz, 1H) and 3.97 (s, 3H). 13C NMR (75 MHz, CDCl3) delta164.5, 151.6, 149.6, 140.0, 126.4, 121.9 and 52.5.
86%
With thionyl chloride; triethylamine; In 1,4-dioxane; methanol; ethyl acetate;
Example 1 Preparation of 2-chloronicotinic acid methyl ester (18) To a mixture of 2-chloronicotinic acid (Aldrich, 100.0 g, 0.63 mol) in 1,4-dioxane (500 mL) was added thionyl chloride (70 mL, 0.96 mol). The suspension was heated under reflux for 22 h with a gas trap to absorb hydrogen chloride gas. After evaporation of the solvent, the residue was dissolved in methanol (300 mL). To the solution was added dropwise triethylamine (TEA, 120 mL, 1.26 mol) at 0° C. over 2 h. The solvents were evaporated and the residue was suspended in ethyl acetate. The precipitate was removed by filtration. The filtrate was concentrated to afford the ester 18 (92.3 g, 86percent) as an oil: Rf (1:5 v/v ethyl acetate-hexane) 0.38. 1H NMR (300 MHz, CDCl3) delta 88.53 (dd, 4.8 Hz, 1H), 8.19 (dd, 7.6 Hz, 1H), 7.37 (dd, 7.7 Hz, 1H) and 3.97 (s, 3H). 13C NMR (75 MHz, CDCl3) delta 164.5, 151.6, 149.6, 140.0, 126.4, 121.9 and 52.5.
3-Bromo-11-oxo-11H-pyrido[2,1-b]quinazoline-6-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; In ethanol; water;
EXAMPLE 13 3-Bromo-11-oxo-11H-pyrido[2,1-b]quinazoline-6-carboxylic acid A solution of <strong>[135484-83-2]methyl 2-amino-4-bromo-benzoate</strong> (460 mg; 2 mmol), 2-chloronicotinic acid (315 mg; 2 mmol), concentrated hydrochloric acid (5 drops), ethanol (5 ml) and water (25 ml) was heated at reflux for 18 hours, cooled to 0 C. and filtered. The solid filter cake obtained was dried in vacuo and used directly in the next example.
With hydrogenchloride; In ethanol; water;
Example 13 3-Bromo-11-oxo-11H-pyrido[2,1-b]quinazoline-6-carboxylic acid A solution of <strong>[135484-83-2]methyl 2-amino-4-bromo-benzoate</strong> (460 mg; 2 mmol), 2-chloronicotinic acid (315 mg; 2 mmol), concentrated hydrochloric acid (5 drops), ethanol (5 ml) and water (25 ml) was heated at reflux for 18 hours, cooled to 0C and filtered. The solid filter cake obtained was dried in vacuo and used directly in the next example.
EXAMPLE 6 STR15 Methyl 2-chloronicotinate: 15.75 g (0.1 mole) of commercially available 2-chloronicotinic acid was dissolved in 100 mL of N,N-dimethylformamide. After cooling this solution to 0° under nitrogen, 4.4 g (0.11 mole) of 60percent sodium hydride was added portionwise while evolved hydrogen gas was vented. The reaction mixture was stirred until the effervescence stopped (~2 hrs.). 7.5 mL (0.112 mole) of methyl iodide was then added dropwise. Stirring of the reaction mixture was continued overnight. Solvent was removed in vacuo at r.t. The resulting brown residue was partitioned between ethyl acetate and water. The organic phase was then washed with brine and dried over anhydrous magnesium sulfate. Solvent removal gave a residue which was purified on silica gel using 1:1 ethyl acetate:hexane mixture to give the desired methyl 2-chloronicotinate as oil.
With caesium carbonate; In N,N-dimethyl-formamide;
A. 2-Chloronicotinic acid methyl ester To a solution of 2-chloronicotinic acid (2.00 g, 12.69 mmol) in DMF (40 mL) at 0° C. is added cesium carbonate (4.96 g, 15.23 mmol) followed by iodomethane (0.95 mL, 15.23 mmol). The reaction mixture is warmed to room temperature and stirred 16 h. Dilution with ethyl acetate is followed by washing with water, 8percent NaHCO3 solution and brine. The organic layer is dried (MgSO4) and concentrated under reduced pressure to give 2-chloronicotinic acid methyl ester as an oil: 1H NMR (CDCl3, 300 MHz): delta 8.55 (1H, dd), 8.18 (1H, dd), 7.33 (1H, dd), 3.97 (3H, s).
With potassium carbonate; dimethyl sulfate; In acetone;
2-Chloro-3-pyridinecarboxylic acid, methyl ester (11) is prepared as follows: a suspension of 22.06 g (140 mmol) of 2-chloro-3-pyridinecarboxylic acid (10), 14.6 mL (154 mmol) of dimethyl sulfate, 29 g (210 mmol) of anhydrous powdered potassium carbonate, and 140 mL of acetone is stirred at room temperature overnight. The mixture is filtered, the salts are washed with acetone, and the filtrate is concentrated to an oil that is diluted with dichloromethane. The organic phase is washed with saturated aqueous sodium dicarbonate, dried, then filtered through a pad of silica gel (230-400 mesh). The filtrate is concentrated to give an oil that is distilled at 90-91 C./0.17 mm to leave 19.9 g of the pure product as a colorless oil.
With perfluorosulfonic acid resin; In water; at 65 - 70℃; for 2.5h;Industrial scale;
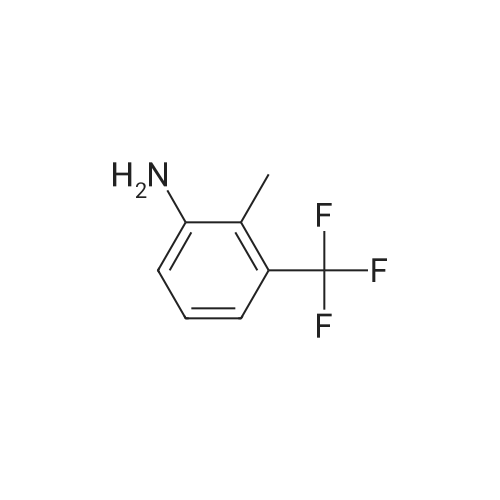
300 L of water was added to a 1000 L reactor, 131.9 kg (837.5 mol) of 2-chloronicotinic acid and 140 kg of <strong>[54396-44-0]2-methyl-3-trifluoromethylaniline</strong> (800 mol) were added separately, stirred, 1.4kg perfluorosulfonated resin was added, the temperature was raised to 65 °C -70 °C, and reacted for 2.5h.Filtered, the filtrate was adjusted to pH 4.5 with a 60percent sulfuric acid solution, stirred for 15-20min, filtered , filter cake was washed with 30kg of water, filter cake was dried to obtain 230.1 kg of flunixin, the yield was 97.1percent, and the purity was 99.9percent, purity was tested according to the method of USP38, the same below.
83%
With potassium hydroxide; toluene-4-sulfonic acid; In water;
iv.) A mixture of <strong>[54396-44-0]2-methyl-3-trifluoromethylaniline</strong> (368 g, 2.1 moles) and 2-chloronicotinic acid (158.0 g, 1.0 mole) in 400 ml of water is heated at 100° C. for 24 hours together with p-toluenesulfonic acid (15.0 g) monohydrate as the acid catalyst. Potassium hydroxide (ca. 145 g) in water (255 ml) is added and the pH is maintained above 11. After diluting the reaction mixture to 1.2 liters with water, the mixture is cooled to 50° C., adjusted to pH 11, treated with 7 g of a decolorizing charcoal and 15 g of a filter aid, and clarified by filtration. The flitrate is diluted with 750 ml of water and the pH is adjusted to 5.0 with concentrated sulfuric acid. Agitation of the suspension for 10 minutes and filtration gives crude, precipitated 2-[[2-methyl-3-(trifluoromethyl)phenyl]amino]-3-pyridinecarboxylic acid (flunixin) (83percent yield). The compound can be further purified by crystallization in methanol and washing with water.
With toluene-4-sulfonic acid; copper(II) oxide; sodium hydroxide; In 1,2-dimethoxyethane; water; at 45℃; for 0.6h;
(1) 2-chloronicotinic acid and <strong>[54396-44-0]2-methyl-3-trifluoromethylaniline</strong> are stirred and added to an aqueous sodium hydroxide solution, 2-chloronicotinic acid and 2-methyl-3-trifluoromethyl The molar ratio of aniline is 3:1; the molar ratio of 2-chloronicotinic acid to sodium hydroxide used to prepare aqueous sodium hydroxide solution is 1:2; ethylene glycol and chain polyethylene glycol dialkyl are added. The ether and the catalyst A, the chain polyethylene glycol dialkyl ether is used in an amount of 3percent of the mass of 2-chloronicotinic acid; the catalyst A is p-toluenesulfonic acid and copper oxide, the pair The molar ratio of toluenesulfonic acid to copper oxide is 3:1; the temperature is controlled at 45 ° C, after 0.6 hours of reaction, the pH of the solution is adjusted, stirred and allowed to stand for stratification, stirred, filtered, and the filter cake is washed and dried to obtain flunixin.;
With potassium fluoride;bis(benzonitrile)palladium(II) chloride; In N,N,N,N,N,N-hexamethylphosphoric triamide; dichloromethane; water; N,N-dimethyl-formamide;
EXAMPLE 18 3-Acetyl-2-chloropyridine The following is the preparation of a compound of Formula 2 in which t is 0, L is chloro, Y is N, Z is CH and R3 is acetyl. A mixture of 2-chloronicotinic acid (20 g, 0.127 mmol) and oxalyl chloride (13.3 mL, 0.152 mmol) in 2 drops of DMF and 200 mL of methylene chloride was stirred at room temperature for approximately 14 hours. The mixture was stirred at reflux temperature for 1 hour. The solution was cooled and partitioned between 100 mL of ice-cold aqueous sodium bicarbonate and 300 mL of methylene chloride. The organic layer was washed with brine, dried (Na2 SO4) and concentrated to give 2-chloronicotinic acid chloride (8.8 g, 49.5 mmol). A mixture of 2-chloronicotinic acid chloride (8.8 g, 49.5 mmol), tetramethyltin (5.5 mL, 40 mmol) and bis(benzonitrile)palladium(II) chloride (0.385 g) in 20 mL of hexamethylphosphoramide was stirred at room temperature for 20 hours. The mixture was stirred with 50 mL of 10percent potassium fluoride and then poured into 50 mL of water. The mixture was extracted with diethyl ether (4*50 mL) and the combined extracts were washed with water, saturated sodium bicarbonate and sodium chloride, dried (Na2 SO4) and concentrated. The residue was purified on silica gel by column chromatography eluding with hexanes/ethyl acetate (3:1) to give 3-acetyl-2-chloropyridine (4.2 g, 27 mmol).
With 4-methyl-morpholine; benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In N,N-dimethyl-formamide; at 20℃; for 18h;
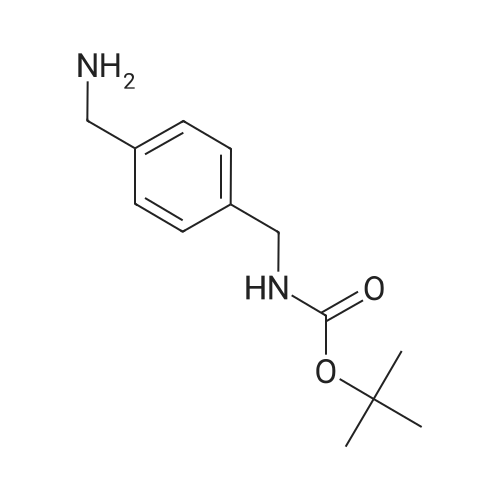
2-Chloro-nicotinic acid (2.86 g, 18.2 mmol), 1-hydroxybenzotriazole (3.0 g, 21.8 mmol) and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (4.18 g, 21.8 mmol) were dissolved in N,N-dimethylformamide (50 ml) at room temperature and <strong>[108468-00-4](4-aminomethyl-benzyl)-carbamic acid tert-butyl ester</strong> (4.29 g, 18.2 mmol) (see Preparation 1) added followed by addition of N-methyl morpholine (4 ml, 36.3 mmol). The reaction mixture was stirred under an atmosphere of nitrogen at room temperature for 18 hours, then partitioned between ethyl acetate (100 ml) and water (100 ml) and the organic layer separated. The organic layer was then washed with a saturated aqueous solution of sodium chloride (100 ml), dried over anhydrous magnesium sulphate and the solvent removed in vacuo. The residue was then triturated with diethylether (2-fold 10 ml) to give (4-[(2-chloro-pyridine-3-carbonyl)-amino]-methyl}-benzyl)-carbamic acid tert-butyl ester (6.71 g) as a white solid. [0236] <1>H NMR (400 MHz, DMSO-d<6>): [delta]=9.01-9.08 (1H, t), 8.43-8.47 (1H, m), 7.89-7.93 (1H, d), 7.45-7.50 (1H, m), 7.30-7.37 (1H, m), 7.26-7.31 (2H, d), 7.17-7.21 (2H, d), 4.39-4.43 (2H, d), 4.03-4.10 (2H, d), 1.37 (9H, s) ppm. [0237] LRMS (electrospray): m/z [M-H]<+> 374.
2-<(1-methyl-3-pyrrolidinyl)oxy>-3-pyridinecarboxylic acid sodium salt[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
In dimethyl sulfoxide; ethyl acetate;
Preparation 6 Sodium 2-[(1-methyl-3-pyrrolidinyl)oxy]-3-pyridinecarboxylate To a stirred suspension of 6.4 g (0.13 mole) of 50percent sodium hydride (mineral oil) in 50 ml of dimethylsulfoxide was added dropwise 6.4 g (0.063 mole) of <strong>[13220-33-2]1-methyl-3-pyrrolidinol</strong>. During addition, the temperature rose from 25° C. to 31° C. After 10 minutes, a solution of 10 g (0.063 mole) of 2-chloronicotinic acid in 50 ml of dimethylsulfoxide was added dropwise causing the temperature to rise. When the temperature reached 55° C., it was maintained there by the intermittent use of an ice bath until addition was complete. The mixture was then heated to 55°-60° C. for 1.5 hr., cooled and filtered. The filter cake was suspended in 100 ml of ethyl acetate and filtered. The solid was recrystallized from ethyl acetatemethanol. Yield of product was 5 g., dec. 240° C. The NMR analysis showed that the compound contained 1/3 mole of sodium acetate as impurity. Analysis: Calculated for C11 H13 N2 O3 Na.1/3C2 H3 O2 Na: C, 51.62; H, 5.20; N, 10.32. Found: C, 51.81; H, 5.15; N, 10.39.
In dimethyl sulfoxide; ethyl acetate;
PREPARATION 6 Sodium-2-[(1-methyl-3-pyrrolidinyl)oxy]-3-pyridinecarboxylate To a stirred suspension of 6.4 g (0.13 mole) of 50percent sodium hydride (mineral oil) in 50 ml of dimethylsulfoxide was added dropwise 6.4 g (0.063 mole) of <strong>[13220-33-2]1-methyl-3-pyrrolidinol</strong>. During addition, the temperature rose from 25° C. to 31° C. After 10 minutes, a solution of 10 g (0.063 mole) of 2-chloronicotinic acid in 50 ml of dimethylsulfoxide was added dropwise causing the temperature to rise. When the temperature reached 55° C., it was maintained there by the intermittent use of an ice bath until addition was complete. The mixture was then heated to 55°-60° C. for 1.5 hr., cooled and filtered. The filter cake was suspended in 100 ml of ethyl acetate and filtered. The solid was recrystallized from ethyl acetatemethanol. Yield of product was 5 g., dec. 240° C. The NMR analysis showed that the compound contained 1/3 mole of sodium acetate as impurity. Analysis: Calculated for C11 H13 N2 O3 Na.1/3C2 H3 O2 Na: C, 51.62; H, 5.20; N, 10.32. Found: C, 51.81; H, 5.15; N, 10.39.
With hydrogenchloride; triphenylphosphine; In tetrahydrofuran; tetrachloromethane; dichloromethane; mineral oil;
(R)-2-(2-Chloroethyl)-2,3-dihydro-4-methylpyrido[3,2-f]1,4-oxazepin-5-(4H)-one hydrochloride [1:1] A solution of 47.4 g (0.3 mole) of 2-chloronicotinic acid and 30 g (0.3 mole) of <strong>[104641-60-3](R)-1-methyl-3-pyrrolidinol</strong> in 400 ml of tetrahydrofuran was added over a period of 1 hr to a stirred suspension of 26.4 g (0.66 mole) of 60% sodium hydride/mineral oil in 500 ml of tetrahydrofuran at 55-60 C. The mixture was stirred at reflux for 2.5 hr and allowed to cool to 25 C. About 400 ml of methylene chloride was added to the slurry followed by a dropwise addition of 34.5 g (0.36 mole) of methane sulfonic acid in 100 ml of methylene chloride. The mixture was stirred for 10 min and 157 g (0.6 mole) of triphenylphosphine was added followed by 200 ml of carbon tetrachloride. The mixture was heated to reflux for 4 hr. To the cooled (25 C.) solution was added 100 ml of triethylamine at a rapid drop. The solution was concentrated on the rotary evaporator and the residue was partitioned between methylene chloride and dilute hydrochloric acid. The methylene chloride was extracted 6 times with dilute hydrochloric acid. The acid extracts were combined, made basic with sodium hydroxide and extracted with chloroform which was dried and concentrated. The residue was dissolved in isopropyl alcohol and treated with a solution of hydrogen chloride in isopropyl alcohol. The resulting hydrochloride weighed 25 g (30%). One gram was recrystallized from isopropyl alcohol, m.p. 152-154 C.; [alpha]D25 =+36.2 (water). Analysis: Calculated for C11 H14 N2 O2 Cl2: C, 47.67; H, 5.09; N, 10.11. Found: C, 47.65; H, 5.20; N, 9.03.
With hydrogenchloride; triphenylphosphine; In tetrahydrofuran; tetrachloromethane; dichloromethane; mineral oil;
(R)-2-(2-Chloroethyl)-2,3-dihydro-4-methylpyrido[3,2-f]-1,4-oxazepin-5-(4H)-one hydrochloride [1:1] A solution of 47.4 g (0.3 mole) of 2-chloronicotinic acid and 30 g (0.3 mole) of <strong>[104641-60-3](R)-1-methyl-3-pyrrolidinol</strong> in 400 ml of tetrahydrofuran was added over a period of 1 hr to a stirred suspension of 26.4 g (0.66 mole) of 60% sodium hydride/mineral oil in 500 ml of tetrahydrofuran at 55-60 C. The mixture was stirred at reflux for 2.5 hr and allowed to cool to 25 C. About 400 ml of methylene chloride was added to the slurry followed by a dropwise addition of 34.5 g (0.36 mole) of methane sulfonic acid in 100 ml of methylene chloride. The mixture was stirred for 10 min and 157 g (0.6 mole) of triphenylphosphine was added followed by 200 ml of carbon tetrachloride. The mixture was heated to reflux for 4 hr. To the cooled (25 C.) solution was added 100 ml of triethylamine at a rapid drop. The solution was concentrated on the rotary evaporator and the residue was partitioned between methylene chloride and dilute hydrochloric acid. The methylene chloride was extracted 6 times with dilute hydrochloric acid. The acid extracts were combined, made basic with sodium hydroxide and extracted with chloroform which was dried and concentrated. The residue was dissolved in isopropyl alcohol and treated with a solution of hydrogen chloride in isopropyl alcohol. The resulting hydrochloride weighed 25 g (30%). One gram was recrystallized from isopropyl alcohol, m.p. 152-154 C.; [alpha]D25 =+36.2 (water). Analysis: Calculated for C11 H14 N2 O2 Cl2: C, 47.67; H, 5.09; N, 10.11. Found: C, 47.65; H, 5.20; N, 9.03.
PREPARATION 2 2-Methoxy-3-pyridinecarboxylic Acid A stainless steel stirred autoclave was charged sequentially with methanol (2.8 l.), sodium methoxide (259 g.) (in portions, keeping the temperature less than 35 C.), and 2-chloro-3-pyridinecarboxylic acid (190 g.). The autoclave was sealed and the reaction mixture heated at 110 C. (50 psig) for 48 hours. The reaction mixture was cooled to 25 C. and discharged from the autoclave. Solids were recovered by filtration. Concentration of the filtrate gave a second crop. These process steps were repeated until virtually all of the methanol had been removed. The several crops of solids were combined, taken up in 2.5 liters of water and acidified with conc. hydrochloric acid to pH 2.7 keeping the temperature below 20 C. The precipitated product was granulated for 30 minutes at 15 C. and recovered by filtration (141 g.). Purified title product was obtained by recrystallization from ethyl acetate-hexane (120.5 g., m.p. 148-150 C.).
2-(1-methylethyl)-11-oxo-11H-pyrido[2,1-b]quinazoline-6-carboxylic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium bromide; In Triethylene glycol dimethyl ether; ethanol; acetic acid;
EXAMPLE 71 Preparation of 2-(1-methylethyl)-11-oxo-11H-pyrido[2,1-b]quinazoline-6-carboxylic acid A mixture of 13 g of 2-chloronicotinic acid, 9.9 g of <strong>[68701-22-4]5-isopropylanthranilic acid</strong> and 0.1 g of potassium bromide in 10 ml of triglyme was heated gradually to a bath temperature of 140 C. After 2 hours a orange solid had separated, the reaction mixture was allowed to cool as 10 ml of ethanol were added, and the crude product was collected. This material was crystallized from acetic acid-water to give 9.3 g of a solid mp 173-193 C., which was dissolved in 100 ml of acetic acid and heated to reflux for 1 hour. The solvent was evaporated and the residue was crystallized from isopropylalcohol to give 5.8 g (37%) of 2-(1-methylethyl)-11-oxo-11H-pyrido[2,1-b]quinazoline-6-carboxylic acid, mp 206-210 C. The analytical sample gave mp 221-223 C.
Step A Preparation of 3-(2-chloronicotinoylamino)-2-Pyridone The acid chloride of 2-chloronicotinic acid was prepared by refluxing 11 g. (0.07 mole) of 2-chloronicotinic acid in 80 ml. of thionyl chloride for 1.5 hours. After evaporating to dryness, the oily residue was dissolved in 50 ml. of ether. The ether solution was added to a stirred ice-cold solution of 7.7 g. (0.07 mole) of <strong>[33630-99-8]3-amino-2-pyridone</strong> in 80 ml. of dry pyridine during 15 minutes. After 30 more minutes at ice temperature and 4 hours at room temperature, it was allowed to stand overnight in an open dish. The crystalline residue was extracted with water and the solid residue (12.9 g.) was collected and recrystallized from dimethyl formamide-ether to give gray crystals of 3-(2-chloronicotinoylamino)-2-pyridone, m.p. 295 C.
2-chloronicotinic acid 1(2-morpholinoethyl)ester[ No CAS ]
[ 554-68-7 ]
Yield
Reaction Conditions
Operation in experiment
90%
With thionyl chloride; triethylamine; In toluene;
EXAMPLE 1: Preparation of 2-chloronicotinic acid 1(2-morpholinoethyl)ester (3) 409,5 g of thionyl chloride are slowly added to 100 g of 2-chloronicotinic acid (1), under stirring, and the resulting suspension is stirred for about 5 minutes. The reaction mixture is heated and kept to ebollition for 60 minutes, to obtain a clear solution which is evaporated under vacuum. The residue is taken up into 700 ml of anhydrous toluene and evaporated again under vacuum to a 250 ml final volume. The obtained solution is dropped, during 30 minutes, into a solution of 91,5 g of 2-morpholinoethanol (2) and 73 g of triethylamine in 500 ml of anhydrous toluene, always keeping temperature at about 20C. The mixture is stirred for 2 hours, then cooled to +5C and the precipitated triethylamine hydrochloride is filtered and washed with 50 ml of toluene. The organic solution is washed with 250 ml of 5% p/v aqueous sodium carbonate, then with 2 x 200 ml of water. The solution is dried over sodium sulfate, filtered and evaporated under vacuum to completely remove the solvent. 154 g of a straw-colored oil are obtained, consisting of 2-chloronicotinic acid (2-morpholinoethyl) ester, having boiling point 250C, the structure of which is confirmed by IR and NMR spectra. Yield about 90% by mole.
In tetrahydrofuran; methanol; hexane; at 20℃; for 1.5h;
Protocol using trimethylsilyldiazomethane: To a solution of 2-chloronicotinic acid 5 (2.0 g, 12.7 mmol) in a mixture THF/MeOH (3:1) (100 mL) was added the solution of trimethylsilyldiazomethane in hexane (2.0 M, 7.6 ml, 15.2 mmol). After 1.5 h at room temperature, 0.2 equiv. of acetic acid was added, then the reaction mixture was concentrated under reduced pressure to give the crude product, which was engaged directly to the following step without any purification. The ester 14 was isolated as yellow oil (2.16 g, 99%). Protocol using dimethyl sulfate: To a suspension of 2-chloronicotinic acid 5 (5.0 g, 32 mmol) in acetone (40 mL) were added the potassium carbonate (6.63 g, 48 mmol, 1.5 equiv.) and the dimethyl sulfate (3.35 mL, 35 mmol, 1.1 equiv.). After 20 h at room temperature, the reaction mixture was filtered and rinsed with acetone. The filtrate was then concentrated under reduced pressure and diluted with CH2Cl2. The solution was then washed with a saturated aqueous solution of sodium carbonate, dried over Na2SO4, filtered and concentrated under reduced pressure, to give the almost clean crude mixture which was purified by distillation under reduce pressure (4.0 g, 71%). 1H NMR (300 MHz, CD3OD) delta 8.50 (dd, J = 4.8, 2.0 Hz, 1H), 8.23 (dd, J = 7.7, 2.0 Hz, 1H), 7.48 (dd, J = 7.7, 4.8 Hz, 1H), 3.94 (s, 3H); 13C NMR (75 MHz, CD3OD) delta 166.3, 153.0, 150.4, 141.8, 128.6, 124.0, 53.4.
85.42%
In methanol; hexanes; toluene; at 20℃;
Intermediate 3. 2-Chloronicotinic acid methyl ester.; To a room temperature solution of2-chloronicotinic acid (5.00 g, 31.7 mmol) in 20% MeOH/toluene (100 mL) was added TMS diazomethane (trimethylsilyl diazomethane) (2.0M in hexanes, 31.7 mL) dropwise and the reaction was monitored by TLC (thin-layer chromatography) until completion. The reaction was concentrated down under reduced pressure. The residue was purified by silica chromatography using a gradient of hexanes:EtOAc (100:0) to hexanes:EtOAc (80:20) to yield 4.65 g (85.42%) of Intermediate 3 as a yellow oil. EPO <DP n="36"/>
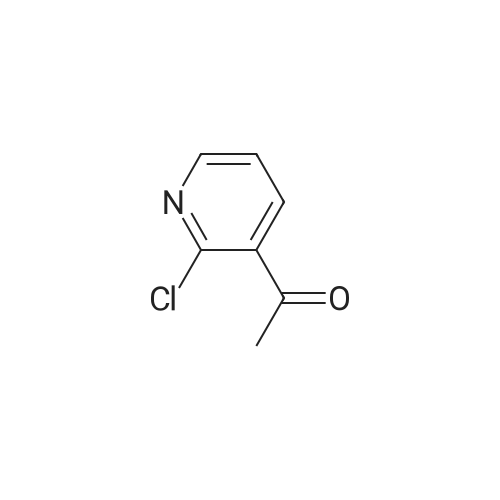
To a solution of 2-chloronicotinic acid (B-1) (7.88 g, 50.0 mmol) in THF (100 mL) was added methyl magnesium bromide (42 mL, 3M ethyl ether solution) dropwise under 0 °C. Upon completion of the addition, the reaction mixture was stirred at 0 °C for 0.5 h and then at room temperature overnight. The reaction mixture was added into ice/water (150 mL), and extracted with ethyl acetate. The combined organic layers were dried over Na2S04 and concentrated to afford the title compound 1 -(2-chloropyridin-3-yl)ethanone (B-2). MS (m/z): 156 (M+1 )+.
In tetrahydrofuran; ethyl methyl ether; at 0 - 20℃; for 0.5h;
1-(2-Cloropyridin-3-yl)ethanone (B-2)To a solution of 2-chloronicotinic acid (B-1) (7.88 g, 50.0 mmol) in THF (100 mL) was added methyl magnesium bromide (42 mL, 3-Methyl ether solution) dropwise under 0° C.Upon completion of the addition, the reaction mixture was stirred at 0° C. for 0.5 h and then at room temperature overnight.The reaction mixture was added into ice/water (150 mL), and extracted with ethyl acetate.The combined organic layers were dried over Na2SO4 and concentrated to afford the title compound 1-(2-chloropyridin-3-yl)ethanone (B-2). MS (m/z): 156 (M+1)+.
With dmap; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; In dichloromethane; at 70 - 80℃;
General procedure: The synthesis route of compounds 1-24 followed the general pathway outlined in Scheme 1. The substituted nicotinic acid (1 mmol) mixed with benzenesulfonamide (1 mmol) through by using 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl) (1.2 mmol) and 4-dimethylaminopyridine (DMAP) (1.2 mmol) in anhydrous CH2Cl2 for 6-8 h at 70-80 C. The reaction was monitored by TLC. The products are extracted with ethyl acetate. The extract is washed successively with 1 N HCl, water, 1 M NaHCO3, and water, dried over MgSO4, filtered and evaporated. The residue is purified by column chromatography using petroleum ether and ethyl acetate (1:1).
With triethylamine; HATU; In N,N-dimethyl-formamide; at 20℃;
Step 1 :Exp-4-g1Step 1 : preparation of 2-Chloro-N-(4-chloro-3-methoxyphenyl)pyridine-3- carboxamide (Exp-4-g3)[0100] Into a vessel containing a solution prepared by dissolving 1 g of 2- chloropyridine-3-carboxylic acid (Exp-4-gl, 6.37 mmol) and 1 g of 4-chloro-3- methoxy-phenylamine (Exp-4-g2, 6.4 mmol) in 30 mL of DMF was added 6.05 g 0- 7- Azabenzotriazol-l-yl)-N,N,N'N'-tetramethyluronium hexafluorophosphate (HATU, 15.9 mmol), and 1.93 g triethylamine (TEA, 19.1 mmol). The reaction mixture thus provided was stirred at ambient temperature (RT) overnight then the mixture was poured into cool water and the resulting mixture extracted with sufficient amounts of EtOAc to obtain the product from the reaction mixture. The organics were combined, washed with brine, dried over MgSC>4, filtered and concentrated under reduced pressure. The residue thus provided was purified on silica column chromatogram (PE:EtOAc = 50: 1-1 : 1) to give 1.1 g of Exp-4-g3 (calculated yield 58%).
58%
With triethylamine; HATU; In N,N-dimethyl-formamide; at 20℃;
Into a vessel containing a solution prepared by dissolving 1 g of 2- chloropyridine-3-carboxylic acid (Exp-4-gl, 6.37 mmol) and 1 g of 4-chloro-3- methoxy-phenylamine (Exp-4-g2, 6.4 mmol) in 30 mL of DMF was added 6.05 g O-(7- Azabenzotriazol-l-y^-A .A.N'.N'-tetramethyluronium hexafluorophosphate (HATU, 15.9 mmol), and 1.93 g tnethylamine (TEA, 19.1 mmol). The reaction mixture thus provided was stirred at ambient temperature (RT) overnight then the mixture was poured into cool water and the resulting mixture extracted with sufficient amounts of EtOAc to obtain the product from the reaction mixture. The organics were combined, washed with brine, dried over MgS04, filtered and concentrated under reduced pressure. The residue thus provided was purified on silica column chromatogram (PE:EtOAc = 50: 1-1 :1) to give 1.1 g of Exp-4-g3 (calculated yield 58%).
2-[(2-chloropyridin-3-oyl)amino]-4-(pyridin-2-yl)thiazole[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
82%
General procedure: 5.1.3. General procedure C. To the carboxylic acid substrate (0.42 mmol, 1.2 equiv) in DMF (1.5 mL) was added CDI (0.50 mmol, 1.1equiv) and DBU (0.67 mmol, 1.9 equiv). The reaction was stirred for 30 min at 23 C. Next, 2-amino-4-(2-pyridyl)thiazole 20 (0.35 mmol, 1.0 equiv) was added and the reaction mixture was stirred for 16 h at 23 C. A small amount of silica gel was added to the reaction mixture, which was concentrated in vacuo under reduced pressure. The crude product impregnated on the silica gel was purified by silica gel flash chromatography (0-10% MeOH-CH2Cl2) to afford the title compound.
N'-[4-(benzyloxy)phenyl]-2-chloropyridine-3-carbohydrazide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
5 g
With benzotriazol-1-ol; 1-ethyl-(3-(3-dimethylamino)propyl)-carbodiimide hydrochloride; N-ethyl-N,N-diisopropylamine; In N,N-dimethyl-formamide; at 20℃; for 12h;
To a solution of 2-chloropyridine-3-carboxylic acid (3.30 g) in DMF (50 ml) were added <strong>[51145-58-5][4-(benzyloxy)phenyl]hydrazine</strong> (5.0 g), HOBt (3.7 g), EDCI (4.6 g) and diisopropylethylamine (9.0 g), and the mixture was stirred at room temperature for 12 hr. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The organic layers were combined, dried over anhydrous magnesium sulfate and filtered. The filtrate was concentrated under reduced pressure, and the obtained residue was purified by silica gel column chromatography (hexane/ethyl acetate) to give the title compound (5.0 g). [0471] 1H NMR (400 MHz, CDCl3) delta 5.01 (2H, s), 6.90-6.94 (4H, brs), 7.31-7.43 (7H, m), 8.11 (1H, dd, J=7.6, 2.0 Hz), 8.32-8.40 (1H, brs), 8.50 (1H, dd, J=4.6, 1.6 Hz)
To a solution of 2-chloronicotinic acid (5g, 31.84 mmol) in dry DCM under nitrogen, oxalyl chloride (3.3 mL, 38.21 mmol) and catalytic amount of N,N-dimethylformamide were added carefully with stirring. The reaction was stirred for 3 h. After completion of the reaction, the reaction mixture was concentrated under vaccum to yield 2-chloronicotinyl chloride as solid which was used for next step without purification. 2-Chloronicotinyl chloride (4.6 g, 26.1 mmol) was dissolved in dry DCM, cooled to 0 C, <strong>[15430-52-1]propargylamine hydrochloride</strong> (26.1 mmol) and triethylamine (9.04 mL, 65.25 mmol) were added. The reaction was warmed to room temperature. After stirring overnight, the reaction mixture was diluted with water and extracted with DCM. The combined organic extracts were washed with aq NaHCO3 and dried with Na2SO4 and concentrated in vacuo. H NMR (300 MHz, CDCl3) delta 8.46 (dd, J = 3.0, 2.2 Hz, 1H), 8.10 (dd, J = 6.2, 3.0 Hz, 1H), 7.36 (d, J = 3.0 Hz, 1H), 6.94 (br s, 1H), 4.25 (m, 2H), 2.32 (t, J = 2.2 Hz, 1H).
8-chloro-3-(2-chloropyridin-3-yl)-6-(trifluoromethyl)-[1,2,4]triazolo[4,3-a]pyridine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
56%
With trichlorophosphate; In ethanol; at 140℃; for 0.25h;Microwave irradiation;
A CEM-designed 10-mL pressure-rated vial was charged with POCl3(2 mL), <strong>[89570-82-1]3-chloro-2-hydrazinyl-5-(trifluoromethyl)pyridine</strong> (211 mg,1mmol), 4-methoxylbenzoic acid or analogous acid (1mmol). The mixture was irradiated in a CEM Discover Focused Synthesiser (150 W, 140C, 200 psi, 15min). The mixture was cooled to room temperature by passing compressed air through the microwave cavity for 2 min. It was poured into cold ice (40 mL) and the formed precipitate filtered. The crude solid was recrystallised from EtOH to give the title compound 2a and others. All the other compounds were synthesised according to the same procedure.
With water; palladium diacetate; triethylamine; triphenylphosphine; In 1,4-dioxane; at 110℃; under 11251.1 Torr; for 2h;Flow reactor;
General procedure: For a typical reaction, a Vapourtec 2R+ Series was used as the platform with a Vapourtec Gas/Liquid Membrane Reactor to load the carbon monoxide. The HPLC pump were both set at 0.125 mL/min, temperature of the reactor at 110 C, pressure of CO at 15 bar with a back pressure regulator of 250 psi (17.24 bar). The system was left running for 2 h to reach steady state after which time the flow streams were switched to pass from the loops where the substrates and catalysts were loaded. The first loop (5 mL) was filled with a solution of palladium acetate (20 mg, 0.08 mmol), triphenylphosphine (48 mg, 0.168 mmol) in 6 mL of 1,4-dioxane while the second loop (5 mL) was filled with a solution made from the ortho-substituted iodoarene substrate (1.68 mmol), triethylamine (0.272 g, 0.374 mL, 2.69 mmol) and water (0.505 g, 28 mmol) in 5.8 mL of 1,4-dioxane. An Omnifit column filled with 1.71 cm3 (r = 0.33 cm, h = 5.00 cm) of cotton was positioned just before the back pressure regulator to trap any particulate matter formed to avoid blocking of the back pressure regulator. After the substrates were passed through the system, the outlet of the flow stream was directed into a receptacle where the excess carbon monoxide gas was vented off in the fume cupboard. The reaction mixture was then evaporated to dryness, ethyl acetate (25 mL) and sodium carbonate solution (2 M, 10 mL) were added and transferred to a separating funnel. After collecting the aqueous layer, the organic layer was extracted with sodium carbonate solution (2 M, 2 × 10 mL). The combined aqueous layers were acidified by the addition of 2 M HCl solution which was then extracted with ethyl acetate (3 x 25 mL). The organic layer was dried over sodium sulfate, and the solvent evaporated under vacuum to give the crude product as a solid. The crude product was then recrystallised from the appropriate solvent.
In a 200 mL reaction flask,Put 111.6g dioxane,12.8 g of carbonyldiimidazole was added with stirring,And then slowly11.3 g of 2-chloronicotinic acid,Under argon atmosphere, heated to 90 C for 2 hours,During the exhaust gas argon several times,15.0 g of compound II-B was added,Insulation reaction for 6 hours.After the reaction,The dioxane was removed by distillation under reduced pressure,The remaining oil was rinsed with 46.8 g of acetone to 467.3 g of water to precipitate a yellow solid,filter,Wash the solid until the lotion was neutral,Vacuum drying,23.8 g of a yellow solid.The resulting solid was recrystallized from 55.4 g of isopropanol,To obtain 18.4 g of compound II-A,Yield 70%.(TLC detection, chloroform: methanol = 10: 1, raw material Rf = 0.1, amino compound Rf = 0.9, product Rf = 0.7).
General procedure: To the mixture of an appropriately substituted phenylamine 16a-h (0.054mol) and glacial acetic acid (100mL), 2-chloronicotinic acid 15 (0.032mol) was added at room temperature. Upon the completion of addition, the reaction mixture was stirred at 100C for 3-6h and monitored by thin-layer chromatography (TLC). The reaction mixture was cooled to room temperature and basified with potassium hydroxide to pH 12, and filtered. The filtrate was acidified with hydrochloric acid to pH 3, and stirred for 0.5h. The precipitated was collected by filtration and dried to give the corresponding 2-(substitutedphenylamino)nicotinic acid 17a-h as white solids.
With acetic acid; at 100℃; for 5h;
General procedure: Corresponding anilines 18a-h (0.112 mol) and 2-chloronicotinic acid (0.064 mol) in glacial acetic acid (75 mL) were added at roomtemperature and the reaction mixture was stirred at 100 C for 3-6 h and analyzed by thin layer chromatography (TLC). The reactionmixture was cooled to room temperature, added with theappropriate amount of water, basified with potassium hydroxide topH = 12 and filtered. The filtrate was acidified to pH 3 with hydrochloricacid and stirred for 0.5 h. The precipitate was collectedby filtration and dried to give the corresponding products 19a-h asa white solid.
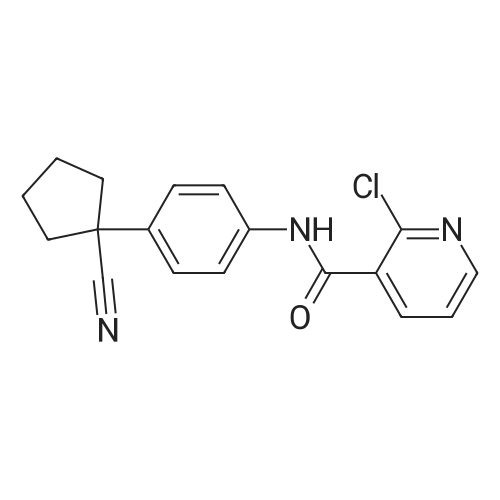
2-chloro-N-(9-ethyl-9H-carbazol-3-yl)nicotinamide[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
73%
In a 50 mL three-neck round-bottom flask was charged 2-chloropyridine-3-carboxylic acid 1 (0.4726 g, 3.0 mmol), HOBT (0.4189 g, 3.1 mmol), EDAC (0.5943 g, 3.1 mmol). The solution was dissolved in DMF (5 mL) and 3-Amino-9-ethylcarbazole 2 (0.6308g, 3.0 mmol) was added. After 15 min, Et 3 N (0.859 mL, 6.0 mmol) was added and the mixture stirred at room temperature for 16 h. After completion of the reaction, water was added (30 mL) and the product was extracted using dichloromethane (30mL). The organic layer was washed with brine and dried with Na 2 SO 4 , filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and the product obtained as a white solid for the precursor 2-chloro-N-(9-ethyl-9H-carbazol-3-yl)nicotinamide 3 (0.7565g, 2.2 mmol, 73%). TLC analysis in CH 2 Cl 2 -MeOH (9:1). Rf = 0.61. 1 H NMR (400 MHz, CDCl 3 ) delta 1.46 (3H, t, J = 7.3 Hz), 4.40 (2H, q, J = 7.1 Hz), 7.43, (1H, t, J = 8.6 Hz), 7.51 (1H, t, J = 7.6 Hz), 7.64 (1H, d, J = 2.3 Hz), 7.66 (1H, d, J = 2.0Hz), 8.13 (1H, d, J = 7.3Hz), 8.26 (1H, d, J = 2.0 Hz), 8.28 (1H, d, J = 1.8 Hz), 8.38 (1H, s), 8.47 (1H, d, J = 1.8 Hz), 8.53 (1H, d, J = 2.0 Hz), 8.54 (1H, d, J = 2.0 Hz); 13 C NMR (100 MHz, CDCl 3 ) 14.1, 38.0, 108.9, 113.6, 119.3, 121.1, 123.0, 123.2, 123.5, 126.4, 129.2, 132.0, 138.0, 140.3, 140.8, 147.4, 151.4, 162.9.
73%
In a 50 mL three-neck round-bottom flask was charged 8 2-chloropyridine-3-carboxylic acid 9 1 (0.4726 g, 3.0 mmol), 10 HOBT (0.4189 g, 3.1 mmol), 11 EDAC (0.5943 g, 3.1 mmol). The solution was dissolved in 12 DMF (5 mL) and 13 3-Amino-9-ethylcarbazole 2 (0.6308 g, 3.0 mmol) was added. After 15 min, 14 Et3N (0.859 mL, 6.0 mmol) was added and the mixture stirred at room temperature for 16 h. After completion of the reaction, 15 water was added (30 mL) and the product was extracted using 16 dichloromethane (30 mL). The organic layer was washed with brine and dried with Na2SO4, filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and the 17 product obtained as a white solid for the precursor 2-chloro-N-(9-ethyl-9H-carbazol-3-yl)nicotinamide (0.7565 g, 2.2 mmol, 73%). TLC analysis in CH2Cl2-MeOH (9:1). Rf=0.61. 1H NMR (400 MHz, CDCl3) delta 1.46 (3H, t, J=7.3 Hz), 4.40 (2H, q, J=7.1 Hz), 7.43, (1H, t, J=8.6 Hz), 7.51 (1H, t, J=7.6 Hz), 7.64 (1H, d, J=2.3 Hz), 7.66 (1H, d, J=2.0 Hz), 8.13 (1H, d, J=7.3 Hz), 8.26 (1H, d, J=2.0 Hz), 8.28 (1H, d, J=1.8 Hz), 8.38 (1H, s), 8.47 (1H, d, J=1.8 Hz), 8.53 (1H, d, J=2.0 Hz), 8.54 (1H, d, J=2.0 Hz); 13C NMR (100 MHz, CDCl3) 14.1, 38.0, 108.9, 113.6, 119.3, 121.1, 123.0, 123.2, 123.5, 126.4, 129.2, 132.0, 138.0, 140.3, 140.8, 147.4, 151.4, 162.9.
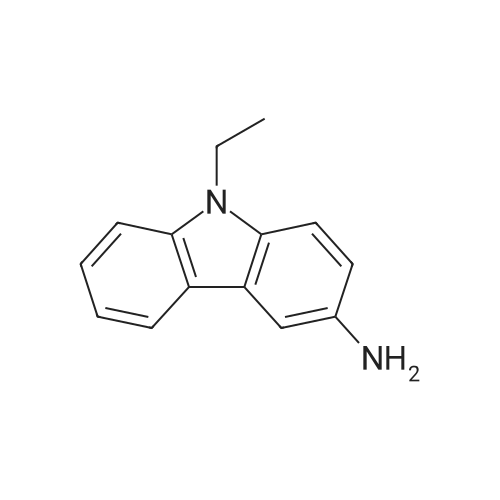
2-((9-ethyl-9H-carbazol-3-yl)amino)nicotinic acid[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
39%
With N-ethyl-N,N-diisopropylamine; In water; at 140℃; for 5h;Inert atmosphere; Microwave irradiation;
Method A. Step (i) A microwave tube was charged with 2-chloronicotinic acid 1 (0.1576 g, 1.0 mmol), <strong>[132-32-1]9-ethyl-9H-carbazol-3-amine</strong> 2 (0.6318 g, 3.0 mmol), DIPEA (0.522 mL, 3.0 mmol), and water (1.5 mL). The solution was heated at 140 C for 5 h under microwave conditions (Power level set to 200W; Caution Pressure may develop). After completion of the reaction, the mixture was allowed to cool to room temperature and transferred to a separatory funnel and diluted with dichloromethane (30 mL). The solution was washed with water (3 x 20 mL). The organic phase was separated and dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure. The crude oil product was purified via column chromatography over silica gel and the product intermediate 2-((9-ethyl-9H-carbazol-3-yl)amino)nicotinic acid was obtained as a white solid (0.1286g, 0.39 mmol, 39%). 1 H NMR (400 MHz, CDCl 3 ) delta 1.32 (t, 3H, J = 7.18 Hz), 4.44 (q, 2H, J = 6.98 Hz ), 6.81 (q, 1H, J = 4.8 Hz), 7.17 (t, 1H, J = 7.3 Hz), 7.44 (t, 1H, J = 8.6 Hz), 7.58 (q, 2H, J = 5.1 Hz), 7.66 (d, 1H, J = 1.8 Hz), 7.68 (d, 1H, J = 1.5 Hz), 8.13 (d, 1H, J = 7.8 Hz), 8.23 (d, 1H, J = 1.8 Hz), 8.25 (d, 1H, J = 2.0 Hz), 8.35 (d, 1H, J = 1.8 Hz), 8.38 (d, 1H, J = 2.0 Hz), 8.42 (s, 1H), 10.37 (s, 1H); 13 C NMR (100 MHz, CDCl 3 ) delta 13.7, 37.0, 106.8, 108.9, 109.0, 113.0, 113.1, 118.4, 120.4, 121.1, 122.1, 122.1, 125.2, 131.5, 136.1, 139.9, 140.4, 153.0, 156.4, 169.2. Step (ii) In a 50 mL three neck round bottom flask equipped with a reflux condenser was charged with 2-((9-ethyl-9H-carbazol-3-yl)amino)nicotinic acid (0.0634 g, 0.2 mmol), HOBt (0.0405 g, 0.2 mmol) and EDAC (0.0575 g, 0.3 mmol) in DMF (3 mL). To the solution was added 3-morpholinopropylamine (0.031 mL, 0.21 mmol). The solution is stirred at room temperature for 8 h until completed by TLC analysis. The reaction mixture is washed with water (10 mL) and extracted with ethyl acetate (3 X 20 mL). The combined organic phases are washed with brine and dried over anhydrous Na 2 SO 4 , filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and the product 4a was obtained as a white solid (0.0458 g, 0.1 mmol, 50%). TLC analysis in CH 2 Cl 2 -MeOH (9:1), R f =0.35. 1 H NMR (400 MHz, CDCl 3 ) delta 1.43 (3H, t, J = 7.6 Hz), 1.83 (3H, t, J = 5.8 Hz), 2.54 (3H, s), 2.60 (2H, t, J = 5.3 Hz), 3.60 (2H, q, J = 5.8 Hz), 3.74 (4H, t, J = 4.6 Hz), 4.37 (2H, q, J = 7.3 Hz), 6.66 (1H, q , J = 4.5 Hz), 7.19 (1H, t, J = 7.8 Hz), 7.36 (1H, d, J = 4.0 Hz), 7.39 (1H, d, J = 3.5 Hz), 7.44 (1H, t, J = 7.1 Hz), 7.69 (1H, d, J = 1.5 Hz), 7.71 (1H, d, J = 2.0 Hz), 7.74 (1H, d, J = 1.5 Hz), 7.76 (1H, d, J = 1.5 Hz), 8.08 (1H, d, J = 7.8 Hz), 8.31 (1H, d, J = 1.8 Hz), 8.32 (1H, d, J = 1.7 Hz), 8.35 (1H, d, J = 1.7 Hz), 10.53 (1H, s); 13 C NMR (100 MHz, CDCl 3 ) delta 13.9, 23.65, 4.56, 40.9, 53.9, 59.0, 67.0, 108.4, 111.8, 113.7, 118.3, 120.6, 121.2, 125.40, 131.90, 135.4, 136.7, 140.4, 151.6, 156.3, 168.4. GC-MS m/z (rel%): [M] 443 (54.8), [M-C 24 H 27 N 5 O] 401 (11.1), [M-C 13 H 9 N 3 ] 207 (100), [M-C 10 H 13 N 3 O] 191 (14.6).
39%
With N-ethyl-N,N-diisopropylamine; In water; at 140℃; for 5h;Microwave irradiation;
Step (i) A microwave tube was charged with 8 2-chloronicotinic acid 9 1 (0.1576 g, 1.0 mmol), 13 <strong>[132-32-1]9-ethyl-9H-carbazol-3-amine</strong> 2 (0.6318 g, 3.0 mmol), 20 DIPEA (0.522 mL, 3.0 mmol), and 15 water (1.5 mL). The solution was heated at 140 C. for 5 h under microwave conditions (Power level set to 200 W; Caution Pressure may develop). After completion of the reaction, the mixture was allowed to cool to room temperature and transferred to a separatory funnel and diluted with dichloromethane (30 mL). The solution was washed with water (3×20 mL). The organic phase was separated and dried over anhydrous Na2SO4, filtered and concentrated under reduced pressure. The crude oil product was purified via column chromatography over silica gel and the product intermediate 2-((9-ethyl-9H-carbazol-3-yl)amino)nicotinic acid was obtained as a white solid (0.1286 g, 0.39 mmol, 39%). 1H NMR (400 MHz, CDCl3) delta 1.32 (t, 3H, J=7.18 Hz), 4.44 (q, 2H, J=6.98 Hz), 6.81 (q, 1H, J=4.8 Hz), 7.17 (t, 1H, J=7.3 Hz), 7.44 (t, 1H, J=8.6 Hz), 7.58 (q, 2H, J=5.1 Hz), 7.66 (d, 1H, J=1.8 Hz), 7.68 (d, 1H, J=1.5 Hz), 8.13 (d, 1H, J=7.8 Hz), 8.23 (d, 1H, J=1.8 Hz), 8.25 (d, 1H, J=2.0 Hz), 8.35 (d, 1H, J=1.8 Hz), 8.38 (d, 1H, J=2.0 Hz), 8.42 (s, 1H), 10.37 (s, 1H); 13C NMR (100 MHz, CDCl3) delta 13.7, 37.0, 106.8, 108.9, 109.0, 113.0, 113.1, 118.4, 120.4, 121.1, 122.1, 122.1, 125.2, 131.5, 136.1, 139.9, 140.4, 153.0, 156.4, 169.2.
tert-butyl 4-(2-(2-chloronicotinamido)ethyl)piperazine-1-carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Method B. (i) In a 100 mL three-neck round-bottom flask was charged 2-chloronicotinic acid 1 (0.3151 g, 2.0 mmol), HOBt (0.4054 g, 3.0 mmol), and EDAC (0.5751 g, 3.0 mmol) were dissolved in CH 2 Cl 2 (10 mL). After stirring for 10 min, 4-(2-aminoethyl)-1-boc-piperazine (0.4586 mL, 2.0 mmol) was added followed by Et 3 N (0.5733 mL, 4.0 mmol) and the reaction mixture stirred at room temperature for 16 h. After completion of the reaction, the solution was washed with water (30 mL), and the product was extracted with CH 2 Cl 2 (3 X 20 mL). The combined organic phases were washed with brine, and dried over anhydrous Na 2 SO 4 , filtered, and concentrated under reduced pressure. The concentrated crude oil tert-butyl 4-(2-(2-chloronicotinamido)ethyl)piperazine-1-carboxylate intermediate was obtained (0.63 g, 1.71 mmol, 86%). The product was used in the next step without further purification. Step (ii) In a 100 mL three neck round bottom flask equipped with a stirring bar and reflux condenser, tert-butyl 4-(2-(2 chloronicotinamido)ethyl)piperazine-1-carboxylate (0.630 g, 1.71 mmol) and 9-ethyl-9H-carbazol-3-amine (0.0361 g, 1.72 mmol) were dissolved in 5 mL of DMSO. To the solution, CuI (0.0651 g, 0.3 mmol) and Cs 2 CO 3 (1.11 g, 3.42 mmol) were added and heated at 90 C for 24 hr. After the reaction was complete (analyzed by TLC), the mixture was allowed to reach room temperature. The mixture was washed with water (30 mL), and extracted with CH 2 Cl 2 (3 X 30 mL). The combined organic phases were washed with brine and dried over anhydrous Na 2 SO 4 , filtered, and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and the product 4b was obtained as a white solid (0.08g, 0.15 mmol, 9%). TLC analysis in CH 2 Cl 2 -MeOH (9:1), R f =0.44. 1 H NMR (400 MHz, CDCl 3 ) delta 1.26 (4H, t, J = 7.1 Hz), 1.42 (3H, t, J = 7.3 Hz), 1.46 (9H, s), 1.65 (3H, s), 2.04 (2H, s), 2.48 (3H, t, J = 4.6 Hz), 2.65 (2H, t, J = 5.9 Hz), 3.47 (4H, t, J = 5.3 Hz), 3.56 (2H, q, J = 5.6 Hz), 4.11 (1H, q, J = 7.1 Hz), 4.34 (2H, q, J = 7.6 Hz) 6.64(1H, q, J = 4.8 Hz), 7.19 (1H, t, J = 6.3 Hz), 7.26 (1H, s,), 7.35 (1H, d, J = 4.3 Hz), 7.39 (1H, d, J = 3.8 Hz), 7.43 (1H, t, J = 7.6 Hz), 7.65 (1H, d, J = 1.8 Hz), 7.67 (1H, t, J = 2.3 Hz), 7.69 (1H, d, J = 2.0 Hz), 8.08 (1H, d, 7.6 Hz), 8.31 (1H, d, J = 1.5 Hz), 8.32 (1H, d, J = 1.8 Hz), 8.33 (1H, d, J = 2.0 Hz), 10.40 (1H, s); 13 C NMR (100 MHz, CDCl 3 ) delta 13.8, 37.5, 42.2, 44.8, 45.5, 66.6, 108.3, 112.8, 115.3, 118.3, 120.5, 120.7, 122.8, 123.2, 125.4, 131.6, 132.8, 136.5, 132.0, 140.3, 147.6, 167.8. GC-MS m/z (rel%): [M] 542 (0.01), [M-C 17 H 19 N 3 O] 281 (67.5), [M-C 12 H 23 N 3 O 3 ] 257 (2.6), [M-C 11 H 17 N 3 O] 207 (100).
(i) In a 100 mL three-neck round-bottom flask was charged 8 2-chloronicotinic acid 9 1 (0.3151 g, 2.0 mmol), 10 HOBt (0.4054 g, 3.0 mmol), and 11 EDAC (0.5751 g, 3.0 mmol) were dissolved in 16 CH2Cl2 (10 mL). After stirring for 10 min, 40 4-(2-aminoethyl)-1-boc-piperazine (0.4586 mL, 2.0 mmol) was added followed by 14 Et3N (0.5733 mL, 4.0 mmol) and the reaction mixture stirred at room temperature for 16 h. After completion of the reaction, the solution was washed with water (30 mL), and the product was extracted with CH2Cl2 (3×20 mL). The combined organic phases were washed with brine, and dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The concentrated crude oil tert-butyl 4-(2-(2-chloronicotinamido)ethyl)piperazine-1-carboxylate intermediate was obtained (0.63 g, 1.71 mmol, 86percent). The product was used in the next step without further purification.
With potassium carbonate; In N,N-dimethyl-formamide; at 60 - 65℃; for 5h;
To a 500 ml four-necked flask equipped with a stirring, thermometer, and reflux condenser, 120 g of N,N-dimethylformamide, 15.7 g (0.1 mol) of 2-chloronicotinic acid, 30.0 g of potassium carbonate, 6.3 g were placed. (0.11 mol) of cyclopropylamine, stirred at 60-65 C for 5 hours, cooled to 20-25 C, and added 17.5 g (0.1 mol) of 2-chloro-3-nitro-4-methylpyridine (IV). Stir the reaction at 90-95 C for 5 hours, cool to 20-25 C, pour the residue into 300 g of ice water, adjust the pH of the system with 3.0% hydrochloric acid to 3.0, filter, filter cake with 30 g of water and 20 g of different Washed with propanol and dried to give 29.5 g of 2-[N-cyclopropyl-N-(3-nitro-4-methylpyridin-2-yl)]aminonicotinic acid (VI) in a yield of 93.9%. The phase purity was 99.6%.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping