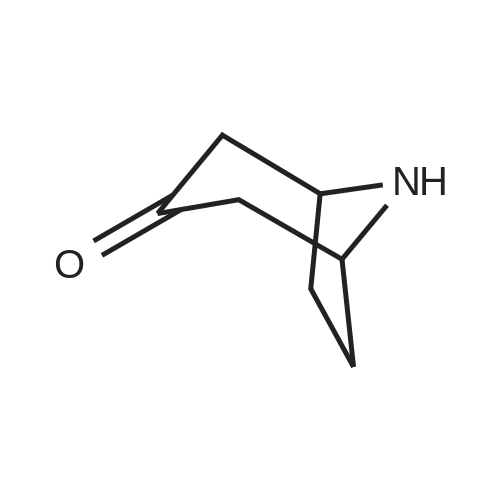
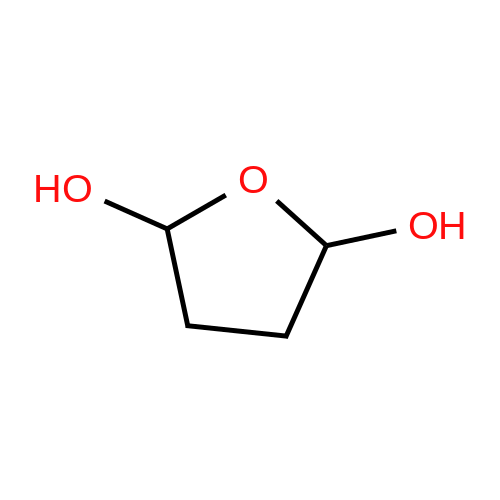
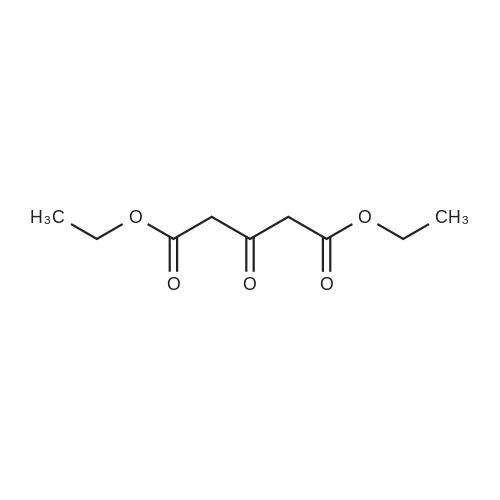
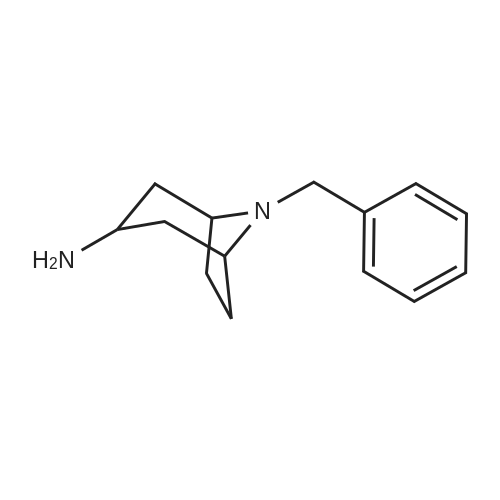
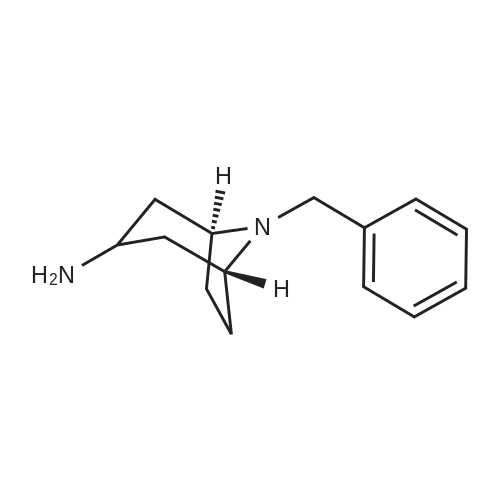
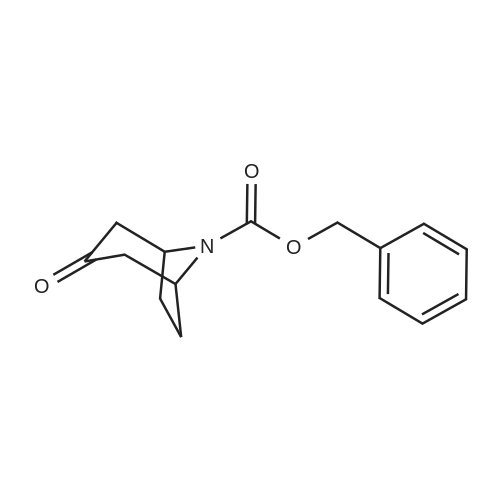
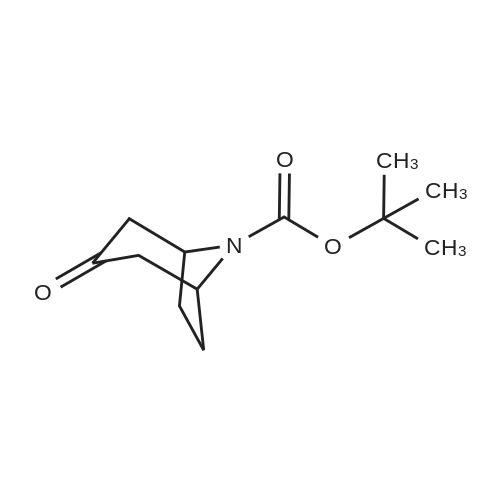
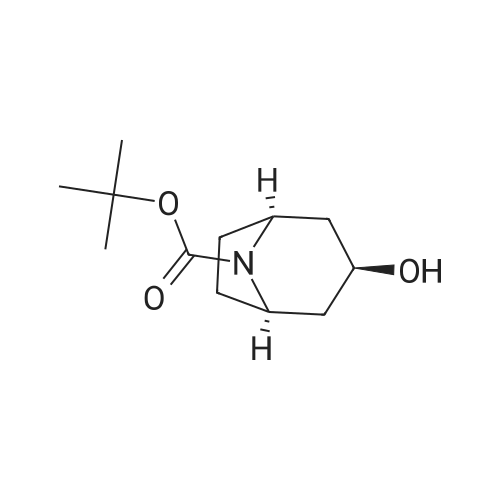
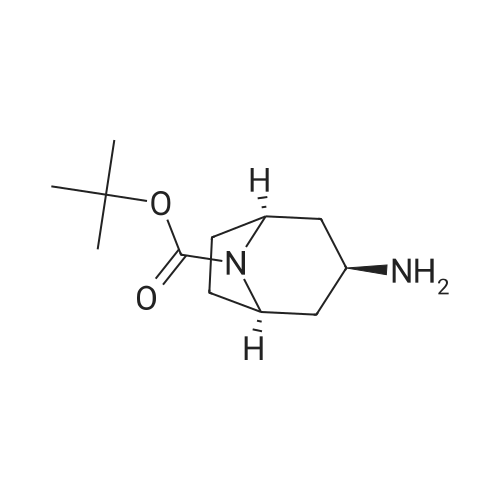
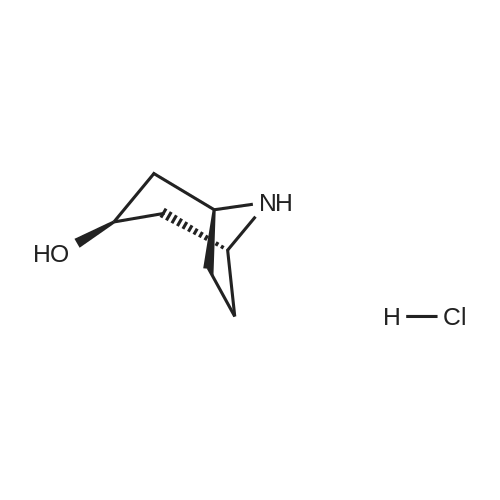
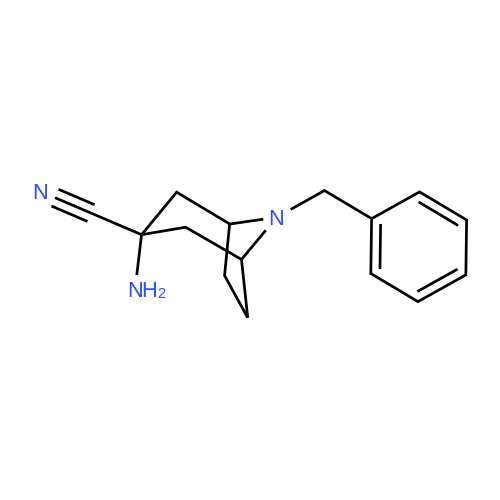
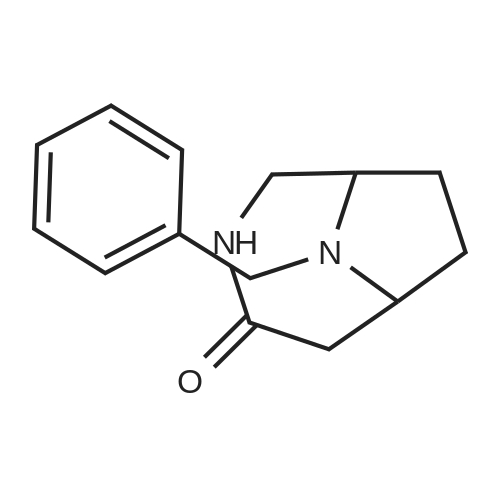
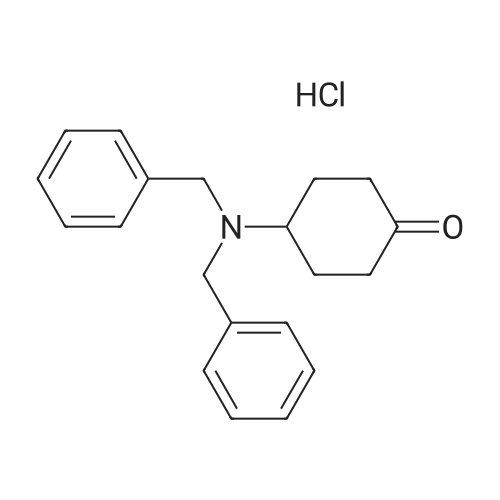
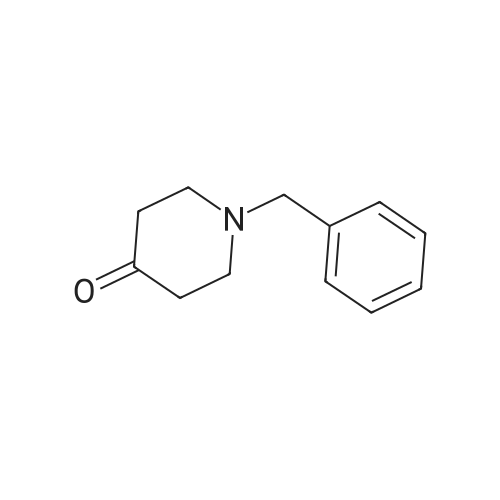
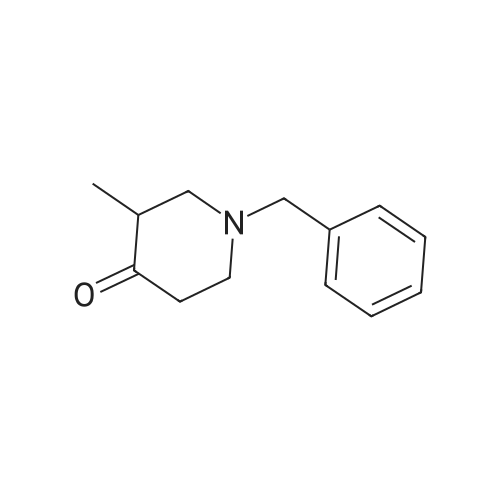
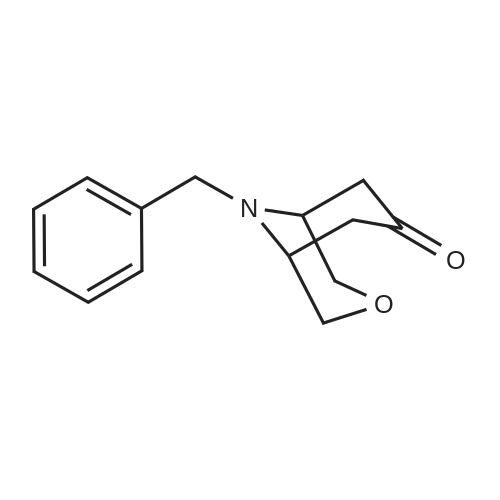
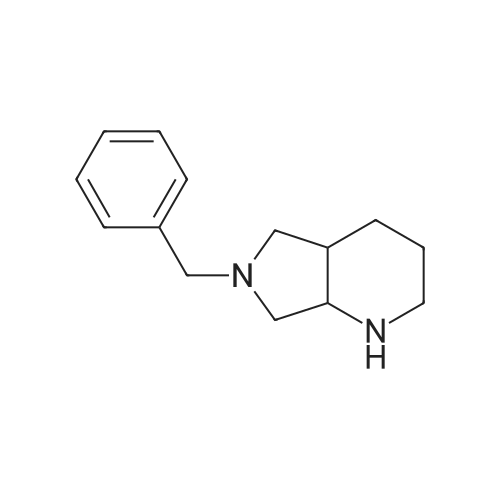
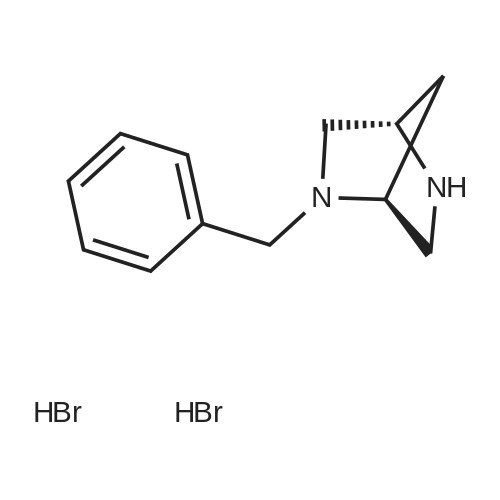
* All experimental methods are cited from the reference, please refer to the original source for details. We do not guarantee the accuracy of the content in the reference.
Stage #1: With hydrogenchloride In water at 0℃; for 0.333333 h; Stage #2: With sodium hydroxide; disodium hydrogenphosphate In water for 2 h;
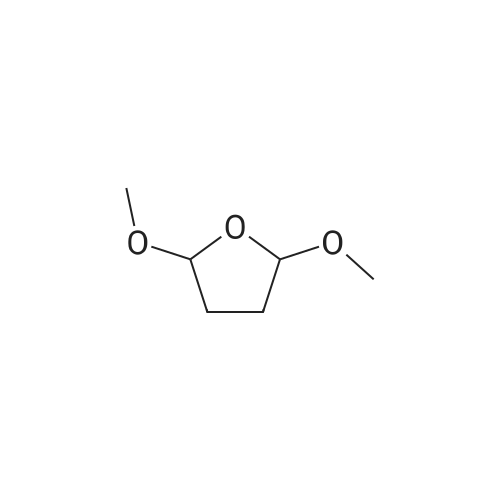
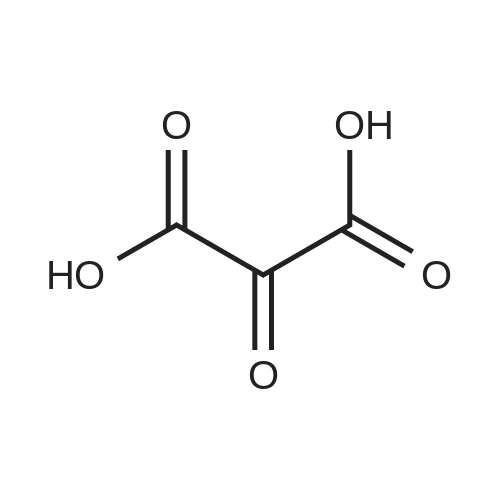
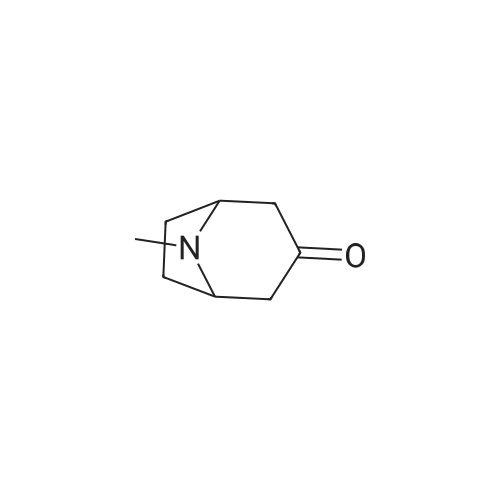
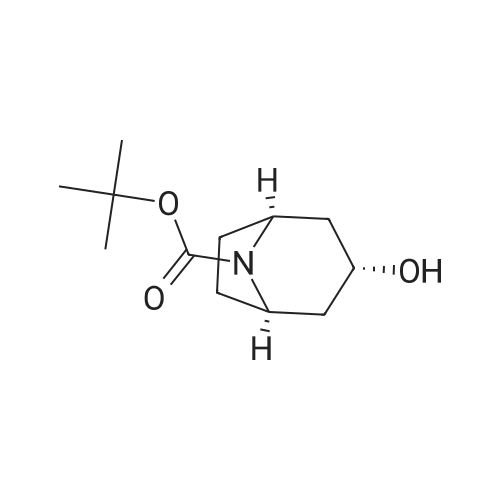
Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0° C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH ~4.5 using 40percent NaOH. The resulting cloudy and pale yellow solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50percent hydrochloric acid, heated to 85° C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40percent NaOH, and extracted with DCM (3*500 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to afford the title intermediate in quantitative yield as a viscous brown oil (~95percent purity based on analytical HPLC). 1H-NMR (CDCl3) δ (ppm) 7.5-7.2 (m, 5H, C6H5), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (mn, 2H, CH2CH2). (m/z): [M+H]+ calcd for C14H17NO 216.14; found, 216.0.
Stage #1: at 70℃; for 1 h; Stage #2: With hydrogenchloride; sodium acetate In water at 0 - 20℃;
Preparation of 8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one; A solution of 2,5-dimethoxy tetrahydro furan (15 g, 113 mmol) in 1N HCI (250 mL) was heated to 700C and maintained for 1 h. The reaction mixture was cooled to O0C, acetone di carboxylic acid (18.2 g, 125 mmol), conc.HCI (11 mL), sodium acetate (18.5 g, 136 mmol) and benzyl amine (14 mL, 125 mmol) were added sequentially, then the reaction <n="103"/>mixture was allowed to rt slowly and maintained over night. The reaction mass was filtered through celite bed, aqueous layer of the filtrate was basified with NaOH solution and extracted with EtOAc (2 x 400 ml_). The organic layer was dried over anhydrous sodium sulfate and concentrated to obtain the crude product as brown colored liquid (21.5 g, 88percent). 1HNMR(CDCI3): δ 7.25-7.5(m, 5H), 3.8(s, 2H), 3.54(s, 2H), 2.66-2.8(m, 2H), 2.02-2.3(m, 4H), 1.6-1.7(m, 2H). Mass: (M+1) 216 calculated for C14H17NO.
72%
Stage #1: With hydrogenchloride In water at 80℃; for 2 h; Stage #2: With hydrogenchloride; sodium acetate In water at 0 - 55℃; for 14 h; Stage #3: With sodium hydroxide In water at 20℃;
A solution of 2,5-dimethoxytetrahydrofuran (19.8 g, 149.8 mmol) in 0.1M hydrochloric acid (180 ml, 18.0 mmol) was stirred at 80°C for 2 hours. The reaction solution was cooled to 0°C, acetonedicarboxylic acid (19.8 g, 149.8 mmol), concentrated hydrochloric acid (13.8 ml, 164.8 mmol), sodium acetate (14.8 g, 179.8 mmol), and benzylamine (17.7 g, 164.8 mmol) were added to the reaction solution, and the mixture was stirred at the same temperature for 12 hours and at 55°C for 2 hours. The reaction solution was cooled to room temperature, and an aqueous solution of 4M sodium hydroxide (70 ml) was added, followed by extraction with chloroform. The organic layer was dried over sodium sulfate and then concentrated under a reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane/ethyl acetate) to give a title compound (23.3 g, 108 mmol, 72percent) as a light yellow oil product. 1H-NMR (400 MHz, CDCl3) δ:1.61-1.66 (2H, m), 2.10-2.13 (2H, m), 2.19-2.23 (2H, m), 2.67-2.72 (2H, m), 3.50 (2H, m), 3.75 (2H, s), 7.26-7.43 (5H, m). ESI-MS: m/z = 216 (M+H+).
61%
Stage #1: for 1 h; Reflux Stage #2: With sodium acetate In water at 0 - 50℃; for 6 h; Stage #3: With sodium hydroxide In water
Step 1: 8-benzyl-8-azabicyclo[3.2.1]octan-3-one A solution of 2,5-dimethoxytetrahydrofuran (2.2 mL) in hydrochloric acid (0.1N, 20 mL) was stirred under refluxing for 1 hour and then cooled to 0° C. 1,3-acetone-dicarboxylic acid (2.5 g), benzylamine (2.25 mL) and 10percent sodium acetate solution (10 mL) were added therein. The reaction mixture was stirred for 1 hour at room temperature, and additionally stirred for 5 hours at 50° C., and then cooled under ice bath. The reaction mixture was alkalized to pH 12 using a 2N sodium hydroxide solution. After layer-separated, the aqueous phase is diluted with ethyl acetate. The combined organic phase was washed with water, dried with anhydrous sodium sulfate, filtered and vaporized under reduced pressure. The concentrate was separated through column chromatography (petroleum ether/ethyl acetate=4/1, v/v) to obtain the product as brown oil (2239 mg, yield: 61percent). 1HNMR (CDCl3, 300 MHz) δ: 7.43-7.24 (m, 5H), 3.75 (s, 2H), 3.49-3.48 (m, 2H), 2.72-2.66 (m, 2H), 2.23 (s, 1H), 2.18-2.16 (m, 1H), 2.14-2.09 (m, 2H), 1.66-1.59 (m, 2H).
60%
Stage #1: With hydrogenchloride In water for 0.333333 h; Stage #2: With disodium hydrogenphosphate; sodium hydroxide In water at 0 - 20℃; Stage #3: With hydrogenchloride In water at 85℃; for 2 h;
Intermediate I-25 (8-benzyl-8-azabicyclo[3.2.1]octan-3-one)Concentrated hydrochloric acid (3 mL) was added to a stirred suspension of intermediate I-24 (8.2 g, 62 mmol, 1 eq) in water (17 mL), the reaction mixture was stirred for additional 20 minutes and diluted with water (25 mL). In a separate flask cooled to 0° C. on an ice bath, concentrated hydrochloric acid (9 mL) was added slowly to a solution of benzyl amine (10 g, 93 mmol, 1.5 eq) in water (35 mL), and this solution was added to the above solution of I-24. A solution of 1,3-acetonedicarboxylic acid (10 g, 68 mmol, 1.1 eq) in water (40 mL) was added followed by a solution of sodium hydrogen phosphate (4.4 g, 31 mmol, 5.0 eq) in water (20 mL). The acidity was adjusted from pH 1 to pH 4.5 using a solution of NaOH (40percent in water). The resulting cloudy and pale- yellow solution was stirred overnight at room temperature. The reaction mixture was acidified to pH 3 from pH 7.5 using aqueous HCl solution (50percent in water) and stirred at 85° C. for 2 hours. The crude reaction mixture was cooled to room temperature, basified to pH 12 using a solution of NaOH (40percent in water) and extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous MgSO4, the solids were removed by filtration and the filtrate was concentrated. The crude reaction product was purified by silica gel chromatography to give I-25 as a yellow oil (8.0 g, 60percent). 1H-NMR (CDCl3) δ : 7.59 (d, J=7.2 Hz, 2H), 7.31-7.40 (m, 3H), 4.04 (s, 2H), 3.75 (s, 2H), 3.13 (d, J=12.0 Hz, 2H), 2.22-2.30 (m, 4H), 1.75-1.80 (m, 2H). MS (ESI): ink 216 (M+H+).
Stage #1: With sodium acetate In water at 3 - 50℃; for 7.75 h; Stage #2: With hydrogenchloride In water at 20 - 25℃;
Under N2, a solution of benzylamine (36.1 ml, 0.33 mol) in water (435 ml) was added within 45 min at 3 to 8° C. to a solution of 1,4-butane dialdehyde (1.15 equiv.), 1,3-acetone dicarboxylic acid (1.5 equiv.) and sodium acetate trihydrate (2 equiv.) in 330 ml water as described in Example 1, Step (a). The mixture was warmed up to 50° C. over 5 h and kept at this temperature for 2 h. After cooling to 20-25° C., 80 ml conc. HCl was added and the solution was washed with TBME (2.x.240 ml). The pH of the aqueous phase was adjusted to 7-8 with NaOH and the product layer was separated. The aqueous phase was extracted with TBME (3.x. with a total of 240 ml). The combined product phases were dried over Na2SO4 and concentrated as completely as possible under vacuum at 500. Yield: 68.7 g (90percent abs.). Assay (HPLC): 93percent pure vs. standard.
Stage #1: With hydrogenchloride In water at 0℃; Stage #2: With sodium acetate In water at 0 - 50℃; for 3.66667 h; Stage #3: With sodium hydroxide In water at 0℃;
To a solution of 0.025 M aqueous hydrochloric acid (100 ml) at [0°C] was added 2, [5-DIMETHOXY-TETRAHYDROFURAN] (30 ml 231 [MMOL).] The reaction was stirred at [0°C] overnight. The reaction was then diluted with water (200 ml) and benzyl amine hydrochloride (40 grams, 278 [MMOL),] 3-oxo-pentanedioic acid (33.7 grams, 231 [MMOL),] and sodium acetate (10.7 grams, 130 [MMOL)] were added. The reaction was stirred for 5 minutes at [0°C,] warmed to ambient temperature and stirred for 90 minutes, then heated to [50°C] for two hours, cooled to [0°C] and basified to pH = 10 with 50 [percent] aqueous sodium hydroxide (14 [ML).] The reaction mixture was extracted with ethyl acetate (3 times) and the organic layers were combined and washed with a saturated sodium chloride solution, dried over magnesium sulfate, filtered and concentrated in vacuo to give a brown oil. Silica gel chromatography gave the title compound (33.46 grams, 67 percent yield
With triethylamine In tetrahydrofuran; methanol; ethyl acetate; 1,2-dichloro-ethane
8-Benzyl-8-Aza-Bicyclo[3.2.1]Octan-3-One To a stirred solution of 29.2 g (209 mmol) tropinone in 300 mL 1,2-dichloroethane was added 45.5 mL (419 mmol) 1-chloroethyl chloroformate, and the resulting solution was warmed to 80° C. The reaction was monitored by TLC on a SiO2 plate eluding with EtOAc/2M NH3:MeOH (5:1). After stirring for 18 h, the solvent was evaporated, 300 mL MeOH was added, and the reaction was heated to reflux. After 45 min, the solvent was evaporated, then 300 mL THF, 38.83 g (227 mmol) benzyl bromide, and 33 mL (24.0 g, 237 mmol) triethylamine was added, and the resulting mixture was stirred at 23° C. After 69 h, the mixture was transferred to a separatory funnel containing 200 mL sat. NaHCO3 solution. The aqueous layer was extracted with EtOAc (2*300 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4 filtered and evaporated to a brown oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2Cl2/ EtOAc (40:1 to 20:1 to 8:1 to 4:1). The appropriate fractions were combined and evaporated to afford 19.91 g (92 mmol, a 44percent yield) of the title compound as a yellow-orange oil. MS (ES) m/z: 216 (MH)+.
44%
With triethylamine In tetrahydrofuran; methanol; ethyl acetate; 1,2-dichloro-ethane
8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one To a stirred solution of 29.2 g (209 mmole) tropinone in 300 mL of 1,2-dichloroethane was added 45.5 mL (419 mmole) 1-chloroethyl chloroformate, and the resulting solution was warmed to 80° C. The reaction was monitored by thin layer chromatography on a silica gel plate eluding with EtOAc/2M NH3:MeOH (5:1). After stirring for 18 h, the solvent was evaporated, 300 mL MeOH was added, and the reaction was heated to reflux. After 45 min, the solvent was evaporated, then 300 mL THF, 38.83 g (227 mmol) benzyl bromide, and 33 mL (24.0 g, 237 mmol) triethylamine was added, and the resulting mixture was stirred at 23° C. After 69 h, the mixture was transferred to a separatory funnel containing 200 mL sat. NaHCO3 solution. The aqueous layer was extracted with EtOAc (2*300 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4 filtered and evaporated to a brown oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2Cl2/ EtOAc (40:1 to 20:1 to 8:1 to 4:1). The appropriate fractions were combined and evaporated to afford 19.91 g (92 mmol, a 44percent yield) of the title compound as a yellow-orange oil. MS (ES) m/z: 216 (MH)+.
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 7, p. 2917 - 2929
9
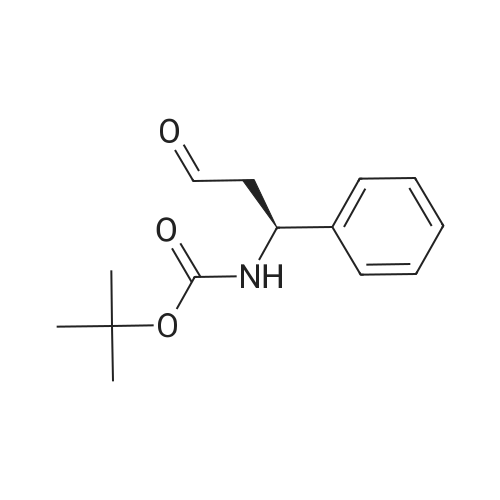
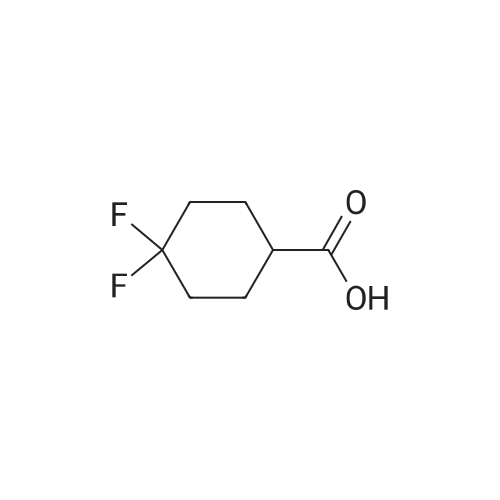
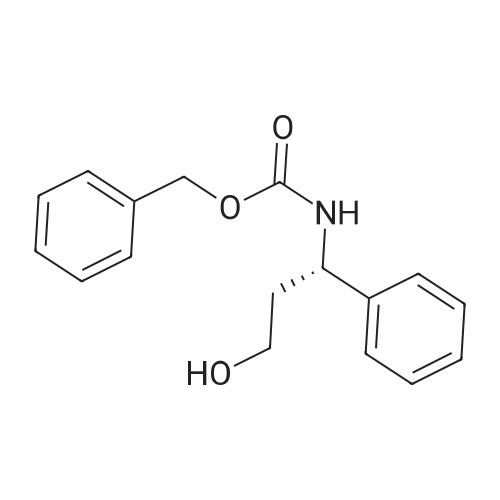
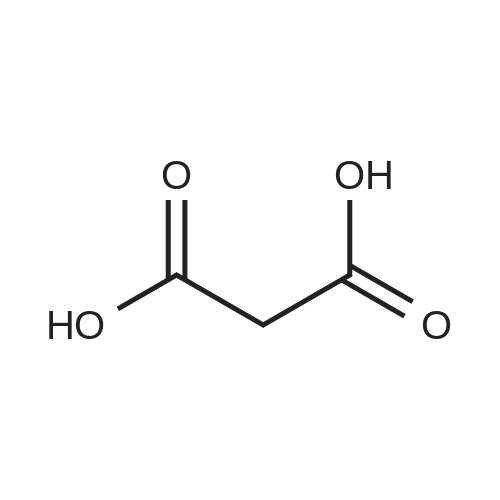
[ 542-05-2 ]
[ 6132-04-3 ]
[ 100-46-9 ]
[ 28957-72-4 ]
Yield
Reaction Conditions
Operation in experiment
44%
With hydrogenchloride In water
Following the procedure described in example 1, 57 ml of 2.5-dimethoxy-tetrahydrofuran (0.440 moles) in 64 ml of water and 0.12 ml of HCl are reacted with 62.9 g of acetonedicarboxylic acid (0.431 moles), 44 g of tribasic sodium citrate bihydrate (0.149 moles) in 64 ml of water, 25 g of ice and 48.15 g of benzylamine (0.450 moles) to give the required product. 41 g of 8-benzyl-8-azabicyclo-[3.2.1]-octan-3-one are obtained. Yield: 44percent with respect to the acetonedicarboxylic acid; boiling point: 135°-140° C. at 0.5 mmHg. Endo-8-benzyl-8-azabicyclo-[3.2.1]-octan-3-ol:
With hydrogen In di-<i>tert</i>-butyl dicarbonate; ethyl acetate
3-Oxo-8-aza-bicyclo[3.2.1]octane-8-carboxylic Acid Tert-Butyl Ester To a solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (33 g, 0.153 mmol) in ethyl acetate (100 ml) in a par bottle is added di-tert-butyl-dicarbonate (40.15 g, 0.184 mmol) and palladium hydroxide on carbon (20percent, 20 g). The reaction mixture is subjected to hydrogen gas (50 psi) at ambient temperature for 5 hours, filtered through a pad of celite, and the filter cake is washed with ethyl acetate. The filtrate is concentrated in vacuo and silica gel chromatography gave the title compound (29.37 g, 85percent).
Reference:
[1] Patent: US2002/119961, 2002, A1,
26
[ 28957-72-4 ]
[ 24424-99-5 ]
[ 143557-91-9 ]
Reference:
[1] Patent: US5968929, 1999, A,
27
[ 28957-72-4 ]
[ 194222-05-4 ]
[ 143557-91-9 ]
Reference:
[1] Patent: US2012/53180, 2012, A1,
28
[ 28957-72-4 ]
[ 423165-07-5 ]
Reference:
[1] Patent: US2011/251192, 2011, A1,
29
[ 28957-72-4 ]
[ 96901-92-7 ]
Reference:
[1] European Journal of Medicinal Chemistry, 2011, vol. 46, # 7, p. 2917 - 2929
[2] Bioorganic and Medicinal Chemistry, 2016, vol. 24, # 18, p. 3994 - 4007
30
[ 28957-72-4 ]
[ 744183-20-8 ]
Reference:
[1] Journal of Medicinal Chemistry, 2011, vol. 54, # 1, p. 67 - 77
Intermediate I-26 (9-benzyl-3,9-diazabicyclo[4.2.1]nonan-4-one)Intermediate I-25 (1.5 g, 7.0 mmol, 1.0 eq) was dissolved in chloroform (15 mL) at -5 C., and concentrated H2SO4 (3.5 mL) was added drop wise to maintain the reaction temperature below 5 C. Solid NaN3 (0.91 g, 13.9 mmol, 2.0 eq) was added carefully, and the mixture was stirred at 20 C. overnight and at 50 C. for an additional 2 hours. The reaction mixture was cooled to room temperature, and slurry of ice in water (12 mL) was slowly added. The reaction mixture was neutralized with solid NaOH to pH 7 and stirred overnight at 25 C. A solution of NaOH (4 mL, 4 M in water) was added, and the reaction mixture was extracted with dichloromethane. The combined organic layers were dried over anhydrous MgSO4, the solids were removed by filtration and the filtrate was concentrated by evaporation to give I-26 as a brown solid (1.3 g, 81%). 1H-NMR (DMSO-d6) delta : 7.24-7.39 (m, 5H), 3.58-3.46 (m, 3H), 3.31 (t, J=5.6 Hz, 1H), 3.24 (t, J=5.6 Hz, 1H), 2.82-2.92 (m, 2H), 2.46-2.52 (m, 2H), 2.03-2.17 (m, 2H), 1.83-1.87 (m, /H), 1.71-1.82 (m, /H). MS (ESI): m/z 231.0 (M+H+).
3-methyl-3,9-diazabicyclo[4.2.1]nonane. 3-Methyl-3,9-diazabicyclo[4.2.1]nonane was prepared by following the procedure described in reference: J. Org. Chem., 1960, 637. Conc. H2SO4 (3.25 ml, 0.061 mol) was added dropwise to a stirred cold (-5 C.) solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (2.153 g, 0.01 mol) in CHCl3 (15 ml) while keeping the temperature below 15 C. After cooling to 0-5 C., neat sodium azide (1.30 g, 0.020 mol) was added in small portions while maintaining pot temperature below 35 C. The mixture was stirred at rt overnight and then heated at 50 C. for 2 h. Rxn mixture was poured into ice and neutralized with Na2CO3 and then basify with 50% NaOH. Organic layer separated and the aq. layer re-extracted with CHCl3 (2×25 ml). Combined organic lyers were washed with water (10 ml), brine and dried (MgSO4). Evaporation of CHCl3 gave 9-benzyl-3,9-diazabicyclo[4.2.1]nonan-4-one as a beige semi-solid. LC/MS: m/e 231 (MH+).
A solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (7.28 g, 33.8 mmol) in dry THF (113 mL) was cooled to -78 C. under nitrogen. To this cold solution was added a 1.0 M solution of 3-methoxyphenyl magnesium bromide in THF (44 mL) via a dropping funnel. The resulting mixture was warmed to room temperature and stirred for about 20 minutes. The reaction was cooled to 0 C. and additional 3-methoxyphenyl magnesium bromide (30 mL, 30.0 mmol) in THF was added. The reaction was warmed to room temperature again after addition and stirred for 30 minutes. The reaction was quenched with saturated ammonium chloride and the product extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated. The residue was purified by column chromatography and eluted with 5% (200 mL), 10% (200 mL), 15% (200 mL), 20% (200 mL), 30% (200 mL), and 100% ethyl acetate/hexanes. Desired fractions were combined and concentrated to give the title intermediate as a light yellowish oil (4.0 g). Starting material (3.87 g) was recovered. (m/z): [M+H]+ calcd for C21H25NO2 324.20; found, 324.5.
8-benzyl-3-exo-(3-bromophenyl)-8-azabicyclo[3.2.1]octan-3-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a solution of 1,3-dibromobenzene (7.4 g, 31.3 mmol) in anhydrous THF (80 mL) at -78 C. under nitrogen was added a solution of 1.6M n-butyllithium in hexanes (20 mL, 31.4 mmol) dropwise. The resulting mixture was stirred at -78 C. for 30 minutes before a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (4.5 g, 20.9 mmol) in anhydrous THF (20 mL) was added dropwise. The reaction mixture was allowed to slowly warm to -40 C. over one hour and then to room temperature over 30 minutes. The reaction was quenched with saturated aqueous NH4Cl and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, concentrated, and further purified by flash chromatography. The product was eluted with 20% (300 mL) to 30% (600 mL) ethyl acetate/hexanes. Desired fractions were combined and concentrated to give the title intermediate as a yellowish oil (6.1 g). (m/z): [M+H]+ calcd for C20H22BrNO 372.10; found 372.3, 374.2 (isotope).
8-benzyl-3-exo-(3-methoxyphenyl)-8-azabicyclo[3.2.1]octan-3-ol acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
To a 3 L-3-necked flask fitted with an overhead stirrer and flushed with dry nitrogen was added cerous chloride powder (88.2 g, 0.35 mol). The solid was diluted with anhydrous tetrahydrofuran (500 mL) and cooled to 0 C. To the suspension was added 1M 3-methoxyphenyl magnesium bromide in THF (360 mL, 0.36 mol) dropwise while the temperature was maintained below 10 C. The resulting solution was stirred at 0 C. for 1.5 hours. A solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (54.5 g, 0.25 mol) in tetrahydrofuran (50 mL) was then added dropwise, while maintaining the internal temperature below 5 C. The resulting solution was stirred at 0 C. for 2 hours. The reaction was quenched with 10% aqueous acetic acid (400 mL) and stirred for 30 minutes at room temperature. Saturated sodium chloride solution (400 mL) was then added and the resulting suspension was stirred at room temperature for 20 hours to allow complete crystallization of product as the acetate salt. The crystals were filtered and washed with cold water (200 mL) followed by isopropyl acetate (200 mL) and dried under vacuum to give the title intermediate as a white crystalline powder (91.1 g, 93% yield). (m/z): [M+H]+ calcd for C21H25NO2 324.20; found, 324.5.
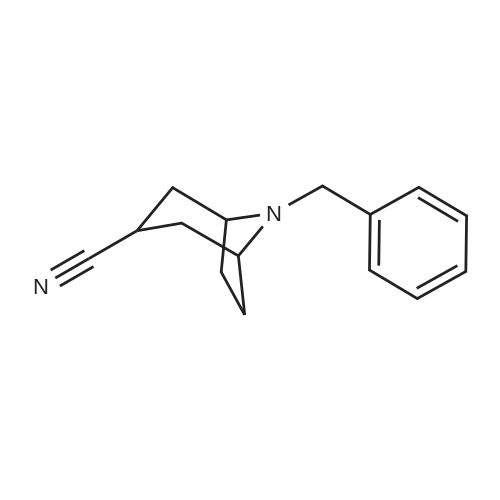
3β-amino-8-benzyl-8-aza-bicyclo[3.2.1]-octane-3-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
95%
With ammonium acetate; In methanol; at 20℃; for 28.5h;
Preparation 2; l-Isopropyl-3- (tetrahydropyran-4-ylmethyl) -bicyclo [3.2.1] - lbeta,3,8-triaza-spiro[4.5] dodecan-2-one; Step 1:; To 400 g (1.846 mol) of 8-benzyl-8-aza- bicyclo [3.2.1] octan-3-one dissolved in 2000 mL of methanol was added successively 1149 g (14.768 mol, 8 eq.) of ammonium acetate and 110 g (2.25 mol, 1.15 eq.) of sodium cyanide. The reaction was stirred for 28.5 hours at room temperature. Then, 2000 mL of DCM and 1000 mL of water were added successively. The mixture was stirred for 5 minutes. After separation of the organic layer, the aqueous layer was extracted with 2000 mL of DCM. The organic layers were dried over sodium sulfate, filtered and evaporated in vacuo to yield 450 g (95%) of pale yellow solid of Sbeta-ammo-S-benzyl-S-aza-bicyclo [3.2.1] -octane-3- carbonit?le and 8-benzyl-8-aza-bicyclo- [3.2.1] octan-3 -one, respectively, in a ratio 90:10 as determined by IH NMR.; Preparation 9; l-Isopropyl-3- (4-methoxy-phenyl) -bicyclo [3.2.1] -lbeta,3,8-t?aza- spiro [4.5] dodecan-2 , 4-dione; Step 1:; To 400 g (1.846 mol) of 8-benzyl-8-aza- bicyclo [3.2.1] octan-3-one m 2000 mL of methanol was added successively 1149 g (14.768 mol, 8 eq.) of ammonium acetate and 110 g (2.25 mol, 1.15 eq.) of sodium cyanide. The reaction was stirred for 28.5 hours at room temperature. Then, 2000 mL of DCM and 1000 mL of water was added successively. The mixture was stirred for 5 minutes. After separation of the organic layer, the aqueous layer was extracted with 2000 mL of DCM. The organic layers were dried over sodium sulfate, filtered and evaporated in vacuo to yield' 450 g (95%) of pale yellow solid containing 3beta-ammo-8 -benzyl -8 -aza-bicyclo [3.2.1] -octane-3 -carbonitrile and 8-benzyl-8-aza-bicyclo- [3.2.1] octan-3-one, respectively, m a ratio of 90:10 as determined by IH NMR.
With hydrogen;palladium hydroxide on carbon; In ethyl acetate; under 3102.97 Torr;
To a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83.6 g, 0.383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon, 50% wet with water; e.g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of H2 gas. The reaction solution was agitated for two days and recharged with H2 as needed to keep the H2 pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The black solution was then filtered through a pad of Celite and concentrated under reduced pressure to yield the title intermediate quantitatively as a viscous, yellow to orange oil. It was used in the next step without further treatment. 1H NMR (CDCl3) delta(ppm) 4.5 (broad, 2H, CH-NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2.3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1.6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
With hydrogen;palladium(II) hydroxide/carbon; In ethyl acetate; under 3102.97 Torr; for 48h;
b. Preparation of 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester To a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83.6 g, 0.383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon, ~50% wet with water; e.g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of H2 gas. The reaction solution was agitated for two days and recharged with H2 as needed to keep the H2 pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The solution was then filtered through a pad of Celite and concentrated under reduced pressure to yield the title intermediate quantitatively as a viscous, yellow to orange oil. It was used in the next step without further treatment. 1H NMR (CDCl3) delta(ppm) 4.5 (broad, 2H, CH-NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2.3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1.6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
With hydrogen;20% Pd(OH)2 on carbon; In ethyl acetate; under 3102.97 Torr; for 48h;Parr hydrogenation vessel;
To a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83.6 g, 0.383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon, 50% wet with water; e.g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of H2 gas. The reaction solution was agitated for two days and recharged with H2 as needed to keep the H2 pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The solution was then filtered through a pad of Celite and concentrated under reduced pressure to yield the title intermediate quantitatively as a viscous, yellow to orange oil. It was used in the next step without further treatment. 1H NMR (CDCl3) delta(ppm) 4.5 (broad, 2H, CH-NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2.3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1.6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
With hydrogen;palladium on carbon; In ethyl acetate; under 3102.97 Torr;
To a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83.6 g, 0.383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon, 50% wet with water; e.g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of H2 gas. The reaction solution was agitated for two days and recharged with H2 as needed to keep the H2 pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The solution was then filtered through a pad of Celite and concentrated under reduced pressure to yield the title intermediate quantitatively as a viscous, yellow to orange oil (51 g). It was used in the next step without further treatment. 1H NMR (CDCl3) delta(ppm) 4.5 (broad, 2H, CH-NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2.3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1.6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
With hydrogen;palladium(II) hydroxide/carbon; In ethyl acetate; under 3102.97 Torr; for 48h;
b. Preparation of 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester To a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83.6 g, 0.383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon, ~50% wet with water; e.g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of H2 gas. The reaction solution was agitated for two days and recharged with H2 as needed to keep the H2 pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The black solution was then filtered through a pad of Celite and concentrated under reduced pressure to provide the title intermediate as a viscous, yellow to orange oil. It was used in the next step without further treatment. 1H NMR (CDCl3) delta(ppm) 4.5 (broad, 2H, CH-NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2.3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1.6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
With sodium hydrogencarbonate;palladium-carbon; In ethyl acetate;
a. Preparation of 3-oxo-8-azabicyclo[3.2.1]octane-8-carboxylic acid tert-butyl ester To a 1 L hydrogenation vessel was added 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (86.1 g, 400 mmol), di-tert-butyl dicarbonate (98.2 g, 450 mmol), 10 wt% Pd/C (24 g, 11 mmol) and EtOAc (400 mL). The suspension was stirred and purged with nitrogen for 10 min. The reaction mixture was stirred under 50 psi hydrogen (3.4 atmospheres) at 20 C. for 28 h. The reaction mixture was then filtered through Celite. The wet solid cake was washed with EtOAc (100 mL). The filtrate and washes were combined and saturated NaHCO3/brine (1:1 mixture, 400 mL) was added. The EtOAc solution was separated, dried over Na2SO4, and concentrated to yield a light yellow sticky oil which solidified upon standing to give the title intermediate (93 g, quantitative yield). 1H NMR (CDCl3, 400 MHz): delta (ppm) 4.47 (s, br, 2H), 2.63 (s, br, 2H), 2.32 (d, J=16.4, 2H), 2.08 (m, 2H), 1.65 (t, J=8, 2H), 1.49 (s, 9H).
With hydrogen;palladium(II) hydroxide/carbon; In ethyl acetate; at 20℃; under 2017.7 Torr; for 4h;
Preparation 2 tert-Butyl 3-oxo-8-azabicyclo[3.2.1]octan-8-carboxylate A mixture of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (Preparation 1) (15.0 g, 69.7 mmol), di-tert-butyl dicarbonate (18.2 g, 83.4 mmol) and 20% w/w palladium hydroxide on carbon (3.0 g) in ethyl acetate (165 ml) was stirred for 4 hours at room temperature under an atmosphere of hydrogen at 269 KPa. The mixture was filtered through Arbocel and the solvent removed under reduced pressure.The residue was purified by column chromatography on silica gel using an elution gradient of hexane:ether (100:0 to 50:50) to afford the title compound as a colourless oil which crystallized on standing (1 6.2 g). 1H NMR (400 MHz, CDCl3): delta:1.48 (9H, s), 1.60-1.68 (2H, m), 2.00-2.11 (2H, m), 2.26-2.34 (2H, m), 2.48-2.82 (2H, m), 4.35-4.58 (2H, m) ppm.
With hydrogen;palladium dihydroxide; In ethyl acetate; under 3102.97 Torr; for 48h;
b. Preparation of 3-oxo-8-azabicyclo [3. 2. 1] octane-8-carboxvlic acid tert-butyl ester; To a solution of 8-benzyl-8-azabicyclo [3.2. 1] octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83. 6 g, 0. 383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon,-50% wet with water; e. g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of Ha gas. The reaction solution was agitated for two days and recharged with Ha as needed to keep the Ha pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The black solution was then filtered through a pad of CeliteNo. and concentrated under reduced pressure to yield the title intermediate quantitatively as a viscous, yellow to orange oil (51 g). It was used in the next step without further treatment. 1H NMR (CDC13) 6 (ppm) 4.5 (broad, 2H, CH- NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2. 3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1. 6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
With hydrogen;palladium hydroxide on carbon; In ethyl acetate; under 3102.97 Torr; for 48h;
To a solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (75 g, 0.348 mol) in EtOAc (300 mL) was added a solution of di-tert-butyl dicarbonate (83.6 g, 0.383 mol, 1.1 eq) in EtOAc (300 mL). The resulting solution and rinse (100 mL EtOAc) was added to a 1 L Parr hydrogenation vessel containing 23 g of palladium hydroxide (20 wt. % Pd, dry basis, on carbon, 50% wet with water; e.g. Pearlman's catalyst) under a stream of nitrogen. The reaction vessel was degassed (alternating vacuum and N2 five times) and pressurized to 60 psi of H2 gas. The reaction solution was agitated for two days and recharged with H2 as needed to keep the H2 pressure at 60 psi until the reaction was complete as monitored by silica thin layer chromatography. The black solution was then filtered through a pad of Celite and concentrated under reduced pressure to provide the title intermediate as a viscous, yellow to orange oil which was used in the next step without further treatment. 1H NMR (CDCl3) delta(ppm) 4.5 (broad, 2H, CH-NBoc), 2.7 (broad, 2H, CH2CO), 2.4-2.3 (dd, 2H, CH2CH2), 2.1 (broad m, 2H, CH2CO), 1.7-1.6 (dd, 2H, CH2CH2), 1.5 (s, 9H, (CH3)3COCON)).
Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0 C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH ~4.5 using 40% NaOH. The resulting cloudy and pale yellow solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with DCM (3*500 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to afford the title intermediate in quantitative yield as a viscous brown oil (~95% purity based on analytical HPLC). 1H-NMR (CDCl3) delta (ppm) 7.5-7.2 (m, 5H, C6H5), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (mn, 2H, CH2CH2). (m/z): [M+H]+ calcd for C14H17NO 216.14; found, 216.0.
Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0 C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH4.5 using 40% NaOH. The resulting cloudy and pale yellow solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with DCM (3×500 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to produce the crude title intermediate as a viscous brown oil (52 g). To a solution of the crude intermediate in methanol (1000 mL) was added di-tert-butyl dicarbonate (74.6 g, 0.342 mol) at 0 C. The solution was allowed to warm to room temperature and stirred overnight. The methanol was removed under reduced pressure and the resulting oil was dissolved in dichloromethane (1000 mL). The intermediate was extracted into 1 M H3PO4 (1000 mL) and washed with dichloromethane (3×250 mL) The aqueous layer was basified to pH 12 using aqueous NaOH, and extracted with dichloromethane (3×500 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure to produce the title intermediate as a viscous, light brown oil. 1H-NMR (CDCl3) delta (ppm) 7.5-7.2 (m, 5H, C6H5), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (m, 2H, CH2CH2). (m/z): [M+H]+ calcd for C14H17NO 216.14; found, 216.0.
Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0 C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH 4.5 using 40% NaOH. The resulting cloudy and pale yellow solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with dichloromethane (3×500 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to produce the crude title intermediate as a viscous brown oil (52 g). To a solution of the crude intermediate in methanol (1000 mL) was added di-tert-butyl dicarbonate (74.6 g, 0.342 mol) at 0 C. The solution was allowed to warm to room temperature and stirred overnight. The methanol was removed under reduced pressure and the resulting oil was dissolved in dichloromethane (1000 mL). The intermediate was extracted into 1 M H3PO4 (1000 mL) and washed with dichloromethane (3×250 mL). The aqueous layer was basified to pH 12 using aqueous NaOH, and extracted with dichloromethane (3×500 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure to produce the title intermediate as a viscous, light brown oil. 1H-NMR (CDCl3) delta (ppm) 7.5-7.2 (m, 5H, C6H5), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (m, 2H, CH2CH2). (m/z): [M+H]+ C14H17NO 216.14; found, 216.0.
Method N Preparation of 3-(3-isopropyl-5-methyl-4H-1,2,4-triazol-4-yl)-exo-8-azabicyclo[3.2.1]octane Step 1: Preparation of 8-benzylbicyclo[3.2.1.]octan-3-one; A solution of 2,5-dimethoxytetrahydrofuran (22.2 ml) in 0.1M HCl was refluxed for 1 hour and then cooled to 0 C. 1,3-Acetonedicarboxylic acid (25 g), benzylamine (15.6 ml) and 10% sodium acetate (95 ml) were added in one portion and the resulting mixture was stirred at room temperature for 1 hour, followed by heating to 50 C. for 5 hours. The reaction mixture was cooled, basified with 2M sodium hydroxide and washed with water. The organics were dissolved in 1M hydrochloric acid and washed with dichloromethane. The aqueous layer was rebasified with 2M sodium hydroxide and extracted with ethyl acetate (3×100 ml). The organic extracts were dried and evaporated to dryness to give the title compound as a brown oil which was used without further purification. Yield 13.66 g, M+H 216.
A mixture of 2,5-dimethoxy tetrahydrofuran (20.5 g, 155.1 mmol), water (42.5 mL) and concentrated HCl (30 mL) was stirred at room temperature for about 30 minutes. To this stirring mixture was sequentially added water (62.5 mL), a premixed solution of benzylamine (17.9 mL, 162.9 mmol), water (87.5 mL) and concentrated HCl (23 mL), a premixed solution of 1,3-acetonedicarboxylic acid (25 g) and water (100 mL), and a solution of Na2HPO4 (16.5 g) in water (50 mL). The resulting mixture was adjusted to pH 4 to 5 with 40% aqueous NaOH and stirred overnight at room temperature. The mixture was acidified to pH 3 with concentrated HCl and heated to 85 C. for 3 hours. After cooling to room temperature, the mixture was basified with 20% aqueous NaOH, saturated with solid sodium chloride and extracted with dichloromethane. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated to give a dark oil which was purified by flash column chromatography. The product was eluted with 20% ethyl acetate/hexanes. Desired fractions were combined and concentrated to give the title intermediate as a yellowish oil (16.17 g).
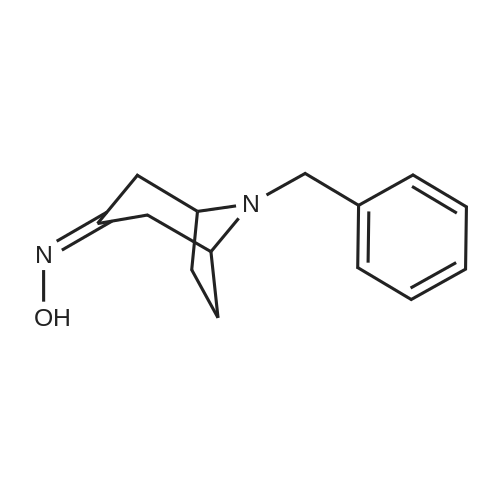
With hydroxylamine hydrochloride; sodium acetate; In methanol; at 20℃;
Preparation of 8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one oxime; To a solution of 8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one (10.5 g, 48.8 mmol) in methanol, sodium acetate (26.5 g, 195.3 mmol) and NH2OH-HCI (12.8 g, 185.5 mmol) were added sequentially at rt and maintained over night. The reaction mixture was concentrated to obtain a residue; residue was diluted with water (500 mL) and basified with 3N NaOH solution till pH was 8, resulted in the formation of white solid. The solid was filtered, washed with water (2 x 100 mL), hexane (2 x 100 mL) and dried to obtain crude product. The crude was recrystallised in ethanol to obtain product as white solid (7.4 g, 66%). 1HNMR(CDCI3): delta 8.5-8.7(broad, 1H), 7.25-7.45(m, 5H), 3.65(s, 2H), 3.3- 3.42(m, 2H), 2.98(d, 1H), 2.5-2.65(m, 1H), 2.1-2.32(m, 2H), 1.96-2.1(m, 2H) and 1.5- 1.7(m, 2H). LC-MS APCI (m/z) = 231 (M+1)+ .
65%
With hydroxylamine hydrochloride; sodium carbonate; In methanol; at 0 - 20℃; for 5h;
A solution of ethyl 3-oxo-8-azabicylo[3.2.1]octane-8-carboxylic acid (2.3 mmol, 0.46 g) in ethanol (1 ml) was mixed with KOH (0.353 g) in water (5 mL) and heated at 100 C for 3 h. After cooling down to room temperature, the solution was diluted with 20 mL of dichloromethane. The organic layer was dried and concentrated in vacuo to give crude 8-aza-bicyclo[3.2.1]octan-3-one (39). 39 was reacted with benzyl chloride as in Section 6.6 to give 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (40). To obtain the oxime 41, hydroxylamine hydrochloride (1.7 mmol, 0.117 g) was stirred in methanol (7 mL) at 0 C. The slurry was treated with Na2CO3 (0.09 g) and stirred for 5 min after which 40 (1.36 mmol, 0.29 g) in methanol (1 mL) was added. After stirring for 5 h at room temperature, methanol was removed in vacuo, the residue was treated with dichloromethane and brine, the organic fractions were combined, dried (anhydrous Na2SO4), and concentrated to give 0.2 g of the oxime 41 as a white solid (Yield 65%). 1H NMR (300 MHz, CDCl3) delta 1.52 (t, J = 9.6, 1H), 1.63 (t, J = 9.6, 1H), 2.04 (m, 2H), 2.14 (d, J = 14.7, 1H), 2.26 (dd, J1 = 2.7, J2 = 12.6), 2.62 (d, J = 14.7, 1H), 2.99 (d, J = 15.6, 1H), 3.36 (b, 2H), 3.66 (s, 2H), 7.25-7.42 (m, 5H). 13C NMR (75 MHz, CDCl3) delta 26.6, 27.5, 31.3, 37.1, 55.5, 57.8, 58.4, 127.0, 128.2 (2C), 128.7 (2C), 138.9, 156.3. MS(ESI) m/z [M + H+] 231.1. The oxime 41 was reduced with Adam?s catalyst as described in Section 6.6 to give the title compound 42 as a yellow oil (76%) which was not characterized and reacted immediately to give 19 (Section 6.3).
59%
With pyridine; hydroxylamine hydrochloride; In ethanol; for 18h;Reflux;
Step 2: 8-benzyl-8-azabicyclo[3.2.1]octan-3-one oxime A solution of the product from step 1 (1809 mg, 8.4 mmol), hydroxylamine chloride (642 mg, 9.25 mmol) and pyridine (0.72 mL) in ethanol (40 mL) was stirred under refluxing for 18 hour, then cooled to room temperature and vaporized off the solvent under reduced pressure. The residue was diluted with dichloromethane. The organic extracted phase was washed with water, dried with anhydrous sodium sulfate, filtered and vaporized under reduced pressure. The concentrate was separated through column chromatography (petroleum ether/ethyl acetate=3/1 to 1/1, v/v) to provide the product as a light-brown solid (1151 mg, yield: 59%). 1HNMR (CDCl3, 300 MHz) delta: 7.41-7.23 (m, 5H), 3.65 (s, 2H), 3.35 (s, 2H), 3.00-2.95 (d, 1H, J=15.9 Hz), 2.63-2.57 (dd, 1H, J=3.3 Hz, 11.7 Hz), 2.27-2.21 (dd, 1H, J=3.6 Hz, 12.0 Hz), 2.16-2.11 (d, 1H, J=14.4 Hz), 2.03-2.01 (m, 2H), 1.65-1.59 (m, 1H), 1.55-1.49 (m, 1H).
0.35 g
With hydroxylamine hydrochloride; sodium acetate; In ethanol; for 2h;Reflux;
The crude product of the previous step denture product N- benzyl-one, 0.19g (2.8mmol) hydroxylamine hydrochloride and 0.23g (2.8mmol) of anhydrous sodium acetate was dissolved in 15mL anhydrous ethanol, the reaction was heated at reflux for 2 hours, TLC tracking, end of the reaction, the solvent was removed, the reaction mixture was dissolved in 20mL (2.5M) sodium hydroxide aqueous solution, ethyl acetate (3 × 10mL) and the combined organic phase was dried over anhydrous magnesium sulfate, and the solvent removed under reduced pressure, flash column to give 0.35 g of the title compound purified by chromatography, 86% yield, as a yellow solid
With hydroxylamine hydrochloride; for 0.666667h;
660g (5 mol) of2,5-dimethoxytetrahydrofuran were heated at 80C in 6 L of 0.1 N HCl for 1h.After cooling to 10C, 803g (5.5mol)of acetone dicarboxylic acid and 460 ml of12N HCl and 492g (6 mol) of sodium acetate were added with stirring. Then 589g(5.5 mol) of benzylamine were introduced and stirred overnight. Insoluble materials were filtered off and (5.5 mol) ofhydroxylamine hydrochloride were added to the filtrate with stirring.After 40 min., the reaction mixture was cooledand pH was adjusted to 7-8 with aqueous KOH under vigorous stirring. Theprecipitated oxime (8) was collectedby filtration, washed with water and dried to give oxime (8) m.p. 124C.A solution of 2.1 mol of this oxime in 6L ofamyl alcohol was heated to 80C with stirring and 420g Na were carefully addedto keep on refluxing. The mixture was poured into 3L of water cooled to 0C.The aqueous layer was decanted, the organic layer was twice extracted withaqueous HCl. Basification with KOH and extraction with CH2Cl2 gave,after drying (Na2SO4) and evaporation compound 9, b.p. 100-115C (0.05mm). 1H NMR(CDCl3) 7.1-7.5 (m, 5, ArH), 3.5 (s, 2, CH2Ar), 3.15 (m,2, H1 and H5), 2.6-3.25 (m, 1, J=40 Hz, axial H3),1.1-2.2 (m, 10, NH2 and 4(CH2))
With hydroxylamine hydrochloride; sodium carbonate; In methanol; at 0 - 20℃; for 16h;Inert atmosphere;
A mixture of hydroxylamine hydrochloride (12.9 g, 181 mmol) in methanol (60 mL) at 0 C was treated with Na2CCh (13.4 g, 123 mmol) and stirred for 5 minutes. To the mixture was added a solution of intermediate Bl-2 (32 g, 145 mmol) in methanol (420 mL) and stirred under nitrogen at 20 C for 16 hours. The reaction mixture was concentrated under reduced pressure. The residue was treated with brine (200 mL) and extracted with dichloromethane (700 mL x 2). The organic extracts were combined, dried over anhydrous sodium sulfate, filtered, concentrated, and washed with ethyl acetate to afford intermediate Bl-3: LC-MS (ESI) m/z: 231 [M+H]+; 1H-NMR (CDCb, 400 MHz): d (ppm) 1.49-1.64 (m, 2H), 1.96-1.98 (m, 2H), 2.11-2.15 (m, 1H), 2.21-2.26 (m, 1H), 2.57-2.62 (m, 1H), 2.96-3.00 (m, 1H), 3.33-3.36 (m, 2H), 3.61-3.69 (s, 2H), 7.23-7.27 (m, 1H), 7.30-7.34 (m, 2H), 7.35- 7.41 (m, 2H), 9.26 (s, 1H).
With sodium tris(acetoxy)borohydride; acetic acid; In dichloromethane; at 20℃; for 96h;
To a solution of 8-benzyl-8-aza-bicyclo [3.2. 1] octan-3-one (3.09 grams, 14.35 [MMOL)] in [DICHLOROETHANE] was added 4-fluoro-phenyl amine (1.4 ml, 14.78 [MMOL),] acetic acid (1.2 ml, 20.96 [MMOL)] and sodium [TRIACETOXYBOROHYDRIDE] (4.64 grams, 21.89 [MMOL).] The reaction was stirred at ambient temperature for four days. The reaction was then quenched with 1 M aqueous sodium hydroxide and stirred for 10 minutes. The reaction mixture was extracted with [DICHLOROMETHANE] (2 times), the combined organic layers were dried over magnesium sulfate, filtered and concentrated in vacuo to give a yellow solid. Silica gel chromatography gave the title compound (3. 28 grams, 73 % yield).
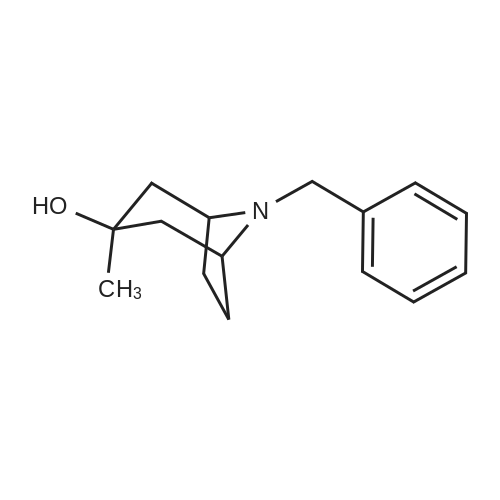
8-benzyl-3-methyl-8-azabicyclo[3.2.1]octan-3-exo-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
35%
In tetrahydrofuran; at -20 - 20℃; for 16.5h;
(i) 8-BENZVL-3-METHVL-8-AZABICYCLO [3. 2. 1LOCTAN-3-EXO-OL; To the solution of 16.00 g (74 mmole) of 8-BENZYL-8-AZABICYCLO [3.2. 1] octan-3-one (J. C. S. Perkin 1. 1997,1307) in 270 ml of dry tetrahydrofurane, under nitrogen atmosphere are dropped 38 ml (330 mmole) of methyl magnesium bromide, at-20 C with syringe. The reaction mixture is stirred AT-20C for 30 minutes, and then allowed to warm up to room temperature and stirring is continued for 16 hours. To the mixture 900 ml of saturated ammonium chloride solution and 300 ml of diethyl ether are added. After mixing and separation the aqueous phase is extracted with 3x 100 ml of dichloromethane, the dichloromethane phase is washed with 100 ml of saturated sodium chloride. The combined organic phase is dried over sodium sulphate and evaporated. The residue is chromatographed on silica gel (chloroform). The product is 5.95 g (35%) oily material. (MH+) = 232.
With ammonium formate;Pd/C; In ethanol; for 4h;Heating / reflux;
To a solution of 8-BENZYLBICYCLO [3.2. 1. ] octan-3-one (8.48g, Method W, step 1) in ethanol (100ML) was added 30 % palladium on carbon (850mg) followed by ammonium formate (8. 5G) and the resulting mixture was refluxed for 4 hours. The mixture was cooled, filtered and evaporated to dryness. The residue was dissolved in THF (50ML) and water (50ML) and d-tert-butyl dicarbonate (8.61g) was added. The resulting mixture was stirred at room temperature for 18 hours. The reaction mixture was concentrated and then partitioned between dichloromethane and 1M citric acid. The organic extracts were washed with saturated sodium bicarbonate, brine, dried and evaporated to dryness. The residue was purified by chromatography on silica eluting with ethyl acetate/isohexane (20: 80) to give the title compound as an oil which solidified on standing. Yield 4.43g. NMR CDCl3: 1.5 (s, 9H), 1.7 (m, 2H), 2.1 (m, 2H), 2.35 (m, 2H), 2.7 (m, 2H), 4.5 9m, 2H).
With pyridine; hydroxylamine hydrochloride; In ethanol; for 18h;Heating / reflux;
Step 2: Preparation of 8-benzylbicyclo[3.2.1.]octan-3-one-O-methyloxime; To a solution of 8-benzylbicyclo[3.2.1.]octan-3-one (13.66 g) in ethanol (250 ml) was added pyridine (5.69 ml) followed by hydroxylamine hydrochloride (4.85 g) and the resulting mixture was refluxed for 18 hours. The reaction mixture was allowed to cool to room temperature and then partitioned between water and dichloromethane. The organic layer was dried and evaporated to give the title compound as a brown solid which was used without further purification. Yield 10.79 g. M+H 231.
With pyridine; hydroxylamine hydrochloride; In ethanol; for 18h;Heating / reflux;
To a solution of 8-BENZYLBICYCLO [3.2. 1. ] octan-3-one (13.66g) in ethanol (250ML) was added pyridine (5. 69ML) followed by hydroxylamine hydrochloride (4.85g) and the resulting mixture was refluxed for 18 hours. The reaction mixture was allowed to cool to room temperature and then partitioned between water and dichloromethane. The organic layer was dried and evaporated to give the title compound as a brown solid which was used without further purification. Yield 10.79g. MS 231 MH+. Step 3: Preparation of 8-benzyl-8-azobicyclo [3.2. 1] octan-3-exo-amine A solution of 8-benzylbicyclo [3.2. 1. ] OCTAN-3-ONE-0-METHYLOXIME (27.78g) in pentanol (500ML) was heated to 165C. Sodium (LOG) was added portionwise over 6 hours. The reaction was heated for a further 4 hours and then cooled to 5 C. The reaction was acidified with 6M hydrochloric acid and the phases separated. The aqueous extracts were basified with sodium hydroxide and extracted with ethyl acetate (3X100 ML). The combined organic extracts were dried and evaporated to dryness to give the title compound as a pale brown oil. Yield 20. 21G. MS 217 MH+.
In tetrahydrofuran; diethyl ether; at -5 - 20℃; for 18h;
IP 9a. endo-8-benzyl-3-methyl-8-azabicyclo[3.2.1]octan-3-ol 16 mL (25.55 mmol) of MeLi (1.6 M in diethyl ether) was added to a solution of 5.00 g (23.22 mmol) of N-benzylnortropinone in 50 mL of THF under argon at -5 C. and the mixture was stirred for 2 hours at RT. Another 16 mL (25.55 mmol) of MeLi (1.6 M in diethyl ether) was added and the mixture was stirred for 16 hours at RT. The reaction mixture was combined with water while cooling with ice, the aqueous phase was extracted with EtOAc, the combined organic extracts were dried over sodium sulfate and evaporated down in vacuo. The crude product was purified by MPLC (Alox, DCM/EtOAc 9:1). Yield: 1.40 g (26% of theoretical); C15H2NO (M=231.333); calc.: molpeak (M+H)+: 232; found: molpeak (M+H)+: 232; Rf value: 0.33 (Alox, DCM/EtOAc 85:15).
In tetrahydrofuran; diethyl ether; at 40℃; for 3h;
IP 10a. endo-8-benzyl-3-ethyl-8-azabicyclo[3.2.1]octan-3-ol 15.5 mL (46.45 mmol) of ethylmagnesium bromide (3M in diethyl ether) was added dropwise to a solution of 5.00 g (23.22 mmol) of N-benzylnortropinone in 50 mL of THF under argon and the mixture was stirred for 3 hours at 40 C. The reaction mixture was combined with water while cooling with ice, the aqueous phase was extracted with EtOAc, and the combined organic extracts were dried over sodium sulfate and evaporated down in vacuo. The crude product was purified by MPLC (Alox, DCM/EtOAc 9:1). Yield: 2.90 g (51% of theoretical); C16H23NO (M=245.360); calc.: molpeak (M+H)+: 246; found: molpeak (M+H)+: 246.
Under N2, a solution of benzylamine (36.1 ml, 0.33 mol) in water (435 ml) was added within 45 min at 3 to 8 C. to a solution of 1,4-butane dialdehyde (1.15 equiv.), 1,3-acetone dicarboxylic acid (1.5 equiv.) and sodium acetate trihydrate (2 equiv.) in 330 ml water as described in Example 1, Step (a). The mixture was warmed up to 50 C. over 5 h and kept at this temperature for 2 h. After cooling to 20-25 C., 80 ml conc. HCl was added and the solution was washed with TBME (2×240 ml). The pH of the aqueous phase was adjusted to 7-8 with NaOH and the product layer was separated. The aqueous phase was extracted with TBME (3× with a total of 240 ml). The combined product phases were dried over Na2SO4 and concentrated as completely as possible under vacuum at 500. Yield: 68.7 g (90% abs.). Assay (HPLC): 93% pure vs. standard.
Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0 C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH 4.5 using 40% NaOH. The resulting cloudy and pale yellow solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with DCM (3×500 mL). The combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to afford the crude title intermediate as a viscous brown oil (52 g). To a solution of the crude intermediate in methanol (1000 mL) was added di-tert-butyl dicarbonate (74.6 g, 0.342 mol) at 0 C. The solution was allowed to warm to room temperature and stirred overnight. The methanol was removed under reduced pressure and the resulting oil was dissolved in dichloromethane (1000 mL). The intermediate was extracted into 1 M H3PO4 (1000 mL) and washed with dichloromethane (3×250 mL) The aqueous layer was basified to pH 12 using aqueous NaOH, and extracted with dichloromethane (3×500 mL). The combined organic layers were dried (MgSO4), filtered and concentrated under reduced pressure to produce the title intermediate as a viscous, light brown oil. 1H-NMR (CDCl3) delta (ppm) 7.5-7.2 (m,5H, C6H5),3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (m, 2H, CH2CH2). (m/z): [M+H]+ calcd for C1-4H17NO 216.14; found, 216.0.
With triethylamine; In tetrahydrofuran; methanol; ethyl acetate; 1,2-dichloro-ethane;
8-Benzyl-8-Aza-Bicyclo[3.2.1]Octan-3-One To a stirred solution of 29.2 g (209 mmol) tropinone in 300 mL 1,2-dichloroethane was added 45.5 mL (419 mmol) 1-chloroethyl chloroformate, and the resulting solution was warmed to 80 C. The reaction was monitored by TLC on a SiO2 plate eluding with EtOAc/2M NH3:MeOH (5:1). After stirring for 18 h, the solvent was evaporated, 300 mL MeOH was added, and the reaction was heated to reflux. After 45 min, the solvent was evaporated, then 300 mL THF, 38.83 g (227 mmol) benzyl bromide, and 33 mL (24.0 g, 237 mmol) triethylamine was added, and the resulting mixture was stirred at 23 C. After 69 h, the mixture was transferred to a separatory funnel containing 200 mL sat. NaHCO3 solution. The aqueous layer was extracted with EtOAc (2*300 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4 filtered and evaporated to a brown oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2Cl2/ EtOAc (40:1 to 20:1 to 8:1 to 4:1). The appropriate fractions were combined and evaporated to afford 19.91 g (92 mmol, a 44% yield) of the title compound as a yellow-orange oil. MS (ES) m/z: 216 (MH)+.
44%
With triethylamine; In tetrahydrofuran; methanol; ethyl acetate; 1,2-dichloro-ethane;
8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one To a stirred solution of 29.2 g (209 mmole) tropinone in 300 mL of 1,2-dichloroethane was added 45.5 mL (419 mmole) 1-chloroethyl chloroformate, and the resulting solution was warmed to 80 C. The reaction was monitored by thin layer chromatography on a silica gel plate eluding with EtOAc/2M NH3:MeOH (5:1). After stirring for 18 h, the solvent was evaporated, 300 mL MeOH was added, and the reaction was heated to reflux. After 45 min, the solvent was evaporated, then 300 mL THF, 38.83 g (227 mmol) benzyl bromide, and 33 mL (24.0 g, 237 mmol) triethylamine was added, and the resulting mixture was stirred at 23 C. After 69 h, the mixture was transferred to a separatory funnel containing 200 mL sat. NaHCO3 solution. The aqueous layer was extracted with EtOAc (2*300 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4 filtered and evaporated to a brown oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2Cl2/ EtOAc (40:1 to 20:1 to 8:1 to 4:1). The appropriate fractions were combined and evaporated to afford 19.91 g (92 mmol, a 44% yield) of the title compound as a yellow-orange oil. MS (ES) m/z: 216 (MH)+.
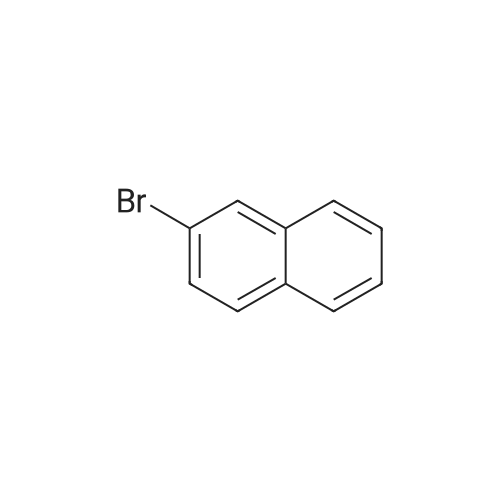
Step 2 8-Benzyl-3-Naphthalen-2-yl-8-Aza-Bicyclo[3.2.1]Octan-3-Ol To a -78 C. solution of 10.75 g (50.35 mmol) 2-bromonaphthalene in 200 mL THF was added 20.1 mL (50.25 mmol) of n-BuLi (2.5 M in hexanes) in drops over 5 min. After 35 min, a solution of 10.51 g (48.82 mmol) 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one in 25 mL THF was added via cannula, and then allowed to warm to room temperature. After 17 h, the mixture was transferred to a separatory funnel containing 200 mL brine. The aqueous layer was extracted with EtOAc (3*150 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4, filtered and evaporated to an orange oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2Cl2/EtOAc (40:1 to 20:1 to 8:1 to 4:1 to 2:1 to 1:1). The appropriate fractions were combined and evaporated to afford 7.07 g (20.6 mmol, a 42% yield) of the title compound as a yellow oil. MS (ES) m/z: 345 (MH)+.
Preparation 5 8-Benzyl-8-azabicyclo[3.2.1]octan-3-one oxime A mixture of the title compound from Preparation 4 (17.72 g, 82 mmol), hydroxylamine hydrochloride (5.72 g, 82 mmol) and pyridine (7.2 ml, 89 mmol), was heated under reflux in ethanol (500 ml) for 20 hours. The reaction was allowed to cool to room temperature and diluted with saturated aqueous sodium carbonate solution. The mixture was filtered and the filtrate evaporated under reduced pressure. The residue was partitioned between dichloromethane and water, the layers separated and.the aqueous layer extracted with dichloromethane (2*). The combined organic extracts were washed with brine, dried (MgSO4), filtered and evaporated under reduced pressure to afford the title compound as a pale brown solid, 18.10 g. 1H NMR (400 MHz, CDCl3): delta [ppm] 1.45-1.56 (1H, m), 1.60-1.67 (1H, m), 1.96-2.07 (2H, bm), 2.12 (1H, m), 2.21 (1H, m), 2.57 (1H, m), 2.97 (1H, m), 3.32 (2H, m), 3.64 (2H, s), 7.06 (1H, s), 7.21-7.28 (1H, m), 7.32 (2H, m), 7.38 (2H, d). LRMS: m/z 231.2 (MH+)
53
[ 28957-72-4 ]
[ 580-13-2 ]
[ 474534-35-5 ]
Yield
Reaction Conditions
Operation in experiment
42%
With n-butyllithium; In tetrahydrofuran; ethyl acetate;
8-Benzyl-3-naphthalen-2-yl-8-aza-bicyclo[3.2.1]octan-3-ol To a -78 C. solution of 10.75 g (50.35 mmol) 2-bromonaphthalene in 200 mL THF was added 20.1 mL (50.25 mmol) of n-BuLi (2.5 M in hexanes) in drops over 5 min. After 35 min, a solution of 10.51 g (48.82 mmol) 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one in 25 mL THF was added via cannula, and then allowed to warm to room temperature. After 17 h, the mixture was transferred to a separatory funnel containing 200 mL brine. The aqueous layer was extracted with EtOAc (3*150 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4, filtered and evaporated to an orange oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2Cl2/ EtOAc (40:1 to 20:1 to 8:1 to 4:1 to 2:1 to 1:1). The appropriate fractions were combined and evaporated to afford 7.07 g (20.6 mmol, a 42% yield) of the title compound as a yellow oil. MS (ES) m/z 345 (MH)+.
8-Benzyl-3-Naphthalen-2-yl-8-Aza-Bicyclo[3.2.1]octan-3-ol To a -78 C. solution of 10.75 g (50.35 mmol) 2-bromonaphthalene in 200 mL THF was added 20.1 mL (50.25 mmol) of n-BuLi (2.5 M in hexanes) in drops over 5 min. After 35 min, a solution of 10.51 g (48.82 mmol) 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one in 25 mL THF was added via cannula, and then allowed to warm to room temperature. After 17 h, the mixture was transferred to a separatory funnel containing 200 mL brine. The aqueous layer was extracted with EtOAc (3*150 mL), then the combined organics were washed with water (100 mL), brine (100 mL), dried over MgSO4, filtered and evaporated to an orange oil. The crude material was purified by flash chromatography on SiO2, using a gradient elution of CH2CI2/EtOAc (40:1 to 20:1 to 8:1 to 4:1 to 2:1 to 1:1). The appropriate fractions were combined and evaporated to afford 7.07 g (20.6 mmol, a 42% yield) of the title compound as a yellow oil. MS (ES) m/z: 345 (MH)+.
tert-butyl 4-(8-benzyl-8-azabicyclo[3.2.1]oct-3-yl)-1-piperazine carboxylate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
14.9 g (45%/o)
With sodium tetrahydroborate;Ti(OiPr)4;
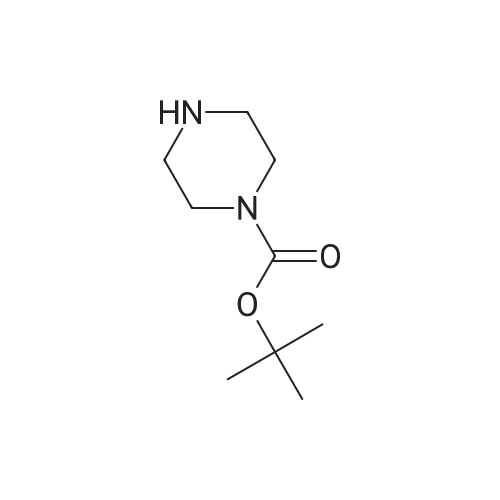
A. tert-Butyl 4-(8-benzyl-8-azabicyclo[3.2.1]oct-3-yl)-1-piperazine carboxylate To a mixture of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (11.9 g, 858 mmol) and tert-butoxycarbonyl piperazine (17.6 g, 943 mmol) was added Ti(OiPr)4 (34 ml, 1.3 mol) at room temperature and the resulting mixure was stirred for 3 h. The mixture was diluted with MeOH (200 ml) and then to this solution was added NaBH4 portionwise at 0 C. The mixture was allowed to warm to room temperature with stirring for 30 min. The reaction was quenched by the addition of sat. NaHCO3 aq. The resulting mixture was filtered through a pad of celite and the filter cake was washed with CH2Cl2. The combined whole mixture was partitioned and the organic layer was separated. The organic layer was washed with brine, dried (MgSO4) and concentrated in vacuo. The residue was purified by column chromatography (SiO2, CH2Cl2/MeOH=10/1) to give 14.9 g (45%/o) of tert-butyl 4-(8-benzyl-8-azabicyclo[3.2.1]oct-3-yl)-1-piperazine carboxylate as a yellow oil. 1H NMR (CDCl3) delta 7.39-7.18 (m, 5H), 3.57 (s, 2H), 3.42 (t, J=4.9 Hz, 4H), 3.24 (br.s, 2H), 2.55-2.43 (m, 5H), 2.00 (t, J=4.3 Hz, 2H),1.75-1.51 (m, 6H), 1.45 (s, 9H).
Preparation 4 8-Benzyl-8-azabicyclo[3.2.1]octan-3-one A solution of 2,5-dimethoxytetrahydrofuran (50 g, 378 mmol) in 0.025 M aqueous hydrochloric acid (160 ml) was cooled to 0 C. and stirred for 16 hours. Benzylamine hydrochloride (65 g, 453 mmol), ketomalonic acid (55 g, 377 mmol) and an aqueous solution of sodium acetate (300 ml, 0.69 M) were added and the reaction stirred at room temperature for 1 hour. The mixture was heated to 50 C. for a further 90 minutes then cooled in an ice bath and basified to pH12 with 2 N aqueous sodium hydroxide solution. The layers were separated, and the aqueous phase extracted with ethyl acetate (3*). The combined organic solutions were washed with water, dried (MgSO4), filtered and evaporated under reduced pressure. The residual brown oil was distilled under reduced pressure (126 C./0.4 kPa) to afford the title compound as an off-white solid, 37.81 g. 1H NMR (400 MHz, CDCl3): delta [ppm] 1.64 (2H, m), 2.06-2.14 (2H, m), 2.18 (1 H, s), 2.23 (1H, s), 2.68 (1H, m), 2.72 (1H, m), 3.48 (2H, s), 3.73 (2H, s), 7.20-7.29 (1H, m), 7.32 (2H, m), 7.42 (2H, d). LRMS: m/z 216.3 (MH+).
Preparation 1 8-Benzyl-8-azabicyclo[3.2.1]octan-3-one A solution of 2,5-dimethoxytetrahydrofuran (50 g, 378 mmol) in hydrochloric acid (0.025 N, 160 ml) was cooled to 0 C. for 16 hours.benzylamine hydrochloride (65 g, 453 mmol), ketomalonic acid (55 g, 377 mmol) and an aqueous solution of sodium acetate (300 ml, 0.69 M) were added and the reaction stirred at room temperature for one hour.The mixture was heated to 50 C. for further 90 minutes, then cooled in an ice bath and basified to PH12 with 2N sodium hydroxide solution.The layers were separated and the aqueous phase extracted with ethyl acetate (3*300 ml).The combined organic extracts were washed with water, dried (MgSO4), filtered and evaporated under reduced pressure.The residual brown oil was distilled under reduced pressure (126/3 MmHg) to afford the title compound as an off-white solid (37.81 g). 1H NMR (400 MHz, CDCl3): delta: 1.64 (2H, m), 2.06-2.14 (2H, m), 2.18 (1H, s), 2.23 (1H, s), 2.68 (1H, m), 2.72 (1H, m), 3.48 (2H, s), 3.73 (2H, s), 7.20-7.29 (1H, m), 7.32 (2H, m), 7.42 (2H, d) ppm. LRMS: m/z 216.3 (MH+).
With hydrogen;palladium dihydroxide; In di-tert-butyl dicarbonate; ethyl acetate;
3-Oxo-8-aza-bicyclo[3.2.1]octane-8-carboxylic Acid Tert-Butyl Ester To a solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (33 g, 0.153 mmol) in ethyl acetate (100 ml) in a par bottle is added di-tert-butyl-dicarbonate (40.15 g, 0.184 mmol) and palladium hydroxide on carbon (20%, 20 g). The reaction mixture is subjected to hydrogen gas (50 psi) at ambient temperature for 5 hours, filtered through a pad of celite, and the filter cake is washed with ethyl acetate. The filtrate is concentrated in vacuo and silica gel chromatography gave the title compound (29.37 g, 85%).
A. 8-Benzyl-8-azabicyclo[3.2.1]octan-3-one This was prepared according to the reported method (Chem. Abs., 1958, 53, 432e). A mixture of 2,5-dimethoxytetrahydrofuran (13 ml, 0.1 mol), 2 drops of c-HCl and water (10 ml) was stirred at room temperature for 1 h, then treated with water(10 ml), acetone-1,3-dicarboxylic acid (14.6 g, 0.1 mol), and benzylamine (10.9 ml, 0.1 mol). After the initial foaming had subsided, the pH was adjusted to 5 with 1N HCl, and stirring was continued for 14 h. The reaction mixture was adjusted to pH 1, washed with ethyl acetate, filtered through celite, and then adjusted to pH 12 with NaOH, and extracted with ethyl acetate. The extract was dried over Na2 SO4 and concentrated to afford 14.5 g (67% crude) of an oil, which was used for next reaction without further purification. 1 H NMR (CDCl3) delta 1.60 (m, 2H), 2.0-2.2 (m, 4H), 2.65 (m, 2H), 3.46 (br. s, 2H), 3.71 (s, 2H), 7.2-7.4 (m, 5H).
2-(3,4-dichlorophenyl)-4-(3,5-dimethylbenzoyl)-1-[[[8-(phenylmethyl)-8-azabicyclo[3.2.1]oct-3-yl]amino]acetyl]piperazine[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
EXAMPLE 18 Preparation of 2-(3,4-dichlorophenyl)-4-(3,5-dimethylbenzoyl)-1-[[[8-(phenylmethyl)-8-azabicyclo[3.2.1]oct-3-yl]amino]acetyl]piperazine STR121 By an analogous method to that described in Example 16, the product from Example 16, compound 3 (185 mg, 0.44 mmol) was combined with 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (97 mg, 0.45 mmol) and Ti(O-i Pr)4 (105 mL, 0.50 mmol) and left stirring for 1 h. To the thick reaction mixture was added NaBH3 CN (59.5 mg, 0.95 mmol) and the mixture was stirred overnight. To the reaction mixture was added H2 O (1 mL) and it was filtered. The filtrate was washed with EtOH, concentrated and purified by silica gel chromatography, eluding with 30:1:0.1 to 15:1:0.1 CH2 Cl2 --MeOH--NH3 aq. to give the title product as a white foam. HRMS (FAB, M+H+); m/e calc'd ?C35 H41 Cl2 N4 O2!+: 619.2607, found 619.2594.
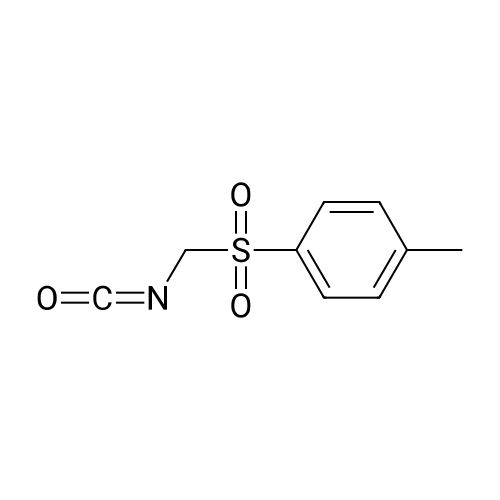
1!octan-3-one (11.2 g). Potassium t-butoxide (2.5 g) was added portionwise to a stirred mixture of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (2.0 g), tosylmethyl isocyanide (2.36 g) and ethanol (2 ml) in dimethoxyethane (50 ml) at 0 C. under nitrogen. The mixture was stirred at 0 C. for 0.5 hours and then overnight at room ternperature. The mixture was then filtered and the solid residue washed with dimethoxyethane. The combined filtrates were evaporated under reduced pressure and chromatographed ?SiO2; hexane:ethyl acetate[(80:20)] to give 3-cyano-8-benzyl-8-azabicyclo[3.2.1]octane (0.87 g).
With sodium tetrahydroborate;Pd(OH)2; In methanol; dichloromethane; ethyl acetate;
EXAMPLE 52 1,1-Dimethylethyl 3-exo-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate (3) and 1,1-dimethylethyl 3-endo-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate (4) STR209 To a solution of N-benzyl-3-oxo-8-azabicyclo[3.2.1]-octane (20 g, 93 mmol) in EtOAc (220 mL) were added t-BOC anhydride (24.2 g, 112 mmol) and 20% Pd(OH)2 /C (4 g). The mixture was hydrogenolyzed at 38.5 psi. After the reaction was complete, catalyst was filtered and filtrate was evaporated to give a solid crude product (21 g). The crude material (19 g, 84 mmol) was dissolved in CH3 OH (100 mL) and NaBH4 (4.8 g, 127 mmol) was added portion wise at 0 C. The reaction was stirred at 0 C. and gradually warmed to RT. After 3 h, the reaction was quenched with acetic acid (8 mL) and CH3 OH was evaporated. The residue was redissolved in CH2 Cl2 (300 mL) and washed with saturated NaHCO3 solution, dried (Na2 SO4), filtered and evaporated to give a solid.
With sodium tris(acetoxy)borohydride; sodium hydrogencarbonate; In tetrahydrofuran; acetic acid;
EXAMPLE XXXV endo-8-Benzyl-3-benzylamino-8-aza-bicyclo[3.2.1]octane 23 g of sodium triacetoxyborohydride are added in portions to a mixture of 17.8 g of <strong>[28957-72-4]N-<strong>[28957-72-4]benzyltropinone</strong></strong>, 8.9 g of benzylamine, 4.8 ml of glacial acetic acid and 300 ml of absolute tetrahydrofuran at room temperature, and the mixture is stirred for 12 hours. The solvent is then distilled off in a rotary evaporator, sodium bicarbonate solution is added to the residue, and the mixture is extracted three times with ethyl acetate. The combined organic phases are then dried over sodium sulphate, and the solvent is distilled off in a rotary evaporator. A 7 g portion of the total residue of 25 g is purified by column chromatography on silica gel with methylene chloride/methanol/concentrated ammonia (30:1:0.1). Yield: 3.1 g (43% of theory), Rf: 0.31 (silica gel; methylene chloride/methanol/concentrated ammonia=9:1:0.1); Melting point: 49-51 C. The following compound is obtained in analogy to Example XXXV:
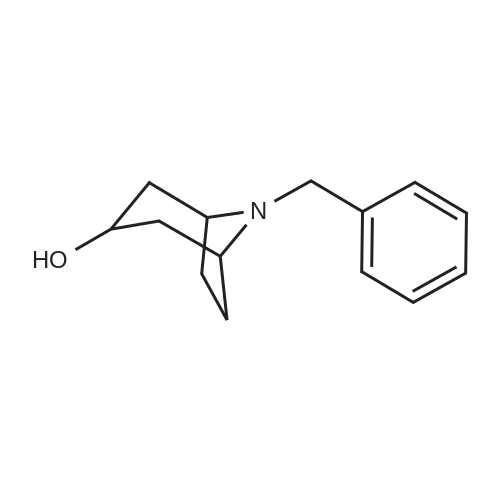
8b) 8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-olTo a solution of ketone (8a) (16.8 g) in THF (95 ml) cooled to -78C, was added L-selectride (94 ml). The reaction mixture wasstirred for 90 minutes, warmed to room temperature and stirredfor 1 hour at room temperature. The mixture then was cooled to0C, 20% sodium hydroxide solution (81 ml) were added, then 30%hydrogen peroxide (41 ml) and stirred for 1 hour at roomtemperature. The aqueous layer was extracted three times withdichloromethane. The combined organic layers were washed withbrine, dried over magnesium sulfate, filtered and evaporated.The crude material was purified by flash chromatography (silicagel, dichloromethane/(methanol/ammonia 9:1) 19:1,dichloromethane/methanol 9:1 + 1% ammonia) to give the desiredproduct (8.12 g).^-H NMR (300MHz, CDC13) : 6: 7.34-7.06 (m, 5H) , 4.05-3.95 (m, 1H) ,3.49 (s, 2H), 3.19-3.05 (m, 2H), 2.13-1.90 (m, 6H), 1.68-1.52(m, 2H), 1.38-1.21 (m, 1H)
8-benzyl-8-azabicyclo[3.2.1]octane-3-carbonitrile[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With potassium tert-butylate; In 1,2-dimethoxyethane; ethanol; at 0 - 50℃; for 5h;
To a solution of 332 mg of 8-benzyl-8-azabicyclo[3.2.1]oct-3-one in 9 ml of dimethoxyethane, 550 mg of tosylmethyl isocyanate, 0.25 ml of ethanol and potassium t-butoxide were successively added at 0 C., followed by 5 hours' stirring at 50 C. The reaction liquid was diluted with ethyl acetate, washed with saturated brine and dried over anhydrous sodium sulfate. Distilling the solvent off under reduced pressure, the resulting residue was purified on silica gel column chromatography (eluent: hexane/ethyl acetate=1/1, to provide 236 mg of the title compound
3-(endo-amino)-8-azabicyclo[3.2.1]octane dihydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
With hydrogenchloride; acetic acid;rhodium;
3-(Exo-amino)-8-azabicyclo[3.2.1]octane, dihydrochloride A mixture of 4.6 g (20 mmole) of 8-(phenylmethyl)-8-azabicyclo[3.2.1]octan-3-one, oxime [J. R. Bagley and T. N. Riley, J. Heterocyclic Chem., 19, 485 (1982)], 0.5 g of 10% rhodium on carbon, and 100 ml of acetic acid was hydrogenated until the requisite amount of hydrogen was taken up. The reaction mixture was filtered and two equivalents of HCl was added. The solid was filtered to yield 2.80 g of the title compound, mp >300 C.
With hydrogenchloride; acetic acid;rhodium;
EXAMPLE H 3-(Exo-amino)-8-azabicyclo[3.2.1]octane, dihydrochloride A mixture of 4.6 g (20 mmole) of 8-(phenylmethyl)-8-azabicyclo[3.2.1]octan-3-one, oxime [J. R. Bagley and T. N. Riley, J. Heterocyclic Chem., 19, 485 (1982)], 0.5 g of 10% rhodium on carbon, and 100 ml of acetic acid was hydrogenated until the requisite amount of hydrogen was taken up. The reaction mixture was filtered and two equivalents of HCl was added. The solid was filtered to yield 2.80 g of the title compound, mp>300 C.
With hydrogenchloride; acetic acid;rhodium;
EXAMPLE H 3-(Exo-amino)-8-azabicyclo[3.2.1]octane, dihydrochloride A mixture of 4.6 g (20 mmole) of 8-(phenylmethyl)-8-azabicyclo[3.2.1]octan-3-one, oxime [J. R. Bagley and T. N. Riley, J. Heterocyclic Chem., 19 , 485 (1982(], 0.5 g of 10% rhodium on carbon, and 100 ml of acetic acid was hydrogenated until the requisite amount of hydrogen was taken up. The reaction mixture was filtered and two equivalents of HCl was added. The solid was filtered to yield 2.80 g of the title compound, mp >300C.
Following the procedure described in example 1, 57 ml of 2.5-dimethoxy-tetrahydrofuran (0.440 moles) in 64 ml of water and 0.12 ml of HCl are reacted with 62.9 g of acetonedicarboxylic acid (0.431 moles), 44 g of tribasic sodium citrate bihydrate (0.149 moles) in 64 ml of water, 25 g of ice and 48.15 g of benzylamine (0.450 moles) to give the required product. 41 g of 8-benzyl-8-azabicyclo-[3.2.1]-octan-3-one are obtained. Yield: 44% with respect to the acetonedicarboxylic acid; boiling point: 135-140 C. at 0.5 mmHg. Endo-8-benzyl-8-azabicyclo-[3.2.1]-octan-3-ol:
Reference Example 14 8-Benzyl-8-azabicyclo[3.2.1] octan-3-one oxime Hydroxyamine hydrochloride (3.87 g) and sodium acetate (4.57 g) were added to 50 ml of methanol, and stirred for 30 min. Then, 8-benzyl-8-azabicyclo[3.2.1] octan-3-one (10 g) was added and stirred for 5 hours. The reaction mixture was concentrated, alkalized with potassium carbonate and water and then extracted with chloroform. The organic layer was washed with saturated brine, dried and concentrated. The resultant crude crystals were washed with n-hexane to provide white crystals (7.8 g).
With hydrogenchloride; sodium hydroxide; sulfuric acid; sodium acetate;
Reference Example 13 8-Benzyl-8-azabicyclo[3.2.1] octan-3-one 2,5-Dimethoxy tetrahydrofuran (25 g) was added to 227 ml of 0.1N hydrochloric acid, and stirred with heat at 80C for an hour. The mixture was cooled to 10C, to which acetone dicarboxylic acid(30.39 g), sodium acetate (18.61 g) and concentrated sulfuric acid (17.4 ml) were added and stirred. Further, benzylamine (22.28 g) was added dropwise to the resultant mixture, and stirred overnight at room temperature. The reaction mixture was alkalized with aqueous solution of sodium hydroxide, and then was extracted with chloroform. The organic layer was washed with water, dried and concentrated to provide the objective compound as brown-oil (41.4g).
With hydrogenchloride; sodium hydroxide; sulfuric acid; sodium acetate;
Reference Example 13 8-Benzyl-8-azabicyclo[3.2.1] octan-3-one 2,5-Dimethoxy tetrahydrofuran (25 g) was added to 227 ml of 0.1N hydrochloric acid, and stirred with heat at 80C for an hour. The mixture was cooled to 10C, to which acetone dicarboxylic acid (30.39 g), sodium acetate (18.61 g) and concentrated sulfuric acid (17.4 ml) were added and stirred. Further, benzylamine (22.28 g) was added dropwise to the resultant mixture, and stirred overnight at room temperature. The reaction mixture was alkalized with aqueous solution of sodium hydroxide, and then was extracted with chloroform. The organic layer was washed with water, dried and concentrated to provide the objective compound as brown oil (41.4 g).
Example 45 (+-)-4-(8-Azabicyclo[3.2.1]oct-2-en-3-yl)-2-(4-fluorophenyl)-3-(pyridin-4-yl)-1 H -pyrrole (Compound No. 1-2543) In a similar manner to the procedures described in Examples 9(i), 9(ii) and 9(iii) above, coupling, debenzylation, dehydration and desilylation reactions were carried out using 8-benzyl-8-azabicyclo[3.2.1]octane-3-one as a starting material instead of 1-benzylpiperidine-4-one to give the title compound (yield 35%) as a pale yellow powder. Melting point: 212 - 213C 1H-Nuclear magnetic resonance spectrum (500 MHz, DMSO-d6) delta ppm: 11.32 (1H, broad singlet); 8.45 (2H, doublet, J=6 Hz); 7.17-7.07 (6H, multiplet); 6.81 (1H, doublet, J=3 Hz); 5.45 (1H, doublet, J=6 Hz); 3.53-3.46 (1H, multiplet); 3.38-3.32 (1H, multiplet); 2.55-2.48 (1H, broad multiplet); 1.86-1.69 (3H, multiplet); 1.66-1.55 (1H, multiplet); 1.52-1.44 (1H, multiplet).
Preparation of 8-Benzyl-8-aza-bicyclo[3.2.1]octan-3-one; A solution of 2,5-dimethoxy tetrahydro furan (15 g, 113 mmol) in 1N HCI (250 mL) was heated to 700C and maintained for 1 h. The reaction mixture was cooled to O0C, acetone di carboxylic acid (18.2 g, 125 mmol), conc.HCI (11 mL), sodium acetate (18.5 g, 136 mmol) and benzyl amine (14 mL, 125 mmol) were added sequentially, then the reaction <n="103"/>mixture was allowed to rt slowly and maintained over night. The reaction mass was filtered through celite bed, aqueous layer of the filtrate was basified with NaOH solution and extracted with EtOAc (2 x 400 ml_). The organic layer was dried over anhydrous sodium sulfate and concentrated to obtain the crude product as brown colored liquid (21.5 g, 88%). 1HNMR(CDCI3): delta 7.25-7.5(m, 5H), 3.8(s, 2H), 3.54(s, 2H), 2.66-2.8(m, 2H), 2.02-2.3(m, 4H), 1.6-1.7(m, 2H). Mass: (M+1) 216 calculated for C14H17NO.
72%
A solution of 2,5-dimethoxytetrahydrofuran (19.8 g, 149.8 mmol) in 0.1M hydrochloric acid (180 ml, 18.0 mmol) was stirred at 80C for 2 hours. The reaction solution was cooled to 0C, acetonedicarboxylic acid (19.8 g, 149.8 mmol), concentrated hydrochloric acid (13.8 ml, 164.8 mmol), sodium acetate (14.8 g, 179.8 mmol), and benzylamine (17.7 g, 164.8 mmol) were added to the reaction solution, and the mixture was stirred at the same temperature for 12 hours and at 55C for 2 hours. The reaction solution was cooled to room temperature, and an aqueous solution of 4M sodium hydroxide (70 ml) was added, followed by extraction with chloroform. The organic layer was dried over sodium sulfate and then concentrated under a reduced pressure. The residue was purified by flash chromatography (silica gel, n-hexane/ethyl acetate) to give a title compound (23.3 g, 108 mmol, 72%) as a light yellow oil product. 1H-NMR (400 MHz, CDCl3) delta:1.61-1.66 (2H, m), 2.10-2.13 (2H, m), 2.19-2.23 (2H, m), 2.67-2.72 (2H, m), 3.50 (2H, m), 3.75 (2H, s), 7.26-7.43 (5H, m). ESI-MS: m/z = 216 (M+H+).
61%
Step 1: 8-benzyl-8-azabicyclo[3.2.1]octan-3-one A solution of 2,5-dimethoxytetrahydrofuran (2.2 mL) in hydrochloric acid (0.1N, 20 mL) was stirred under refluxing for 1 hour and then cooled to 0 C. 1,3-acetone-dicarboxylic acid (2.5 g), benzylamine (2.25 mL) and 10% sodium acetate solution (10 mL) were added therein. The reaction mixture was stirred for 1 hour at room temperature, and additionally stirred for 5 hours at 50 C., and then cooled under ice bath. The reaction mixture was alkalized to pH 12 using a 2N sodium hydroxide solution. After layer-separated, the aqueous phase is diluted with ethyl acetate. The combined organic phase was washed with water, dried with anhydrous sodium sulfate, filtered and vaporized under reduced pressure. The concentrate was separated through column chromatography (petroleum ether/ethyl acetate=4/1, v/v) to obtain the product as brown oil (2239 mg, yield: 61%). 1HNMR (CDCl3, 300 MHz) delta: 7.43-7.24 (m, 5H), 3.75 (s, 2H), 3.49-3.48 (m, 2H), 2.72-2.66 (m, 2H), 2.23 (s, 1H), 2.18-2.16 (m, 1H), 2.14-2.09 (m, 2H), 1.66-1.59 (m, 2H).
60%
Intermediate I-25 (8-benzyl-8-azabicyclo[3.2.1]octan-3-one)Concentrated hydrochloric acid (3 mL) was added to a stirred suspension of intermediate I-24 (8.2 g, 62 mmol, 1 eq) in water (17 mL), the reaction mixture was stirred for additional 20 minutes and diluted with water (25 mL). In a separate flask cooled to 0 C. on an ice bath, concentrated hydrochloric acid (9 mL) was added slowly to a solution of benzyl amine (10 g, 93 mmol, 1.5 eq) in water (35 mL), and this solution was added to the above solution of I-24. A solution of 1,3-acetonedicarboxylic acid (10 g, 68 mmol, 1.1 eq) in water (40 mL) was added followed by a solution of sodium hydrogen phosphate (4.4 g, 31 mmol, 5.0 eq) in water (20 mL). The acidity was adjusted from pH 1 to pH 4.5 using a solution of NaOH (40% in water). The resulting cloudy and pale- yellow solution was stirred overnight at room temperature. The reaction mixture was acidified to pH 3 from pH 7.5 using aqueous HCl solution (50% in water) and stirred at 85 C. for 2 hours. The crude reaction mixture was cooled to room temperature, basified to pH 12 using a solution of NaOH (40% in water) and extracted with dichloromethane. The combined organic layers were washed with brine, dried over anhydrous MgSO4, the solids were removed by filtration and the filtrate was concentrated. The crude reaction product was purified by silica gel chromatography to give I-25 as a yellow oil (8.0 g, 60%). 1H-NMR (CDCl3) delta : 7.59 (d, J=7.2 Hz, 2H), 7.31-7.40 (m, 3H), 4.04 (s, 2H), 3.75 (s, 2H), 3.13 (d, J=12.0 Hz, 2H), 2.22-2.30 (m, 4H), 1.75-1.80 (m, 2H). MS (ESI): ink 216 (M+H+).
a. Preparation of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0 C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH~4.5 using 40% NaOH. The resulting solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with DCM (3*500 mL). The combined organic layers were washed with brine, dried, filtered and concentrated under reduced pressure to produce the crude title intermediate as a viscous brown oil (52 g). To a solution of the crude intermediate in methanol (1000 mL) was added di-tert-butyl dicarbonate (74.6 g, 0.342 mol) at 0 C. The solution was allowed to warm to room temperature and stirred overnight. The methanol was removed under reduced pressure and the resulting oil was dissolved in dichloromethane (1000 mL). The intermediate was extracted into 1 M H3PO4 (1000 mL) and washed with dichloromethane (3×250 mL) The aqueous layer was basified to pH 12 using aqueous NaOH, and extracted with dichloromethane (3×500 mL). The combined organic layers were dried, filtered and concentrated under reduced pressure to provide the title intermediate as a viscous, light brown oil. 1H-NMR (CDCl3) delta(ppm) 7.5-7.2 (m, 5H, C6H5), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (m, 2H, CH2CH2). (m/z): [M+H]+ calcd for C14H17NO 216.14; found, 216.0.
A solution of 2, 5-dimethoxytetrahydrofuran (22. 2ML) in O. 1M HCL was refluxed for 1 hour and then cooled to 0 C. 1.3-Acetonedicarboxylic acid (25g), benzylamine (15. 6ML) and 10% sodium acetate (95ML) was added in one portion and the resulting mixture was stirred at room temperature for 1 hour and then heated to 50 C for 5 hours. The reaction mixture was cooled, basified with 2M sodium hydroxide, extracted with dichloromethane and washed with water. The organics were extracted with 1M hydrochloric acid and washed with dichloromethane. The aqueous layer was basified with 2M sodium hydroxide and extracted with ethyl acetate (3XLOOML). The organic extracts were dried and evaporated to dryness to give the title compound as a brown oil which was used without further purification. Yield 13.66g. MS 216 MH+.
Example 1: Synthesis of l-isopropyl-lH-indazole-3-carboxylic acid {(lS, 3R, 5R)-8-[2-(4-acetylpiperazin-1-yl) ethyl]-8-azabicyclo [3.2. 1] oct-3- yl} amide; a. Preparation of 8-benzyl-8-azabicyclo [3. 2. 1] octan-3-one; Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2, 5-dimethoxy tetrahydrofuran (82. 2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0C (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5- dimethoxytetraliydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH-4. 5 using 40% NaOH. The resulting cloudy and pale yellow solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with dichloromethane (3 x 500 mL). The combined organic layers were washed with brine, dried (MgS04), filtered and concentrated under reduced pressure to produce the crude title intermediate as a viscous brown oil (52 g). To a solution of the crude intermediate in methanol (1000 mL) was added di-tert- butyl dicarbonate (74.6 g, 0.342 mol) at 0 C. The solution was allowed to warm to room temperature and stirred overnight. The methanol was removed under reduced pressure and the resulting oil was dissolved in dichloromethane (1000 mL). The intermediate was extracted into 1 M H3PO4 (1000 mL) and washed with dichloromethane (3 x 250 mL) The aqueous layer was basified to pH 12 using aqueous NaOH, and extracted with dichloromethane (3 x 500 mL). The combined organic layers were dried (MgS04), filtered and concentrated under reduced pressure to produce the title intermediate as a viscous, light brown oil (54 g). 1H-NMR (CDC13) 8 (ppm) 7.5-7. 2 (m, 5H, C6Hs), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2. 6 (dd, 2H, CH2CO), 2.2-2. 1 (dd, 2H, CH2CO), 2.1-2. 0 (m, 2H, CH2CH2), 1.6 (m, 2H, CH2CH2). (m/z) : [M+H] + calcd for C14Hl7NO 216.14 ; found, 216.0. b.
Method P; Preparation of (3-enrfo)-3-(l-methyl-l^-imidazol-2-yl)-8-azabicyclo[3.2.1]octan-3-ol; OHStep 1: Preparation of 8-benzylbicyclo[3.2.1.]octan-3-one; A solution of 2,5-dimethoxytetrahydrofuran (22.2ml) in 0.1M HC1 was refluxed for 1hour and then cooled to 0C. 1.3-Acetonedicarboxylic acid (25g), benzylamine (15.6ml) and10% sodium acetate (95ml) was added in one portion and the resulting mixture was stirred atroom temperature for 1 hour and then heated to 50C for 5 hours. The reaction mixture wascooled, basified with 2M sodium hydroxide, extracted with dichloromethane and washed withwater. The organics were extracted with 1M hydrochloric acid and washed withdichloromethane. The aqueous layer was basified with 2M sodium hydroxide and extractedwith ethyl acetate (3x100ml). The organic extracts were dried and evaporated to dryness togive the sub-title compound as a brown oil which was used without further purification (yield13.66g,MS216MH+)
Concentrated hydrochloric acid (30 mL) was added to a heterogeneous solution of 2,5-dimethoxy tetrahydrofuran (82.2 g, 0.622 mol) in water (170 mL) while stirring. In a separate flask cooled to 0 C. (ice bath), concentrated hydrochloric acid (92 mL) was added slowly to a solution of benzyl amine (100 g, 0.933 mol) in water (350 mL). The 2,5-dimethoxytetrahydrofuran solution was stirred for approximately 20 min, diluted with water (250 mL), and then the benzyl amine solution was added, followed by the addition of a solution of 1,3-acetonedicarboxylic acid (100 g, 0.684 mol) in water (400 mL) and then the addition of sodium hydrogen phosphate (44 g, 0.31 mol) in water (200 mL). The pH was adjusted from pH 1 to pH 4.5 using 40% NaOH. The resulting solution was stirred overnight. The solution was then acidified to pH 3 from pH 7.5 using 50% hydrochloric acid, heated to 85 C. and stirred for 2 hours. The solution was cooled to room temperature, basified to pH 12 using 40% NaOH, and extracted with DCM (3×500 mL). The combined organic layers were washed with brine, dried, filtered and concentrated under reduced pressure to produce the crude title intermediate as a viscous brown oil (52 g). To a solution of the crude intermediate in methanol (1000 mL) was added di-tert-butyl dicarbonate (74.6 g, 0.342 mol) at 0 C. The solution was allowed to warm to room temperature and stirred overnight. The methanol was removed under reduced pressure and the resulting oil was dissolved in dichloromethane (1000 mL). The intermediate was extracted into 1 M H3PO4 (1000 mL) and washed with dichloromethane (3×250 mL) The aqueous layer was basified to pH 12 using aqueous NaOH, and extracted with dichloromethane (3×500 mL). The combined organic layers were dried, filtered and concentrated under reduced pressure to provide the title intermediate as a viscous, light brown oil. 1H-NMR (CDCl3) delta (ppm) 7.5-7.2 (m, 5H, C6H5), 3.7 (s, 2H, CH2Ph), 3.45 (broad s, 2H, CH-NBn), 2.7-2.6 (dd, 2H, CH2CO), 2.2-2.1 (dd, 2H, CH2CO), 2.1-2.0 (m, 2H, CH2CH2), 1.6 (m, 2H, CH2CH2). (m/z): [M+H]+ calcd for C14H17NO 216.14; found, 216.0.
660g (5 mol) of2,5-dimethoxytetrahydrofuran were heated at 80C in 6 L of 0.1 N HCl for 1h.After cooling to 10C, 803g (5.5mol)of acetone dicarboxylic acid and 460 ml of12N HCl and 492g (6 mol) of sodium acetate were added with stirring. Then 589g(5.5 mol) of benzylamine were introduced and stirred overnight. Insoluble materials were filtered off and (5.5 mol) ofhydroxylamine hydrochloride were added to the filtrate with stirring.After 40 min., the reaction mixture was cooledand pH was adjusted to 7-8 with aqueous KOH under vigorous stirring. Theprecipitated oxime (8) was collectedby filtration, washed with water and dried to give oxime (8) m.p. 124C.A solution of 2.1 mol of this oxime in 6L ofamyl alcohol was heated to 80C with stirring and 420g Na were carefully addedto keep on refluxing. The mixture was poured into 3L of water cooled to 0C.The aqueous layer was decanted, the organic layer was twice extracted withaqueous HCl. Basification with KOH and extraction with CH2Cl2 gave,after drying (Na2SO4) and evaporation compound 9, b.p. 100-115C (0.05mm). 1H NMR(CDCl3) 7.1-7.5 (m, 5, ArH), 3.5 (s, 2, CH2Ar), 3.15 (m,2, H1 and H5), 2.6-3.25 (m, 1, J=40 Hz, axial H3),1.1-2.2 (m, 10, NH2 and 4(CH2))
With sodium hydroxide; In tetrahydrofuran; water; ethyl acetate;
a. Preparation of toluene-4-sulfonic acid 8-benzyl-8-azabicyclo[3.2.1]oct-2-en-3-yl ester To a 1 L flask was added 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (12.9 g, 60 mmol) and THF (100 mL). The mixture was stirred at room temperature until complete dissolution was observed, and then was cooled to -30 C. Lithium-bis-(trimethylsilyl)amide (LiHMDS) solution (1 M in THF, 69 mL, 69 mmol) was added to the reaction solution over 30 min. The solution was stirred at -25+-5 C. for 1 h. A solution of p-toluene sulfonic acid anhydride (22.5 g, 69 mmol) in THF (150 mL) was added to the reaction mixture at a temperature of -7+-3 C. The resulting solution was stirred at -10 C. to 10 C. for 1 h. To the solution was added EtOAc (250 mL) and 1 N NaOH (200 mL) and the solution was stirred at room temperature for 5 min. The organic layer was separated and washed with 0.5 N NaOH (200 mL) followed by 1:1 brine:water (200 mL). The organic layer was dried over Na2SO4, and concentrated to give a yellow solid residue. The solids were slurried in hexane (40 mL)/trace EtOAc(<5mL) and filtered to provide the title intermediate (19.1 g, 86% yield). (m/z): [M+H]+ calcd for C21H23NO3S 370.15; found 370.2. 1H NMR (CDCl3, 400 MHz): delta (ppm) 7.84-7.86 (m, 2H), 7.21-7.37 (m, 7H), 5.4 (dt, J=5.6, 1.6, 1H), 3.58 (d, J=1.2, 2H), 3.24-3.32 (m, 2H), 2.58-2.63 (m, 1H), 2.46 (s, 3H), 2.05-2.13 (m, 1H), 1.89-1.95 (m, 1H), 1.70-1.77 (m, 2H), 1.41-1.49 (m, 1H).
<strong>[28957-72-4]N-<strong>[28957-72-4]benzyltropinone</strong></strong> (available from Crescent Chemical Co. Inc., Hauppage, N.Y.) is deprotonated with lithium diisopropylamide (LDA; generated from n-buLi and diisopropylamine) in THF under N2 atmosphere at -78 C. CO2 (gas or solid) is added, and the reaction is quenched by addition of iodomethane. The reaction mixture is then allowed to warm to ambient temperature, and after standard workup, is hydrogenated over Pd on carbon with trifluoroacetic acid in MeOH. The catalyst is filtered, and the solvent evaporated. Methylisocyanate and diisopropylethylamine in dichloromethane are then reacted with the hydrogenated product to provide, after standard workup, 8-[(methylamino)carbonyl]-3-oxo-8-azabicyclo[3.2.1]octane-2-carboxylic acid, methyl ester.
To a solution of 0.025 M aqueous hydrochloric acid (100 ml) at [0C] was added 2, [5-DIMETHOXY-TETRAHYDROFURAN] (30 ml 231 [MMOL).] The reaction was stirred at [0C] overnight. The reaction was then diluted with water (200 ml) and benzyl amine hydrochloride (40 grams, 278 [MMOL),] 3-oxo-pentanedioic acid (33.7 grams, 231 [MMOL),] and sodium acetate (10.7 grams, 130 [MMOL)] were added. The reaction was stirred for 5 minutes at [0C,] warmed to ambient temperature and stirred for 90 minutes, then heated to [50C] for two hours, cooled to [0C] and basified to pH = 10 with 50 [%] aqueous sodium hydroxide (14 [ML).] The reaction mixture was extracted with ethyl acetate (3 times) and the organic layers were combined and washed with a saturated sodium chloride solution, dried over magnesium sulfate, filtered and concentrated in vacuo to give a brown oil. Silica gel chromatography gave the title compound (33.46 grams, 67 % yield
a. Preparation of 8-benzyl-3-exo-(3-methoxyphenyl)-8-azabicyclo[3.2.1]octan-3-ol To a 3 L-3-necked flask fitted with an overhead stirrer and flushed with dry nitrogen was added cerous chloride powder (88.2 g, 0.35 mol). The solid was diluted with anhydrous tetrahydrofuran (500 mL) and cooled to 0 C. To the suspension was added 1M 3-methoxyphenyl magnesium bromide in THF (360 mL, 0.36 mol) dropwise while the temperature was maintained below 10 C. The resulting solution was stirred at 0 C. for 1.5 hours. A solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (54.5 g, 0.25 mol) in tetrahydrofuran (50 mL) was then added dropwise, while maintaining the internal temperature below 5 C. The resulting solution was stirred at 0 C. for 2 hours. The reaction was quenched with 10% aqueous acetic acid (400 mL) and stirred for 30 minutes at room temperature. Saturated sodium chloride solution (400 mL) was then added and the resulting suspension was stirred at room temperature for 20 hours to allow complete crystallization of product as the acetate salt. The crystals were filtered and washed with cold water (200 mL) followed by isopropyl acetate (200 mL) and dried under vacuum to give the title intermediate as a white crystalline powder (91.1 g, 93% yield). (m/z): [M+H]+ calcd for C21H25NO2 324.20; found, 324.5.
8-benzyl-3-exo-(3-methoxyphenyl)-8-azabicyclo[3.2.1]octan-3-ol hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
To a 3 L flask was added cerous chloride powder (194 g, 0.79 mol). The flask was flushed with nitrogen and THF (800 mL) was added. The reaction mixture was stirred at 25 C. for 1 h. To the mixture was added ~1M 3-methoxyphenyl magnesium bromide in THF (800 mL, 0.87 mol) dropwise. The resulting slurry was stirred at 3 C. for 1.5 hours. A solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (120.4 g, 0.56 mol) in THF (200 mL) was then added dropwise, while maintaining the internal temperature at -5 C. The resulting solution was stirred for 15 min. The reaction mixture was added to a flask containing 6 N HCl (800 mL) maintaining the temperature at 10 C. After solvent was removed by rotary evaporation, the reaction mixture was stirred at room temperature overnight. The solids were isolated by filtration, washed with 6N HCL (70 mL) and acetonitrile (3*70 mL), and dried to provide the HCl salt of the title intermediate as an off-white solid (161 g).
8-benzyl-3-exo-(3-methoxyphenyl)-8-azabicyclo[3.2.1]octan-3-ol acetate[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
93%
To a 3 L-3-necked flask fitted with an overhead stirrer and flushed with dry nitrogen was added cerous chloride powder (88.2 g, 0.35 mol). The solid was diluted with anhydrous tetrahydrofuran (500 mL) and cooled to 0 C. To the suspension was added 1M 3-methoxyphenyl magnesium bromide in THF (360 mL, 0.36 mol) dropwise while the temperature was maintained below 10 C. The resulting solution was stirred at 0 C. for 1.5 hours. A solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (54.5 g, 0.25 mol) in tetrahydrofuran (50 mL) was then added dropwise, while maintaining the internal temperature below 5 C. The resulting solution was stirred at 0 C. for 2 hours. The reaction was quenched with 10% aqueous acetic acid (400 mL) and stirred for 30 minutes at room temperature. Saturated sodium chloride solution (400 mL) was then added and the resulting suspension was stirred at room temperature for 20 hours to allow complete crystallization of product as the acetate salt. The crystals were filtered and washed with cold water (200 mL) followed by isopropyl acetate (200 mL) and dried under vacuum to give the title intermediate as a white crystalline powder (91.1 g, 93% yield). (m/z): [M+H]+ calcd for C21H25NO2 324.20; found, 324.5.
8-benzyl-3-exo-(3-methoxyphenyl)-8-azabicyclo[3.2.1]octan-3-ol hydrochloride[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Preparation 2: Synthesis of 3-endo-(8-azabicyclo[3.2.1]oct-3-yl)-benzamidea. Preparation of 8-benzyl-3-exo-(3-methoxyphenyl)-8-azabicyclo[3.2.1]octan-3-olTo a 3 L flask was added cerous chloride powder (194 g, 0.79 mol). The flask was flushed with nitrogen and THF (800 mL) was added. The reaction mixture was stirred at 25 C. for 1 h. To the mixture was added 1M 3-methoxyphenyl magnesium bromide in THF (800 mL, 0.87 mol) dropwise. The resulting slurry was stirred at 3 C. for 1.5 hours. A solution of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-one (120.4 g, 0.56 mol) in THF (200 mL) was then added dropwise, while maintaining the internal temperature at -5 C. The resulting solution was stirred for 15 min. The reaction mixture was added to a flask containing 6 N HCl (800 mL) maintaining the temperature at 10 C . After solvent was removed by rotary evaporation, the reaction mixture was stirred at room temperature overnight. The solids were isolated by filtration, washed with 6N HCL (70 mL) and acetonitrile (3×70 mL), and dried to provide the HCl salt of the title intermediate as an off-white solid (161 g).
To a flame dried 100 mL RBF under N2 was added 8.0 mmol (1.722 gram) of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (1) in 5 mL of anhydrous THF. The flask was cooled to 0 C. and 1.1 equivalents of the requisite lithium reagent were slowly dripped into the THF solution. The mixture was stirred for 1 hour at which time another 1.1 equivalents of lithium reagent was added. The flask was allowed to slowly warm to room temperature over two hours. The reaction mixture was then cooled to 0 C. and slowly quenched with ice water. The crude product was evaporated to near dryness, taken up in methanol and purified by prep HPLC (using a Shimadzu preparative HPLC employing methanol/water and 0.1% trifluoroacetic acid buffer with a Phenomenex Luna, C 18, 21 mm×100 mm, 10 mum column at a gradient of 0-50% B (where A=10% HPLC grade methanol/0.1% trifluoroacetic acid/90% HPLC grade water and B=90% HPLC grade methanol/0.1% trifluoroacetic acid/10% HPLC grade water ) and a flow rate of 25 mL/min. over 10 minutes with a 5-10 minute hold, to give the benzyl protected tropanol (2) as a clear colorless oil. Post-purification LC/MS data was obtained on a Shimadzu analytical LC/Micromass Platform LC (ESI+) at 220 nm using the following set of conditions: Column I (Phenomenex 10 mum C18, 4.6×30 mm), Solvent system I (gradient of 0-100% B where A=10% HPLC grade methanol/0.1% trifluoroacetic acid/90% HPLC grade water and B=90% HPLC grade methanol/0.1% trifluoroacetic acid/10% HPLC grade water), in 2 minutes with a 1 minute hold at a flow rate of 5 mL/minute. To tropanol (2) in a 100 mL RBF equipped with a balloon of H2 was added 10 mL of methanol along with a catalytic amount of 10% Palladium on Carbon. The heterogeneous mixture was stirred overnight at room temperature. The crude product was then filtered through celite and evaporated to dryness on the rotovap to give tropanol (3) as a colorless oil. 8-benzyl-3-methyl-8-azabicyclo[3.2.1]octan-3-ol. 1H NMR (300 MHz, CD3OD) delta ppm: 1.08 (s, 3 H), 1.86 (m, 2 H), 1.99 (m, 2 H), 2.22 (m, 2 H), 2.50 (m, 2 H), 3.77 (br s, 2 H), 4.11 (s, 2 H), 7.40 (m, 5 H). LC/MS: m/z 232.10 (MH+), Rf 0.58 min., 98% purity; 36% isolated yield.
Step a: Preparation of 3-methylidene-8-(phenylmethyl)-8-azabicyclo[3.2.1]octane; A 500 ml flask with side arm, stirring bar, N2 inlet, and septum stopper was charged with a solution of potassium tert-butoxide in THF (82 ml, 1M) and methyltriphenyl phosphonium bromide (29.2 g, 82 mmol). It was cooled to 0 C. under dry N2, and anhydrous THF (140 ml) was added via syringe at 0 C. The yield solution was stirred for 20 min. 8-(Phenylmethyl)-8-azabicyclo[3.2.1]octane-3-one (14.0 g, 65 mmol) in anhydrous THF (ml) was added via syringe at 0 C. and the solution was stirred 1 h at room temperature then quenched with water (6 ml). The mixture was acidified to pH 1 and THF was removed in vacuo at 30 C. The residue was diluted with water (450 ml) and Ph3PO was extracted with toluene (3×200 ml). The aqueous solution was basified with 6N NaOH (35 ml), and extracted with ethyl acetate (3×200 ml). The organic layers were combined, washed with saturated NaCl (3×100 ml), dried over NaSO4, and evaporated to yield a crude product which was purified by flash chromatography (400 g of silica, ethyl acetate containing 0.1% TEA). 3-Methylidene-8-(phenylmethyl)-8-azabicyclo[3.2.1]octane was recovered as a yellow oil (11.3 g, 81.5%). LC/MS ESI RT, 1.27 min, MH+ 214; NMR (CDCl3, 400 MHz; delta): 1.58 ppm (q, 2H), 1.80-2.05 ppm (m, 4H), 2.55 ppm (d, 2H), 3.28 ppm (s, 2H), 3.65 ppm (s, 2H), 4.80 ppm (s, 2H), 7.29 ppm (t, 1H), 7.35 ppm (t, 2H), 7.46 ppm (d, 2H).
1-Bromo-4-fluoro-benzene (22.7 g) dissolved in diethylether (100 ml.) is added to a solution of n-butyllithium (1.7 mol/L in pentane, 86.8 ml.) in diethylether (200 ml.) cooled to -35 0C. The combined solutions are stirred at -35 - -40 0C for 1 h, before 8-benzyl-8-aza- bicyclo[3.2.1]octan-3-one (22.5 g) dissolved in diethylether (150 ml.) is added quickly. The solution is warmed to -10 0C within 1 h and then quenched by the addition of aqueous NH4CI solution. The resulting mixture is extracted with ethyl acetate, the combined extracts are washed with brine, and then 4 M hydrochloric acid is added to the extract phase. The organic phase is separated from the aqueous phase and an oily precipitation formed after the addition. The oily and aqueous phase are basified with 4 M aqueous NaOH solution and the resulting mixture is extracted with ethyl acetate. The organic extracts are dried (Na2SO4) and the solvent is evaporated to give the title compound. Yield: 21.O g (75% of theory) Mass spectrum (ESI+): m/z = 312 [M+H]+
75%
1-Bromo-4-fluoro-benzene (22.7 g) dissolved in diethylether (100 ml.) is added to a solution of n-butyllithium (1.7 mol/L in pentane, 86.8 ml.) in diethylether (200 ml.) cooled to -35 0C. The combined solutions are stirred at -35 to -40 0C for 1 h, before 8-benzyl-8-aza- bicyclo[3.2.1]octan-3-one (22.5 g) dissolved in diethylether (150 ml.) is added quickly. The solution is warmed to -10 0C within 1 h and then the reaction is quenched by the addition of aqueous NH4CI solution. The resulting mixture is extracted with ethyl acetate, the combined extracts are washed with brine, and 4 M hydrochloric acid is added to the organic phase. The organic phase is separated from the aqueous phase and an oily precipitation formed after the addition. The oily and aqueous phase are combined and basified with 4 M aqueous NaOH solution and the resulting mixture is extracted with ethyl acetate. The organic extracts are dried (Na2SO4) and the solvent is evaporated to give the title compound. Yield: 21.O g (75% of theory) Mass spectrum (ESI+): m/z = 312 [M+H]+
75%
Example XLIIdelta-Benzyl-S-^-fluoro-phenvD-delta-aza-bicvclorS^.?octan-S-o; 1-Bromo-4-fluoro-benzene (22.7 g) dissolved in diethylether (100 ml_) is added to a solution of n-butyllithium (1.7 mol/L in pentane, 86.8 ml_) in diethylether (200 ml_) cooled to -35 C. The combined solutions are stirred at -35 - -40 0C for 1 h, before 8- benzyl-8-aza-bicyclo[3.2.1]octan-3-one (22.5 g) dissolved in diethylether (150 ml_) is added quickly. The solution is warmed to -10 0C within 1 h and then quenched by the addition of aqueous NH4CI solution. The resulting mixture is extracted with ethyl acetate, the combined extracts are washed with brine and 4 M hydrochloric acid is added. The organic phase is separated from the aqueous phase and an oily precipitation formed after the addition. The oily and aqueous phase are basified with 4 M NaOH solution and the resulting mixture is extracted with ethyl acetate. The organic phase and extracts are dried (Na2SO4) and the solvent is evaporated to give the title compound. Yield: 21.0 g (75% of theory)Mass spectrum (ESI+): m/z = 312 [M+H]+
8-benzyl-(3-endo)-(1-methyl-1H-imidazol-2-yl)-8-azabicyclo[3.2.1]octan-3-ol[ No CAS ]
Yield
Reaction Conditions
Operation in experiment
Step 2: Preparation of 8-benzyl-(3-e«do)-(l-methyl-l//-imidazol-2-yl)-8-azabicyclo [3.2.1 ]octan-3 -ol; OH1-Methylimidazole (0.385g) was dissolved in dry THF (20ml) under an argonatmosphere and cooled to -78C. A 1.6M solution of butyl lithium in hexane (3.125ml) wasadded slowly and the resulting mixture was stirred at -78C for 90 minutes. A solution of 8-benzyl-8-azabicyclo[3.2.1]octan-3-one (1.05g) in dry THF (5ml) was added. The mixture wasallowed to reach ambient temperature and stirred overnight. The reaction was quenched withsaturated ammonium chloride solution and extracted with diethyl ether. The organic phasewas dried over MgSO4, filtered and evaporated to an oil, which was purified by columnchromatography using a 40g SCX column eluting with CHiCh/CHsOH/O.SSO ammonia(9/1/0.1) to yield 8-benzyl-(3-enrfo)-(l-methyl-l//-imidazol-2-yl)-8-azabicyclo[3.2.1]octan-3-ol as a white solid. Yield 342mg.NMR (CDC13): 7.4(m, 2H), 7.25 (m, 3H), 6.85 (s, 1H), 6.8 (s, 1H), 3.85 (s, 3H), 3.6 (s,2H), 3.3 (s, 2H), 2.55(m, 2H), 2.25 (m, 2H), 2.05 (m, 2H), 1.85 (m, 2H); MH+ 298.34.






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science






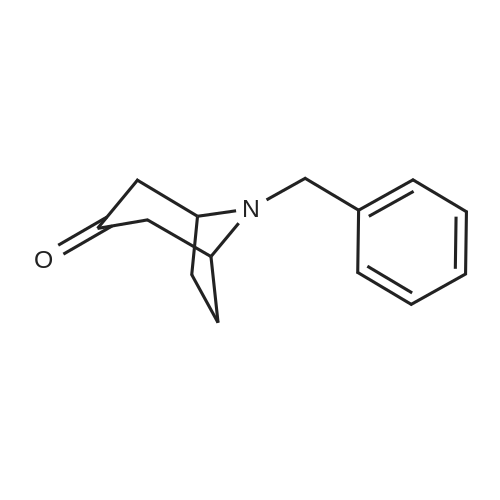

 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping