| 67.7% |
Stage #1: With sodium hexamethyldisilazane In tetrahydrofuran at -76 - -74℃; for 1.16667 h;
Stage #2: at -76 - 10℃; for 2.08333 h; |
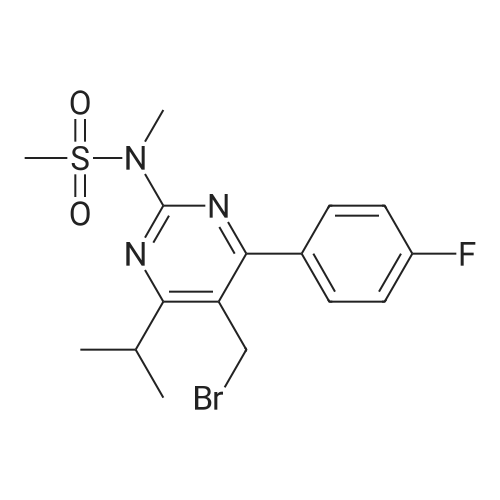
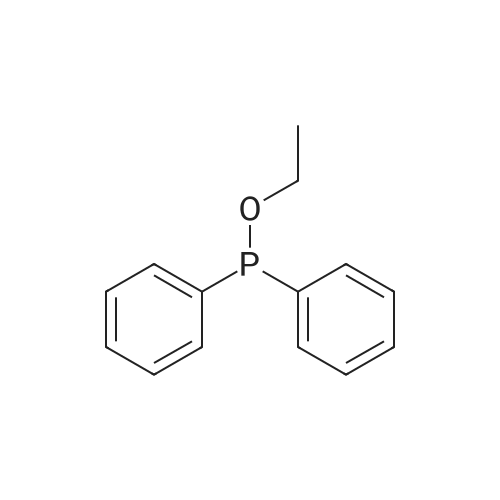
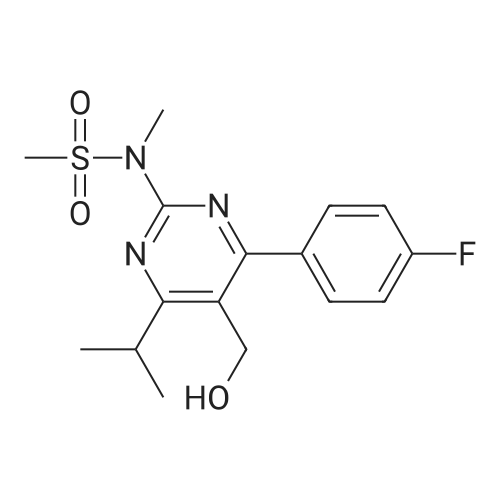
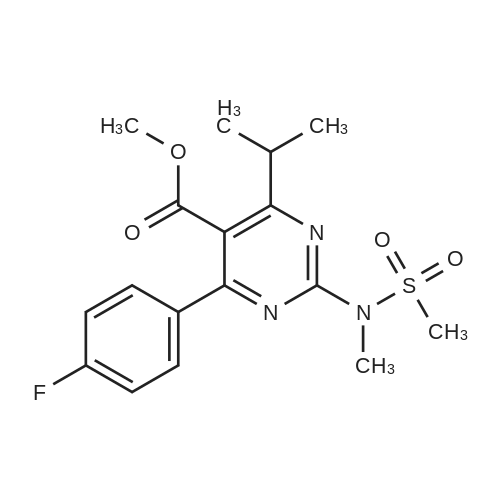
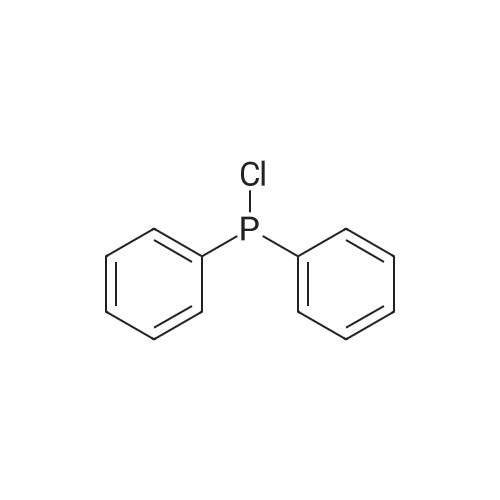
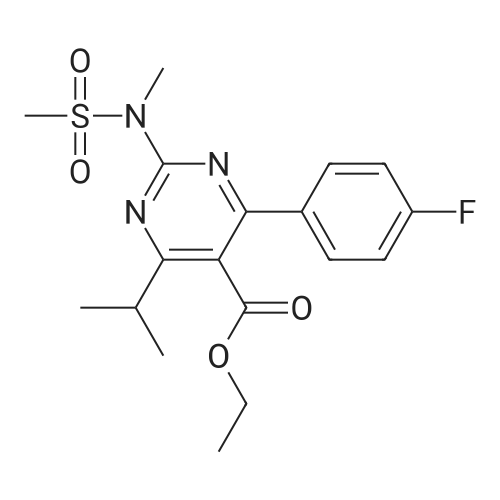
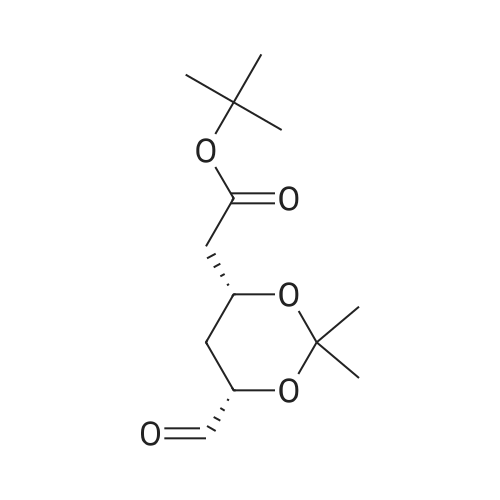
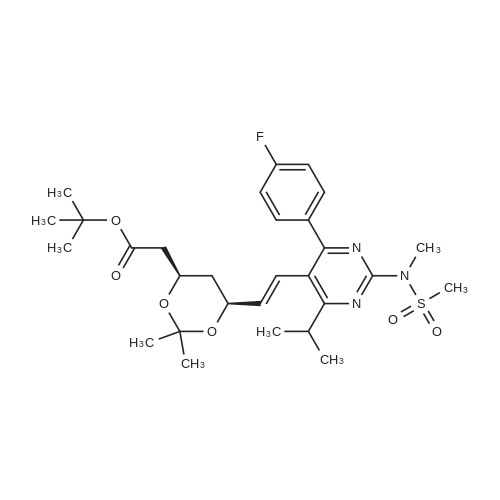
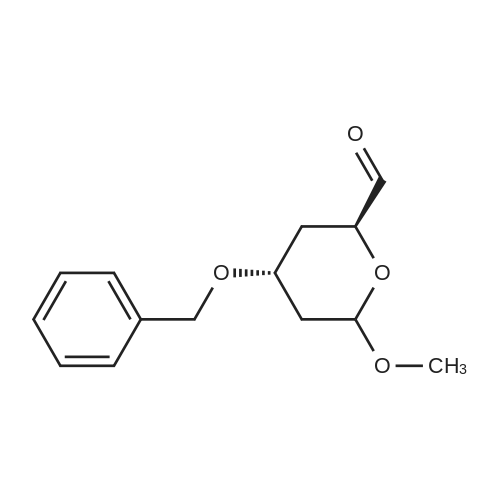
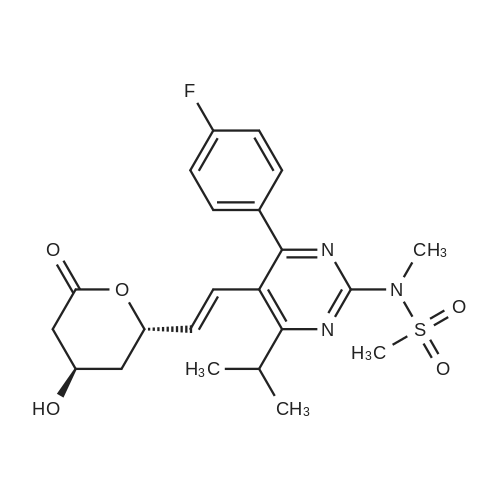
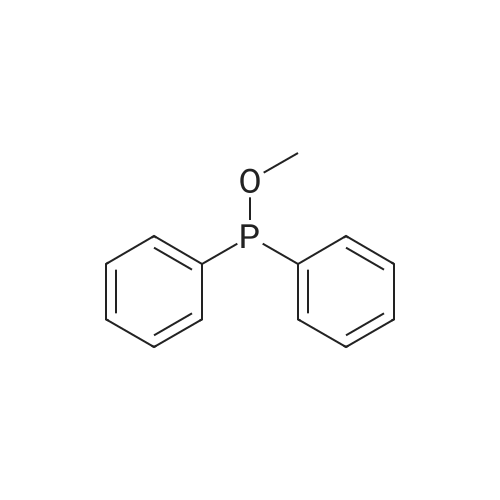
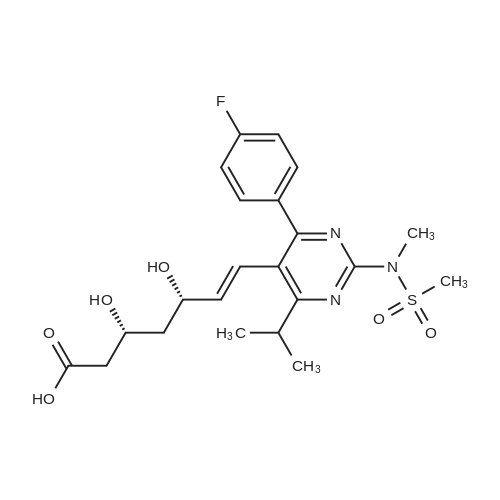
A mixture of DPPO (19.17 g) and THF (227 ml) were warmed briefly to 40 C. until a clear solution had formed then inerted by the sequential application of vacuum and nitrogen (5 cycles). The mixture was immersed in an acetone/CO2 bath cooling the contents to -75 C. Sodium bis(trimethylsilyl)amide (37.4 ml of 1.0 M solution in THF) was added to the reaction mixture over 10 minutes from a pressure equalising dropping funnel maintaining the temperature below -74 C and forming a red solution of the anion. THF (10 ml) was rinsed through the dropping funnel into the mixture and the mixture stirred a further 1 hour at -76 C forming a red suspension. BFA (80 ml of 13.5percent w/w toluene solution) was added in portions to the suspension over 20 minutes from a pressure equalising dropping funnel maintaining the temperature below -73 C. Toluene (20 ml) was rinised through the dropping funnel into the mixture and the mixture stirred a further 15 minutes at -76 C. The chilling bath was lowered and the suspension allowed to warm to 10 C over 1.5 hours. Glacial acetic acid (3.21 g) in water (15 g) was added in one portion raising the temperature to 18 C and dissolving all solids and the mixture was stirred a further 5 minutes. The mixture was concentrated by distillation at atmospheric pressure (jacket 110 C) to a temperature of 94 C collecting a total of 274 ml distillates. The concentrated mixture was cooled to 40 C, water (40 ml) was added and the mixture stirred for 5 minutes then allowed to settle for 15 minutes. The lower aqueous phase was discarded. Sodium hydrogen carbonate (2.99 g) in water (40 ml) was added and the mixture stirred for 5 minutes then allowed to settle for 15 minutes. The lower aqueous phase was discarded. Water (30 ml) was added and the mixture stirred for 5 minutes then allowed to settle for 15 minutes. The lower aqueous phase was discarded. The organic phase was transferred to a distillation apparatus with toluene (20 ml) and concentrated by distillation at atmospheric pressure (jacket 125-130 C) to a temperature of 116 C collecting 85 ml distillates. Vacuum was applied (400-500 mbar) and a further 16.5 ml distillates collected to a temperature of 111 C. The vacuum was released and the concentrated mixture allowed to cool to 80 C. Warm MeOH (140 ml, 50 C) was added with rapid stirring and the batch allowed to self-cool to 20 C over 30 minutes during which time a solid was deposited. The suspension was further cooled to 2 C for 30 minutes then the solid was collected by filtration on a sinter and pulled as dry as possible. The solid was washed with cold MeOH (60 ml, 2C.) and again pulled as dry as possible then transferred to a vacuum oven and dried overnight (50 C, 200 mbar); giving BEM (14.01 g, 67.7percent). 1H NMR (CDCl3, 270 MHz) 7.65 [m, 2H, ArH], 7.09 [m, 2H, ArH], 6.52 [dd, 1H, ArCH=CH], 5.47 [dd, 1H, ArCH=CH], 3.57, 3.50 [2 x s, 6H, NCH3, SO2CH3], 3.38 [hept., 1H, ArCHMe2], 2.45, 2.30 [2x dd, 2H, CH2CO2tBu], 1.55, 1.13 [dt, dd, 2H, acetonide CH2], 1.50, 1.40 [2x s, 6H, acetonide C(CH3)2], 1.45 [s, 9H, CO2C(CH3)3], 1.27 [dd 6H, ArCH(CH3)2] |
| 61% |
Stage #1: With sodium hexamethyldisilazane In tetrahydrofuran at -74℃; for 1 h;
Stage #2: at -74 - 10℃; for 3 h;
Stage #3: With acetic acid In tetrahydrofuran; water; toluene at 10 - 40℃; for 0.166667 h; |
Sodium bis (trimethylsilyl) amide (1 M in tetrahydrofuran, 23 mi) is added dropwise, AT-74°C, to a suspension of the compound of formula (38) (12 g, 22.3 MMOL) in tetrahydrofuran (130 M .). STIRRING is carried out at-74°C for 1 hour and then a solution of the compound of formula (40) (6.9 g, 26.8 MMOI) in toluene (28 ML) is added dropwise. Stirring is then carried out AT-74°C for 1 hour, then warming to 10°C over the course of 1 hour and stirring for a further 1 hour at that temperature. A mixture of acetic acid (2 ml) and water (8.4 ML) is added, at 10°C, to the resulting yellow suspension and stirring is carried out at room temperature for 5 minutes. The tetrahydrofuran is then distilled off, and, at 40°C, 45 ml of water are added to the reaction mixture and vigorous stirring is carried out for 5 minutes. The aqueous phase is separated off and a solution of sodium hydrogen carbonate (2.27 G) in water (45 ml) is added to the organic phase. Vigorous stirring is again carried out for 5 minutes and then the aqueous phase is removed again. The organic phase is diluted with 250 ML of toluene, washed successively with water and saturated sodium chloride solution and dried (using NA2SO4). The salt mixture is filtered off and the filtrate is concentrated by evaporation. The concentrated residue is then purified by column chromatography on silica gel (hexane: ethyl acetate 8: 1). 2.59 G (61 percent) of the desired product (39) can be obtained in the form of colourless crystals. 'H NMR (300 MHz, CDCI3) : 0.91-1. 08 (m, 1H) ; 1.20 (d, J = 6. 7 HZ, 6H); 1. 24 (S, 3H); 1. 38 (S, 9H); 1.41 (S, 3H); 1.41-1. 56 (m, 1 H) ; 2.21 (dd, J = 15. 2, 7. 9, 1 H) ; 2. 35 (dd, J = 15. 0, 5. 0 HZ, 1H) ; 3. 27-3. 37 (m, 1H) ; 3. 43 (S, 3H); 3.52 (S, 3H); 4.17-4. 24 (m, 1H) ; 4. 47-4. 53 (m, 1H) ; 5.43 (dd, J = 16.4, 5.5 Hz, 1H) ; 6.55 (dd, J = 16.1, 0.8 Hz, 1H) ; 7.24 (dd, J = 8.8, 8.8 HZ, 2H); 7.65 (dd, J = 8.8, 5.6 Hz, 2H). 13C NMR (75 MHz, CDCI3) : 18.7, 20.6, 20.7, 27.0, 29.0, 30.9, 32.0, 35.0, 41.3, 41.4, 64.8, 68.1, 79.6, 97.7, 113.7 (JCF = 21.7 Hz), 120.0, 122.0, 131. 0 (JCF = 8.4 Hz), 133.2 (JCF = 3.2 Hz), 136.3, 156.0, 162.0 (JCF = 249 Hz), 162.2, 168.8, 173.6. |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping