| 92% |
With Oxone; In methanol; water; at 20℃; for 20h;Cooling with ice; |
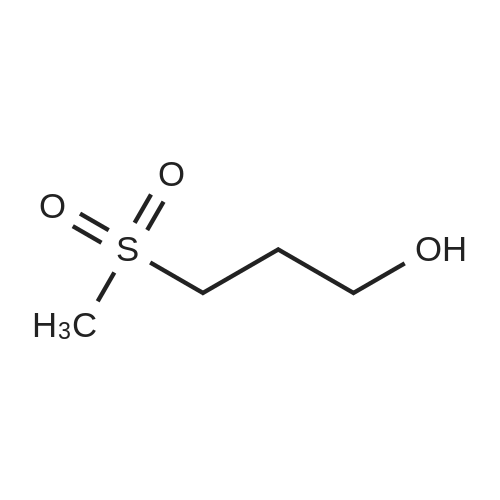
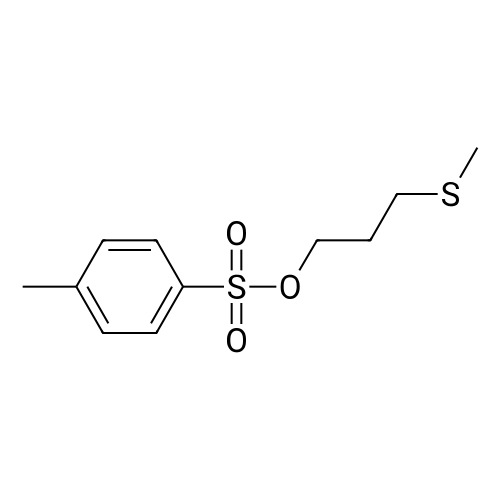
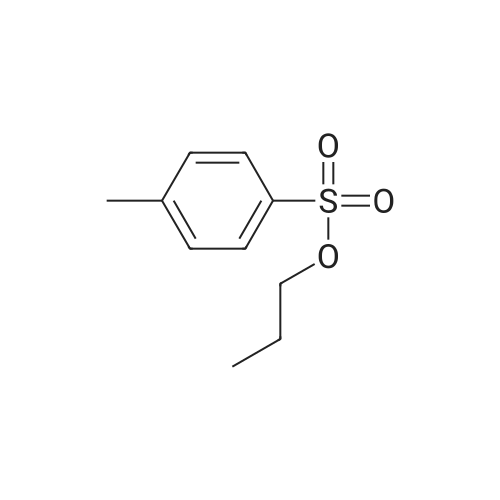
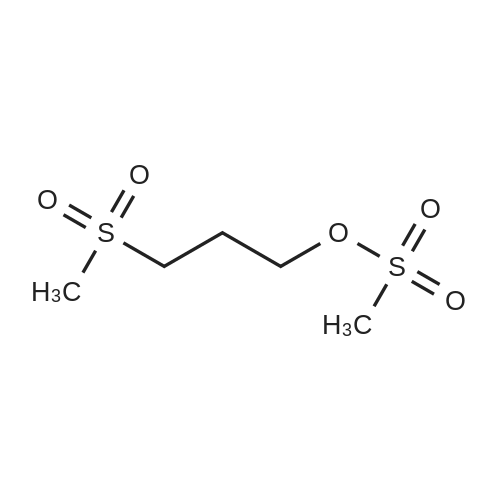
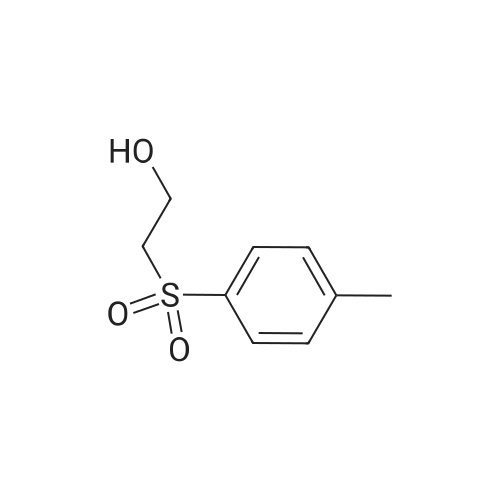
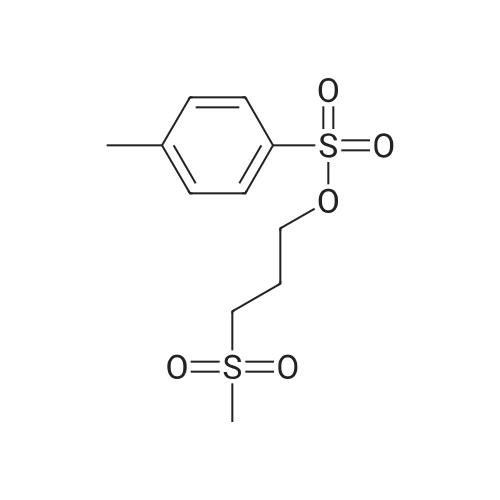
(B) To an ice-cooled solution of 3-(methylthio)propyl 4-methylbenzenesulfonate (3.6 g; 13.8 mmol) in MeOH (70 mL) was added a suspension of monopersulfate compound (17 g; 27.4 mmol) in water (70 mL) in a portion-wise fashion. Upon completion of the addition, the mixture was allowed to warm to rt, and stirring was continued for 20 h. The reaction was partially concentrated to remove the MeOH and the mixture was further diluted with water and extracted with EtOAc (2x). The combined organic extracts were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 0- 67% EtOAc in hexanes to afford 3- (methylsulfonyl)propyl 4-methylbenzene-sulfonate (3.72 g, 92%) as a white solid. LC/MS: mass calcd. for C11H16O5S2: 292.38, found 315.1 [M+Na]+. |
| 92% |
With Oxone; In methanol; water; at 20℃; for 20h;Cooling with ice; |
(B) To an ice-cooled solution of 3-(methylthio)propyl 4-methylbenzenesulfonate (3.6 g; 13.8 mmol) in MeOH (70 mL) was added a suspension of monopersulfate compound (17 g; 27.4 mmol) in water (70 mL) in a portion-wise fashion. Upon completion of the addition, the mixture was allowed to warm to rt, and stirring was continued for 20 h. The reaction was partially concentrated to remove the MeOH and the mixture was further diluted with water and extracted with EtOAc (2*). The combined organic extracts were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 0-67% EtOAc in hexanes to afford 3-(methylsulfonyl)propyl 4-methylbenzene-sulfonate (3.72 g, 92%) as a white solid. LC/MS: mass calcd. for C11H16O5S2: 292.38, found 315.1 [M+Na]+. |
| 92% |
With monopersulfate; In methanol; water; at 20℃; for 20h; |
(B) To an ice-cooled solution of 350 3-(methylthio)propyl 4-methylbenzenesulfonate (3.6 g; 13.8 mmol) in 21 MeOH (70 mL) was added a suspension of monopersulfate compound (17 g; 27.4 mmol) in 12 water (70 mL) in a portion-wise fashion. Upon completion of the addition, the mixture was allowed to warm to rt, and stirring was continued for 20 h. The reaction was partially concentrated to remove the MeOH and the mixture was further diluted with water and extracted with EtOAc (2×). The combined organic extracts were dried (Na2SO4), filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography eluting with 0-67% EtOAc in hexanes to afford 352 3-(methylsulfonyl)propyl 4-methylbenzene-sulfonate (3.72 g, 92%) as a white solid. LC/MS: mass calcd. for C11H16O5S2: 292.38, found 315.1 [M+Na]+. |
| 90% |
With oxone; In methanol; at 20℃;Cooling with ice; |
3-(methylthio)propyl-4-methylbenzenesulfonate (12.5 g, 48 mmol) was added to 250 mL in an ice bathStirring in a methanol solution, adding Oxone (59 g, 96 mmol) dropwise to the reaction solution, returning to room temperature, and stirring overnight. thenThe reaction solution was concentrated under reduced pressure, and a large amount of a white solid precipitated, which was filtered and washed with water to give a white solid (12.6 g) in a yield of 90%. |
|
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; |
152 (l-r2-(lH-Indazol-4-ylV4-morpholin-4-yl-thienor3.2-d1pyrimidin-6-ylmethyl1- piperidin-4-vU-(3-methanesulfonyl-propylVmethyl-amine.Via [ 1 -(2-chloro-4-morpholin-4-yl-thieno[3 ,2-d]pyrimidin-6-ylmethyl)- piperidin-4-yl]-(3-methanesulfonyl-propyl)-methyl-amine, prepared from (3- methanesulfonyl-propyl)-methyl-piperidin-4-yl-amine. Amine preparation: Toluene-4-sulfonic acid 3-methylsulfanyl-propyl ester was prepared from 3-(methylthio)-l-propanol using standard conditions. Treatment with mCPBA in DCM yielded toluene-4-sulfonic acid 3-methanesulfonyl-propyl ester. A mixture of 4-methylamino-piperidine- 1-carboxylic acid tert-butyl ester and toluene-4-sulfonic acid 3-methanesulfonyl-propyl ester was heated in MeCN in the prescence of potassium carbonate to yield 4- [(3-methanesulfonyl-propyl) -methyl- EPO <DP n="82"/>amino]-piperidine-l-carboxylic acid tert-butyl ester. Treatment of this compound with HCl in DCM/MeOH yielded the desired amine, isolated as the hydrochloride salt.1H NMR (400MHz, CDCl3) 1.50-1.70 (m, 4H, 2 x CH2), 1.90-1.97 (m, 2H, CH2), 2.00-2.05 (m, 2H, CH2), 2.21 (s, 3H, CH3), 2.38 (m, H, CH), 2.55 (m, 2H, CH2), 2.74 (s, 3H, CH2), 2.96-3.04 (m, 4H, 2 x CH2), 3.75 (s, 2H, CH2), 3.83-3.89 (m, 4H, 2 x CH2), 4.00-4.02 (m, 4H, 2 x CH2), 7.28 (s, H, CH), 7.41 (t, H, ArH, J=7.74Hz), 7.50 (d, H, ArH, J=8.24Hz), 8.18 (d, H, ArH, J=7.05Hz), 8.93 (s, H, ArH); MS (ESI+) 584.39 (MH+). |
|
With Oxone; In methanol; at 6 - 20℃; for 16h;Product distribution / selectivity; |
Reference Example 6Synthesis of 3- (methylsulfonyl) propyl 4-methylbenzenesunfonate; [0414][0415]3- (Methylthio) propyl 4-methylbenzenesunfonate (29.4 g) was dissolved in methanol, and the mixture was cooled to 6C or below. While cooling to 6C or below, oxone (registered trade mark; 105.2 g) dissolved in water (400 mL) was added dropwise over 1 hr. After stirring at 6C or below for 1 hr, the mixture was stirred at room temperature for 14 hr. Water (800 mL) was added, and the mixture was stirred at 6C or below for 1 hr. The precipitated solid was collected by filtration, and washed twice with water (400 mL) . The solid was suspended in methanol (150 mL) , and the suspension was heated to 65C.Water (150 mL) was added dropwise over 30 min, and the mixture was cooled to room temperature and stirred at 6C or below for 1 hr. The solid was collected by filtration, and washed twice with water (150 mL) . The solid was vacuum-dried at 50C to give white title compound (25.21 g) .1H NMR (300MHz, CDC13) : delta 2.17-2.28 (2H, m) , 2.46 (3H, s) , 2.91 (3H, s), 3.07-3.15 (2H, m) , 4.18 (2H, t, J=5.9 Hz), 7.34-7.37 (2H, d, J=8.0 Hz), 7.78-7.80 (2H, d, J=8.3 Hz). |
|
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃; for 21h; |
Step B: 3 -(methylsulfonyl)propyl 4-methylbenzenesulfonate (34-2)To a solution of 34-1 (35 g, 135 mmol) in dry DCM (400 mL) with ice-bath cooling was added MCPBA (46.5 g, 270 mmol) portionwise. The resulting mixture was stirred at 0 C for lh, and then warmed to the room temperature and stirred for 20 h. The reaction was quenched byaddition of aqueous solution of NaHSO3 and the DCM layer was washed with Na2CO3 (aq.), water and brine, respectively, and concentrated to afford a residue, which was purified by chromatography on silica gel (petroleum ether:ethyl acetate = 3/1) to give compound 34-2. |
|
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃; for 21h; |
To a solution of34-1 (35 g, 135 mmol) in dry DCM (400 mL) with ice-bath cooling was addedMCPBA(46.5 g, 270 mmol) portionwise. The resulting mixture was stirred at 0 oc for 1h, andthen warmed to the room temperature and stirred for 20 h. The reaction was quenched by5 addition of aqueous solution ofNaHS03 and the DCM layer was washed with Na2C03 (aq.),water and brine, respectively, and concentrated to afford a residue, which was purified bychromatography on silica gel (petroleum ether:ethyl acetate= 3/1) to give compound 34-2. |
|
With oxone; In methanol; water; at 20℃; for 20h;Cooling with ice; |
To a solution of product 18 (7.32g, 28.1mmol) in methanol (150mL) was added dropwise a solution of potassium peroxysulfate (34.6g, 56.3mmol) in water (150mL) under ice-cooling. After completion of the dropwise addition, the mixture was stirred for 20 hr, during which the mixture was allowed to gradually warm to room temperature. Methanol was evaporated under reduced pressure, and the mixture was diluted with water, and the organic material was extracted with ethyl acetate. The extract was washed with saturated brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The precipitated crystals were washed with ethyl acetate-heptane to give a colorless crystal product 19 (7.86g, yield 95%). |
|
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃; for 21h; |
To a solution of product from Step A (35 g, 135 mmol) in dry DCM (400 mL) in an ice-bath was added MCPBA (46.5 g, 270 mmol) portionwise. The resulting mixture was stirred at 0 C for lh, and then warmed to the room temperature and stirred for 20 h. The reaction was quenched by addition of aqueous solution of NaHS( and the DCM layer was washed with Na2C( (aq.), water and brine, respectively, and concentrated to afford a residue, which was purified by chromatography on silica gel (eluting with PE:EA=3: 1) to give the title compound. |
|
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0 - 20℃; for 21h; |
To a solution of product from Step A (35 g, 135 mmol) in dry DCM (400 mL) in an ice-bath was added MCPBA (46.5 g, 270 mmol) portionwise. The resulting mixture was stirred at 0for 1h, and then warmed to the room temperature and stirred for 20 h. The reaction was quenched by addition of aqueous solution of NaHSO3 and the DCM layer was washed with Na2CO3 (aq. ) , water and brine, respectively, and concentrated to afford a residue, which was purified by chromatography on silica gel (eluting with PE:EA3:1) to give the title compound. |
|
With Oxone; In tetrahydrofuran; water; at 20℃; for 12h;Inert atmosphere; |
62 g of 3-(methylthio)propyl 4-methylbenzenesulfonate obtained in step 1) was loaded in THF/distilled water (150/100 ml) in a flask in nitrogen atmosphere, followed by stirring for dissolving them. Then, 310 g of oxone was added thereto. The mixture was stirred for 12 hours at room temperature. Upon completion of the reaction, distilled water was slowly added thereto, followed by extraction using ethylacetate. The extract was washed with brine, dried over anhydrous MgSO4, and concentrated to give the target compound. (0288) 1H NMR (400 MHz, CDCl3): delta 7.81 (2H, d), 7.38 (2H, d), 4.20 (2H, m), 3.13 (2H, m), 2.93 (3H, s), 2.48 (3H, s), 2.23 (2H, m). |
|
With Oxone; In methanol; at 20 - 30℃; for 2h;Inert atmosphere; |
A solution of 3- (methylthio) propyl p-toluenesulfonate 24a (10.0 g, 38.40 mmol) was dissolved in 100 mL of methanol,100 mL of a solution containing Oxone reagent (35.42 g, 57.60 mmol), and the reaction was stirred at room temperature for 2 hours.The reaction mixture was concentrated under reduced pressure, 100 mL of water and 100 mL of ethyl acetate were added to the reaction solution. The aqueous phase was extracted with ethyl acetate (50 mL of X 2) and the combined organic layers were washed with water (30 mL of X 3) and saturated sodium chloride solution Dried over anhydrous sodium sulfate, filtered, and the filtrate was concentrated under reduced pressure to give the crude title product 3- (methylsulfonyl) propyl p-toluenesulfonate 24b (8.0 g, white solid) The purification was carried out directly to the next reaction. |
| 1.95 g |
With oxone; In methanol; water; at 0 - 20℃; for 20h; |
Step 1: Preparation of 3-methylsulfonylpropyl 4-methylbenzenesulfonate To a solution of 3-methylsulfanylpropyl 4-methylbenzenesulfonate (2 g, 7.7 mmol) in methanol (50 mL) was added a solution of oxone (9.47 g, 15.4 mmol) in water (50 mL) dropwise at 0 oC. After being warmed to rt and stirred at rt for 20 hrs, the mixture was filtered and the filtrate was extracted with EA (50 mL) for three times. The combined organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated in vacuo to give 3- methylsulfonylpropyl 4-methylbenzenesulfonate as a white solid (1.95 g) which was used in the next step directly without further purification. |
| 38 g |
With 3-chloro-benzenecarboperoxoic acid; In dichloromethane; at 0℃; |
To a solution of 3-(Methylthio)-1-propanol (20g, 0.19mol) and TEA (66ml, 0.48mol) in DCM (800mL) cooled at 0C was added p-toluenesulfonyl chloride (44g, 0.23mol, dissoloved in 200mL DCM) dropwise. After addtion, the mixture was then stirred at r.t. overnight. After the reaction was complete, the resluting solution was washed with saturated Na2CO3 solution, brine, dried over MgSO4 and concentrated in vacuo to give the crude of 3-methylsulfanylpropyl 4-methylbenzenesulfonate, which was used in the next step without further purification. To the solution of the crude of 3-methylsulfanylpropyl 4-methylbenzenesulfonate (prepared above) in DCM (200 mL) cooled at 0oC was added m-CPBA (66g, 0.39mol) slowly and the mixture was then stirred at rt overnight. After the reaction was complete, the mixture was washed with saturated sodium thiosulfate (300mL) 3 times , saturated sodium bicarbonate (300mL) 3 times, dried over MgSO4 and then concentrated in vacuo to give 3-methylsulfonylpropyl 4-methylbenzenesulfonate (11b, 38g, 69% yield over 2 steps) as a white solid. To a solution of tert-butyl 4-chloro-2-nitrophenylcarbamate (580 mg, 2.1 mmol) in DMF(5mL) cooled at 0oC was added NaH( 169 mg, 4.4 mmol). The mixture was then stirred at rt for 30mins. Then to the resulting solution was added 3-methylsulfonylpropyl 4-methylbenzenesulfonate (11b, 750 mg, 2.6 mmol) and the mixture was stirred at 50oC overnight. After the reaction was complete, to the mixture was added water (30 mL) and the resulting mixture was extracted with DCM (50 mL) 3 times. The combined organic layer was washed with brine, dried over MgSO4 and concentrated in vacuo. The residue was purified by column chromatography on silica gel (elution with DCM:MeOH 20 :1) to give tert-butyl N-(4-chloro-2-nitro-phenyl)-N-(3-methylsulfonylpropyl)carbamate(11c, 360 mg, 69% yield). To a soltuion of tert-butyl N-(4-chloro-2-nitro-phenyl)-N-(3-methylsulfonylpropyl)carbamate (11c, 360 mg, 1.45 mol) in MeOH (10 mL) was added 2% Pt/C (50mg) under N2 atmosphere, the mixture wa then hydrogenated at rt for 3 hours. After the reaction was complete, the mixture was filtred and the filtrate was concentrated in vacuo to give the crude of tert-butyl N-(2-amino-4-chloro-phenyl) -N-(3-methylsulfonylpropyl)carbamate (11d, 330 mg, 100% yield), which was used in the next step directly without further purificaton. A mixture of tert-butyl N-(2-amino-4-chloro-phenyl)-N-(3-methylsulfonylpropyl)carbamate (11d, 320 mg, 0.92mmol) and sodium chloroacetate (130 mg, 1.1 mmol) in 4N HCl (15 mL) was stirred to 100oC overnight. After the reaction was complete, The reaction was concentrated in vacuo and the residue was redissoved in DCM (100 mL). The resulting soltuion was washed with saturated NaHCO3 (50 mL), brine (50 mL), dried over MgSO4 and concentrated in vacuo. The residue was purified by column crhomatography on silica gel (elution with DCM/EtOAc=3/1) to give 5-chloro-2-(chloromethyl) -1-(3-methylsulfonylpropyl)benzimidazole (11e, 130 mg, 44% yield). To a mixture of 2-(methylsulfonyl)-1H-indole (72 mg, 0.37 mmol) and 5-chloro-2-(chloromethyl) -1-(2- (methylsulfonyl)ethyl)-1H-benzo[d]imidazole (11e, 120 mg, 0.37mmol) in DMF (3 mL) was added K2CO3 (104 mg, 0.74 mmol) and the mixture was stirred at rt overnight. After the reaction was completed, the mixture was filtered and the filtrate was purified by preparative HPLC to give 5-chloro-2-[(3-methylsulfonylindol-1-yl)methyl]-1-(3-methylsulfonylpropyl)benzimidazole (11, 100 mg, 78%). MS: calcd (MH+) 480.1, exp (MH+) 480.1. 1H NMR(DMSO-d6, 400MHz):delta8.29(s, 1H), 7.80-7.86(m, 1H), 7.65-7.73(m, 3H), 7.26-7.34(m, 3H), 5.92(s, 2H), 4.86(t, J=7.6Hz, 2H), 3,73(br, 3H), 3.19(t, J=8.0Hz, 2H), 2.96(s, 3H), 2.08(t, J=7.6Hz, 2H); 13C NMR (101MHz, DMSO-d6) delta151.4, 143.2, 137.3, 134.7, 134.6, 126.9, 124.2, 123.8, 123.3, 122.6, 119.5, 119.2, 115.5, 112.4, 112.3, 51.1, 45.6, 43.6, 42.3, 23.1. |
|
With Oxone; In tetrahydrofuran; water; at 0 - 20℃; for 12h;Inert atmosphere; |
Under a nitrogen atmosphere, 62 g of 3-(methylthio)propyl 4-methylbenzenesulfonate obtained in step 1 was added in THF/distilled water (150/100 mL) in a flask and stirred to dissolve, and then 310 g of oxone was added dropwise at 0?. After stirring at room temperature for 12 hours or longer, upon completion of the reaction, distilled water was slowly added dropwise, extracted with ethyl acetate, washed with brine, and dried over anhydrous magnesium sulfate to give the title compound. (0182) 1H NMR (400MHz, CDCl3) : delta 7.81 (2H, d) , 7. 38 (2H, d) , 4.20(2H, m), 3.13(2H, m), 2.93(3H, s), 2.48(3H, s), 2.23(2H, m). |






 Chemistry
Chemistry
 Pharmaceutical Intermediates
Pharmaceutical Intermediates
 Inhibitors/Agonists
Inhibitors/Agonists
 Material Science
Material Science







 For Research Only
For Research Only
 110K+ Compounds
110K+ Compounds
 Competitive Price
Competitive Price
 1-2 Day Shipping
1-2 Day Shipping